

Abstract
The dendritic cell-specific ICAM-3 non-integrin (DC-SIGN) and its close relative DC-SIGNR recognize various glycoproteins, both pathogenic and cellular, through the receptor lectin domain-mediated carbohydrate recognition. While the carbohydrate-recognition domains (CRD) exist as monomers and bind individual carbohydrates with low affinity and are permissive in nature, the full-length receptors form tetramers through their repeat domain and recognize specific ligands with high affinity. To understand the tetramer-based ligand binding avidity, we determined the crystal structure of DC-SIGNR with its last repeat region. Compared to the carbohydrate-bound CRD structure, the structure revealed conformational changes in the calcium and carbohydrate coordination loops of CRD, an additional disulfide bond between the N and the C termini of the CRD, and a helical conformation for the last repeat. On the basis of the current crystal structure and other published structures with sequence homology to the repeat domain, we generated a tetramer model for DC-SIGN/R using homology modeling and propose a ligand-recognition index to identify potential receptor ligands.
Keywords: DC-SIGN, HIV gp120, C-type lectin, dendritic cell, X-ray structure
Abbreviations used: DC-SIGN, dendritic cell-specific ICAM-3 non-integrin; DC-SIGNR, related DC-SIGN; DC-SIGN/R, refers to both DC-SIGN and DC-SIGNR; CRD, carbohydrate recognition domain; HIV, human immunodeficiency virus; PDB, Protein Data Bank
Introduction
The dendritic cell-specific ICAM-3 non-integrin (DC-SIGN) and its close relative DC-SIGNR are members of the C-type lectin family. Originally discovered as a human immunodeficiency virus (HIV)-binding protein, DC-SIGN has been shown to bind carbohydrates on various pathogens, including Ebola, Mycobacterium tuberculosis, hepatitis C virus and cytomegalovirus.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 In the case of HIV-1 infection, DC-SIGN and DC-SIGNR (together referred to as DC-SIGN/R) have been proposed to facilitate the viral infection of T-cells in trans through binding with HIV gp120.5, 12 However, recent evidence suggests that DC-SIGN/R function as antigen capturing receptors to facilitate the presentation of HIV-1 antigen by dendritic cells.13 DC-SIGN/R each consist of four domains: a cytoplasmic domain with a di-leucine motif for internalization, a single-spanning transmembrane region, a region with seven and one-half 23 amino acid residue repeats, and a carbohydrate-recognition domain (CRD). DC-SIGNR is 77% identical with DC-SIGN in amino acid sequence, and differs mainly in tissue and cellular expression patterns although recent reports indicate that it may differ in binding and processing of pathogens.14, 15, 16
It has been established that DC-SIGN/R recognize specifically high-mannose carbohydrates.17 Previous structural studies have shown the molecular details of this interaction.18 More recently, however, DC-SIGN/R were shown to recognize terminal fucose and galactose-containing carbohydrates, such as blood group antigens B Lewisa, and Lewisx structures in addition to mannose.18, 19 While recognition between various carbohydrate substructures gives insight into how various carbohydrate model compounds are recognized by the CRD of the receptor, the overall receptor binding affinity appears to depend on the multivalent nature of the ligand.20 For example, while DC-SIGN/R bind to model carbohydrate with millimolar affinity, the receptors recognize HIV gp120, which carries multiple high-mannose-based, N-linked glycosylations with nanomolar affinity. This carbohydrate valency-dependent avidity effect was shown to be the result of DC-SIGN/R tetramerization through its repeat region.17, 21 To understand the nature of the receptor-carbohydrate interaction, we have determined the crystal structure of DC-SIGNR with a portion of the repeat domain. We propose a tetramer model for the intact extracellular receptor, and formulate a scheme to predict the potential ligands.
Results
Description of the overall structure
The crystals of DC-SIGNR CRD with its last repeat belong to the orthorhombic space group P212121. The protein solution was supplemented with 10 mM mannose and 5 mM CaCl2 prior to crystallization. Crystals were obtained under several conditions that include polyethylene glycol with various molecular masses (2000–8000 Da) and buffers with a pH between 6.0 and 8.0. Crystals contained one molecule per asymmetric unit with a solvent content of 32.8% (v/v) (Matthew's coefficient of 1.8). Molecular replacement rotation and translation correlation coefficients were ranked and yielded a single solution well above the background. The initial phased map had clear electron density for both the main chains and the side-chains. After rebuilding of loops, the electron density was continuous throughout the structure and the final structure consists of residues 260–398, with Met282 modeled in two alternative conformations. The final refined crystallographic R-factors are 17.75% and 19.3% for R work and R free, respectively, at 1.41 Å resolution (Table 1 ). The overall structure is a typical long-form C-type lectin, and the CRD portion superimposes with a root-mean-square (r.m.s) deviation of 0.67 Å (for 126 Cα atoms) to the CRD-only structure (Figure 1 (b)).18 Although both mannose and calcium were present in the crystallization solution, only calcium was observed bound in the canonical calcium binding site. This structure contains additional residues at both the amino and carboxyl termini including a disulfide bond linking the two termini as well as a short α-helix at the beginning of the repeat domain.
Table 1.
Crystallographic data collection and refinement statistics
| A. Data collection | |
| Space group, cell lengths (Å) | P212121 |
| Resolution (Å) | 20.0–1.41 (1.45–1.41)a |
| Cell dimensions | |
| a (Å) | 38.23 |
| b (Å) | 54.88 |
| c (Å) | 62.32 |
| No. observations | 25,340 (2102) |
| Completeness (%) | 97.5 (99.2) |
| Rsymb (%) | 6.9 (22.2) |
| I/σI | 37.1 (5.6) |
| B. Refinement statistics | |
| Rwork/Rfreec (%) | 17.7/19.3 |
| No. atoms | |
| Protein | 1143 |
| Water | 103 |
| Other | 1 |
| Ramachandran plot | |
| Most favored (%) | 91.9 |
| Allowed (%) | 8.1 |
| Generously allowed (%) | 0 |
| Forbidden (%) | 0 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.015 |
| Bond angles (deg.) | 1.37 |
Values within parentheses are for the highest-resolution shell.
Rsym=Σ|Ih−〈Ih〉|/ΣIh,where 〈Ih〉 is the mean intensity of multiple measurements of symmetry-equivalent reflections.
Rwork or , where Fc is the calculated and Fo is the observed structure factor amplitude of reflection h for the working or 5% free set, respectively.
Figure 1.
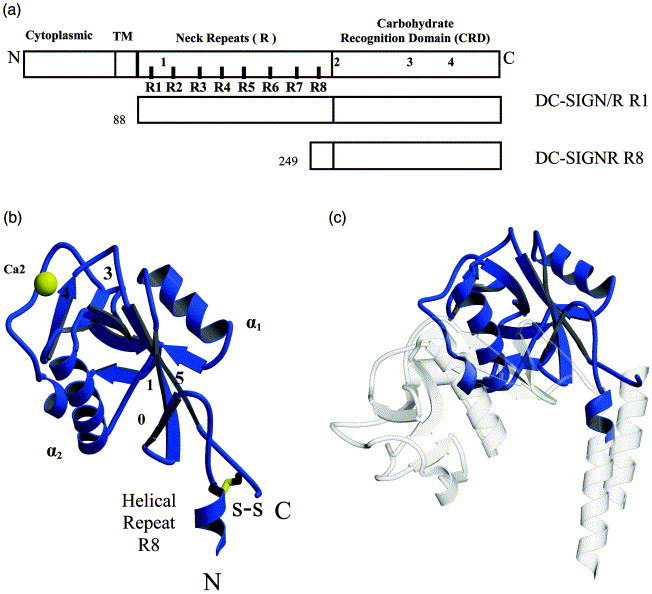
The structure of DC-SIGNR with repeat domain R8. (a) A representation of the four domains of DC-SIGN and DC-SIGNR including cytoplasmic, transmembrane (TM), repeats 1–8 (R1–R8) and the carbohydrate recognition domain (CRD). The portion of DC-SIGNR crystallized, DC-SIGNR R8, is shown (amino acid residues 249–399). The four trypsin digestion sites are indicated as 1, 2, 3, and 4. (b) The crystal structure of DC-SIGNR R8 (blue) showing the position of the carbohydrate-binding calcium (yellow, Ca2), the disulfide that links the N and the C terminus, and the visible portion of the R8 forming a helical repeat. (c) Superposition of DC-SIGNR R8 (blue) with the DC-SIGNR R7 (PDB code1SL6; gray). Carbohydrate has been omitted for clarity.
The N-terminal disulfide and R8 repeat
Additional amino acids were present in both the N and C termini compared with the CRD-only structure. At the C-terminus we observed additional density for amino acids 395–398, including a disulfide bond between Cys395 and Cys265, which links both the N-and C-termini into close proximity (Figure 1). As a result, the ring of the N-terminal histidine residue (His267) stacks against the ring of the C-terminal Phe396. The helical repeat domain has been shown to be responsible for tetramerization of the receptor. Our DC-SIGNR R8 construct contains the last repeat that immediately precedes the CRD domain. This repeat region encompasses residues Gln249-Cys265. A portion of this repeat, Ala260-Cys265, is ordered in the structure and forms a short α-helix. Although the rest of the R8 repeat appears disordered in our crystal, presumably due to the proximity to the N terminus of the expressed recombinant R8 construct, the presence of a short helix is consistent with the secondary structure prediction that the repeat domain is mainly α-helical. The N-terminal CRD disulfide bond (Cys265-Cys395) and the helical repeat conformation was observed recently in the structure of DC-SIGNR R7 (CRD with its last two repeats).19, 22 The r.m.s deviation between the CRD domain of DC-SIGNR R8 and that of R7 is 0.76 Å for 129 Cα atoms. However, the hinge angle between the CRD and the repeat domain differs by about 40° (100° and 60°, respectively) between the two structures, indicating a domain flexibility between the CRD and the repeat domain of the receptor (Figure 1).
The calcium and carbohydrate-binding sites
The primary calcium site involved in binding carbohydrate (Ca2) has a well-ordered calcium ion in this structure. The amino acid residues involved directly in coordinating the calcium ion are Glu359, Asn361, Glu366, and Asp378, and a water molecule (W19). With the exception of Asn377, which is rotated out of the calcium coordination, the ligand positions are well conserved between the R8 and CRD-only structures of DC-SIGNR (Figure 2 (a) and Table 2 ). In contrast, no attributable electron density was found near the two auxiliary calcium ions (Ca1 and Ca3) binding site and the two residues coordinating the auxiliary calcium, Asn362 and Asn365, moved 4.0 Å and 1.9 Å, respectively, compared to the CRD-only structure (1K9J). The movement of Asn362 and Asn365 effectively disrupts the coordination of Ca1 and Ca3, further evidence that both auxiliary calcium ions are absent from the DC-SIGNR R8 structure. Despite the presence of mannose in the crystallization buffer and the existence of additional electron density at the putative carbohydrate-binding site, attempts to fit mannose were not satisfactory, and instead, water molecules were built throughout the carbohydrate-binding site.
Figure 2.
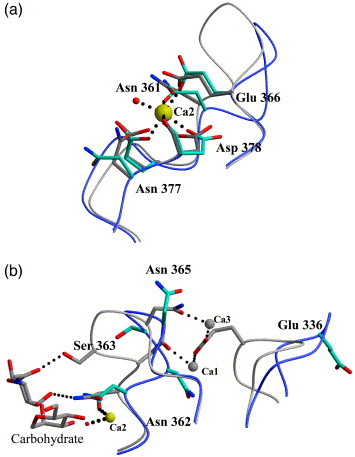
Carbohydrate-binding and calcium-binding sites. (a) Cα traces of the DC-SIGNR R8 (blue) and DC-SIGNR CRD (gray) primary calcium-binding sites showing side-chains involved in coordinating calcium. In the absence of carbohydrate, Asp377 is not involved in calcium coordination. The calcium ion, seen in nearly the same position in both structures is shown in yellow (Ca2). A water molecule (red) is present in the location where normally ligand binds (DC-SIGNR R8 structure). (b) The secondary calcium-binding site shows the Cα loop in DCSIGNR R8 and DC-SIGNR CRD Loop movement is observed between ligand-bound and apo structures from closed to open, respectively. Calcium ions present in the structure of DC-SIGNR CRD only are shown in gray (Ca1 and Ca3). Side-chain movements between each structure are summarized in Table 2.
Table 2.
Comparison of the calcium ligand distances
| Distance (Å) |
|||||
|---|---|---|---|---|---|
| Calcium ion | Residue | Atom | RR8 | 1K9J | Change Δ (Å) |
| Ca2 | Glu 359 | OE2 | 2.14 | (OE1) 2.63 | 0.49 |
| Ca2 | Asn 361 | OD1 | 2.04 | 2.44 | 0.4 |
| Ca2 | Glu 366 | OE1 | 2.03 | 2.43 | 0.4 |
| Ca2 | Asn 377 | OD1 | 5.79 | 2.47 | 3.32 |
| Ca2 | Asp 378 | O | 2.10 | 2.42 | 0.32 |
| Ca2 | Asp 378 | OD1 | 2.02 | 2.34 | 0.32 |
| Ca2 | W 19 | 2.04 | N/A | ||
| Ca2 | Man C2 | O4 | N/A | 2.57 | |
| Ca2 | Man C2 | O3 | N/A | 2.49 | |
| Ca2–Ca2 | 0.29 | ||||
| Ca3 | Glu 336 | OE1 | 12.94 | 2.28 | 10.66 |
| Ca3 | Asn 365 | OD1 | 4.32 | 2.46 | 1.86 |
| Ca3 | Asp 367 | OD2 | 3.33 | 2.54 | 0.79 |
| Ca3 | Asp 367 | OD1 | 2.35 | 2.58 | 0.23 |
| Ca1 | Asp 332 | OD1 | 3.01 | 2.58 | 0.43 |
| Ca1 | Asp 332 | OD2 | 1.99 | 2.49 | 0.5 |
| Ca1 | Glu 336 | OE2 | 13.09 | 2.58 | 10.51 |
| Ca1 | Asn 362 | OD1 | 6.44 | 2.43 | 4.01 |
| Ca1 | Glu 366 | O | 3.27 | 2.45 | 0.82 |
| Ca1 | Asp 367 | OD1 | 2.35 | 2.33 | 0.02 |
The comparison between the current apo-DC-SIGNR R8 and the mannose-containing CRD structure showed both the primary calcium/carbohydrate-binding loop (residues 361–366) and the secondary calcium-binding loop (residues 332–339) assumed an “open” conformation in the apo state while adopting a “closed” conformation in the carbohydrate-bound state of the receptor (Figure 2). In the presence of carbohydrate, the conformation of the primary carbohydrate-binding loop (residues 361–366), the closed conformation, is defined by the coordinating hydrogen bonds between the side-chains of Asn361 and Ser363, and the bound N-acetyl-d-glucosamine (GlcNAc1) as well as between Asn362 and Asn365, and their bound Ca1 and Ca3. In the absence of carbohydrate, however, Ser363 moved 4.9 Å toward the solvent, resulting in a more open conformation for the primary carbohydrate-binding loop. In conjunction with this loop movement, Asn362 and Asn365 lost their coordination geometry for the auxiliary calcium sites. The ejection of the auxiliary Ca1 and Ca3, in turn, resulted in the displacement of another calcium coordination residue, Glu336 from the secondary calcium-binding loop, toward the solvent and thus adopting an open conformation for the loop (Table 2 and Figure 2). Interestingly, an arginine residue from a symmetry-related molecule, Arg397, is found near the putative Ca1 and Ca3 sites, forming a hydrogen bond with the secondary calcium-binding loop to neutralize, as a surrogate to the missing calcium ion, the partial negative charges of the region.
Despite the presence of 5 mM CaCl2 in the crystallization setup, both Ca1 and Ca3 appear to be absent, suggesting that these auxiliary calcium sites are of low affinity compared to that of the primary calcium-binding site (Ca2), and that their occupancies are coupled to the binding of the carbohydrate ligand. Namely, they are glycan-induced calcium-binding sites. In the absence of the bound carbohydrate, both calcium coordination loops adopt an open, conformation ejecting the auxiliary calcium ions and become less ordered. The result suggests the function of these glycan-induced auxiliary calcium is to stabilize the conformation of the glycan-binding loops synergistically to the bound glycan rather than to pre-conform the glycan-binding loop.23, 24, 25
Modeling of the DC-SIGN/R tetramer
A homology search was performed using sequences corresponding to various lengths of the repeat domain of DC-SIGNR against known structures in the Protein Data Bank (PDB). The resulting sequence identities between segments of known structures and portions of DC-SIGNR repeats are 32% between residues 32–117 of the focal adhesion kinase (PDB code 1K05) and repeats R1–R3 of DC-SIGNR (Figure 3 ), 31% between residues 328–390 of Muts (PDB code 1NNE) and repeats R5–R7, 33% between residues 23–67 of the large ribosomal subunit from Deinococcus radiodurans (PDB code 1NKW) and repeats R6 and R7, 37% between residues 60–106 of the monomeric isocitrate dehydrogenase (PDB code 1ITW) and repeats R6–R8. All homologous structures are helical in nature.
Figure 3.

Alignment of homologous repeat sequences. CLUSTAL W(1.74) sequence alignment of the ecto-domain of DC-SIGNR repeats 1–8 (indicated by R1–R8) sequence with homologous sequences used to predict the boundary of helical regions. Helical regions are denoted by the letter H and coil or turn regions are indicated by the letter C. Because repeats are nearly identical, differing by only a single amino acid residue in some cases, most of the homologous matches can be translated along the repeats.
Both homology modeling and sequence-based secondary structure prediction resulted in similar secondary structure assignment, including the boundary of helices, turns and loops throughout the R1–R8 repeat domain of DC-SIGNR. Additional structural information derived from gel-filtration experiments on truncated receptors showing that receptor tetramerization requires R5–R8 repeats and analytical ultracentrifugation observations suggesting an elongated shape of the tetramer were included in the modeling of the tetramer.21 Based on the overlapping homologous structures and the biophysical shape consideration and using the focal adhesion kinase (PDB code 1K05) as a template (Supplementary Data Figure 1), a polyalanine model of DC-SIGNR tetramer was built manually using the crystallographic program O and subjected to energy minimization using CNS (Figure 4 ). The tetramer model displays a 4-fold symmetry, with the core tetramerization domain adopting a four-helix bundle structure similar to that of the focal adhesion kinase (see Supplementary Data for a more detailed description of the model). The arrangement of the R7 and R8 helices in this model agree with the recently deposited structure of DC-SIGNR containing both R7 and R8 repeats (PDB code 1SL6).22 The dimensions of the model proposed here are ∼80 Å×80 Å×190 Å with individual CRD separated by ∼50 Å. On the basis of the model, the tetrameric CRD head encompasses an area of approximately 6400 Å2.
Figure 4.
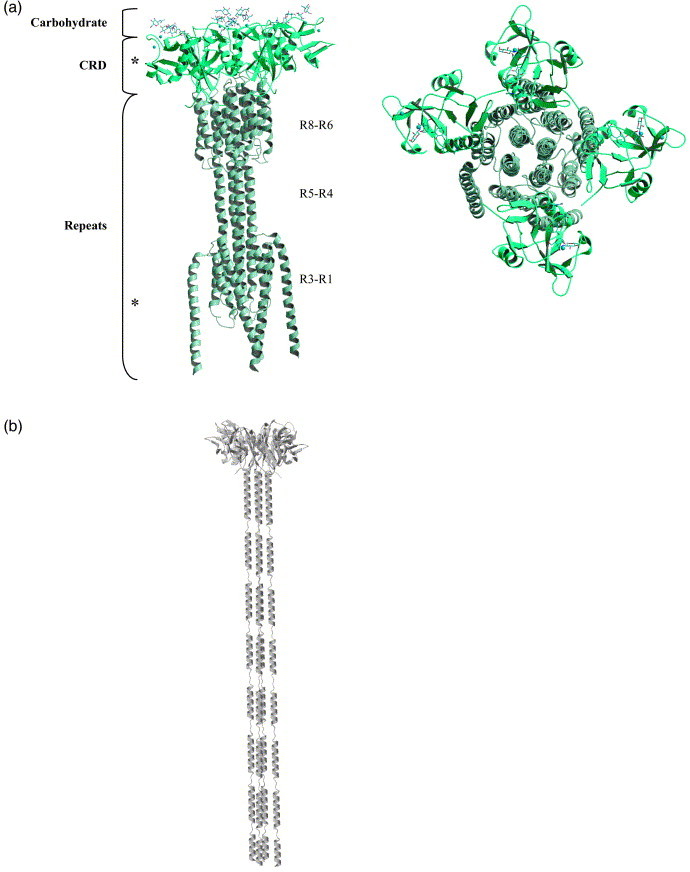
Model of the extracellular portion of the DC-SIGN/R tetramer. (a) A side-view of the model tetramer. The boundary of the repeat domain, CRD and carbohydrate (from DC-SIGNR CRD PDB code 1K9J) are shown as well as a view of the model looking down onto the top of the tetramer. (b) A single helical tetramer model is shown with helical breaks in the region near proline residues.
We carried out limited proteolysis using trypsin to explore the likelihood of the proposed model of helical repeat bundles for the tetrameric DC-SIGNR versus a model consisting of an elongated linear concatenation of helical repeats (Figure 4). Since identical trypsin digestion sites are found within each repeat region, a differential use of each potential trypsin site would suggest differential protection from the protease. The tight packing of the proposed model predicts a biased protease-sensitivity for the different repeats with the core tetramer packing repeats less accessible than the peripheral repeats, while the linear helical concatenation model predicts an equal protease-sensitivity for each repeat. The digestion with trypsin was carried out using a recombinant expressed and refolded full extracellular DC-SIGNR, termed DC-SIGNR R1, that has been characterized to be a tetramer.21 Digestion of DC-SIGNR R1 with trypsin resulted in four major fragments F1∼25 kDa, F2∼9 kDa 8, F3∼19 kDa, and F4∼7 kDa, with the F1 and F2 appearing before F3 and F4 in time-based digestions (Supplementary Data Figure 3). No other intermediate fragment could be identified. The amino-terminal sequencing revealed fragments F1 (residues 14–237) and F2 (residues 238–313) resulting from cleavages at trypsin sites between repeats R1 and R2 (site 1) and within the CRD (site 3). Fragments F3 (residues 14–179) and F4 (residues 270–313) appeared to be derived from F1 and F2 by further digesting at site 3 and 4, respectively (Figure 1). These results indicate that most of the tetramerization repeats (R2–R8) remain resistant to digestion by trypsin, consistent with it being a compact tetramer unit rather than an elongated linear helical tetramer in which all repeats appear equally susceptible to protease. Digestion experiments with subtilisin are consistent with these results, indicating protease-sensitive sites being primarily between repeats R1 and R2, and after the helical repeat domain at the beginning of the CRD region.22
Evaluating potential DC-SIGN/R ligands
Earlier studies of the DC-SIGN/R CRD binding to model carbohydrate compounds suggest that the receptors prefer a high-mannose type of carbohydrate.17, 18, 26 More recently, the receptors were shown to recognize also sialyl-Lewis-like carbohydrates.19 The dissociation constant (K d) between DC-SIGN/R CRD and the model compounds, however, are millimolar at best, while the functional ligand recognition by the receptor has better than micromolar affinity. Thus, much of the receptor-ligand binding affinity appears to be derived from an avidity effect of the DC-SIGN/R tetramer. The requirement of tetramer binding for ligand recognition would, in turn, impose limitations to its ligand selection. Namely, ligands carrying multiple glycosylations capable of engaging the multimeric DC-SIGN/R CRD simultaneously would be preferred by the receptor. The surface area encompassed by the tetrameric CRD in our current DC-SIGN/R model is approximately 6400 Å2, or 1600 Å2 per CRD molecule. This requires the potential ligands of DC-SIGN/R to possess a surface glycosylation level exceeding one glycan molecule per 1600 Å2 of its surface area. This enables us to formulate a potential ligand index i to evaluate potential ligands of DC-SIGN/R on the basis of their surface glycosylation density:
| (1) |
where N is the number of predicted potential glycosylation sites and M is the molecular mass of the candidate protein. A potential DC-SIGN/R ligand would possess an index greater than 1.0 and proteins with the indices less than 1.0 are less likely to be ligands of the receptor.
The calculation of this potential ligand index for a number of viral envelope glycoproteins as well as for some cell-surface glycoproteins is summarized in Table 3 . Of the potential viral targets of DC-SIGN/R, HIV-1, coronavirus and Marburg virus are known to bind DC-SIGN. In addition, HRSV, influenza and human foamy viruses appear to be good candidates for DC-SIGN/R. Among the cellular targets, in addition to the known ICAM-3 ligand, several surface glycoproteins also score favorably for DC-SIGN binding.
Table 3.
Probability index of potential DC-SIGN/R ligands
| Glycoprotein | Description | Mass (kDa) | Number of potential glycosylationsa | Potential ligand index i |
|---|---|---|---|---|
| A. Viral proteins | ||||
| gp120 | HIV-1 | 54.0 | 24 | 4.8 |
| GP | Marburg virus | 74.4 | 23 | 3.7 |
| Spike glycoprotein E2 | Coronavirus-229E | 128.6 | 30 | 3.4 |
| Glycoprotein G | HRSV-A2 | 32.5 | 7 | 1.9 |
| Hemagglutinin | Influenza A | 39.6 | 8 | 1.9 |
| Env polyprotein | Human foamy virus | 113.7 | 15 | 1.8 |
| Env polyprotein | Spuma retrovirus | 113.4 | 15 | 1.8 |
| GH | Herpes simplex1 | 91.1 | 10 | 1.5 |
| GB | Herpes simplex1 | 100.3 | 10 | 1.3 |
| GD | Herpes simplex1 | 43.3 | 3 | 0.7 |
| Envelope glycoprotein | HTLV | 34.6 | 5 | 1.3 |
| Envelope glycoprotein | Dengue type 3 | 49.7 | 3 | 0.6 |
| Glycoprotein E | Hemorrhagic fever | 22.4 | 1 | 0.4 |
| Envelope glycoprotein | West Nile virus | 18.4 | 1 | 0.4 |
| B. Cellular targets | ||||
| Mucin (Muc-1) | Tumor marker | 108.0 | 100 (O) | 12.9 |
| Bovine Mucin (BSM) | Mucosal secretion | 158.4 | 5(N)/171(O) | 17.2 |
| CD24 | Adhesion molecule mucin-like | 8.08 | 2/15(O) | 10.9 |
| CD43 | Leukosialin, mucin | 40.3 | 26(O) | 6.4 |
| ICAM-3 | Adhesion molecule | 49.1 | 15 | 2.3 |
| CD45 | Tyr phosphatase | 63.4 | 16 | 3 |
| CD16 | Fc Receptor | 21.0 | 6 | 2.4 |
| ICAM-2 | Adhesion molecule | 22.5 | 6 | 2.2 |
| ICAM-1 | Adhesion molecule | 49.2 | 8 | 1.8 |
| CD47 | Integrin-associated protein | 35.2 | 6 | 1.6 |
| CD44 | Hermes antigen | 81.5 | 11(O) | 1.6 |
| CD31 | PECAM-1 | 82.5 | 10(O) | 1.5 |
| IgG | Antibody | 150.0 | 8 | 0.8 |
| KIR 2DL2 | NK receptor | 21.6 | 2 | 0.7 |
| HLA-CW3 | MHC I | 44.8 | 2 | 0.4 |
The O-linked glycans are indicated as (O). Otherwise, the numbers indicate N-linked glycans.
Discussion
DC-SIGN and DC-SIGNR are part of an antigen-capturing network of receptors expressed on dendritic cells. Previously, the structures of a mannose-bound and a Lewisx-bound form of the receptor showed critical residues involved in both calcium and carbohydrate interactions.18, 19 Our current structure of DC-SIGNR R8 represents an apo form of the receptor. The structure revealed that much of the CRD adopts a conformation very similar to that observed in the carbohydrate-bound receptor, with the exception of two loops that are involved in the coordination of carbohydrate (residues 361–366) and auxiliary calcium ions (residue 332–339) in the bound-form. In the absence of the bound carbohydrate, both loops adopt open conformations that are likely attributed to the loss of interactions with the putative carbohydrate and calcium. The absence of two bound auxiliary calcium ions compared with the structure of the carbohydrate-bound receptor suggests that the auxiliary calcium sites are of low affinity compared to the primary calcium site, and their presence appears to be ligand-induced.
The multivalent nature of DC-SIGN/R indicates that recognition of small carbohydrate compounds by individual CRD alone is not sufficient to achieve the high-affinity interactions of DC-SIGN and DC-SIGNR with pathogens like HIV-1 gp120. The functional receptors have been shown to be tetramers.17, 21 In addition, biochemical studies with repeat domain deletion mutants have shown that a minimum of three repeats are necessary to form tetramers, with additional repeats functioning to stabilize the tetramer.21 On the basis of the current crystal structures and available biophysical data, a tetramer for the entire extracellular DC-SIGNR receptor was constructed by homology modeling in which the repeat regions form helical bundles to bring together their CRDs in a 4-fold related symmetry. This helical bundle-mediated oligomerization resembles superficially the trimer of rat mannose-binding protein.20 While the receptor repeat domain is conserved in most species, a notable exception is that of Old World Rhesus monkey, whose DC-SIGNR gene (CD209L2) is missing all the repeats and DC-SIGN gene is missing the fourth repeat.27 CD209L2 is predicted to be a monomer and has been shown to be less efficient in binding to both ICAM-3 and HIV gp120. The fourth repeat in our model serves as a connecting helix between the two helical bundles. Deletion of this repeat would most likely shorten this connecting helix but may not affect the formation of the helical bundles (R6–R8 and R1–R3). The results of trypsin digestion studies appear to support a model in which the helical repeats are protected from protease by forming tightly packed helical bundles rather than by forming a single elongated helical domain (Figure 4).
Using this DC-SIGNR tetramer model and the assumption that high-affinity ligand binding requires simultaneous engagement of multiple CRD of the tetrameric receptor, we formulated a prediction scheme for potential ligands of DC-SIGN/R based on their predicted gross glycosylation density. The results show that several viral envelope glycoproteins, including HIV-1 gp120, Marburg virus GP, coronavirus spike protein, and HRSV glycoprotein G, possess high ligand indices. Among them, gp120 of HIV, GP of Ebola, and the spike protein of coronavirus are known ligands of DC-SIGN. Of the potential cellular targets, in addition to the known ICAM-3 ligand, mucins are notably ranked high in our scoring scheme. The low-scoring molecules, such as IgG, KIR2DL2 and HLA-CW3 did not exhibit binding to DC-SIGN/R (data not shown).
It should be noted that the receptor-ligand binding will also depend on the geometrical constraint, including the distance between and the orientation of the CRDs. The distance between glycans, in general, should correlate with their surface density. Situations in which local spacing variation resulting in the distance between glycans either too close or too far apart to simultaneously engage the multimeric CRD would clearly affect the recognition by the receptor. Nevertheless, the known flexibility of glycans and the observed variation in the hinge angle between the receptor CRD and repeat domains of DC-SIGNR illustrate the built-in flexibilities in both the receptor and ligands, and thus lend some degree of freedom to the receptor-ligand recognition. These intrinsic flexibilities would lead to greater variability in distance and orientation, and marginalize the geometric constraint. Nonetheless, the most obvious reasons to use the surface area of CRD instead of the distance are (1) to enable us to derive a prediction scheme based on surface glycan density of a potential ligand, and (2) to have the prediction less dependent on the precise conformation, thus, the degree of correctness, of the tetramer model.
In addition, equation (1) assumes a globular shape for proteins and a uniform distribution of their glycosylation. Clearly, both the local distribution of glycans and the actual shape of the protein presenting the glycans influence the receptor recognition. For example, despite a low score for Dengue and hemorrhagic fever, evidence suggests that these viruses are recognized by DC-SIGN/R.28, 29 Although the Dengue virus envelope protein is not heavily glycosylated, the crystal structures of both the type 2 and type 3 Dengue envelope protein E showed that the two conserved glycosylation sites are located at the protein dimer interface, resulting in four glycans distributed symmetrically at ∼32 Å apart across the interface.30, 31 This generates four closely packed glycan residues, which enables the recognition by the tetrameric DC-SIGN. This equation is thus a first-order approximation that does not reflect variations in protein shape or distribution of glycosylation.
In conclusion, the mechanism of receptor-carbohydrate recognition may be more complicated than previously thought. The high-affinity binding strategy employed by these receptors appears to be twofold. First, the structure of each individual CRD determines the preference of the receptor for particular carbohydrate structures. Secondly, and perhaps more importantly, the high-affinity interaction as well as ligand specificity rely on receptor oligomerization, which would increase the affinity of ligand binding and impose constraints on the density and distribution of carbohydrates found on target pathogens.
Materials and Methods
Protein expression, purification and crystallization
DNA encoding amino acid residues 250–399 of the human DC-SIGNR, which includes the last repeat (R8) and the CRD, referred as DC-SIGNR R8, was inserted into the pET 22b vector (Figure 1(a)). The expression of the full-length extracellular domain of DC-SIGN and DC-SIGNR has been described.21 Proteins were expressed as inclusion bodies in Escherichia coli BL21 (DE3) and reconstituted in vitro. Refolded DC-SIGNR R8 was loaded onto a Source 15Q column (Amersham) and further purified by size-exclusion chromatography using a Superdex S200 column (Amersham). The peak fractions were then concentrated to 10 mg/ml and characterized using SDS-PAGE, N-terminal sequencing and mass spectrometry.
Initial crystallization screening trials were carried out by microbatch experiments using an automated crystallization robot (Douglas Instruments Oryx 6).32, 33 Repeated attempts to crystallize the entire ectodomain of either DC-SIGN or DC-SIGNR did not yield any diffraction-quality crystals. In contrast, rod-like crystals of the DC-SIGNR R8 construct appeared in many conditions within six hours of setup. Optimization of crystal growth conditions was performed by fine-screening of pH and precipitant concentration. Crystals used for X-ray data collection were grown by the hanging-drop, vapor-diffusion method in a well solution of 100 mM MgCl2, 100 mM sodium cacodylate (pH 6.5), 12% (w/v) polyethylene glycol 3000.
X-ray data collection and structure determination
Crystals of DC-SIGNR R8 were briefly transferred into well solution supplemented with 20% (v/v) glycerol and flash-frozen in a liquid nitrogen stream at 100 K. The X-ray diffraction data were collected on a 3X3 charge-coupled device detector at the Structural Biology Center Collaborative Access Team beamline 19ID and processed using HKL2000.34 The crystals diffracted to 1.41 Å and were indexed to the orthorhombic space group P212121 with cell dimensions a=38.2 Å, b=54.8 Å, and c=62.3 Å.
Molecular replacement using the coordinates for DC-SIGNR CRD (PDB accession code 1K9J) provided phase information.18 Diffraction data from 41–3.0 Å were used for the rotation and translation functions with the program AMoRe.35 After rigid body refinement using program packages AMoRe and CNS.36 A complete model was built with the occupancies for disordered side-chains and loops set to zero. Initial refinement in CNS included simulated annealing, conjugate gradient minimization and individual temperature factor refinement. Further refinement using maximum likelihood methods was performed with the program Refmac 5.35 The final geometry of the structure was evaluated using the program PROCHECK.37 Least-squares superpositions were performed using the program LSQMAN.38
Modeling of the DC-SIGN tetramer
A protein search using the program BLASTp for sequences corresponding to one, two, three, four and all eight repeat domains of DC-SIGNR in various combinations was used to query the PDB.39 From this search we identified a representative set of structures that includes focal adhesion kinases, Taq Muts and DNA-binding proteins, with sequence identity of 30–70% (PDB accession codes 1K05, 1P85, 1IOM, 1NNE, 1NKW, 1EWR, 1ITW and 1HP7). The homologous portions of these structures were aligned on the basis of sequence homology to each corresponding repeat subunit of DC-SIGNR, and their secondary structure was viewed using the program O.40 The structure of the focal adhesion kinase (PDB code 1K05) was used as a template for tetramer formation, with the additional structures being used primarily to predict location of turns. The final model was refined in CNS using rigid body and energy minimization. Ribbon diagrams were prepared using the program MOLSCRIPT.41
Evaluating potential ligands of DC-SIGN/R
Since both receptors use multiple CRD domains to modulate avidity-mediated binding to various carbohydrates found on a variety of pathogens, we derived a formula to evaluate and identify potential receptor ligands. Let the surface area encompassed by the tetramer of DC-SIGNR CRD be S o, the surface area of a protein of interest be S, then the binding of DC-SIGN/R requires the number of glycosylations N satisfying:
Thus, an index for potential ligands can be defined as:
| (2) |
When the likelihood index i is greater than 1, the protein of interest possess, on average, higher glycosylation density than is required for binding to DC-SIGN/R and, conversely, when i is less than 1, the target protein is under glycosylated for DC-SIGN/R binding.
Assuming a spherical nature for proteins, which is only a crude approximation but will nonetheless result in a correct power-dependence on the molecular mass, the surface area S of a given protein can be calculated, to the first approximation, from its molecular mass by:
| (3) |
where N A is Avogadro's constant (6.022×1023), M is the molecular mass (in Da), and D is the average density of a protein, which has a value of 1.3–1.4 g/ml.42
If D=1.4 g/ml is taken and equation (3) is substituted in equation (2), then:
| (4) |
where S o is in Å2.
Protein Data Bank accession code
Coordinates have been deposited with the Protein Data Bank under accession code 1XPH.
Acknowledgements
We thank C. Hammer for mass spectrometry, M. Garfield for N-terminal sequencing, B. Hagos for assistance with protein expression, and C. Foster, A. Johnson, Z. Lu, S. Ginell, and N. Duke for assistance with synchrotron data collection. We thank J. Arthos for helpful discussions. We thank C. Foster, S. Garman and S. Radaev for helpful comments with structure and manuscript. Use of the Argonne National Laboratory Structural Biology Center beamlines at the Advanced Photon Source was supported by the US Department of Energy, Office of Energy Research, under Contract no. W-31-109-ENG-38. This work was supported by NIAID intramural funding.
Edited by I. Wilson
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2005.01.063.
The Supplementary Data consists of three Figures. The first Figure shows how the template focal adhesion kinase structure (PDB code 1K05) was used as a template to build the tetramer model. The second Figure shows the flexibility of CRD and how this flexibility could augment ligand binding. The third Figure shows trypsin digestion of DC-SIGNR R1. Four trypsin sites were located in the first repeat and within the CRD domain. The core repeats remained relatively protease resistant.
Appendix. Supplementary data
References
- 1.Alvarez C.P., Lasala F., Carrillo J., Muniz O., Corbi A.L., Delgado R. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 2002;76:6841–6844. doi: 10.1128/JVI.76.13.6841-6844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cole G., Coleman N., Soilleux E. HCV and HIV binding lectin, DC-SIGNR, is expressed at all stages of HCV induced liver disease. J. Clin. Pathol. 2004;57:79–80. doi: 10.1136/jcp.57.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curtis B.M., Scharnowske S., Watson A.J. Sequence and expression of a membrane-associated C-type lectin that exhibits CD4-independent binding of human immunodeficiency virus envelope glycoprotein gp120. Proc. Natl Acad. Sci. USA. 1992;89:8356–8360. doi: 10.1073/pnas.89.17.8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gardner J.P., Durso R.J., Arrigale R.R., Donovan G.P., Maddon P.J., Dragic T., Olson W.C. L-SIGN (CD 209L) is a liver-specific capture. receptor for hepatitis C virus. Proc. Natl Acad. Sci. USA. 2003;100:4498–4503. doi: 10.1073/pnas.0831128100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geijtenbeek T.B., Kwon D.S., Torensma R., Van Vliet S.J., Van Duijnhoven G.C., Middel J. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]
- 6.Halary F., Amara A., Lortat-Jacob H., Messerle M., Delaunay T., Houles C. Human cytomegalovirus binding to DC-SIGN is required for dendritic cell infection and target cell trans-infection. Immunity. 2002;17:653–664. doi: 10.1016/s1074-7613(02)00447-8. [DOI] [PubMed] [Google Scholar]
- 7.Lozach P.Y., Lortat-Jacob H., de Lacroix d.L., Staropoli I., Foung S., Amara A. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J. Biol. Chem. 2003;278:20358–20366. doi: 10.1074/jbc.M301284200. [DOI] [PubMed] [Google Scholar]
- 8.Lozach P.Y., Amara A., Bartosch B., Virelizier J.L., Arenzana-Seisdedos F., Cosset F.L., Altmeyer R. C-type Lectins L-SIGN and DC-SIGN capture and transmit infectious hepatitis C virus pseudotype particles. J. Biol. Chem. 2004;279:32035–32045. doi: 10.1074/jbc.M402296200. [DOI] [PubMed] [Google Scholar]
- 9.Pohlmann S., Zhang J., Baribaud F., Chen Z., Leslie G.J., Lin G. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J. Virol. 2003;77:4070–4080. doi: 10.1128/JVI.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmons G., Reeves J.D., Grogan C.C., Vandenberghe L.H., Baribaud F., Whitbeck J.C. DC-SIGN and DC-SIGNR bind Ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology. 2003;305:115–123. doi: 10.1006/viro.2002.1730. [DOI] [PubMed] [Google Scholar]
- 11.Van Kooyk Y., Geijtenbeek T.B. DC-SIGN: escape mechanism for pathogens. Nature Rev. Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- 12.Geijtenbeek T.B., Van Kooyk Y. DC-SIGN: a novel HIV receptor on DCs that mediates HIV-1 transmission. Curr. Top. Microbiol. Immunol. 2003;276:31–54. doi: 10.1007/978-3-662-06508-2_2. [DOI] [PubMed] [Google Scholar]
- 13.Moris A., Nobile C., Buseyne F., Porrot F., Abastado J.P., Schwartz O. DC-SIGN promotes exogenous MHC-I-restricted HIV-1 antigen presentation. Blood. 2004;103:2648–2654. doi: 10.1182/blood-2003-07-2532. [DOI] [PubMed] [Google Scholar]
- 14.Pohlmann S., Soilleux E.J., Baribaud F., Leslie G.J., Morris L.S., Trowsdale J. DC-SIGNR, a DC-SIGN homologue expressed in endothelial cells, binds to human and simian immunodeficiency viruses and activates infection in trans. Proc. Natl Acad. Sci. USA. 2001;98:2670–2675. doi: 10.1073/pnas.051631398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soilleux E.J., Barten R., Trowsdale J. DC-SIGN; a related gene, DC-SIGNR; and CD23 form a cluster on 19p13. J. Immunol. 2000;165:2937–2942. doi: 10.4049/jimmunol.165.6.2937. [DOI] [PubMed] [Google Scholar]
- 16.Bashirova A.A., Geijtenbeek T.B., Van Duijnhoven G.C., Van Vliet S.J., Eilering J.B., Martin M.P. A dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN)-related protein is highly expressed on human liver sinusoidal endothelial cells and promotes HIV-1 infection. J. Expt. Med. 2001;193:671–678. doi: 10.1084/jem.193.6.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitchell D.A., Fadden A.J., Drickamer K. A novel mechanism of carbohydrate recognition by the C-type lectins DC-SIGN and DC-SIGNR. Subunit organization and binding to multivalent ligands. J. Biol. Chem. 2001;276:28939–28945. doi: 10.1074/jbc.M104565200. [DOI] [PubMed] [Google Scholar]
- 18.Feinberg H., Mitchell D.A., Drickamer K., Weis W.I. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science. 2001;294:2163–2166. doi: 10.1126/science.1066371. [DOI] [PubMed] [Google Scholar]
- 19.Guo Y., Feinberg H., Conroy E., Mitchell D.A., Alvarez R., Blixt O. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nature Struct. Mol. Biol. 2004;11:591–598. doi: 10.1038/nsmb784. [DOI] [PubMed] [Google Scholar]
- 20.Taylor M.E., Bezouska K., Drickamer K. Contribution to ligand binding by multiple carbohydrate-recognition domains in the macrophage mannose receptor. J. Biol. Chem. 1992;267:1719–1726. [PubMed] [Google Scholar]
- 21.Snyder G.A., Ford J.M., Torabi-Parizi P., Arthos J.A., Schuck P., Colonna M., Sun P.D. Characterization of DC-SIGN/R interaction with HIV-1 gp120 and ICAM molecules favors the receptor's role as an antigen capturing rather than adhesion receptor. J. Virol. 2005;79 doi: 10.1128/JVI.79.8.4589-4598.2005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feinberg H., Guo Y., Mitchell D.A., Drickamer K., Weis W.I. Extended neck regions stabilize tetramers of the receptors DC-SIGN and DC-SIGNR. J. Biol. Chem. 2004;280:1327–1335. doi: 10.1074/jbc.M409925200. [DOI] [PubMed] [Google Scholar]
- 23.Drickamer K. C-type lectin-like domains. Curr. Opin. Struct. Biol. 1999;9:585–590. doi: 10.1016/s0959-440x(99)00009-3. [DOI] [PubMed] [Google Scholar]
- 24.Feinberg H., Park-Snyder S., Kolatkar A.R., Heise C.T., Taylor M.E., Weis W.I. Structure of a C-type carbohydrate recognition domain from the macrophage mannose receptor. J. Biol. Chem. 2000;275:21539–21548. doi: 10.1074/jbc.M002366200. [DOI] [PubMed] [Google Scholar]
- 25.Hakansson K., Reid K.B. Collectin structure: a review. Protein Sci. 2000;9:1607–1617. doi: 10.1110/ps.9.9.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frison N., Taylor M.E., Soilleux E., Bousser M.T., Mayer R., Monsigny M. Oligolysine-based oligosaccharide clusters: selective recognition and endocytosis by the mannose receptor and dendritic cell-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin. J. Biol. Chem. 2003;278:23922–23929. doi: 10.1074/jbc.M302483200. [DOI] [PubMed] [Google Scholar]
- 27.Bashirova A.A., Wu L., Cheng J., Martin T.D., Martin M.P., Benveniste R.E. Novel member of the CD209 (DC-SIGN) gene family in primates. J. Virol. 2003;77:217–227. doi: 10.1128/JVI.77.1.217-227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navarro-Sanchez E., Altmeyer R., Amara A., Schwartz O., Fieschi F., Virelizier J.L. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003;4:723–728. doi: 10.1038/sj.embor.embor866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tassaneetrithep B., Burgess T.H., Granelli-Piperno A., Trumpfheller C., Finke J., Sun W. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Expt. Med. 2003;197:823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Modis Y., Ogata S., Clements D., Harrison S.C. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J. Virol. 2005;79:1223–1231. doi: 10.1128/JVI.79.2.1223-1231.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Modis Y., Ogata S., Clements D., Harrison S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl Acad. Sci. USA. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chayen N.E. Comparative studies of protein crystallization by vapour-diffusion and microbatch techniques. Acta Crystallog. sect. D. Biol. Crystallog. 1998;54:8–15. doi: 10.1107/s0907444997005374. [DOI] [PubMed] [Google Scholar]
- 33.Chayen N.E. The role of oil in macromolecular crystallization. Structure. 1997;5:1269–1274. doi: 10.1016/s0969-2126(97)00279-7. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z.a.M.W. In: Macromolecular Crystallography, Part A. Carter C.W., Sweet R.M., editors. vol. 276. 1997. pp. 307–326. (Methods in Enzymology). [Google Scholar]
- 35.Collaborative Computional Project 4 The CCP4 suite: programs for protein crystallography. Acta Crystallog. sect. D Biol. Crystallog. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallog. sect. D Biol. Crystallog. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 37.Laskowski R.A., MacArthur M.W., Moss D.S., Thorton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallog. 1993;26:283–291. [Google Scholar]
- 38.Kleywegt G.J., Read R.J. Not your average density. Structure. 1997;5:1557–1569. doi: 10.1016/s0969-2126(97)00305-5. [DOI] [PubMed] [Google Scholar]
- 39.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 40.Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog. sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 41.Kraulis P.J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 1991;24:946–950. [Google Scholar]
- 42.Klapper M. On the nature of the protein interior. Biochim. Biophy. Acta. 1971;229:557–566. doi: 10.1016/0005-2795(71)90271-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.