

Abstract
Rapid development of diagnostic immunoassays against novel emerging or genetically modified pathogens in an emergency situation is dependent on the timely isolation of specific antibodies. Non-immune antibody phage display libraries are an efficient in vitro method for selecting monoclonal antibodies and hence ideal in these circumstances. Such libraries can be constructed from a variety of sources e.g. B cell cDNA or synthetically generated, and use a variety of antibody formats, typically scFv or Fab. However, antibody source and format can impact on the quality of antibodies generated and hence the effectiveness of this methodology for the timely production of antibodies. We have carried out a comparative screening of two antibody libraries, a semi-synthetic scFv library and a human-derived Fab library against the protective antigen toxin component of Bacillus anthracis and the epsilon toxin of Clostridium botulinum. We have shown that while the synthetic library produced a diverse collection of specific scFv-phage, these contained a high frequency of unnatural amber stops and glycosylation sites which limited their conversion to IgG, and also a high number which lost specificity when expressed as IgG. In contrast, these limitations were overcome by the use of a natural human library. Antibodies from both libraries could be used to develop sandwich ELISA assays with similar sensitivity. However, the ease and speed with which full-length IgG could be generated from the human-derived Fab library makes screening this type of library the preferable method for rapid antibody generation for diagnostic assay development.
Keywords: Phage display, Monoclonal antibody, Antibody library
Highlights
► Phage libraries ideal for rapid isolation of diagnostic antibodies in an emergency. ► We compared utility of synthetic scFv and natural Fab libraries for such purposes. ► All antibodies from Fab library could be rapidly expressed as full-length IgG. ► Majority of scFv library antibodies carry amber stops or CDR glycosylations. ► Screening Fab library therefore preferred method for rapid antibody development.
1. Introduction
Antibody phage display is an in vitro screening technique that allows for rapid selection of multiple monoclonal antibodies of high affinity and specificity (Hoogenboom, 2005). It involves the display of a polyclonal collection of antibody fragments containing the variable regions, numbering up to 1011 different clones, on the surface of filamentous phage carrying the genetic sequence of the displayed fragment. Antibody library repertoires can be obtained primarily by two methods, either from cDNA antibody sequences derived from the B cells of immunized or non-immune animal or human donors, or synthetically generated using random nucleotide sequences within selected CDRs (complementarity-determining regions) in combination with one or multiple framework regions to replicate the diversity of a natural antibody repertoire (Conrad and Scheller, 2005). These sequences are then fused to the sequence encoding the gene III (gIII) coat protein of the phage, enabling expression and incorporation of the antibody fragment on its surface. This collection of phage-displayed antibodies is then panned repeatedly against the antigen of interest, producing a polyclonal phage collection enriched for antigen-specific antibodies, from which individual monoclonal clones can then be identified and characterized.
The rapidity of this technique makes it ideal for generating novel antibodies for diagnostic and potentially therapeutic responses to outbreaks of emerging infectious disease such as SARS coronavirus and H5N1 Avian Influenza or genetically modified pathogens released in a bioterrorism incident. Since antibodies are highly specific and are capable of recognizing virtually every class of pathogen, including toxins, viruses, bacteria and fungi, they enable easy and rapid identification of pathogens (Nowakowski et al., 2002, Hayhurst et al., 2003, Paoli et al., 2004, Steiniger et al., 2007, Cabezas et al., 2008). Indeed, sandwich ELISAs utilizing high affinity mouse monoclonal antibodies have been developed against likely bioterrorist threats such as epsilon toxin of Clostridium botulinum and protective antigen, a toxin component of anthrax, with demonstrated toxin detection limits of 1–2 ng/ml (el Idrissi and Ward, 1992, Mabry et al., 2006). In addition, their effectiveness against all classes of pathogens, low toxicity and minimal severe or long-term side effect make them ideal as prophylactic or therapeutics drugs that have to be rushed into use (Casadevall et al., 2004).
However, while traditional hybridoma techniques isolate clones that secrete full-length immunoglobulins, the initial phage library screen typically utilizes bacterially expressed clones that are in the scFv or Fab format. Therefore for functional use as an antibody, the clone must be transferred to an appropriate vector for expression of recombinant full-length immunoglobulin in eukaryotic cell culture (Kohler and Milstein, 1975, Jostock et al., 2004). However, some antibody clones may fail to express properly or lose affinity during this conversion from a bacterial to a eukaryotic expression system, as well as a change of antibody structural format. This may be caused by the presence of amber stops within coding sequences generated from random synthetic nucleotide sequences (Marcus et al., 2006), or improper folding. This is a particular problem for synthetic libraries as they have not undergone positive selection within the immune system to remove antibodies that are unable to express or fold properly, unlike libraries generated from natural repertoires. On the other hand, synthetic libraries may have advantages over natural ones in that they possess non-natural sequences that may code for antibodies with unusual specificities that are not present in natural repertoires (Griffiths et al., 1994).
Poor conversion of the isolated antibody fragment-phage clones to a soluble antibody format impairs the suitability of phage display as a method for the rapid generation of antibodies. Therefore, in this study, we compared the relative ability of a semi-synthetic scFv library and a natural non-immune Fab library to generate useful diagnostic antibodies that can be converted and expressed as soluble recombinant antibodies to be used as a detection reagent. Our choice of antigens was two commonly cited bioterrorism threats, epsilon toxin (Etox) and protective antigen (PA). While screening of the scFv library produced multiple clones that bound as phage, only a small percentage of clones were successfully expressed as either soluble scFv or full-length IgG and retained specificity and functionality. On the other hand, screening of the Fab library produced multiple high affinity clones against Etox and PA, all of which functioned as soluble Fab. Upon conversion to full-length IgG, they were capable of sub-microgram levels of detection in a sandwich ELISA. Many of the synthetic scFv clones were found to contain amber stops in their CDRs or N-linked glycosylation sites. However, removal of these sequences did not render the majority of these clones amenable to expression as soluble recombinant antibody, suggesting that improper folding may also be a factor. The results of the study suggest that non-immune naturally derived antibody libraries may be preferable to synthetic scFv libraries for rapid isolation of antibodies against typical foreign or pathogenic protein antigens.
2. Material and methods
2.1. Panning of scFv library
The Tomlinson I and J scFv-phage library (Goletz et al., 2002) (Source BioScience LifeSciences , UK) was panned as follows. Briefly, 80 μg of purified protein in 4 ml of PBS was coated onto Maxisorb Immunotubes (Nunc, Denmark) overnight at 4 °C then blocked with 2% skim milk in 1× PBS (MPBS) for 2 h at room temperature, followed by 3 washes with 1× PBS. The phage library (in 4 ml of MPBS) was applied for 2 h then discarded. The immunotube was washed with PBS/0.1% Tween 10 times for the first pan and 20 times for all subsequent pans. Elution of the remaining bound phage was carried out by digestion with 500 μl of 1 mg/ml trypsin solution for 10 min at room temperature. Eluted phage was re-infected into 1.75 ml of TG1 E. coli at OD600 0.4 for 30 min at 37 °C and grown up on a 2TYE plate overnight. To recover the library, the plate was scraped and grown up in 50 ml 2TYAG broth (100 μg/ml Ampicillin, 2% glucose) until log growth phase (OD600 0.5); The phage library was rescued by adding 5 × 1010 pfu of KM13 helper phage to 10 ml of this culture followed by incubation for 30 min at 37 °C and grown up in 50 ml 2TYAK (100 μg/ml Ampicillin and 50 μg/ml Kanamycin) liquid culture overnight at 30 °C. PEG precipitation was used to recover the phage the next day.
2.2. Panning of Fab library
The Fab phage display library was obtained from Humanyx Pte Ltd, Singapore which was constructed from the peripheral blood lymphocytes of non-immune human donors by the method of de Haard (de Haard et al., 1999). The library was enriched for binders to antigen as previously described via selection using biotinylated PA bound to Stretavidin-coated magnetic Dynabeads (Invitrogen, US) or immunotubes for Etox (Harrison et al., 1996). Briefly, 100 pmol of full-length PA previously biotinylated using sulfo-NHS-Biotin (Thermo Scientific, US) was incubated for 1 h at room temperature with 70 μl of Dynabeads in 500 μl of casein block (Thermo Scientific, US). Unbound PA was removed by 3 washes of casein block. For Etox, the antigen was coated onto immunotubes as described for screening of the scFv library.
To select for positive binders, 1 ml (~ 1013 phage) of pre-blocked phage library was incubated with either pre-coated beads or immunotube for 2 h at room temperature followed by 3 washes of 2% MPBS/T (containing 0.05% Tween-20), 3 washes with PBS/T and 2 washes with 1× PBS. For subsequent pans, beads or immunotube was washed 7 times with 2%MPBS/T, 7 times with PBS/T and twice with PBS. Bound phage was eluted with 500 μl of Triethylamine over 10 min, neutralized with 250 μl 1 M Tris pH7.4 and re-infected into TG1 at log phase growth; this was then rescued with helper phage M13KO7 (New England Biolabs, US) at an MOI of 20 and plated out onto 2YTAK plates (with 100 μg/ml Ampicillin and 25 μg/ml Kanamycin). Phage was recovered the next day as previously described using PEG precipitation and subsequent pans were carried out as above (Harrison et al., 1996).
2.3. Recovery of monoclonal clones, sequencing and BstN1 digest
To recover monoclonal clones, eluted phage was re-infected into log phase growth TG1 and then serially diluted in 2YT before plating out onto 2YTAG plates (1% or 2% glucose for scFv or Fab libraries respectively). Individual colonies were picked and inoculated into 2TYAG starter culture in 96-well U-bottom plates and grown up in shaking culture overnight at 37 °C. For the scFv library, 10 μl of starter culture was used to inoculate 200 μl of 2TYAG the next day in 96-deep well plates and grown for 1 1/2 to 2 h before infection with the respective helper phages for 1 h. Cultures were pelleted and re-suspended in 300 μl of fresh 2TYAK media and grown overnight to produce monoclonal phage for screening.
For the Fab library, clones were screened directly as soluble Fab. Briefly, clones from overnight culture were replicated into 120 μl 2TYAg (0.1% glucose) culture media in a 96-well U-bottom plate using a 96-pin replicator (Nunc, Denmark). The culture was grown up at 37 °C and Fab expression was induced overnight at 30 °C with 30 μl of 2TYAI (final concentration of 1 mM IPTG) at OD600 0.5; and the Fab-bearing supernatant recovered by centrifugation. Colony PCR for Fab library clones was carried out on starter culture of positive clones with the following primers HX01-F/R (5′-AGCGGATAACAATTTCACACAGG-3′/5′-TTTGTCGTCTTTCCAGACGTT AGT-3′). The PCR product was digested with BstN1 (New England Biolabs, US) and ran on a 3% agarose gel to identify unique patterns. Unique clones were sequenced using these same primers or LMB3/pHEN primers (5′-CAGGAAACAGCTATGAC-3′/5′-CTATGCGGCCCCATTCA-3′ for the scFv library.
2.4. Production of soluble scFv, phage, purified Fab and IgG
To express soluble scFv , the heavy and light chain coding sequences were excised via NcoI and NotI restriction sites from the library phagemid vector and cloned into modified pGEX-4T1 expression vector (GE Healthcare, US) carrying an N-terminal signal sequence for periplasmic expression and a C-terminal Helix–Loop–Helix dimerization motif which enables formation of scFv dimers with equivalent avidity as a full-length IgG (Pack et al., 1993). Each scFv monomer is connected to the dimerization motif via the flexible hinge of murine IgG3. Due to the presence of amber stop codons within the antibody CDRs which would prevent expression of the scFv in non-suppressor E. coli strains, overlapping PCR was carried out to introduce a single point mutation to convert the TAG stop codon to GAG, hence retaining the correct amino acid sequence. The sequence was then cloned into the expression vector via NcoI and NotI. Expression of scFv dimers was performed in the non-suppressor E. coli strain HB2151. Inoculated cultures were induced with 0.2 mM IPTG at late log growth phase (OD600 0.8) and then grown overnight at room temperature. To extract the scFv dimers from the periplasm, the cell pellet was re-suspended in 1/10 volume BBS buffer (200 mM borate pH8.0, 160 mM NaCl), incubated at 4 °C for 90 min, sonicated briefly for 20 s and then clarified by centrifugation for 30 min at 16 000 g. The extract was used for subsequent ELISA experiments.
Purified Fab was obtained by transforming the Fab encoding plasmid into E coli HB2151, since it enables expression of Fab without the gIII fusion protein due to the amber stop codon which is suppressed in E. coli TG1. Fab was expressed in a small scale 50 ml 2YTAg culture induced with a final concentration of 1 mM IPTG at OD600 0.5, and extracted by resuspending the pellet in 2 ml TES buffer (0.2 M Tris pH8.0, 0.5 mM EDTA, 0.5 M sucrose, 1 mM PMSF and 1 μg/ml pepstatin); followed by purification with 100 μl Talon beads (Clontech, US). Periplasmic extract was first conditioned with 5 μl 1 M MgCl2 and incubated with the beads for 1 h at 4 °C prior to elution with 250 μl elution buffer (50 mM Tris pH8.0, 100 mM NaCl, 100 mM imidazole, 20% glycerol). Full-length IgG was constructed by cloning the Fab or scFv variable heavy and light chain sequences into a pCMV promoter-based vector carrying a IgG1 constant region, separated by an internal ribosome entry sequence (Lim et al., 2008); followed by expression in HEK293T cells using 293fectin transfection reagent (Invitrogen, US) according to manufacturers protocol. IgG was purified from the culture supernatant using Protein A (Thermo Scientific, US), eluted with IgG elution buffer (Thermo Scientific, US) and then buffer exchanged into 1× PBS/20% glycerol. Biotinylation and conjugation was carried out using EZ-link sulfo-NHS biotin (Thermo Scientific, US) and Lightning Link HRP conjugation kit (Innova Biosciences, UK) respectively according to manufacturer's instructions.
2.5. ELISA
For direct binding ELISAs, pre-blocked PEG-precipitated phage (diluted 1/10 in 2% MPBS); phage culture supernatant (mixed 1/1 with 5% MPBS); antibody (Fab / scFv / IgG) culture supernatants or periplasmic extracts (mixed 1/1 with 5% BPBS–5% BSA in PBS) was applied to antigen-coated Maxisorb 96-well ELISA plates (Nunc, Denmark). Plates were prepared by coating overnight at 4 °C with 10 μg/ml antigen in 1× PBS and blocked with 360 μl of 4%MPBS (for phage) or 5%BPBS (for scFv/Fab/IgG) for 2 h. Secondary antibody-αM13-HRP (GE Healthcare, US) for phage; α-c-myc-HRP (Roche, Switzerland) for Fab/scFv; and α-HuFc-HRP for IgG, was added in blocking agent (2.5% MPBS for phage, 5% BPBS for scFv / Fab / IgG). The detection was visualized with the addition of TMB substrate (Thermo Scientific, US) for 10 min. For sandwich ELISAs, capture Fab or IgG was coated overnight at 5 μg/ml and the plates were blocked with 360 μl of 5% BPBS as above. Antigen was applied in 5%BPBS, followed by detector phage mixed 1/1 with 5%MPBS or 1 μg/ml biotinylated or directly conjugated IgG in 5%BPBS. Phage or IgG was detected using αM13-HRP (GE Healthcare, US) or Extravidin-HRP (Sigma-Aldrich, US) respectively. Serial dilution sandwich ELISAs were carried out in the same manner using serially diluted antigen. All ELISAs were carried out in a volume of 100 μl and four washes with PBS/T were carried out in between each step, with exception for the final wash. Before development with TMB substrate where 1× PBS was used instead of PBS/T. Incubations were all carried out for 1 h at room temperature unless specified.
2.6. Expression and purification of antigen
PA was dual tagged with an N-terminal His-tag and a C-terminal FLAG-tag in the pCTS-FLAG vector (Sigma-Aldrich, US) and expressed in BL21(DE3) E. coli. Dual tagging enabled a more efficient purification of full-length PA (Unpublished data). Induction was carried out with 1 mM IPTG (final concentration) at OD600 0.6 and grown overnight at room temperature. PA secreted into the supernatant was precipitated with ammonium sulfate. The precipitate was re-suspended in 50 mM sodium phosphate buffer (pH 7.8, 500 mM NaCl) and purified on Ni-NTA column (Qiagen). The eluate containing 250 mM imidazole was buffer exchanged into 50 mM TBS before binding to M2-αFLAG beads. Elution was carried out with 100 μg/ml 3× FLAG peptide and the purified full-length PA was buffer exchanged with TBS/20% glycerol for storage. N-terminal His tagged Etox was expressed and purified with Ni-NTA beads as described above.
3. Results
3.1. Screening of Tomlinson I and J scFv libraries against PA antigen
We carried out screening of the Tomlinson I and J (Tom I/J) library against PA for a total of three successive pans (Fig. 1A). The ELISA of the PEG-precipitated polyclonal phage after each pan showed an increase in signal against its panned antigen for each successive pan for both libraries, indicating successful enrichment. The last pan (3rd Pan) from each library was expanded into monoclonals as they had the highest specific signal and thus most likely to be enriched for antigen-specific clones. Indeed, the majority of selected clones were able to bind PA (Fig. 1B). Analysis of positive clones, however, found that the majority consisted of replicates of a few unique clones. This was not surprising as certain antibody clones which had strong binding to PA and also expressed well as phage would be expected to dominate the enriched library.
Fig. 1.
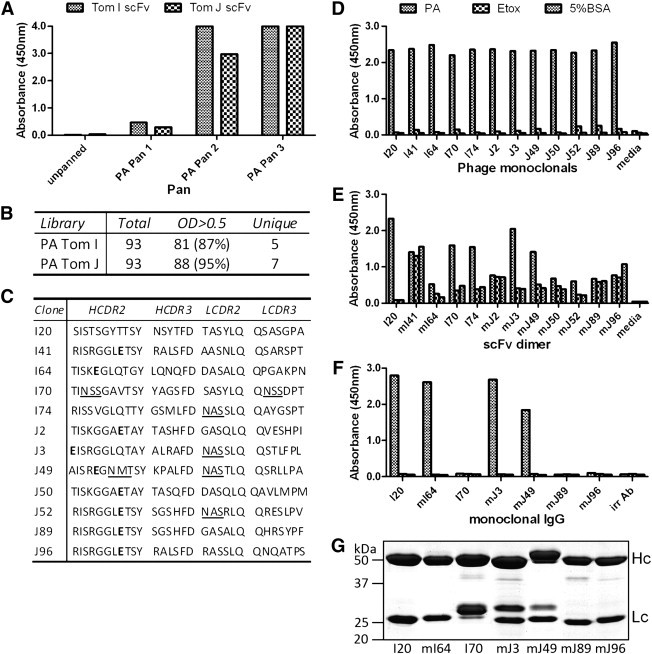
Screening of Tomlinson scFv libraries against PA. (A) Polyclonal ELISA of Tom I and J phage pans against PA showing enrichment for PA-binding clones. (B) Number of total screened, positive (OD > 0.5) and unique clones (as determined by sequencing of variable regions) obtained. (C) CDR sequences of identified unique clones. Bold residues indicate amber stop and underline indicates putative N-linked glycosylation sites. (D–F) Monoclonal ELISA of anti-PA clones isolated from Tomlinson I and J scFv libraries in various formats as (D) scFv-phage particles (E) soluble scFv dimers (F) purified full-length IgG, showing high affinity and specify as phage but loss of either affinity and/or specificity after expression as soluble scFv or IgG. Prefix (m) before the clone name indicates clones which had their amber stop codon changed to glutamic acid so prevent premature termination of translation. 5% BSA is 5% bovine serum albumin in PBS used as an antigen negative control. Culture media or irrelevant antibody was used as a clone negative control. (G) Reduced comassie gel of purified IgG showing heavy (Hc) and light chains (Lc).
From analysis of the CDR regions, it was found that the majority of scFv sequences contained amber stops within their CDRs (9 out of 12 unique clones, Fig. 1C). The presence of amber stops is possible in the synthetic scFv library due to the use of random nucleotides to encode the complementarity-determining region (CDR) 2 and 3 amino acids, which generates all possible codons including stop codons (Griffiths et al., 1994, Goletz et al., 2002). While other stop codons would result in the non-expression of the antibody-gIII fusion protein, in the TG1 E. coli strain the amber stop codon TAG is translated as glutamic acid due to the presence of an amber suppressor tRNA gene (SupE) (Weigert et al., 1965). In addition, several scFv clones (I70, 74, J3 and J49) were also found to have predicted N-linked glycosylation sites (N - X - S / T) (Kaplan et al., 1987) within their heavy and light CDRs (Fig. 1C). This would have an impact on the utility of these antibodies when expressed in eukaryotic systems, as demonstrated below.
To verify that the unique clones isolated were functional, they were expressed as soluble scFv antibodies and tested by ELISA for specific binding to their target antigen. This task was made more complex due to the presence of amber stops that had to be removed by PCR mutagenesis, as expression was carried out in the non-suppressor strain HB2151. When tested as soluble anti-PA scFv dimers, the majority was found to be highly cross-reactive and non-specific, with only one antibody (I20) showing high specificity with no cross-reactivity to other antigens; whereas four others (I70, I74, J3 and J49) were found to show some specificity with mild cross-reactivity (Fig. 1E). This is in sharp contrast to the high affinity and specificity displayed by the same clones when expressed as scFv-phage (Fig. 1D).
To determine whether this loss of specificity was due to their expression in a non-natural scFv format, five clones that retained specificity (I20, I70, I74, J3 and J49) and five clones that were cross-reactive (I41, I64, J50, J89, and J96) were expressed as full-length IgG. Three antibodies were found to express poorly with undetectable yield on purification of culture media (I41, I74 and J50). Of the antibodies that were successfully expressed, three (I70, J89 and J96) had no binding ability, while the remaining four (I20, I64, J3, and J49) retained affinity and specificity, in correlation to the results of the scFv ELISA (Fig. 1F). I64, although initially considered cross-reactive as scFv also had a slight PA-specific signal above background and could be considered a weak binder. On the other hand, the loss of binding of I70 could be attributed to an N-linked glycosylation site on the light chain CDR3, which produced a noticeable shift in mass as observed on SDS-PAGE (Fig. 1G). Glycosylation on the CDR region, which contacts the antigen, is highly likely to interfere with antigen binding. However, glycosylation was also observed in clones that retained binding activity, such as in the light chains of J3 and the light and heavy chains of J49. As these particular chains were observed to have doublet bands on SDS-PAGE, it could be that these two clones undergo incomplete glycosylation, resulting in a portion of expressed antibody retaining binding activity. It is unclear why some clones were unable to express as IgG, but glycosylation was probably not the only determining factor, as only one (I74) out of the three poorly expressing antibodies (I41, I74 and J50) contained a predicted glycosylation site.
To assess if the poor convertibility of the isolated clones were not particular to the antigen used (PA) but inherent in this library, screening of the Tomlinson libraries was carried out against a different antigen, Etox. A similar enrichment profile was observed, although a significant increase in signal was only observed with both Tom I and J libraries by Pan 3, which was expanded into monoclonals (Fig. 2A). This suggested less efficient enrichment compared to the screening against PA and may explain the lower percentage of positive clones (8%). Due to the low number of positive Etox clones (OD > 0.5), additional weaker binders were also selected for further analysis and a total of nine unique clones isolated and sequenced (Fig. 2B). Unfortunately, similar to what was observed earlier, six clones were found to contain amber stops, while two others contained predicted glycosylation sites (Fig. 2C). In view of our previous findings that the presence of amber stops and glycosylation sites could have led to a loss of antibody functionality, we decided not to pursue the conversion of these scFv Etox clones to soluble antibody formats and proceed to the panning with the Humanyx Fab library.
Fig. 2.
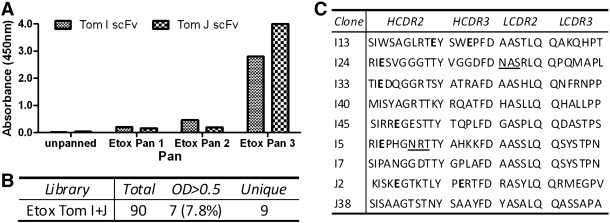
Screening of Tomlinson scFv libraries against Etox. (A) Polyclonal phage ELISA showing enrichment for Etox-binding clones. (B) Number of total screened, positive (OD > 0.5) and unique clones (as determined by sequencing of variable regions) obtained. Due to the low number of positive clones with OD > 0.5, additional weaker clones were screened to give additional unique clones (C) CDR sequences of identified unique clones. Bold residues indicate amber stop and underline indicates putative N-linked glycosylation site. Numerous clones contain either stop codons and glycosylation sites.
3.2. Panning of the Humanyx Fab library
As with the Tomlinson libraries, successful enrichment of the Humanyx Fab library for both PA- and Etox-specific binders was achieved and the pans which gave the highest signal (3rd Pan) were expanded into monoclonals (Fig. 3A). To avoid the confounding effects that may occur when screening as ‘antibody-displayed on phage’ format as observed with the scFv library, the Fab library clones were screened as soluble Fab antibodies. In addition, due to possible domination of the pan by a few clones as observed in the scFv library screen, a greater number of clones (~ 400) were picked for the Humanyx library screen in order to identify more unique clones. The Fab libraries contained a high percentage of positive clones, 22% for PA and 60% for Etox (Fig. 3B). In order to highlight duplicate clones, BstN1 digestion was carried out to identify unique restriction fragment length patterns. The Fab sequences were subsequently confirmed by DNA sequencing. The panning against Etox yielded 16 unique clones; while the panning against PA yielded 17 unique clones, all of which were functional as soluble Fab (Fig. 3B).
Fig. 3.
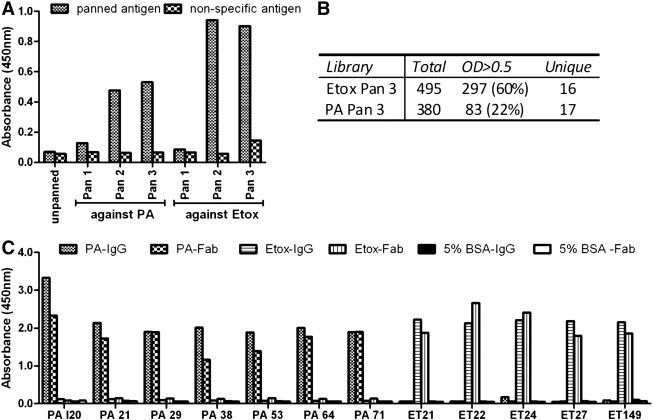
Screening of Humanyx library against PA and Etox. (A) Polyclonal phage ELISA showing specific enrichment with subsequent pans for both PA and Etox antigens (B) number of total screened, positive (OD > 0.5) and unique clones (as determined by sequencing of variable regions) obtained. For the Etox library only clones OD > 1.0 were selected for further screening due to the large number of positives. (C) Monoclonal ELISA of selected anti-PA and Etox clones showing 100% retention of high affinity and specificity after conversion to IgG from Fab.
In order to demonstrate the suitability of the Humanyx library-derived antibodies for diagnostic assays, six anti-PA antibodies (PA21, 39, 38, 53, 64, and 71) and five anti-Etox antibodies (ET21, 22, 24, 27, and 62) were selected for further evaluation in a typical diagnostic ELISA. These clones were chosen for their high binding affinity as observed on a preliminary Fab-vs-Phage sandwich screen, as well as their sequence diversity so as to ensure diverse antigenic binding sites. These clones were converted and expressed as full-length recombinant IgG for increased avidity. All antibodies retained their high affinity and specificity on direct binding ELISA to their target antigen on conversion from Fab to IgG (Fig. 3C).
3.3. Screening for antibody pairs by sandwich ELISA
Sandwich ELISA is commonly used as an antibody-based diagnostic tool. To determine whether the antibodies isolated from the scFv and Fab libraries can be used as detection reagents, we evaluated the performance of a panel of library-derived antibodies as capture-detector pairs in a sandwich ELISA. Each antibody was used both as capture and detector for all possible pairings. The choice of capture and detector antibodies is key to achieving high sensitivity in sandwich ELISAs, as pairs that have overlapping binding sites will compete with one another for antigen binding and hence perform poorly in the assay. Therefore, antibody libraries which can produce a large number of unique high affinity clones are advantageous, for they are more likely to generate antibody pairs that target different antigenic sites on the target molecule. As an additional comparison, I20, the best performing PA antibody that remained functional as a soluble scFv and IgG was also included. For the study with antibodies derived from the Fab library screening, purified IgG was used as capture and the biotinylated version was used as detector. In the PA IgG vs IgG-bio sandwich ELISA, clones PA21 and PA29 performed the best as detectors, while clones PA53 and PA64 were identified as the best capture antibodies (Fig. 4A). In contrast, I20, while specific gave very low signals when used as capture antibody, and did not bind antigen when biotinylated as a detector antibody. As biotinylation seemed to diminish the binding of the scFv library-derived I20 antibody, for all anti-PA IgGs derived from the scFv library that retained functionality (I20, mI64, mJ3 and mJ49), directly HRP conjugated antibodies were used for detection. Both I20 and mI64 were found to work well as either capture or detector while mJ3 was found to be a suitable detector antibody (Fig. 4B).
Fig. 4.
Rapid screening of sandwich ELISA antibody pairs. Selected anti-PA IgG antibodies from the Fab (A) and synthetic scFv (B) libraries, as well as anti-Etox IgG antibodies from the Fab library (C) in all possible pairings as capture vs detector antibody (IgG vs IgG-bio or IgG vs IgG-HRP) for use in sandwich ELISA with 3 μg/ml PA or Etox antigen. 5% BSA is 5% bovine serum albumin in PBS used as an antigen negative control.
For Etox, this comparison was only carried out for clones from the Fab library due to the likely loss of functionality of scFv clones when converting from scFv to IgG as shown in the earlier PA screen. Clones ET21, ET2, ET27 and ET149 all worked well as capture antibodies, while clones ET21, ET22 and ET27 also gave high signals as detector antibodies (Fig. 4C).
3.4. Determination of best pair sensitivity
The best pairs of PA and Etox antibodies were used in a sandwich ELISA with serially diluted antigen to determine the limit of detection for each pair. For the Fab library-derived anti-PA antibodies, clones PA64 and PA38 were used as capture antibodies against the best detector clones PA21 and PA29. All pairings gave almost identical results, with a limit of detection of 100–300 ng/ml (Fig. 5A). With the scFv library-derived anti-PA antibodies, the best pairs were I20 as capture along with mI64 and mJ3 as detector and two other moderate pairs using mI64 as capture against I20 and mJ3 as detector were also included. All pairs except I20 vs mJ3 had similar performance with a limit of detection at around 300 ng/ml, a detection limit poorer as compared to the pairs derived from the Fab library (Fig. 5B).
Fig. 5.
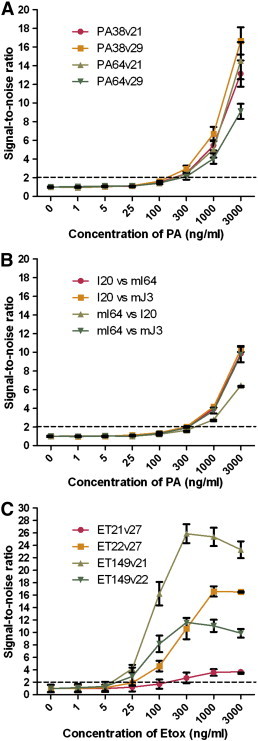
Determination of the limit of sensitivity of selected antibody pairs against PA and Etox. The lower limit of detection was determined for the indicated anti-PA IgG pairs from the Fab (A) and synthetic scFv (B) libraries, as well as anti-Etox pairs (C) from the Fab library in a sandwich ELISA using serially diluted PA or Etox antigen. Results shown are the average of three independent experiments with error bars showing standard error of mean. Signal-to-noise ratio is the ratio of the absorbance at the particular antigen concentration over the absorbance at 0 ng/ml concentration of antigen.
The two best Etox pairs identified in the IgG vs IgG-Bio sandwich ELISA, detector clones ET21 and ET22 against capture clone ET149; along with two moderately performing pairs, detector clone ET27 against capture clones ET21 and ET22, were tested for their limit of detection. Their sensitivity as indicated by the signal curves corresponded fairly well to their performance observed previously in the IgG vs IgG-bio sandwich ELISA. The two best pairs that used Etox 149 as capture had the lowest limit of detection between 5 and 25 ng/ml; while the other two moderately sensitive pairs had a limit of detection between 25 and 100 ng/ml (Fig. 5C). Pair ET21vET27 was noted to have high background in the IgG vs IgG-bio sandwich ELISA and this was reflected in the limit of detection signal curve as it had the poorest signal to noise (background) ratio at any given dilution.
4. Discussion
We have compared the abilities of two types of antibody phage display libraries, synthetic scFv and natural non-immune Fab libraries, to rapidly produce useful diagnostic antibodies. Both libraries have wide diversity estimated at greater than 109 individual clones, which in previous instances have been shown to give multiple high affinity antibodies against a variety of antigens (Griffiths et al., 1994, de Haard et al., 1999, Goletz et al., 2002). The polyclonal ELISA demonstrated a clear pattern of enrichment for both scFv and Fab libraries against both target antigens and resulted in a significant percentage of positive clones.
However, our screening of the scFv library indicated a high percentage of clones that contained amber stops within the CDRs. The presence of amber stop codons has been reported previously in various studies utilizing the Tomlinson I and J libraries (Hirose et al., 1998, Yan et al., 2004, Martinez-Torrecuadrada et al., 2005, Marcus et al., 2006). While this may be attributed to the use of random base insertion during the construction of the semi-synthetic library, the percentage of clones containing the amber stop codon appears disproportionally high. The exact reason for this observation is unclear. However, it has been shown that when using suppressor strains such as TG1, the presence of amber stop codons reduces expression (Oh et al., 2007). If expression of the antibody-gIII fusion protein is toxic to the bacteria, selection for clones with reduced expression of this protein may occur, increasing the likelihood for the selection of clones with amber stop codon as opposed to those with the corresponding classical glutamic acid codon.
While this problem is correctable by DNA mutagenesis techniques and suitable for expressing and analyzing a few high affinity antibodies (Marcus et al., 2006), the large number that would have to be processed in order to have a wide range of antibodies targeting different epitopes, as required in this study for rapid discovery of suitable antibody pairs, render this procedure impractical. On the other hand, this problem is not found in libraries using naturally derived sequences, as any antibody sequences that could not be properly expressed would have been eliminated from the natural immune repertoire during positive selection. The presence of N-linked glycosylation sites on CDRs on the other hand, was also shown to affect functionality in mammalian-expressed IgG in this study. This is yet another artifact present in synthetic repertoires and has to be taken into consideration when using synthetic libraries for rapid antibody selection.
Furthermore, the poor ability of synthetic scFv clones to be expressed as soluble antibody even after the removal of amber stop codons also limited their utility for rapid antibody isolation. Out of twelve unique scFv clones, five could be expressed as functional soluble scFv; and only three of these along with one other weakly binding scFv could be converted into functional recombinant full-length IgG. In contrast, all isolated clones from the Humanyx Fab library worked as soluble Fab and could be expressed as functional IgG upon conversion. The non-natural structure of scFv may have allowed for some synthetic scFv sequences to fold in a way that allowed specific binding when expressed on phage but not when expressed as soluble antibody. Whereas, the screening for specific monoclonal Fab was performed using soluble Fab, therefore improperly folded antibodies would have been automatically eliminated. Furthermore, natural antibodies from the Fab library, unlike synthetic antibodies, are already pre-selected for proper folding and binding activity due to positive selection in the immune system, thus are unlikely to fold improperly when expressed as Fab and possibly even if expressed as scFv.
We were able to isolate multiple pairs of antibodies from the Fab library that were capable of detecting our target antigens at low concentrations. In particular, we were able to identify two pairs of anti-Etox monoclonal antibodies (clone 21 vs clone 149 and clone 22 vs clone 149) that gave limits of detection close to that obtained by antibody pairs isolated by standard hybridoma technology, reported to be 1–2 ng/ml (el Idrissi and Ward, 1992). Given that hybridoma-derived monoclonals have been affinity matured, the remarkable ability of our isolated pairs to achieve a 5–25 ng/ml limit of detection highlights the potential of the phage screening process for generating specific and high affinity antibodies rapidly. While our screening of anti-PA IgGs from the scFv library also identified functional pairs, these had a marginally poorer limit of detection at 300 ng/ml to 1 μg/ml. More importantly, these functional pairs required IgGs that had to be modified to remove their amber stop codons, thus reducing their suitability for rapid development into diagnostic reagents. These pairs also used an IgG that have a significant percentage of polypeptide chains bearing unnatural glycosylations within the CDRs (mJ3), which may render a portion of antibody molecules defective and reduce the functionality of the antibody.
Considering the comparative inefficiency of the synthetic libraries in generating functional antibodies, it might be advisable to use naturally-sourced antibody libraries where clones have been pre-selected by the immune system. The main advantage of synthetic libraries is the possibility of isolating ‘self’ antibodies which would normally be eliminated from natural libraries during negative selection to maintain tolerance. However, this would not be a consideration when screening for antibodies against foreign antigens. In conclusion, our study suggests that natural antibody libraries are preferable to synthetic repertoires for the rapid development of potential diagnostic antibodies to novel pathogens due to the ease and speed at which useful antibodies can be isolated.
Acknowledgements
This work was supported by Defence Research and Technology Office (DRTech), Ministry of Defence, Singapore.
References
- Cabezas S., Rojas G., Pavon A., Alvarez M., Pupo M., Guillen G., Guzman M.G. Selection of phage-displayed human antibody fragments on Dengue virus particles captured by a monoclonal antibody: application to the four serotypes. J. Virol. Methods. 2008;147:235. doi: 10.1016/j.jviromet.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Casadevall A., Dadachova E., Pirofski L.A. Passive antibody therapy for infectious diseases. Nat. Rev. Microbiol. 2004;2:695. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- Conrad U., Scheller J. Considerations on antibody-phage display methodology. Comb. Chem. High Throughput Screen. 2005;8:117. doi: 10.2174/1386207053258532. [DOI] [PubMed] [Google Scholar]
- de Haard H.J., van Neer N., Reurs A., Hufton S.E., Roovers R.C., Henderikx P., de Bruine A.P., Arends J.W., Hoogenboom H.R. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J. Biol. Chem. 1999;274:18218. doi: 10.1074/jbc.274.26.18218. [DOI] [PubMed] [Google Scholar]
- el Idrissi A.H., Ward G.E. Evaluation of enzyme-linked immunosorbent assay for diagnosis of Clostridium perfringens enterotoxemias. Vet. Microbiol. 1992;31:389. doi: 10.1016/0378-1135(92)90131-c. [DOI] [PubMed] [Google Scholar]
- Goletz S., Christensen P.A., Kristensen P., Blohm D., Tomlinson I., Winter G., Karsten U. Selection of large diversities of antiidiotypic antibody fragments by phage display. J. Mol. Biol. 2002;315:1087. doi: 10.1006/jmbi.2001.5314. [DOI] [PubMed] [Google Scholar]
- Griffiths A.D., Williams S.C., Hartley O., Tomlinson I.M., Waterhouse P., Crosby W.L., Kontermann R.E., Jones P.T., Low N.M., Allison T.J. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 1994;13:3245. doi: 10.1002/j.1460-2075.1994.tb06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison J.L., Williams S.C., Winter G., Nissim A. Screening of phage antibody libraries. Methods Enzymol. 1996;267:83. doi: 10.1016/s0076-6879(96)67007-4. [DOI] [PubMed] [Google Scholar]
- Hayhurst A., Happe S., Mabry R., Koch Z., Iverson B.L., Georgiou G. Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. J. Immunol. Methods. 2003;276:185. doi: 10.1016/s0022-1759(03)00100-5. [DOI] [PubMed] [Google Scholar]
- Hirose M., Hayano T., Shirai H., Nakamura H., Kikuchi M. Isolation of anti-glutathione antibodies from a phage display library. Protein Eng. 1998;11:243. doi: 10.1093/protein/11.3.243. [DOI] [PubMed] [Google Scholar]
- Hoogenboom H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005;23:1105. doi: 10.1038/nbt1126. [DOI] [PubMed] [Google Scholar]
- Jostock T., Vanhove M., Brepoels E., Van Gool R., Daukandt M., Wehnert A., Van Hegelsom R., Dransfield D., Sexton D., Devlin M., Ley A., Hoogenboom H., Müllberg J. Rapid generation of functional human IgG antibodies derived from Fab-on-phage display libraries. J. Immunol. Methods. 2004;289:65. doi: 10.1016/j.jim.2004.03.014. [DOI] [PubMed] [Google Scholar]
- Kaplan H.A., Welply J.K., Lennarz W.J. Oligosaccharyl transferase: the central enzyme in the pathway of glycoprotein assembly. Biochim. Biophys. Acta. 1987;906:161. doi: 10.1016/0304-4157(87)90010-4. [DOI] [PubMed] [Google Scholar]
- Kohler G., Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Lim A.P., Chan C.E., Wong S.K., Chan A.H., Ooi E.E., Hanson B.J. Neutralizing human monoclonal antibody against H5N1 influenza HA selected from a Fab-phage display library. Virol. J. 2008;5:130. doi: 10.1186/1743-422X-5-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabry R., Brasky K., Geiger R., Carrion R., Jr., Hubbard G.B., Leppla S., Patterson J.L., Georgiou G., Iverson B.L. Detection of anthrax toxin in the serum of animals infected with Bacillus anthracis by using engineered immunoassays. Clin. Vaccine Immunol. 2006;13:671. doi: 10.1128/CVI.00023-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus W.D., Lindsay S.M., Sierks M.R. Identification and repair of positive binding antibodies containing randomly generated amber codons from synthetic phage display libraries. Biotechnol. Prog. 2006;22:919. doi: 10.1021/bp050420y. [DOI] [PubMed] [Google Scholar]
- Martinez-Torrecuadrada J., Cifuentes G., Lopez-Serra P., Saenz P., Martinez A., Casal J.I. Targeting the extracellular domain of fibroblast growth factor receptor 3 with human single-chain Fv antibodies inhibits bladder carcinoma cell line proliferation. Clin. Cancer Res. 2005;11:6280. doi: 10.1158/1078-0432.CCR-05-0282. [DOI] [PubMed] [Google Scholar]
- Nowakowski A., Wang C., Powers D.B., Amersdorfer P., Smith T.J., Montgomery V.A., Sheridan R., Blake R., Smith L.A., Marks J.D. Potent neutralization of botulinum neurotoxin by recombinant oligoclonal antibody. Proc. Natl. Acad Sci. U. S. A. 2002;99:11346. doi: 10.1073/pnas.172229899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M.Y., Joo H.Y., Hur B.U., Jeong Y.H., Cha S.H. Enhancing phage display of antibody fragments using gIII-amber suppression. Gene. 2007;386:81. doi: 10.1016/j.gene.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Pack P., Kujau M., Schroeckh V., Knüpfer U., Wenderoth R., Riesenberg D., Plückthun A. Improved bivalent miniantibodies, with identical avidity as whole antibodies, produced by high cell density fermentation of Escherichia coli. Biotechnology (N. Y.) 1993;11:1271. doi: 10.1038/nbt1193-1271. [DOI] [PubMed] [Google Scholar]
- Paoli G.C., Chen C.Y., Brewster J.D. Single-chain Fv antibody with specificity for Listeria monocytogenes. J. Immunol. Methods. 2004;289:147. doi: 10.1016/j.jim.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Steiniger S.C., Altobell L.J., III, Zhou B., Janda K.D. Selection of human antibodies against cell surface-associated oligomeric anthrax protective antigen. Mol. Immunol. 2007;44:2749. doi: 10.1016/j.molimm.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Weigert M.G., Lanka E., Garen A. Amino acid substitutions resulting from suppression of nonsense mutations. II. Glutamine insertion by the Su-2 gene; tyrosine insertion by the Su-3 gene. J. Mol. Biol. 1965;14:522. doi: 10.1016/s0022-2836(65)80201-7. [DOI] [PubMed] [Google Scholar]
- Yan J.P., Ko J.H., Qi Y.P. Generation and characterization of a novel single-chain antibody fragment specific against human fibrin clots from phage display antibody library. Thromb. Res. 2004;114:205. doi: 10.1016/j.thromres.2004.06.013. [DOI] [PubMed] [Google Scholar]