

Graphical abstract
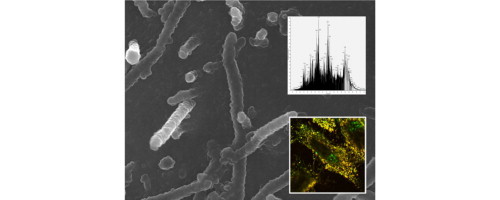
. a) In the background, scanning electron micrograph of RSV infected cells reveals viral filaments budding from the surface of virus infected cells. b) Inserted at the top, MS spectrum represents the characterization of the digested RSV virus particles. c) Inserted at the bottom, RSV infected cells were imaged using immunofluorescence microscopy: red represents virus filaments; green is HSP90; yellow staining represents co-localization of both antigens within the virus filaments.
Highlights
► The current proteomic researches on viruses and hosts are described. ► TAP, IP, SILAC, ICAT, and iTRAQ facilitate sample enrichment and quantification. ► Clinically important viruses are discussed on their interactions with hosts. ► Functional validation is essential to confirm the roles of the identified proteins.
Keywords: MS-based proteomics, Virus proteome, Virus and host interactions
Abstract
In terms of proteomic research in the 21st century, the realm of virology is still regarded as an enormous challenge mainly brought by three aspects, namely, studying on the complex proteome of the virus with unexpected variations, developing more accurate analytical techniques as well as understanding viral pathogenesis and virus–host interaction dynamics. Progresses in these areas will be helpful to vaccine design and antiviral drugs discovery. Mass spectrometry based proteomics have shown exceptional display of capabilities, not only precisely identifying viral and cellular proteins that are functionally, structurally, and dynamically changed upon virus infection, but also enabling us to detect important pathway proteins. In addition, many isolation and purification techniques and quantitative strategies in conjunction with MS can significantly improve the sensitivity of mass spectrometry for detecting low-abundant proteins, replenishing the stock of virus proteome and enlarging the protein–protein interaction maps. Nevertheless, only a small proportion of the infectious viruses in both of animal and plant have been studied using this approach. As more virus and host genomes are being sequenced, MS-based proteomics is becoming an indispensable tool for virology. In this paper, we provide a brief review of the current technologies and their applications in studying selected viruses and hosts.
1. Introduction
One of the major raising threats facing mankind is the challenge of viruses. In their evolutionary lifespan, viruses characterize remarkable potential to change and update to new subtypes of virus families and thereby gain the ability to be transmissible easily among humans as well as animals, triggering outbreaks of dangerous epidemic diseases or even pandemic risks worldwide. The struggle between human and viruses has long been recorded in the history of mankind as well as the medical development since the initial discovery of tobacco mosaic virus in 1898 [1]. Researches on viruses and experimental trials of antiviral therapies have never stopped but greatly facilitated by the use of high throughput sequencing techniques in the past decade, enabling the study of virus genomes in great details. Presently, the number of complete virus genomes in the NCBI database has increased to 2521, covering 118 taxonomy groups (http://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=10239&opt=Virus). As virus genome database continues to grow, it is increasingly important to study viruses at the protein level, including functions and expressions of viral genome-encoded proteins taking part in virus-mediated diseases. This is because that building up the knowledge of protein compositions of infectious viral particles is the prerequisite for functional studies. Nevertheless, only a limited number of viral proteins in a few virus families have been studied due to the challenges of obtaining information on complex viral proteins and the limitations of analytical assays and equipments. Understanding protein–protein interactions (PPIs) between viruses and host cells is another equally or more critical part, of which yet only a small percentage of viruses and their hosts have been studied. A successful virus infection of its host not only needs the participation of the viral particles in the first place but also requires more of the host responses providing the essential substances and spaces for virus development. The infection process usually comprises five stages: attachment and entry, translation, genome replication, assembly, and budding and release from the host cells, each of which is of significance in sub cellular proteomic studies and functional understandings of key proteins altered during infection. However, such five parts of the infectious cycle are not so easy to accomplish because the host cells must be both permissive and sensitive to the virus particles in order for them to survive and replicate successfully. For instance, for influenza virus, the non-permissive recognition of hemagglutinin to sialic acid receptors on different host cell membranes could restrict the host range specificity [2]. For some virus such as the adenovirus, successful survival and maturation of viral particles requires the delivery of virus genome, making it accessible to the nucleus [3]. This process bears certain intracellular high-risk factors, such as extremes of pH, attacks of enzymes and immune responses. As a result, the viral genome has to be encapsulated in a compact core structure as a protective coat, only to be released at the nucleus in order to utilize the intranuclear environment to replicate. Therefore understanding the behaviors of protein–protein interactions within the infectious cycle is invaluable, yet little is known towards the protein networks closely related to virus infectious cycle.
In recent years, rapid development in viral genome sequencing, gene expression analysis and emerging techniques of proteomics has allowed us to gain insight into viral proteins and virus–host interactions at the protein level, yielding fruitful results in an unbiased and comprehensive way. For examples, different MS-based proteomic approaches have played important roles in deciphering the protein composition of vaccinia virus (VACV) virion. Yoder et al. used a combination of fractionation techniques with three types of mass spectrometers to study purified VACV virion. Sixty-three VACV virion proteins were found, including two novel ones, E6R and L3L [4]. In another research endeavors of VACV, Chung et al. identified 75 viral proteins in intracellular mature virion (IMV) particles by using 2D-LC–MS/MS [5]. These included E6R, L3L and five other previously unknown proteins. Twenty-three IMV-associated host proteins were also identified. More recently, by using different purification steps and accurate mass and time (AMT) tag in LCMS, Resch et al. replenished the composition of VACV mature virion to a total of 80 proteins, including 11 previously unreported proteins [6]. In addition, 24 host proteins omitting isoforms were also detected. Different MS-based methods coupled with different virion purification and protein/peptide fractionation techniques resulted in different pools of VACV virion proteins, with overlapping and unique identifications. When combined, these identifications reveal a more complete picture of protein stock in the mature viral particles and help to improve our understanding of their functions.
In this review, we will focus on mass spectrometry based studies of virus proteins and virus–host interactions at the protein level. We will described the different MS methods used, followed by a survey of successful applications in virus proteomics. General techniques for mass spectrometry based proteomics are not covered here, as it can be found in several excellent reviews published recently [7], [8], [9].
2. MS-based methods
Several approaches have been used together with liquid chromatography (LC) separation followed by either electrospray ionization (ESI) or matrix assisted laser desorption/ionization (MALDI) tandem mass spectrometry to study protein–protein interactions. These techniques, namely tandem affinity purification (TAP), immunoprecipitation (IP), and quantification by stable isotope labeling, have been increasingly advanced to put perspectives into the interactions between viruses and hosts at the protein level. As sample preparation strategies differ, sensitivity and subsequent MS identifications also varies. However, each method has its own uniqueness and strength, and could be used complementarily for elucidating the mechanisms viruses use to infect their hosts and host countermeasures to fight the infection.
2.1. TAP-MS based methodology:
TAP tagging is a dual purification technique. The protein of interest is expressed with the TAP tag at either the N- or the C-terminus. The TAP tag contains two affinity tags separated by a protease cleavage site. Usually, the distal tag is Protein A, the proximal tag is a calmodulin-binding peptide, and the tobacco etch virus (TEV) protease cleavage sequence is used to join the two tags [10]. When the TAP-tagged protein is expressed from a chosen cell type, it forms a protein complex with its binding partners. The protein complex can be isolated by first using IgG-sepharose beads, which capture Protein A, followed by washing and cleavage with TEV protease. Non-specific bindings with the complex can be reduced by washing and proteins bound to Protein A and resin are left behind after the cleavage. The protein complex is recaptured with calmodulin sepharose beads by calcium-dependent interaction with calmodulin-binding peptide, washed again to reduce non-specific binding and finally released by calcium chelation with EGTA. Finally, the purified protein complex is digested with trypsin and analyzed using nanoLC coupled with tandem MS for protein identification.
TAP-MS based methodology has two main advantages. Firstly, the successive two-step affinity procedure helps to reduce the amount of contaminating proteins. Secondly, it can be applied to almost any protein without relying on any specific antibody, making it reproducible which contribute to the efficiency of the further robust MS analysis [11]. The TAP tagging strategy has been successfully used in various model organisms. However, in mammalian cells, expression of the tagged proteins is more difficult from their endogenous chromosomal locations. To circumvent this problem, various tag combinations and vector backbones have been introduced to express the cDNA encoding the protein of interest fused in frame with a TAP tag [12], [13], [14]. The expression of the fusion protein must be carefully controlled so that the level is similar to that of the endogenous protein of interest [15]. Several studies have used TAP tagging to identify virus and host factors associated with key virus proteins. In one experiment, a TAP tag was fused with the nucleoprotein (N) of borna disease virus (BDV), a non-segmented negative-strand RNA virus that persistently infects the central nervous system of warm-blooded animals. The TAP-tagged N was efficiently incorporated into the viral ribonucleoprotein complexes (vRNPs), did not interfere with BDV replication and was also packaged into viral particles. Tandem affinity purification of vRNPs resulted in two forms of N (p38 and p39), the phosphoprotein (P, p23), and the matrix protein (M, p16), as well as the viral genomic RNA [16]. In another study, the viral RNA polymerase subunits PB1, PB2 and PA of the influenza A virus were expressed by co-transfection of their cDNAs in human HEK293T cells, with one of the polymerases fused to a TAP tag. The purified protein complex containing all three viral polymerase subunits and their cellular protein partners were separated by SDS–PAGE and identified by MALDI-TOF-MS. The 10 associated human proteins found included nuclear factors involved in RNA synthesis, modification and export, as well as cytosolic proteins involved in translation and transport [17].
2.2. Co-immunoprecipitation (CO-IP) based MS methodology:
Co-immunoprecipitation, also known as pull-down, is an antibody-dependent technique requiring a specific antibody, which recognizes and purifies the target protein from cell lysis. Although there exist some disadvantages of this method, e.g. that the number of antibodies is limited compared to the numerous intracellular proteins, and that the specificity of antibody is required but not always guaranteed [18], CO-IP-MS is still commonly used in proteomic research as an increasing number of related papers have been published. In one study, Noisakran et al. employed co-immunoprecipitation, two-dimensional gel electrophoresis, and quadruple time-of-flight tandem mass spectrometry to study the functions of NS1 protein in dengue and its interacting partners in the infected human embryonic kidney cells (HEK293T). MS analysis successfully identified eight isoforms of NS1 and human heterogeneous nuclear ribonucleoprotein (hnRNP) C1/C2, which were co-purified by immunoprecipitation with anti-NS1 monoclonal antibody. This association between NS1 and hnRNP C1/C2 was suggested to have implications on cellular responses in favor of virus replications [19]. Also, Watanabe et al. utilized immunoprecipitation and MALDI-TOF-MS to analyze the influenza virus M1 protein and its interacting partners. It was found that heat shock cognate protein 70 (Hsc70) would bind to the C-terminal of M1 to repress its function [20].
2.3. HLA peptidome scanning chip based MS methodology:
From the immunological perspectives, human leukocyte antigen (HLA) class I molecules are considered as bio-chips for collecting and displaying peptides degraded and secreted from the multiple cellular compartments [21]. Sharma et al. used an immunoinformatic strategy to characterize the potential peptides from the proteome of H1N1 influenza virus that could be recognized by HLA molecules, resulting in the discovery of 15 and 14 Influenza virus specific peptides matching with HLA class I and HLA class II molecules, respectively [22]. Therefore, these HLA class I molecules may carry invaluable information for viral and host proteome after the virus infection and HLA-chip followed by MS could be used as a strategy for mapping the host proteome changes before and after virus infection. Recently, Wahl et al. used MS for comparative studies of the eluted host peptides from the HLA class I molecules of the native Hela cells and Hela cells infected with three different strains of influenza virus, H1N1 PR8, H1N1 7485, and H3N2 309 [23]. In the infected cells, this approach not only identified 20 unique host peptides, which were strain-specific to these three influenza viruses, but also detected 347 host peptides with increased expressions. All these altered host peptides could be further studied by Ingenuity Pathway Analysis to map the virus–host protein interactions. In another research, Hickman et al. utilized Q-TOF-MS to study peptides binding to HLA-B*0702 molecules produced by human T cells before and after HIV infection, resulting in identification of 15 proteins uniquely presented on the infected T cells [24]. What is more, an elevated level of soluble HLA class I molecules was detected in patients’ serum, whether upon viral infection [25] or during cancer development [26], and these sHLA class I molecules may also hold specific disease-related peptides, which could be further investigated as potential biomarkers for disease diagnostics [27].
2.4. MS-based quantitative proteomics
In the last decade, proteomics has been developed into the realm of high-throughput technologies with quantitative analysis capabilities mainly due to the advancement of MS-based quantification method and the development of computational software.
Various mass spectrometry based methods have been employed for quantitative analysis of protein expression in a complex protein mixture upon particular perturbation, such as prior to and after virus infection. These quantitative approaches utilize the stable isotope-labeling techniques combined with mass spectrometry analysis, including isotope coded affinity tag (ICAT), isobaric tag for relative and absolute quantitation (iTRAQ), and stable isotope labeling of amino acids in cell culture (SILAC).
ICAT is chemical labeling reagent targeting the reduced cysteine residue. Two protein mixtures from two different cell states are treated with the isotopically light and heavy ICAT reagents (e.g. C12 and C13), respectively. The two samples are then combined together and digested by a protease. And the peptides labeled with ICAT reagents are further isolated by affinity chromatography before mass spectrometry analysis [28], [29]. The ICAT approach has been used to identify differentially expressed proteins related to virus–host interactions. In one study, Booy et al. used ICAT, 2D gel electrophoresis and 2D-LC–MS/MS to analyze infectious hematopoietic necrosis virus (IHNV), which could induce infectious hematopoietic necrosis (IHN) and bacterial kidney disease (BKD). In particular, natural killer enhancement factor (NKEF) was observed to be down-regulated by least 2-fold during virus infection and it was considered as a viral resistant factor involved in cell signaling, although its specific role during virus infection needed further investigation [30]. Jiang et al. studied the proteome of SARS infected and uninfected Vero E6 cells by using the same labeling technique followed by LCMS, which enabled them to identify many differentially expressed proteins [31]. In this sense, the quantitative characterization of the whole genome was successful due to several reasons. First, the enrichment of the tagged peptides by affinity capture greatly reduced the degree of the amount of contaminants and improved the detection sensitivity. Second, ICAT approach was capable of detecting proteins regardless of their molecular weights.
iTRAQ is another quantitative approach based on multiplexed isobaric tagging chemistry and the complete tagging molecule is composed of a reporter group, a mass balance group, and a peptide reactive group, which could form an amide linkage to any primary amine [32]. The mass of the reporter group ranges from 114.1 to 117.1 Da, while the mass of the balance group ranges from 28 to 31 Da, and the sum of the reporter group and the balance group remains constant. Therefore, the same peptide with unique iTRAQ reagents from different samples exhibits identical mass and chromatographic property. During collision-induced dissociation (CID) and subsequent MS/MS analysis, the balance group is neutrally lost and the remaining reporter groups are detected quantitatively while the peptide fragments identify protein. Thus, iTRAQ could be employed to analyze quantitative changes in highly multiplexing samples [33], [34]. For instance in one study, Zhang et al. utilized iTRAQ coupled with 2D-LC–MS/MS to comprehensively study the hepatitis B virus (HBV) induced angiogenesis and DNA methylation and effectively identified important proteins and enzymes consistent with HBV replication [35] and epigenetic regulation [36], respectively. Notably, iTRAQ based proteomic approach was efficient to determine the localization of viral proteins. Li et al. utilized iTRAQ combined with LC-MALDI-TOF/TOF-MS to study the proteins of white spot syndrome virus (WSSV). Twenty-three envelope proteins and six nucleocapsid proteins of WSSV were successfully identified. Among them, 14 protein localizations were newly determined by this approach [37]. Also, a recently published paper demonstrated the potential of iTRAQ in combination with 2D-LC–MS/MS to detect the prognostic biomarkers of the hepatocellular carcinoma (HCC) after infection with the hepatitis B virus (HBV) [34], and apolipoprotein A-I and a2-HS glycoprotein were found to be closely related to the HCC pathological state.
SILAC has emerged as a powerful quantitative approach and is well-suited for a wide range of biological applications, such as embryonic stem cells [38], cancer [39], phosphotyrosine signaling networks [40], and virus–host interactions. In SILAC, two growing cells are fed with different culture medium, one containing a light amino acid (e.g. C12, N14) and the other carrying a heavy amino acid (e.g. C13, N15), respectively. After several generations, the cellular proteins can be completely labeled by the respective light and heavy amino acid. Two cell populations are combined with one to one ratio and digested by a protease. The relative intensity of the same peptide with light or heavy label represents the relative abundance of the protein in its original cell population [41], [42]. Edward et al. used SILAC coupled with LC–MS/MS to map the nucleolus proteome upon the coronavirus infectious bronchitis virus infection [43]. A total of 378 cellular proteins were identified and one viral protein, the N protein, was found to be localized in the nucleolus, which had not been reported previously. Also, Emmott et al. employed the same proteomic approach to quantitatively study the influenza A virus–nucleolar interactions in the human embryonic kidney 293-T cells upon infection with H1N1 and H3N2. Four (HA, NP, M1 and NS1) and six (PB2, HA, NP, M1, NS1 and NS2) virus-encoded proteins were identified for H1N1 and H3N2, respectively. Yet only a small number of the identified cellular proteins exhibiting significant alternations were nucleolar proteins [44]. In another research, SILAC approach was combined with 2D gel electrophoresis and MALDI-TOF-MS to gain insight on the virus and host interactions of pseudorabies virus (PrV) infected madin-darby bovine kidney (MDBK) cells. A total of 55 proteins were identified to be modulated with different levels after comparing the infected cells and non-infected cells. Among them, two proteins, pul29 and pul42, were significantly modulated with changes in the transcription level, thus suggesting that the phosphorylation by their specific upstream kinase, the US3 protein, could be considered as a cellular response upon infection [33]. In addition, by combining SILAC, co-immunoprecipitation and RNA interference (RNAi), quantitative immunoprecipitation combined with knockdown (QUICK) has been established and applied to quantitatively screen protein–protein interactions and it may be helpful to study the virus–host interactions [45], [46].
Besides these mass spectrometry based quantitative approaches, the integrated proteomic software, such as Trans-Proteomic Pipeline [47], Scaffold [48], Sorcerer-2 (http://www.sagenresearch.com/products/sorcerer-2/), Integrated Proteomics Pipeline (http://www.integratedproteomics.com/node/5), etc. also plays a critical role in analyzing quantitative mass spectrometric data by searching through the protein database and statistically validating the identification. Other software such as Ingenuity Pathways Analysis [43], [44] can further group the identified proteins into functionally different categories of proteins according to their proportion in order to understand the correlations of the functional proteins from an additional dimension.
3. Virus proteomic studies
Several clinical important viruses and their infections within the host cell partners have been studied using mass spectrometry based proteomic approaches. These studies are summarized as follows.
3.1. Hepatitis C virus (HCV)
Hepatitis C virus (HCV) is a small enveloped positive-strand RNA virus belonging to the Flaviviridae family. The HCV protein complex consists of at least 9 proteins, i.e. four structural proteins – Core, E1, E2, and p7, and five nonstructural proteins [49], [50]. Acute and chronic liver diseases, e.g. chronic hepatitis, cirrhosis, and hepatocellular carcinoma, are closely associated with the infection of HCV. However the pathogenesis remains to be understood in details. Therefore, many proteomic researches are focusing on the roles of these nine proteins in HCV infection with their host cells to expand the knowledge and enhance the understandings of HCV pathogenesis.
Evidences have shown that the HCV core protein is involved in the viral pathogenesis resulting in oxidative stress induction. Also, the locations where HCV core protein have been found are in both of mitochondria and endoplasmic reticulum (ER) coupling to lipid droplets, although the former of which is less reported [51], [52]. Recently, proteomic techniques enabled Tsutsumi et al. to reveal the functions of HCV core protein. By monitoring protein expressions in HepG2 cells using 2D gel electrophoresis and MALDI-TOF-MS before and after infection, a total of 16 proteins were found to be differentially expressed with 13 proteins increasing and 3 proteins decreasing in abundance [53]. In particular, prohibitin, a mitochondrial protein chaperon, was up-regulated after interaction with HCV core protein. The up-regulated prohibitin interacted with mitochondrially encoded subunit II of COX and lead to the impaired function of COX, which may be responsible for the over-expression of the mitochondrial respiratory chain. Thus, this study may enable us to elucidate the pathogenesis related to reactive oxygen species (ROS) in liver dysfunctions. Meanwhile, with respect to the HCV core protein that mostly found in the lipid droplets, Sato et al. utilized 1D SDS–PAGE with MALDI-TOF-MS and direct nano-flow liquid chromatography (DNLC)–MS/MS to analyze the protein contents of the lipid droplets in Hep39 cells and Hepswx cells, which can and can’t express HCV core protein, respectively [54]. They found 38 proteins in the lipid droplets of Hep39 cells and 30 proteins in that of Hepswx cells. One interesting discovery was that DEAD box proteins, DDX1 and DDX3, were exclusively found in Hep39 cells, however their functions related to HCV infection remain elusive. What is more, E2 protein is another important HCV structural component that is known to be highly glycosylated. Iacob et al. used MALDI-TOF-MS and LC–MS/MS to analyze the E2 protein and its glycosylation sites, respectively. The mass spectrometric data confirmed all of the 11 sites of glycosylation. Among them, two-N-linked sites were found to be occupied by complex type oligosaccharides, which played a major role in HCV envelope protein folding, HCV entry strategies, and regulation of the immune response [55].
With respect to the HCV nonstructural proteins, nonstructural protein 5A (NS5A) is known to play a critical part on RNA binding [56] and RNA replication [57], [58], however the precise role of NS5A remains unclear, especially in the virus and host interactions. MS-based proteomic analysis enables researchers to exhibit more detailed perspectives of NS5A upon HCV infection. For instance, Choi et al. used immunoprecipitation to isolate potential host proteins interacting with NS5A and NS5B, which were flag-tagged [59]. The associated host proteins were analyzed by 2D gels followed by MS analysis. A member of low molecular weight heat shock proteins, HSP27, was identified and confirmed by Western blot. The 1–122 amino acid domain of HSP27 was detected to interact exclusively with the N-terminal region of NS5A rather than NS5B and the co-localization of HSP27 and NS5A in ER was also confirmed by immunofluorescence, suggesting its increased role during heat shock. In other studies, nonstructural protein 4A (NS4A) and nonstructural protein 4B (NS4B) were reported to inhibit translations in the culture cells [60], [61], yet the mechanisms of this inhibitory activity were not well understood until proteomic techniques were utilized to identify the key host proteins involved in this repressed translation process. In tone study by Kou's research group [62], GST pull-down assay and LC–MS/MS analysis were employed to identify the host proteins interacting with NS4A or NS4B in Huh7 cells. One host protein, human translation elongation factor 1 alpha-1 (eEF1A) which could signal aminoacyl-tRNA to the ribosome, was found to bind exclusively to NS4A rather than NS4B. Mass spectrometric data further showed that the central domain of NS4A (21–34) could interact with eEF1A, leading to a diminished effect on cap-dependent and HCV IRES-mediated translation activities.
Another proteomic study emphasized on the altered proteins in response to the HCV replication in infected human liver carcinoma Huh7 cells [63]. By using 2D gel electrophoresis, a total of 289 proteins were found to be differentially expressed. Also, by using MALDI-TOF/TOF-MS, 179 proteins were identified and grouped according to their known locations and functions, however their specific functions during HCV infection needed further investigation.
3.2. Dengue
Dengue virus is another ssRNA positive-strand virus that belongs to the family Flaviviridae. Dengue virus infection, a mosquitoes-transmitted epidemic disease mainly in the tropical and subtropical areas, has clinical symptoms including headache, muscle and joint pains, vascular leakage, and hemorrhagic diatheses [64], [65], [66]. Since a more developed dengue vaccine is essential to target the serotypes of dengue virus [66], characterization of the potential vaccine components should be put in priority. Thus, in order to map detailed changes in subcellular proteins upon dengue virus infection and to find target proteins as disease biomarkers, several proteomic researches have been applied to characterize the host and DENV-related proteins during infection, although the pathogenesis of dengue virus infection remains far from clear. In one study, Pattanakitsakul et al. used 2D gel electrophoresis and Q-TOF-MS/MS to study the major changes related to the proteomes of subcellular compartments in DENV infected and non-infected human endothelial (EA.hy926) cells, resulting in a discovery of 35 significantly changed host proteins, most of which were from the cytosolic fraction [67]. Specifically, program cell death 6 interacting protein, also named Alix, was previously reported to be involved in the infectious cycle of HIV-1 budding [68], transportation of nucleocapsid to cytosol [69], and endocytic membrane trafficking [70]. In this paper it was further found to play a key role in the viral replication after interacting with the lysobisphosphatidic acid (LBPA), a late endosome marker in the host cell. In another paper published earlier, the same group used annexin V/propidium iodide double labeling strategy followed by 2D gels and MALDI-TOF-MS to quantitatively analyze the early host responses in human hepatocytes (HepG2) during dengue virus infection [71]. By comparing the host responses at 12, 24, 48 h post-infection, it was shown that host proteins as well as their morphological conditions exhibited an elevated level of alternations at the post-infection time point of 24 h, which could be valuable as an indicator for dengue virus infection and for early diagnosis. Further quantitative proteomic analysis compared infected HepG2 cells and mock-control cells at the incubation time interval between 1 and 24 h after virus infection, 17 host proteins were differentially expressed with 7 proteins down regulated and 10 proteins up-regulated. Some of these proteins participated mainly in RNA processing and translation activities, e.g. ATP-dependent RNA helicase DDX17 (DEAD box protein p72), pre-mRNA processing factor 4 homologue (PRP4), elongation factor Tu (EF-Tu), and mitochondrial precursor. EF-Tu was found earlier to bind to viral RNA-dependent RNA polymerase and further deliver aa-tRNA to ribosome. Pre-mRNA processing factor 4 homologue (PRP4) and elongin C were detected only during dengue infection. In addition to the knowledge of virus and host interactions, one important glycoprotein, dengue virus nonstructural protein 1 (NS1), was found to be interrelated with human heterogeneous nuclear ribonucleoprotein (hnRNP) C1/C2 in dengue virus-infected human embryonic kidney cells (HEK293T), supporting the hypothesis that this association could benefit viral replication [19]. Recently, based on the interactions between dengue-2 virus with mosquito-host cells, a model for dengue-2 virus's receptor-mediated endocytosis and transport has been proposed to interpret specific proteins involved in this procedure, including Hsc70 acting as an attachment molecule in the membrane, actin-binding proteins and actin polymerization taking part in the transportation of virus in the cytoplasm, and prohibitin responsible for activation of immune responses [72].
Although host cells can provide machinery for the translation of viral mRNAs, energy, and enzymes for genome replication, host cells are always like the battlefields initiating the responses to minimize the effects of virus attack. However, successful infections could decrease the immune response by modulating the related proteins. Higa et al. used 1D electrophoresis and LC–MS/MS to comparatively study the liver-secreted proteins in infected and control cells. And the same sample was also analyzed by 2D gel electrophoresis and MALDI-TOF/TOF-MS. Twenty proteins were identified in both of these two MS approaches, and two uniquely identified proteins in infected cells were thioredoxin and tissue inhibitor of metalloproteinase 1 (TIMP-1), the expression of which decreased by some extent in the secretome of infected cell. Therefore, it was highly indicative that this decreased concentration of TIMP-1 compromised the dengue virus infection and triggered the increased effect of endothelial permeability and plasma leakage during infection because of TIMPs’ role in anti-metalloproteinase [73].
3.3. Human immunodeficiency virus type 1 virus (HIV-1 virus)
HIV-1, a RNA virus of the viral family Retroviridae, is one of the most serious viruses and it can induce fatal disease by specifically breaking down the immune functions of T cells. The interaction between the HIV-1 surface glycoprotein, HIV-gp120, and its associating receptors in host cells, e.g. helper T cells (CD4+T cells), macrophages, and dendritic cells, is considered as the key step for virus entry to initiate the infectious cycle. In 1986, Putney et al. suggested that the glycosylation of gp120 was important for its interaction with CD4 [74] and Matthews et al. later demonstrated that gp120 specifically bound to CD4T cell receptor by using cell fusion assay [75]. In 1999, Borchers and Tomer showed that MS-based method was capable to detect this particular binding with much higher sensitivity and reliability at the protein level. They used MALDI-MS to precisely characterize the complex formed by gp120 and CD4 and found a mass value of 145 kDa, corresponding to the mass of one gp120 plus one CD4 molecule [76]. In other protein interactions upon HIV-1 infection, the cellular protein cyclophilin A could not only interact with HIV-1 Gag protein for assembly and virion replication [77], but also bind to HIV-1 capsid protein in order to enhance the virus infectivity [78]. Furthermore, Misumi et al. used 2D gel electrophoresis and MALDI-TOF-MS to study the isoforms of cyclophilin A during infection and successfully found three isoforms of cyclophilin A with isoelectric points at 6.40, 6.53, and 6.88, respectively. Peptide mass fingerprinting data indicated that the isoform with pI 6.53 could relocate from the inside to the outside of the viral membrane in favor of a direct role for attachment to the viral particle surface [79].
These particular protein interactions demonstrated the conserved molecular cooperators of virus and host interactions as well as the importance of the virus entry triggering the initial step of virus development. Understanding this process could largely enhance our knowledge towards successful HIV-1 interventions. Although currently there is no vaccine available against HIV or AIDS, many researches and clinical trials have been funded globally to find alternative solutions to treat HIV-1 infections and augment the effectiveness of pharmaceuticals by targeting these specifically interacting proteins, such as CD4, gp120, etc. In one research, Mamikonyan et al. tried to illustrate the functional component of thymus nuclear protein (TNP) in antivirus treatment of AIDS. As the main contributor of the anti-HIV-1 drug VGV-1™, TNP was experimentally characterized to have high affinities to gp41, gp120, and T cell receptor CD4 [80]. Using immunoaffinity chromatography followed by gel electrophoresis and MS/MS analysis, the components of TNP were identified to contain H1.1, H2B, H4, and H2A histones, which interacted with gp41, gp120 and CD4 molecules. This study suggested that histone components of TNP mediated immune responses and induced antivirus effect by suppressing HIV-1 entry. Nef protein is another CD4 interacting partner located at virus membrane. The association between Nef protein and CD4 is of critical importance due to its special role in down-regulation of CD4, resulting in endocytosis and enhanced HIV-1 virus development in the infected host cells [81], [82]. In one recent study, a strategy for developing an anti-HIV-1 drug was designed based on such interaction [83]. The modification of the specific cysteine residues on Nef protein was performed by adding a functional chemical, N-a-p-tosyl-l-phenylalanine chloromethyl ketone (TPCK), which covalently attached to Cys55 and Cys206 on the Nef protein. This modification effectively reduced the Nef–CD4 binding by nearly 50% in live cells and the strategy was suitable for further pharmaceutical design and investigation.
There are some other important proteins involved in the process of HIV-1 infection of host cells. HIV-1 transactivating protein (Tat) protein is essential for viral replication and may play important roles in HIV-1 associated diseases [84]. One intriguing phenomenon is that upon HIV-1 infection, the infected T cells were protected from apoptosis thus enabled them to survive longer bearing with virions. Although the exact mechanism is unknown, it has been suggested that HIV-1 Tat protein may be responsible for such protection [85]. In the MS-based proteomic study, Tat protein was identified to down-regulate cytoskeletal proteins such as actin, β-tubulin, annexin II, gelsolin, cofilin and Rac/Rho-GDI complex. The reduced expressions of these proteins were able to limit the cytoskeletal changes induced by apoptosis and thereby retaining HIV-1 virion for a long period. At the same time, MS-based approaches have been used to find potential inhibitors of Tat protein for the development of novel therapeutics. For instance, Jayasuriya et al. found durhamycin A as a inhibitor of Tat protein by screening microbial fermentation extracts and characterized the its structure by NMR and MS/MS [86].
In addition, HIV-1 not only breaks down the human immune system by efficiently and specifically attacking the T lymphocytes, but also accelerates the aggravation of other diseases. HIV-associated neurocognitive impairment (HAND) based encephalitis is still prevalent despite the introduction of active antiretroviral therapy [87]. Recently, a proteomic analysis of human synaptosomes was undertaken to shed light on neurocognitive impairment. The results revealed novel synaptic protein changes upon the HIV-1 infection with abnormal concentrations of synapsin 1b, 14-3-3ζ, 14-3-3ɛ, and stathmin that closely related to HIV-1 loading and immunoproteasome subunit expression level, suggesting their increased roles in HAND [88]. Dementia is another representative mental disease aggravated during HIV-1 infection [89]. Two groups used proteomic techniques to investigate the mechanisms of this process and roles of viral proteins. In one study, Dukelow et al. found that the dysfunction of blood–brain barrier (BBB), which separated blood circulation and central nervous system, was mainly associated with the interaction between human brain microvascular endothelial cells (HBMEC) and HIV-1 infected macrophages [90]. Two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) combined with LC/MS/MS enabled them to detect 77 differentially expressed proteins upon infection. In human brain microvascular endothelial cells (HBMEC), HIV-1 infected monocyte-derived macrophages (MDM) could induce the up regulation of these 77 proteins, which were related to cytoskeleton, regulatory, and redox proteins. In another study, Pocernich et al. tried to elucidate the functions of the Tat protein expressed by human astrocytes, though the number of HIV-1 infected astrocytes is limited [91]. Due to its remarkable property that it can leave the cell where it is expressed and further transactivate a number of genes of other cells [92], Tat protein expressed from human astrocytes could redistribute to the extracellular environment and be taken up by neuron, causing dysfunctions in both of the uninfected cells and the infected astrocytes. Ten proteins were detected to be differentially expressed by comparing SVGA-Tat cells that express Tat intracellularly and SVGA-pcDNA cells that do not express Tat. These altered proteins could be critical in understanding HIV-1 infected astrocytes related to Tat expression.
On the other hand, a large number of proteomic studies were conducted to find the biomarkers, which could efficiently improve clinical diagnosis of HIV induced neurodegenerative disorders, such as HIV-1 associated dementia (HAD). As patients’ serum is the most convenient source for clinical diagnosis, Wiederin et al. employed SELDI-TOF-MS analysis, weak cation exchange chromatography, one-dimensional electrophoresis, and LC–MS/MS to target potential serum biomarkers for HAD. The study showed that gelsolin and prealbumin were differentially expressed in demented samples compared to non-demented controls and may serve as candidate biomarkers [93].
3.4. Influenza virus
Influenza virus belongs to the Orthomyxoviridae family. It is a negative-sense RNA virus and has three generas: influenza A virus, influenza B virus, influenza C virus. Due to its powerful mutating ability and potential to result in a new pandemic, influenza A virus poses more threats to human as well as other hosts than the rest two influenza viruses. In the last century, there were some typical outbreaks of influenza A virus subtypes, such as the H1N1 Spanish Flu [94], the H2N2 Asian Flu [95], the H3N2 and H5N1 Hong Kong Flu [96]. In the 21st century, a new influenza A/H1N1 has emerged, which resulted from a triple-reassortment of influenza virus genes from avian, human and swain origin [97]. New warnings and concerns about those pandemic flu viruses were raised, a large number of researches followed, aiming to efficiently produce the updated vaccines and find advanced techniques to characterize the flu viruses. Hemagglutinin (HA) was of critical importance for vaccine design and antivirus drug discovery. As a key viral envelop glycoprotein [98], HA can bind specifically to sialic acid-containing receptors on the host cell surface and enables virus to attach to the targeted host cells [99], [100]. Also in the current influenza vaccines, the HA from H1N1, H3N2, and influenza B are combined as a trivalent complex, which can be simultaneously identified by MS-based approach with high sensitivity. Williams et al. presented a LC/MS/MS approach to quickly identify the HA subtypes in different influenza virus strains so as to improve the efficiency and effectiveness for vaccine preparations and productions [101]. In another study, MALDI-MS was used to locate the antigenic domains of HA of H3N2, which was found to contain residues 109–125, 158–170, 316–326 of the HA1 subunit and residues 159–183 of the HA2 subunit [102]. Also, Roschek et al. used Direct Analysis in Real Time Mass Spectrometry combined with Direct Binding Assay to find the anti-influenza components in elderberry extracts [103]. Two anti-influenza flavonoids were discovered and suggested to be suitable as constraints for interacting with HA binding domain pocket of H1N1, and the antivirus effect of one flavonoide named 5,7,30,40-tetra-O-methylquercetin, was as good as that of Tamiflu. Thus this approach is amendable to provide important information to assist vaccine development and antivirus drug discovery in order to better cope with the next new influenza pandemic.
Apart from hemagglutinin, influenza virus A has 10 known virus-encoded proteins, including neuraminidase (NA), matrix-1 (M1), matrix-2 (M2), nucleoprotein (NP), PA, PB1, PB1-F2, PB2, NS1 and NS2, which is also named nuclear export protein (NEP). These virus proteins play important roles involved in the virus infectious cycle. In one research, Shaw et al. used gel-based shortgun techniques to identify the protein content of influenza A/WSN/33 and host proteins incorporated into the virus particles [104]. Nine of these 11 virus-encoded proteins were successfully identified, including two low-abundant proteins, M2 and NEP, which were later investigated by Western blot. The remaining two viral proteins, NS1 and PB1-F2, were not detected in the viral particles as they were known to be predominantly expressed and localized in the cytoplasm and mitochondria of virus-infected cells, respectively [105], [106]. In another study, Liu et al. used nanoelectrospray Q-TOF-MS/MS to study H5N1 with an emphasis on amino acid substitutions in the M1 viral protein, which exhibits vital functions in the infectious cycle by regulating the transportation of ribonucleoprotein (RNP) [107], [108] and controlling the viral RNA synthesis through binding to RNA. In particular, it was found that there were six amino acid substitutions and two modifications of oxidation and deamidation at the protein level in the mutation-bearing strains of H5N1. The structure features of M1 characterized by these substitutions and modifications were analyzed by bioinformatics [109]. In another newly published paper, the signature peptides of M1 protein, which were highly conserved within the influenza A virus and influenza B virus with the host scope from human to avian and swine, were effectively detected and identified by MALDI-FT-ICR mass spectrometry. And both of the conserved sequences and the unique sequences of the digested M1 peptides were identified by FluAlign algorithm, which further contributed to the precise typing of the influenza virus subtypes [110]. In addition, one developed-software called FluTyper enabled to incorporate signature peptides of four influenza antigens together, e.g. hemagglutinin, neuraminidase, nucleoprotein, and M1 protein, thereby comprehensively improving the sensitivity and confidence of typing the influenza virus [111]. In respect to the role of M1 protein involved in the virus infectious cycle, Watanabe et al. employed immunoprecipitation and MALDI-TOF-MS and was the first to discover that heat shock cognate protein 70 (Hsc70) could bind to the C-terminal domain of M1. The localization of Hsc70 changed drastically to the nucleus in the late stages of virus infectious cycle, and lead to the inhibition of the nuclear export of M1 and NP, thus repressed virus maturation and production [20]. Virus RNA polymerase is a compact protein complex consisting of PA, PB1, PB2 subunits with important roles in RNA synthesis and replication [17], [112]. Jorba et al. used the TAP approach combined with MALDI-TOF-MS to search for the protein partners interacting with influenza A virus RNA polymerase in infected human HEK293T cells, and found 10 associated proteins mainly involved in cellular RNA synthesis, processing and transport.
To shed light on host cellular responses during infection, Liu et al. searched for the differentially expressed proteins upon H9N2 infection and identified 34 differentially expressed host proteins. Among these, cytoplasmic actin and cytokeratin were found to be altered significantly and were given detailed discussions concerning their unique roles in the cytoskeleton network [113]. Vester et al. used 2D gel electrophoresis and nanoLC–MS/MS to quantitatively and qualitatively study madin-darby canine kidney (MDCK) and human lung carcinoma cell lines (A549) during H1N1 virus infection [114]. Several proteins with differential abundance profiles were given detailed discussions. For instance, among those identified proteins, keratin 10 was known to regulate gene expression of the cytoskeleton proteins during infection. Eukaryotic translation elongation factor 1 (EF-1) was known to be responsible for viral protein synthesis. Also, ran GTPase was known to activate protein 1 (RanGAP1) to import vRNP to the host nucleus and proteasome activator hPA28 subunit β (PA28-beta) was known to participate in the protein degradation pathway related to apoptosis. Recently, Coombs et al. used SILAC combined with 2D-LC–MS/MS to explore the cellular proteins and their quantitative differences after infection of the human lung A549 cells with H1N1. A total of 4689 cytosolic proteins were identified and among them, 87 proteins were up-regulated or down-regulated significantly by more than 5-fold. Gene ontology and pathway analysis were used for further functional studies of those proteins [115]. In addition, macrophages are highly associated with the activation of innate immune response upon influenza virus infections. Lietzen et al. used iTRAQ isotope-labeling strategy combined with LC/MS/MS analysis to quantitatively study the human primary macrophages infected with influenza A viruses at three different post-infection time, 6, 12, and 18 h [116]. Mitochondrial, cytoplasmic and nuclear fractions of macrophages as well as secretome of the infected macrophages were selected for subcellular proteome analysis with 1999, 1423, 1230, and 627 proteins identified, respectively. Some apoptosis-related proteins, interferon-inducible proteins, mitochondrial and lysosomal proteins were observed as differentially expressed proteins during the virus infection. Due to the advantages of MS-based proteomic techniques, not only the differentially expressed proteins can be quantified, but also the dynamic changes over the time course can be precisely monitored, therefore augmenting the successful functional studies of virus and host proteins in the infectious cycle and facilitating vaccine production.
3.5. Severe acute respiratory syndrome (SARS)
At the beginning of the 21st century, a novel virus breakout was associated with the severe acute respiratory syndrome-associated coronavirus (SARS-CoV), which is a positive-sense RNA virus belonging to the Coronaviradae family. SARS-CoV genome has five major open reading frames encoding replicase polyprotein, spike (S) glycoprotein, small envelope (E) protein, membrane (M) glycoprotein and nucleocapsid (N) protein [117], [118]. Recently analytical techniques enabled researchers to profile the key host proteins and abnormal responses induced by SARS-CoV particles. In one research, Jiang et al. performed a comprehensive analysis by using 2D gel electrophoresis followed by ESI-MS/MS and ICAT coupled with 2D-LC–MS/MS to quantitatively map the proteome of infected and uninfected Vero E6 cells. Although 2D gel electrophoresis ESI-MS/MS identified 63 proteins, the sensitivity of ICAT 2D-LC–MS/MS was much higher with 322 proteins detected, enabling it to gain insight into the low-abundant proteins such as signal proteins [119]. Among them, 186 proteins were differentially expressed by at least 1.5-fold. Exploring the functions of these proteins could help to better understand the mechanism of SARS-CoV pathogenesis. In respect to the post translational modifications of viral proteins upon infection, Lin et al. analyzed the SARS nucleocapsid (N) protein in infected HEK 293 cells by 2D gel electrophoresis and discovered the N protein spot with distinctive PI values. Further MALDI-TOF/TOF-MS data showed two phosphorylation sites on the N protein [120], however their precise roles remain unclear. In another study, Zeng et al. combined 2D-LC–MS/MS and 1D gel, LC–MS/MS to study the proteomes of SARS-CoV and its infected Vero E6 cells. All of the four structured proteins of SARS – S, M, N, and E, were identified and a novel phosphorylation site on the M protein was also characterized [121]. Moreover, surface enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF-MS) was amendable as a unique proteomic tool profiling multiple disease-specific signatures. Kang et al. employed this methodology combined with a decision tree classification algorithm and analyzed the serum of the SARS patients by using four biomarkers, they could precisely detect 36 out of 37 SARS samples and 987 out of 993 non-SARS samples [122].
What is more, some important viral proteins have been increasingly regarded as the potential drug targets due to their special roles in viral pathogenesis. For instance, the SARS CoV replicase gene encoded 3C-like protease (3CLpro) was known to induce apoptosis, however, its specific role in regulating viral progression during SARS infection was less understood. Lai et al. utilized 2D nanoLC-Q-TOF-MS to comparatively study the host cells transfected with 3CLpro. They discovered that up-regulated proteins were mainly associated with ubiquitin proteasome pathway, mitochondrial mediated apoptosis, ATP synthesis and carbohydrate metabolism [123]. Additionally, proteomics could be applied as a strategy to design antivirus drugs against SARS. For example, Chu et al. used MALDI-TOF-MS to investigate the possible protease cleavage sites of SARS 3CLpro, which might be helpful to design synthetic inhibitors against this SARS viral proteins [117]. Zhang et al. utilized SILAC combined with LC–MS/MS to quantitatively explore the cellular proteome changes related to SARS-CoV replication, and found 74 significantly altered proteins. One of them, BAG3 was further knocked down by siRNA, which strongly inhibited SARS RNA replication, suggesting that BAG3 could be an anti-SARS drug candidate [124].
3.6. Human respiratory syncytial virus (RSV)
Human respiratory syncytial virus (RSV) is a negative-sense single-stranded RNA virus of the family Paramyxoviridae. It is a leading cause of epidemic respiratory disease, yet the vaccine against RSV is not available at present. With respect to the virus–host interactions, RSV polymerase was found to have a tight relationship with the cellular lipid-raft membranes, which facilitated the viral replication and maturation. In particular, the main components of the RSV polymerase, e.g. nucleocapsid protein, matrix protein, phosphoprotein, fusion protein, large protein, M2-1 protein, were identified by 2D-LC–MS/MS with M2-1 protein exhibiting a higher association with lipid-raft membranes of the infected HEp2 cells. Heat shock protein 70 (Hsp70) was also identified and found to have an increased abundance in lipid-raft membranes [125]. Brown et al. further used different validation methods to examine the association between the cellular heat shock protein 70 (Hsp70) and RSV during virus infection. Flotation gradient analysis confirmed the increased level of raft-associated Hsp70 and immunoprecipitation assays validated the interaction between Hsp70 and virus polymerase complex in lipid-rafts [126]. Yeo et al. utilized ESI-Q-TOF-MS and found that there was an increased level of raft-associated and phosphorylated phosphatidylinositol (PI) during RSV infection in favor of successful maturation of viruses [127]. In another paper, Brasier et al. performed 2D gel electrophoresis followed by MALDI-TOF-MS to study the altered nuclear proteome of A549 alveolar type II-like epithelial cells infected by RSV. A total of 24 proteins were identified as significantly changed proteins, including Hsp70, Hsp60, and nuclear domain 10 (ND10). Validation by immunofluorescence microscopy showed that RSV could induce cytoplasmic Hsp70 aggregation and nuclear accumulation during infection [128]. Radhakrishnan et al. used 1D nanoLC–MS/MS and imaging techniques to study the associated cellular partners involved in RSV maturation. Purified viruses were analyzed in search for host factors packaged into the virus particles. Functional validation of those cellular proteins could assist to better understand virus invasion and maturation process. One heat shock protein, Hsp90, was identified and validated as a RSV associated cellular protein playing a key role in virus particle assembly. Also, Hsp90 was treated with geldanamycin and 17-allylaminogeldanamycin, which were specific inhibitors to Hsp90, resulting in impaired formation of RSV particles. Thus it suggested that Hsp90 could serve as a new antivirus target [129]. Recently, Munday et al. employed SILAC coupled to LC–MS/MS to quantitatively study A549 cells infected with human respiratory syncytial virus and identified 1140 cellular proteins and six viral proteins, which were further processed by Ingenuity Pathways Analysis for functional grouping. The subsequent highlighted results were validated by immunefluorescence confocal microscopy, Western blot, and functional assays [130]. In addition, quantitative proteomic analysis of A549 cells infected with HRSV subgroup B was also performed by the same research group. Mitochondrial proteins, cell cycle regulatory molecules, nuclear pore complex proteins and nucleocytoplasmic trafficking proteins were observed for their significantly altered expressions upon infection. Proteins involved in antiviral responses and alternations of subnuclear structures, i.e. nucleolus and ND10, were validated by Western blot and indirect immunofluorescence confocal microscopy analysis, respectively [131].
3.7. Other animal virus
Compared to the numerous virus families, only a small number of viruses have been studied at the protein level. These studies are listed in Table 1 . However later on, an increasing number of proteomic researches are expected to be applied to many other viruses.
Table 1.
Analytical techniques for investigation of the proteome of animal viruses.
| Virus family | Virus | Method | Reference |
|---|---|---|---|
| Arenaviridae | Pichindé virus | SELDI-TOF-MS, tandem MS | [132] |
| Baculoviridae | Autographa californica nucleopolyhedrovirus | MudPIT, 2D-LC–MS/MS | [133] |
| Coronaviridae | Mouse hepatitis virus | SILAC, LC–MS/MS | [134] |
| Flaviviridae | Bovine viral diarrhea virus | 2D-LC–MS/MS | [135] |
| West nile virus | 2D-DIGE, LC–MS/MS | [136] | |
| Swine fever virus | 2D-DIGE, MALDI-TOF-MS/MS | [137] | |
| Hepadnaviridae | Hepatitis B virus | 2D gel, MALDI-TOF/TOF-MS; iTRAQ, 2D-LC–MS/MS | [138], [139], [140] |
| Hepeviridae | Hepatitis E virus | CO-IP, MALDI-TOF-MS | [141] |
| Herpersviridae | Herpesvirus | LC–MS/MS | [142] |
| Epstein-barr virus | SILAC, MALDI-TOF/TOF-MS | [143] | |
| Pseudorabies virus | 2D-LC–MS/MS | [33] | |
| Human cytomegalovirus | 2D gel, LC–MS/MS; 2D gel, MALDI-TOF/TOF-MS | [144] | |
| Marek's disease virus | LC–MS/MS | [132], [145] | |
| Murine cytomegalovirus | iTRAQ, LC–MS/MS | [146] | |
| Iridoviridae | Iridovirus | LC–MS/MS | [147] |
| Chilo iridescent virus | 2D gel, MALDI-TOF-MS | [148] | |
| Tiger frog virus | 2D gel, MALDI-TOF/TOF-MS | [149] | |
| Nimaviridae | White spot syndrome virus | HLA purification, nanoLC–MS/MS | [150] |
| Paramyxoviridae | Measles virus | MALDI-TOF-MS | [151] |
| Poxviridae | Myxoma virus | 2D gel, MALDI-TOF-MS | [152] |
| Fowlpox virus | MALDI-TOF-MS | [153] | |
| Vaccina virus | MALDI-TOF-MS | [154] | |
| Retroviridae | Simian immunodeficiency virus | MALDI-MS and LC–MS | [155] |
| Murine leukemia virus | 2D-DIGE, MALDI-TOF/TOF-MS | [156], [157] | |
| Reovirus | 2D gel, LC–MS/MS | [158] | |
| Rhabdoviridae | Rabies virus | 2D gel, MALDI-TOF-MS, nanoLC–MS/MS | [159] |
| Roniviridae | Yellow head virus | SELDI-TOF-MS, tandem MS | [160] |
3.8. Plant virus
Plant viruses are rarely studied and most of their genomes have not been sequenced. Therefore the approaches to detect the unidentified viruses are yet to be established. MS-based proteomic methods, such as LC–MS/MS, have already been used to successfully identify and characterize unknown viruses in the infected plants [161]. By using high-resolution tandem mass spectrometry and virus database search with pattern-matching and homology comparison, Blouin et al. readily identified four known plant viruses and two novel viruses from five different virus families [162]. Also, the proteome changes in hot pepper nucleus induced by tobacco mosaic virus (TMV) have been reported [163] as well as altered cellular factor expressions in virus-infected Fusarium graminearum [164], both of which were identified by MALDI-TOF-MS and ESI-MS/MS, respectively. What is more, the protein contents and the associated host partners of some plant viruses, such as tomato bushy stunt virus, have been systematically studied, benefited from the experimental data of genomics and proteomics approaches at the global level [165].
4. Conclusions
In the recent decade, the advancement of MS-based proteomics has revolutionized the way how protein interacting partners are investigated, it has enabled us to gain unprecedented knowledge on viral proteomes and cellular protein networks interacting with virus infection. However, the challenge still lies on functional validations of the large number of proteins identified and the even larger number of infectious viruses uninvestigated. The purpose of functional validation is to differentiate direct cellular responses from indirect secondary cellular processes related to virus infection; for instance, some indirect cellular processes during the late stages of HIV or RSV infection, such as syncytium formation, could be detected in proteomic analysis. Therefore, functional validations to highlight the proteins involved in direct cellular responses upon virus infections would provide novel insights to proteomic datasets. In respect to other endeavors, much effect should be put on expanding the virus proteomes of different families, elucidating the functions of the key proteins involved in the viral pathogenesis, and discovering sensitive biomarkers.
Acknowledgements
We thank Xueming Dong for helpful comments Chris Jeffree of University of Edinburgh and Jim Aitken of MRC Virology Unit, Glasgow for assistance in the preparation and imaging of the RSV-infected cells by SEM. KT is supported by Ministry of Health of Singapore and RJS is supported by Ministry of Health of Singapore, National Research Foundation, and Defense Science & Technology Agency.
Biographies

Jie Zheng graduated from China Pharmaceutical University in June 2010. He is currently a Ph.D. student in Nanyang Technological University, Singapore. His research interests include application of proteomic techniques on virus diagnostics, and virus–host interactions.

Richard J. Sugrue obtained a 1st class hon. degree in microbiology from University of London and a PhD in Biochemistry from the University of Kent, followed by research positions at the NIMR and crystallography dept at Birkbeck College in London, and the IMCB in Singapore working on influenza virus retroviral maturation, and Dengue virus replication enzymes. In 1998 he started a group working on RSV at the MRC Virology Unit in Glasgow. In 2005 he returned to Singapore and is currently Head of Division of Molecular and Cell biology. His interests include pathogen–host interactions in influenza, RSV and HMPV.

Kai Tang obtained his B.S. degree from Fudan University in 1986. He did his graduate research on mass spectrometry of nucleic acids in Oak Ridge National Laboratory from 1991 and obtained his Ph.D. degree from Vanderbilt University in 1994. From 1994 to 2004, he was a senior scientist in Sequenom Inc. of San Diego, California. He joined the School of Biological Sciences, Nanyang Technological University in 2005. His current research interests include mass spectrometry applications in biomarker discovery, the analyses of virus mutations and virus–host interactions, and method development for H/D exchange coupled with LC/MS.
References
- 1.Norkin L.C. ASM Press; Washington, DC: 2010. Virology: Molecular Biology and Pathogenesis. [Google Scholar]
- 2.Ito T., Couceiro J.N.S.S., Kelm S., Baum L.G., Krauss S., Castrucci M.R., Donatelli I., Kida H., Paulson J.C., Webster R.G., Kawaoka Y. J. Virol. 1998;72:7367. doi: 10.1128/jvi.72.9.7367-7373.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spector D.J. Virology. 2007;359:116. doi: 10.1016/j.virol.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Yoder J.D., Chen T.S., Gagnier C.R., Vemulapalli S., Maier C.S., Hruby D.E. Virol. J. 2006;3:10. doi: 10.1186/1743-422X-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung C.S., Chen C.H., Ho M.Y., Huang C.Y., Liao C.L., Chang W. J. Virol. 2006;80:2127. doi: 10.1128/JVI.80.5.2127-2140.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Resch W., Hixson K.K., Moore R.J., Lipton M.S., Moss B. Virology. 2007;358:233. doi: 10.1016/j.virol.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 7.Yates J.R., Han X.M., Aslanian A. Curr. Opin. Chem. Biol. 2008;12:483. doi: 10.1016/j.cbpa.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yates J.R., Ruse C.I., Nakorchevsky A. Annu. Rev. Biomed. Eng. 2009;11:49. doi: 10.1146/annurev-bioeng-061008-124934. [DOI] [PubMed] [Google Scholar]
- 9.Kast J., Walsh G.M., Rogalski J.C., Klockenbusch C. Expert Rev. Mol. Med. 2010;12:30–58. doi: 10.1017/S1462399410001614. [DOI] [PubMed] [Google Scholar]
- 10.Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Seraphin B. Methods. 2001;24:218. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 11.Volkel P., Le Faou P., Angrand P.O. Biochem. Soc. Trans. 2010;38:883. doi: 10.1042/BST0380883. [DOI] [PubMed] [Google Scholar]
- 12.Knuesel M., Wan Y., Xiao Z., Holinger E., Lowe N., Wang W., Liu X.D. Mol Cell Proteomics. 2003;2:1225. doi: 10.1074/mcp.T300007-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Burckstummer T., Bennett K.L., Preradovic A., Schutze G., Hantschel O., Superti-Furga G., Bauch A. Nat Methods. 2006;3:1013. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
- 14.Mak A.B., Ni Z., Hewel J.A., Chen G.I., Zhong G., Karamboulas K., Blakely K., Smiley S., Marcon E., Roudeva D., Li J., Olsen J.B., Wan C., Punna T., Isserlin R., Chetyrkin S., Gingras A.C., Emili A., Greenblatt J., Moffat J. Mol. Cell. Proteomics. 2010;9:811. doi: 10.1074/mcp.M000002-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gingras A.C., Aebersold R., Raught B. J. Physiol. 2005;563:11. doi: 10.1113/jphysiol.2004.080440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayer D., Baginsky S., Schwemmle M. Proteomics. 2005;5:4483. doi: 10.1002/pmic.200402095. [DOI] [PubMed] [Google Scholar]
- 17.Jorba N., Juarez S., Torreira E., Gastaminza P., Zamarreno N., Albar J.P., Ortin J. Proteomics. 2008;8:2077. doi: 10.1002/pmic.200700508. [DOI] [PubMed] [Google Scholar]
- 18.Moresco J.J., Carvalho P.C., Yates J.R., 3rd J. Proteomics. 2010;73:2198. doi: 10.1016/j.jprot.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noisakran S., Sengsai S., Thongboonkerd V., Kanlaya R., Sinchaikul S., Chen S.T., Puttikhunt C., Kasinrerk W., Limjindaporn T., Wongwiwat W., Malasit P., Yenchitsomanus P.T. Biochem. Biophys. Res. Commun. 2008;372:67. doi: 10.1016/j.bbrc.2008.04.165. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe K., Fuse T., Asano I., Tsukahara F., Maru Y., Nagata K., Kitazato K., Kobayashi N. FEBS Lett. 2006;580:5785. doi: 10.1016/j.febslet.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 21.Hickman H.D., Luis A.D., Buchli R., Few S.R., Sathiamurthy M., VanGundy R.S., Giberson C.F., Hildebrand W.H. J. Immunol. 2004;172:2944. doi: 10.4049/jimmunol.172.5.2944. [DOI] [PubMed] [Google Scholar]
- 22.Sharma P., Kumar A. J. Proteomics Bioinform. 2010;3:4. [Google Scholar]
- 23.Wahl A., Schafer F., Bardet W., Hildebrand W.H. Hum. Immunol. 2010;71:14. doi: 10.1016/j.humimm.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hickman H.D., Luis A.D., Bardet W., Buchli R., Battson C.L., Shearer M.H., Jackson K.W., Kennedy R.C., Hildebrand W.H. J. Immunol. 2003;171:22. doi: 10.4049/jimmunol.171.1.22. [DOI] [PubMed] [Google Scholar]
- 25.Migliaresi S., Bresciani A., Ambrosone L., Spera M., Barbarulo D., Lombari V., Pirozzi G., Borgia G., Lombardi M.L., Tirri G., Manzo C. Ann. Rheum. Dis. 2000;59:20. doi: 10.1136/ard.59.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leleu X., Le Friec G., Facon T., Amiot L., Fauchet R., Hennache B., Coiteux V., Yakoub-Agha I., Dubucquoi S., Avet-Loiseau H., Mathiot C., Bataille R., Mary J.Y. Clin. Cancer Res. 2005;11:7297. doi: 10.1158/1078-0432.CCR-05-0456. [DOI] [PubMed] [Google Scholar]
- 27.Bassani-Sternberg M., Barnea E., Beer I., Avivi I., Katz T., Admon A. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18769. doi: 10.1073/pnas.1008501107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gygi S.P., Rist B., Gerber S.A., Turecek F., Gelb M.H., Aebersold R. Nat. Biotechnol. 1999;17:994. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 29.Guaragna A., Amoresano A., Pinto V., Monti G., Mastrobuoni G., Marino G., Palumbo G. Bioconjug. Chem. 2008;19:1095. doi: 10.1021/bc800010b. [DOI] [PubMed] [Google Scholar]
- 30.Booy A.T., Haddow J.D., Ohlund L.B., Hardie D.B., Olafson R.W. J. Proteome Res. 2005;4:325. doi: 10.1021/pr049840t. [DOI] [PubMed] [Google Scholar]
- 31.Jiang X.S., Dai J., Sheng Q.H., Zhang L., Xia Q.C., Wu J.R., Zeng R. Mol. Cell. Proteomics. 2005;4:12. doi: 10.1074/mcp.M400079-MCP200. [DOI] [PubMed] [Google Scholar]
- 32.Ross P.L., Huang Y.L.N., Marchese J.N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D.J. Mol. Cell. Proteomics. 2004;3:1154. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Skiba M., Glowinski F., Koczan D., Mettenleiter T.C., Karger A. Vet. Microbiol. 2010;143:14. doi: 10.1016/j.vetmic.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Feng H., Wang M., Chen W.N. Curr. Microbiol. 2010;61:280. doi: 10.1007/s00284-010-9608-3. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J., Niu D., Sui J., Ching C.B., Chen W.N. Proteomics. 2009;9:2836. doi: 10.1002/pmic.200800911. [DOI] [PubMed] [Google Scholar]
- 36.Niu D., Sui J., Zhang J., Feng H., Chen W.N. Proteomics. 2009;9:3856. doi: 10.1002/pmic.200900071. [DOI] [PubMed] [Google Scholar]
- 37.Li Z., Lin Q., Chen J., Wu J.L., Lim T.K., Loh S.S., Tang X., Hew C.L. Mol. Cell. Proteomics. 2007;6:1609. doi: 10.1074/mcp.M600327-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Collier T.S., Sarkar P., Rao B., Muddiman D.C. J. Am. Soc. Mass Spectrom. 2010;21:879. doi: 10.1016/j.jasms.2010.01.031. [DOI] [PubMed] [Google Scholar]
- 39.Yang W., Cai Q., Lui V.W., Everley P.A., Kim J., Bhola N., Quesnelle K.M., Zetter B.R., Steen H., Freeman M.R., Grandis J.R. J. Proteome Res. 2010;9:3073. doi: 10.1021/pr901211j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hammond D.E., Hyde R., Kratchmarova I., Beynon R.J., Blagoev B., Clague M.J. J. Proteome Res. 2010;9:2734. doi: 10.1021/pr100145w. [DOI] [PubMed] [Google Scholar]
- 41.Mann M. Nat. Rev. Mol. Cell Biol. 2006;7:952. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 42.Ong S.E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., Mann M. Mol. Cell. Proteomics. 2002;1:376. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 43.Emmott E., Rodgers M.A., Macdonald A., McCrory S., Ajuh P., Hiscox J.A. Mol. Cell. Proteomics. 2010;9:1920. doi: 10.1074/mcp.M900345-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emmott E., Wise H., Loucaides E.M., Matthews D.A., Digard P., Hiscox J.A. J. Proteome Res. 2010;9:5335. doi: 10.1021/pr100593g. [DOI] [PubMed] [Google Scholar]
- 45.Selbach M., Mann M. Nat. Methods. 2006;3:981. doi: 10.1038/nmeth972. [DOI] [PubMed] [Google Scholar]
- 46.Braun P., Cusick M.E., Vidal M. Nat. Methods. 2007;4:103. doi: 10.1038/nmeth1206-975. [DOI] [PubMed] [Google Scholar]
- 47.Deutsch E.W., Mendoza L., Shteynberg D., Farrah T., Lam H., Tasman N., Sun Z., Nilsson E., Pratt B., Prazen B., Eng J.K., Martin D.B., Nesvizhskii A.I., Aebersold R. Proteomics. 2010;10:1150. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Searle B.C. Proteomics. 2010;10:1265. doi: 10.1002/pmic.200900437. [DOI] [PubMed] [Google Scholar]
- 49.Rehermann B., Nascimbeni M. Nat. Rev. Immunol. 2005;5:215. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 50.Randall G., Rice C.M. Curr. Opin. Infect. Dis. 2001;14:743. doi: 10.1097/00001432-200112000-00013. [DOI] [PubMed] [Google Scholar]
- 51.Schwer B., Ren S.T., Pietschmann T., Kartenbeck J., Kaehlcke K., Bartenschlager R., Yen T.S.B., Ott M. J. Virol. 2004;78:7958. doi: 10.1128/JVI.78.15.7958-7968.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suzuki R., Sakamoto S., Tsutsumi T., Rikimaru A., Tanaka K., Shimoike T., Moriishi K., Iwasaki T., Mizumoto K., Matsuura Y., Miyamura T., Suzuki T. J. Virol. 2005;79:1271. doi: 10.1128/JVI.79.2.1271-1281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsutsumi T., Matsuda M., Aizaki H., Moriya K., Miyoshi H., Fujie H., Shintani Y., Yotsuyanagi H., Miyamura T., Suzuki T., Koike K. Hepatology. 2009;50:378. doi: 10.1002/hep.22998. [DOI] [PubMed] [Google Scholar]
- 54.Sato S., Fukasawa M., Yamakawa Y., Natsume T., Suzuki T., Shoji I., Aizaki H., Miyamura T., Nishijima M. J. Biochem. 2006;139:921. doi: 10.1093/jb/mvj104. [DOI] [PubMed] [Google Scholar]
- 55.Iacob R.E., Perdivara I., Przybylski M., Tomer K.B. J. Am. Soc. Mass Spectrom. 2008;19:428. doi: 10.1016/j.jasms.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang L., Hwang J., Sharma S.D., Hargittai M.R., Chen Y., Arnold J.J., Raney K.D., Cameron C.E. J. Biol. Chem. 2005;280:36417. doi: 10.1074/jbc.M508175200. [DOI] [PubMed] [Google Scholar]
- 57.Tarao K., Fujiyama S., Ohkawa S., Miyakawa K., Tamai S., Hirokawa S., Masaki T., Tanaka K. Cancer Epidemiol. Biomarkers Prev. 2005;14:164. [PubMed] [Google Scholar]
- 58.Targett-Adams P., Boulant S., McLauchlan J. J. Virol. 2008;82:2182. doi: 10.1128/JVI.01565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Choi Y.W., Tan Y.J., Lim S.G., Hong W., Goh P.Y. Biochem. Biophys. Res. Commun. 2004;318:514. doi: 10.1016/j.bbrc.2004.04.052. [DOI] [PubMed] [Google Scholar]
- 60.Stasko D., Hoffmann S.P., Kim K.C., Fackler N.L., Larsen A.S., Drovetskaya T., Tham F.S., Reed C.A., Rickard C.E., Boyd P.D., Stoyanov E.S. J. Am. Chem. Soc. 2002;124:13869. doi: 10.1021/ja012671i. [DOI] [PubMed] [Google Scholar]
- 61.Libbus M.K., Phillips L., Knudson K.J. Public Health Nurs. 2002;19:470. doi: 10.1046/j.1525-1446.2002.19608.x. [DOI] [PubMed] [Google Scholar]
- 62.Kou Y.H., Chou S.M., Wang Y.M., Chang Y.T., Huang S.Y., Jung M.Y., Huang Y.H., Chen M.R., Chang M.F., Chang S.C. J. Biomed. Sci. 2006;13:861. doi: 10.1007/s11373-006-9104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang C., Yi Z., Liu F., Lan S., Wang J., Lu H., Yang P., Yuan Z. Proteomics. 2006;6:519. doi: 10.1002/pmic.200500233. [DOI] [PubMed] [Google Scholar]
- 64.Lindenbach B.D., Rice C.M. Advances in virus research. 2003;59:23. doi: 10.1016/s0065-3527(03)59002-9. [DOI] [PubMed] [Google Scholar]
- 65.Clyde K., Kyle J.L., Harris E. J. Virol. 2006;80:11418. doi: 10.1128/JVI.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hemungkorn M., Thisyakorn U., Thisyakorn C. Biosci. Trends. 2007;1:90. [PubMed] [Google Scholar]
- 67.Pattanakitsakul S.N., Poungsawai J., Kanlaya R., Sinchaikul S., Chen S.T., Thongboonkerd V. J. Proteome Res. 2010;9:4640. doi: 10.1021/pr100357f. [DOI] [PubMed] [Google Scholar]
- 68.Munshi U.M., Kim J., Nagashima K., Hurley J.H., Freed E.O. J. Biol. Chem. 2007;282:3847. doi: 10.1074/jbc.M607489200. [DOI] [PubMed] [Google Scholar]
- 69.Le Blanc I., Luyet P.P., Pons V., Ferguson C., Emans N., Petiot A., Mayran N., Demaurex N., Faure J., Sadoul R., Parton R.G., Gruenberg J. Nat. Cell Biol. 2005;7:653. doi: 10.1038/ncb1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Odorizzi G. J. Cell Sci. 2006;119:3025. doi: 10.1242/jcs.03072. [DOI] [PubMed] [Google Scholar]
- 71.Pattanakitsakul S.N., Rungrojcharoenkit K., Kanlaya R., Sinchaikul S., Noisakran S., Chen S.T., Malasit P., Thongboonkerd V. J. Proteome Res. 2007;6:4592. doi: 10.1021/pr070366b. [DOI] [PubMed] [Google Scholar]
- 72.Paingankar M.S., Gokhale M.D., Deobagkar D.N. Arch. Virol. 2010;155:1453. doi: 10.1007/s00705-010-0728-7. [DOI] [PubMed] [Google Scholar]
- 73.Higa L.M., Caruso M.B., Canellas F., Soares M.R., Oliveira-Carvalho A.L., Chapeaurouge D.A., Almeida P.M., Perales J., Zingali R.B., Da Poian A.T. Biochim. Biophys. Acta. 2008;1784:1607. doi: 10.1016/j.bbapap.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 74.Putney S.D., Matthews T.J., Robey W.G., Lynn D.L., Robert-Guroff M., Mueller W.T., Langlois A.J., Ghrayeb J., Petteway S.R., Jr., Weinhold K.J. Science. 1986;234:1392. doi: 10.1126/science.2431482. [DOI] [PubMed] [Google Scholar]
- 75.Matthews T.J., Weinhold K.J., Lyerly H.K., Langlois A.J., Wigzell H., Bolognesi D.P. Proc. Natl. Acad. Sci. U.S.A. 1987;84:5424. doi: 10.1073/pnas.84.15.5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Borchers C., Tomer K.B. Biochemistry. 1999;38:11734. doi: 10.1021/bi990935w. [DOI] [PubMed] [Google Scholar]
- 77.Matsuoka S., Dam E., Lecossier D., Clavel F., Hance A.J. Retrovirology. 2009;6:21. doi: 10.1186/1742-4690-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mascarenhas A.P., Musier-Forsyth K. FEBS J. 2009;276:6118. doi: 10.1111/j.1742-4658.2009.07315.x. [DOI] [PubMed] [Google Scholar]
- 79.Misumi S., Fuchigami T., Takamune N., Takahashi I., Takama M., Shoji S. J. Virol. 2002;76:10000. doi: 10.1128/JVI.76.19.10000-10008.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mamikonyan G., Kiyatkin A., Movsesyan N., Mkrtichyan M., Ghochikyan A., Petrushina I., Hwang J., Ichim T.E., Keledjian H., Agadjanyan M.G. Curr. HIV Res. 2008;6:318. doi: 10.2174/157016208785132545. [DOI] [PubMed] [Google Scholar]
- 81.Anderson S.J., Lenburg M., Landau N.R., Garcia J.V. J. Virol. 1994;68:3092. doi: 10.1128/jvi.68.5.3092-3101.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aiken C., Konner J., Landau N.R., Lenburg M.E., Trono D. Cell. 1994;76:853. doi: 10.1016/0092-8674(94)90360-3. [DOI] [PubMed] [Google Scholar]
- 83.Jin Y.J., Zhang X., Cai C.Y., Burakoff S.J. AIDS Res. Ther. 2010;7:26. doi: 10.1186/1742-6405-7-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rasheed S., Yan J.S., Hussain A., Lai B. J. Transl. Med. 2009;7:75. doi: 10.1186/1479-5876-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coiras M., Camafeita E., Urena T., Lopez J.A., Caballero F., Fernandez B., Lopez-Huertas M.R., Perez-Olmeda M., Alcami J. Proteomics. 2006;6(Suppl. 1):S63. doi: 10.1002/pmic.200500437. [DOI] [PubMed] [Google Scholar]
- 86.Jayasuriya H., Lingham R.B., Graham P., Quamina D., Herranz L., Genilloud O., Gagliardi M., Danzeisen R., Tomassini J.E., Zink D.L., Guan Z., Singh S.B. J. Nat. Prod. 2002;65:1091. doi: 10.1021/np010642f. [DOI] [PubMed] [Google Scholar]
- 87.McArthur J.C. J. Neuroimmunol. 2004;157:3. doi: 10.1016/j.jneuroim.2004.08.042. [DOI] [PubMed] [Google Scholar]
- 88.Gelman B.B., Nguyen T.P. J. Neuroimmune Pharmacol. 2010;5:92. doi: 10.1007/s11481-009-9168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McArthur J.C., Brew B.J., Nath A. Lancet Neurol. 2005;4:543. doi: 10.1016/S1474-4422(05)70165-4. [DOI] [PubMed] [Google Scholar]
- 90.Ricardo-Dukelow M., Kadiu I., Rozek W., Schlautman J., Persidsky Y., Ciborowski P., Kanmogne G.D., Gendelman H.E. J. Neuroimmunol. 2007;185:37. doi: 10.1016/j.jneuroim.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pocernich C.B., Boyd-Kimball D., Poon H.F., Thongboonkerd V., Lynn B.C., Klein J.B., Calebrese V., Nath A., Butterfield D.A. Brain Res. Mol. Brain Res. 2005;133:307. doi: 10.1016/j.molbrainres.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 92.Deshmane S.L., Mukerjee R., Fan S., Sawaya B.E. PLoS One. 2011;6:e16148. doi: 10.1371/journal.pone.0016148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wiederin J., Rozek W., Duan F., Ciborowski P. Proteome Sci. 2009;7:8. doi: 10.1186/1477-5956-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Taubenberger J.K., Morens D.M. Emerg Infect Dis. 2006;12:15. doi: 10.3201/eid1201.050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xu R., McBride R., Paulson J.C., Basler C.F., Wilson I.A. J. Virol. 2010;84:1715. doi: 10.1128/JVI.02162-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nelson M.I., Holmes E.C. Nat Rev Genet. 2007;8:196. doi: 10.1038/nrg2053. [DOI] [PubMed] [Google Scholar]
- 97.Ramakrishnan M.A., Wang P., Abin M., Yang M., Goyal S.M., Gramer M.R., Redig P., Fuhrman M.W., Sreevatsan S. Emerg. Infect. Dis. 2010;16:728. doi: 10.3201/eid1604.091583. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Lamb R.A., Takeda M. Nat. Med. 2001;7:1286. doi: 10.1038/nm1201-1286. [DOI] [PubMed] [Google Scholar]
- 99.Kash J.C., Basler C.F., Garcia-Sastre A., Carter V., Billharz R., Swayne D.E., Przygodzki R.M., Taubenberger J.K., Katze M.G., Tumpey T.M. J. Virol. 2004;78:9499. doi: 10.1128/JVI.78.17.9499-9511.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hernandez L.E., Brewin N.J., Drobak B.K. Anal. Biochem. 1996;241:59. doi: 10.1006/abio.1996.0378. [DOI] [PubMed] [Google Scholar]
- 101.Williams T.L., Luna L., Guo Z., Cox N.J., Pirkle J.L., Donis R.O., Barr J.R. Vaccine. 2008;26:2510. doi: 10.1016/j.vaccine.2008.03.014. [DOI] [PubMed] [Google Scholar]
- 102.Morrissey B., Streamer M., Downard K.M. J. Virol. Methods. 2007;145:106. doi: 10.1016/j.jviromet.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 103.Roschek B., Jr., Fink R.C., McMichael M.D., Li D., Alberte R.S. Phytochemistry. 2009;70:1255. doi: 10.1016/j.phytochem.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 104.Shaw M.L., Stone K.L., Colangelo C.M., Gulcicek E.E., Palese P. PLoS Pathog. 2008;4:e1000085. doi: 10.1371/journal.ppat.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hale B.G., Randall R.E., Ortin J., Jackson D. J. Gen. Virol. 2008;89:2359. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 106.Yamada H., Chounan R., Higashi Y., Kurihara N., Kido H. FEBS Lett. 2004;578:331. doi: 10.1016/j.febslet.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 107.Martin K., Helenius A. Cell. 1991;67:117. doi: 10.1016/0092-8674(91)90576-k. [DOI] [PubMed] [Google Scholar]
- 108.Bui M., Whittaker G., Helenius A. J. Virol. 1996;70:8391. doi: 10.1128/jvi.70.12.8391-8401.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu N., Song W., Lee K.C., Wang P., Chen H., Cai Z. J. Am. Soc. Mass Spectrom. 2009;20:312. doi: 10.1016/j.jasms.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 110.Schwahn A.B., Wong J.W., Downard K.M. J. Virol. Methods. 2010;165:178. doi: 10.1016/j.jviromet.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 111.Wong J.W., Schwahn A.B., Downard K.M. BMC Bioinformatics. 2010;11:266. doi: 10.1186/1471-2105-11-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Elton D., Amorim M.J., Medcalf L., Digard P. Biol. Lett. 2005;1:113. doi: 10.1098/rsbl.2004.0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu N., Song W., Wang P., Lee K., Chan W., Chen H., Cai Z. Proteomics. 2008;8:1851. doi: 10.1002/pmic.200700757. [DOI] [PubMed] [Google Scholar]
- 114.Vester D., Rapp E., Gade D., Genzel Y., Reichl U. Proteomics. 2009;9:3316. doi: 10.1002/pmic.200800893. [DOI] [PubMed] [Google Scholar]
- 115.Coombs K.M., Berard A., Xu W., Krokhin O., Meng X., Cortens J.P., Kobasa D., Wilkins J., Brown E.G. J. Virol. 2010;84:10888. doi: 10.1128/JVI.00431-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lietzen N., Ohman T., Rintahaka J., Julkunen I., Aittokallio T., Matikainen S., Nyman T.A. PLoS Pathog. 2011;7:e1001340. doi: 10.1371/journal.ppat.1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chu L.H., Choy W.Y., Tsai S.N., Rao Z., Ngai S.M. Protein Sci. 2006;15:699. doi: 10.1110/ps.052007306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Science. 2003;300:1394. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 119.Jiang X.S., Tang L.Y., Dai J., Zhou H., Li S.J., Xia Q.C., Wu J.R., Zeng R. Mol. Cell. Proteomics. 2005;4:902. doi: 10.1074/mcp.M400112-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lien S.P., Shih Y.P., Chen H.W., Tsai J.P., Leng C.H., Lin M.H., Lin L.H., Liu H.Y., Chou A.H., Chang Y.W., Chen Y.M., Chong P., Liu S.J. Biochem. Biophys. Res. Commun. 2007;358:716. doi: 10.1016/j.bbrc.2007.04.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zeng R., Ruan H.Q., Jiang X.S., Zhou H., Shi L., Zhang L., Sheng Q.H., Tu Q., Xia Q.C., Wu J.R. J. Proteome Res. 2004;3:549. doi: 10.1021/pr034111j. [DOI] [PubMed] [Google Scholar]
- 122.Kang X., Xu Y., Wu X., Liang Y., Wang C., Guo J., Wang Y., Chen M., Wu D., Bi S., Qiu Y., Lu P., Cheng J., Xiao B., Hu L., Gao X., Liu J., Song Y., Zhang L., Suo F., Chen T., Huang Z., Zhao Y., Lu H., Pan C., Tang H. Clin. Chem. 2005;51:56. doi: 10.1373/clinchem.2004.032458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lai C.C., Jou M.J., Huang S.Y., Li S.W., Wan L., Tsai F.J., Lin C.W. Proteomics. 2007;7:1446. doi: 10.1002/pmic.200600459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang L., Zhang Z.P., Zhang X.E., Lin F.S., Ge F. J. Virol. 2010;84:6050. doi: 10.1128/JVI.00213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McDonald T.P., Pitt A.R., Brown G., Rixon H.W., Sugrue R.J. Virology. 2004;330:147. doi: 10.1016/j.virol.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 126.Brown G., Rixon H.W.M., Steel J., McDonald T.P., Pitt A.R., Graham S., Sugrue R.J. Virology. 2005;338:69. doi: 10.1016/j.virol.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 127.Yeo D.S., Chan R., Brown G., Ying L., Sutejo R., Aitken J., Tan B.H., Wenk M.R., Sugrue R.J. Virology. 2009;386:168. doi: 10.1016/j.virol.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 128.Brasier A.R., Spratt H., Wu Z., Boldogh I., Zhang Y.H., Garofalo R.P., Casola A., Pashmi J., Haag A., Luxon B., Kurosky A. J. Virol. 2004;78:11461. doi: 10.1128/JVI.78.21.11461-11476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Radhakrishnan A., Yeo D., Brown G., Myaing M.Z., Iyer L.R., Fleck R., Tan B.H., Aitken J., Sanmun D., Tang K., Yarwood A., Brink J., Sugrue R.J. Mol. Cell. Proteomics. 2010;9:1829. doi: 10.1074/mcp.M110.001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Munday D.C., Emmott E., Surtees R., Lardeau C.H., Wu W., Duprex W.P., Dove B.K., Barr J.N., Hiscox J.A. Mol. Cell. Proteomics. 2010;9:2438. doi: 10.1074/mcp.M110.001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Munday D.C., Hiscox J.A., Barr J.N. Proteomics. 2010;10:4320. doi: 10.1002/pmic.201000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bowick G.C., Soman K.V., Wang H., Aronson J.F., Luxon B.A., Lomas L.O., Gorenstein D.G., Herzog N.K. J. Biomed. Biotechnol. 2010;2010:1–9. doi: 10.1155/2010/956823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Braunagel S.C., Russell W.K., Rosas-Acosta G., Russell D.H., Summers M.D. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9797. doi: 10.1073/pnas.1733972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vogels M.W., van Balkom B.W., Kaloyanova D.V., Batenburg J.J., Heck A.J., Helms J.B., Rottier P.J., de Haan C.A. Proteomics. 2011;11:64. doi: 10.1002/pmic.201000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee S.R., Nanduri B., Pharr G.T., Stokes J.V., Pinchuk L.M. Biochim. Biophys. Acta. 2009;1794:14. doi: 10.1016/j.bbapap.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 136.Pastorino B., Boucomont-Chapeaublanc E., Peyrefitte C.N., Belghazi M., Fusai T., Rogier C., Tolou H.J., Almeras L. Mol. Cell. Proteomics. 2009;8:1623. doi: 10.1074/mcp.M800565-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Li S., Qu H., Hao J., Sun J., Guo H., Guo C., Sun B., Tu C. Biochim. Biophys. Acta. 2010;1804:1882. doi: 10.1016/j.bbapap.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 138.Zhao C., Zhang W., Tian X., Fang C., Lu H., Yuan Z., Yang P., Wen Y. J. Med. Virol. 2010;82:14. doi: 10.1002/jmv.21654. [DOI] [PubMed] [Google Scholar]
- 139.Niu D., Feng H., Chen W.N. J. Proteomics. 2010;73:1283. doi: 10.1016/j.jprot.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 140.Feng H., Li X., Niu D., Chen W.N. J. Proteomics. 2010;73:1421. doi: 10.1016/j.jprot.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 141.Zheng Z.Z., Miao J., Zhao M., Tang M., Yeo A.E., Yu H., Zhang J., Xia N.S. J. Gen. Virol. 2010;91:1728. doi: 10.1099/vir.0.019323-0. [DOI] [PubMed] [Google Scholar]
- 142.Favoreel H.W., Enquist L.W., Feierbach B. Trends Microbiol. 2007;15:426. doi: 10.1016/j.tim.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 143.Johannsen E., Luftig M., Chase M.R., Weicksel S., Cahir-McFarland E., Illanes D., Sarracino D., Kieff E. Proc. Natl. Acad. Sci. U.S.A. 2004;101:16286. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Varnum S.M., Streblow D.N., Monroe M.E., Smith P., Auberry K.J., Pasa-Tolic L., Wang D., Camp D.G., Rodland K., 2nd, Wiley S., Britt W., Shenk T., Smith R.D., Nelson J.A. J. Virol. 2004;78:10960. doi: 10.1128/JVI.78.20.10960-10966.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Thanthrige-Don N., Parvizi P., Sarson A.J., Shack L.A., Burgess S.C., Sharif S. Dev. Comp. Immunol. 2010;34:699. doi: 10.1016/j.dci.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 146.Franco C.F., Mellado M.C., Alves P.M., Coelho A.V. Talanta. 2010;80:1561. doi: 10.1016/j.talanta.2009.06.081. [DOI] [PubMed] [Google Scholar]
- 147.Chen L.M., Tran B.N., Lin Q., Lim T.K., Wang F., Hew C.L. J. Gen. Virol. 2008;89:2869. doi: 10.1099/vir.0.2008/003681-0. [DOI] [PubMed] [Google Scholar]
- 148.Ince I.A., Boeren S.A., van Oers M.M., Vervoort J.J., Vlak J.M. Virology. 2010;405:253. doi: 10.1016/j.virol.2010.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Luo Y., Weng S., Wang Q., Shi X., Dong C., Lu Q., Yu X., He J. Virus Res. 2009;144:171. doi: 10.1016/j.virusres.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 150.Xie X., Yang F. Virology. 2005;336:93. doi: 10.1016/j.virol.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 151.Ovsyannikova I.G., Johnson K.L., Naylor S., Poland G.A. J. Immunol. Methods. 2005;297:153. doi: 10.1016/j.jim.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 152.Zachertowska A., Brewer D., Evans D.H. J. Virol. Methods. 2004;122:63. doi: 10.1016/j.jviromet.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 153.Puehler F., Schwarz H., Waidner B., Kalinowski J., Kaspers B., Bereswill S., Staeheli P. J. Biol. Chem. 2003;278:6905. doi: 10.1074/jbc.M207336200. [DOI] [PubMed] [Google Scholar]
- 154.Chen T.S., Yoder J.D., Hruby D.E. Anal. Biochem. 2003;315:277. doi: 10.1016/s0003-2697(03)00030-7. [DOI] [PubMed] [Google Scholar]
- 155.Chackerian B., Rudensey L.M., Overbaugh J. J. Virol. 1997;71:7719. doi: 10.1128/jvi.71.10.7719-7727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kattenhorn L.M., Mills R., Wagner M., Lomsadze A., Makeev V., Borodovsky M., Ploegh H.L., Kessler B.M. J. Virol. 2004;78:11187. doi: 10.1128/JVI.78.20.11187-11197.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Medvedkin V., Moore R., Falick A.M., Conner J., Omelchenko T., Young M., Anderson W.F., Golubkov V.S., Rozenberg-Adler Y. J. Virol. Methods. 2010;169:290. doi: 10.1016/j.jviromet.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 158.Li L., Sevinsky J.R., Rowland M.D., Bundy J.L., Stephenson J.L., Sherry B. J. Proteome Res. 2010;9:2460. doi: 10.1021/pr901151k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zandi F., Eslami N., Soheili M., Fayaz A., Gholami A., Vaziri B. Proteomics. 2009;9:2399. doi: 10.1002/pmic.200701007. [DOI] [PubMed] [Google Scholar]
- 160.Bourchookarn A., Havanapan P.O., Thongboonkerd V., Krittanai C. Biochim. Biophys. Acta. 2008;1784:504. doi: 10.1016/j.bbapap.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 161.Cooper B., Eckert D., Andon N.L., Yates J.R., Haynes P.A. J. Am. Soc. Mass Spectrom. 2003;14:736. doi: 10.1016/S1044-0305(03)00125-9. [DOI] [PubMed] [Google Scholar]
- 162.Blouin A.G., Greenwood D.R., Chavan R.R., Pearson M.N., Clover G.R., MacDiarmid R.M., Cohen D. J. Virol. Methods. 2010;163:49. doi: 10.1016/j.jviromet.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 163.Lee B.J., Kwon S.J., Kim S.K., Kim K.J., Park C.J., Kim Y.J., Park O.K., Paek K.H. Biochem. Biophys. Res. Commun. 2006;351:405. doi: 10.1016/j.bbrc.2006.10.071. [DOI] [PubMed] [Google Scholar]
- 164.Kwon S.J., Cho S.Y., Lee K.M., Yu J., Son M., Kim K.H. Virus Res. 2009;144:96. doi: 10.1016/j.virusres.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 165.Nagy P.D., Pogany J. Adv. Virus Res. 2010;76:123. doi: 10.1016/S0065-3527(10)76004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]