

Abstract
3C-like proteinase of severe acute respiratory syndrome (SARS) coronavirus has been demonstrated to be a key target for drug design against SARS. The interaction between SARS coronavirus 3C-like (3CL) proteinase and an octapeptide interface inhibitor was studied by affinity capillary electrophoresis (ACE). The binding constants were estimated by the change of migration time of the analytes in the buffer solution containing different concentrations of SARS 3CL proteinase. The results showed that SARS 3CL proteinase was able to complex with the octapeptide competitively, with binding constants of 2.44 × 104 M−1 at 20 °C and 2.11 × 104 M−1 at 37 °C. In addition, the thermodynamic parameters deduced reveal that hydrophobic interaction might play major roles, along with electrostatic force, in the binding process. The ACE method used here could be developed to be an effective and simple way of applying large-scale drug screening and evaluation.
Keywords: Binding constants, Capillary electrophoresis, Interface inhibitor, SARS 3C-like proteinase, Thermodynamic parameters
In late 2002, severe acute respiratory syndrome (SARS) 1 infected more than 1000 people, mostly in China, Hong Kong, Taiwan, and Canada, and caused deaths due to the infection by a novel human coronavirus [1], [2], [3]. Then the sequence of the complete genome of this novel human coronavirus, SARS-associated coronavirus (SARS–CoV), was determined. It contained 29,727 nucleotides in length and 11 open reading frames, with a genome organization similar to that of other known coronaviruses, including two human coronaviruses, HCoV–OC43 and HCoV–229E. The phylogenetic analysis and sequence comparisons indicated that SARS–CoV was not closely related to any of the three previously known groups of characterized coronaviruses [4], [5]. The SARS–CoV main proteinase (33.8 kDa), also called the 3C-like (3CL) proteinase, was essential for the life cycle of SARS–CoV, playing a pivotal role in mediating viral replication and transcription functions through extensive proteolytic processing of two replicase polyproteins: pp1a (486 kDa) and pp1ab (790 kDa) [6], [7].
Because of the control of the activities of the coronavirus replication complex and the availability of numerous inhibitors of other coronavirus proteinases and efficient expression, the 3CL proteinase was a preferred target for the task of discovering drugs against SARS. Hilgenfeld’s group determined the crystal structures of human coronavirus (strain 229E) main proteinase and a substrate analogue hexapeptidyl chloromethyl ketone (CMK) inhibitor complex of porcine coronavirus (transmissible gastroenteritis virus, TGEV) main proteinase. The results revealed a remarkable degree of conservation of the substrate-binding site, and a homology model for SARS–CoV main proteinase was constructed [6]. The crystal structures of the SARS–CoV main proteinase at different pH values and binding to a specific inhibitor with a covalent-bonded substrate analogue were reported by Rao’s group, providing insights to the substrate binding site and a structural basis for rational drug design [7]. The virtual screening tested by Xiong et al. [8] indicated that 73 available proteinase inhibitors in the MDDR database might dock into both the binding pockets of the TGEV main proteinase and the SARS–CoV 3CL proteinase. A series of cell-based assays with SARS virus and Vero E6 cells, including enzyme-linked immunosorbent assay (ELISA), Western blot analysis, immunofluorescence, and flow cytometry assays, were applied and reported by Wu et al. [9] to screen existing drugs, natural products, and synthetic compounds to identify effective anti-SARS agents, with the results showing that approximately 50 compounds were active at 10 μM and that 7 compounds were active at 3 μM. Based on the atomic coordinates of the SARS–CoV main proteinase, research was conducted by Chou’s group on KZ7088 (a derivative of AG7088) and the AVLQSGFR octapeptide to the enzyme, which were interacting with the active site of the SARS enzyme through six hydrogen bonds, with KZ7088 having a clear definition of the binding pocket [10]. Kuo et al. [11] prepared a peptide with fluorescence quenching pair (Dabcyl and Edans) at both ends of a peptide substrate and used this fluorogenic peptide substrate to characterize the SARS main proteinase and screen inhibitors.
Capillary electrophoresis (CE) may offer several advantages to the study of the interactions between small ligands and biomacromolecules, including small sample consumption (nanogram), short analysis time, ease of automation, high efficiency, and high resolving power [12], [13], [14]. One company, Cetek, has developed and optimized the CE Assay, an advanced CE assay for high information binding [15], [16]. The weak bindings (K b = 103–104 M−1) of macromolecules and small ligands can be detected easily by affinity capillary electrophoresis (ACE). Another advantage of the ACE method is that all of the interacting components can be studied in solution, thereby eliminating the possibility of denaturation of conformational alteration of the biomacromolecules such as protein and nucleic acid [12]. Various affinity interactions, such as drug–protein, protein–protein, protein–nucleic acid, protein–carbohydrate, peptide–antibiotic, enzyme–cofactors, lectin–sugar, antigen–antibody, and cyclodextrins (CDs)–enantiomer, have been investigated by ACE during recent years [17], [18].
We demonstrated previously that the dimer of SARS 3CL proteinase should be the biologically functional form and play a major role in catalysis [19]. Inhibitors targeting the dimeric interface should be a new strategy to control the activity of SARS 3CL proteinase. Because one monomer of SARS 3CL proteinase binds to the other, specifically at the N-terminal interface [6], [7], to compete with the dimeric interaction of the proteinase, an octapeptide interface inhibitor was designed according to the amino acid sequence of the N terminus of SARS 3CL proteinase. In the current study, a simple and effective CE method with very low sample consumption was developed and applied to study the interaction between SARS 3CL proteinase and the octapeptide interface inhibitor. The binding constants were measured by the changes in the migration time of the octapeptide inhibitor at different concentrations of SARS 3CL proteinase in the running buffer solutions. The octapeptide was found to bind to SARS 3CL proteinase specifically and competitively. Thermodynamic analysis indicates that hydrophobic interactions play a major role, along with electrostatic force, in the binding processes.
Materials and methods
Reagents and materials
SARS 3CL proteinase was expressed in Escherichia coli and purified according to published procedures [17]. The octapeptide (Ser-Gly-Phe-Arg-Lys-Met-Ala-Phe) was synthesized by solid-phase peptide synthesis using the standard 9-fluorenylmethoxycarbonyl/tert-butyl strategy and was purified by reverse-phased HPLC. Bovine serum albumin (BSA, MW = 67,000) and ovalbumin (OVA, MW = 43,000) were purchased from Fluka. All other reagents were of analytical grade without further purification. Deionized water was used throughout.
Apparatus
CE was performed with the Beckman P/ACE 5000 system (Fullerton, CA, USA). The uncoated fused silica capillary (Beckman, 57 cm × 50 μm i.d., 50 cm to the detector) was used. The capillary chamber temperature (± 0.1 °C) was controlled by forced liquid cooling. A run voltage of 15 kV in the normal polarity mode was applied. UV detection was performed at 214 nm. The GS-15R multipurpose refrigerated centrifuge (Beckman) was applied to clear any trapped air bubbles in the running buffer solutions containing SARS 3CL proteinase before use.
Solution and sample preparation
The concentrations of SARS 3CL proteinase stock solution were determined from UV absorbance at 280 nm (concentration/mg ml−1 = A 280 × 1.04). The stock solutions of octapeptide, BSA, and OVA were prepared by accurately weighing defined amounts of the compounds, dissolving them in a defined volume of deionized water, and then storing at 4 °C. The running buffer in the CE experiments was 25 mM sodium phosphate (pH 7.4). The investigated octapeptide and SARS 3CL proteinase solutions in 25 mM sodium phosphate buffer (pH 7.4) were prepared by diluting the defined volume of the stock solutions to the constant volume with buffer solution. The constant concentration of octapeptide in the sample was 1.50 × 10−4 M. Dimethyl sulfoxide (DMSO) served as an electroosmotic flow (EOF) marker and was added to the sample at a concentration of 0.01% (v/v), which proved to have no influence on SARS 3CL proteinase with the concentration less than 5% (v/v). The varying concentrations of SARS 3CL proteinase in the running buffer were prepared from 0 to 10 μM. All solutions and deionized water were stored at 4 °C, being filtered through 0.22-μm cellulose acetate membrane filters (Shanghai Xingya Resin, Shanghai, China) and sonicated for 10 min to remove air from solution prior to use. SARS 3CL proteinase solutions were centrifuged for 5 min with 12,000 rpm at 4 °C to exclude air from solutions before use.
Procedures for CE experiments
New capillary was conditioned for 60 min with 0.2 M NaOH, for 30 min with water, and for 10 min with running buffer. Between measurements, the capillary was flushed for 3 min with 0.2 M NaOH, for 2 min with water, and for 2 min with running buffer. The sample containing octapeptide and DMSO was introduced into the capillary by high-pressure injection (15 psi for 10 s). The electrophoresis was carried out using phosphate buffer solutions containing different concentrations of SARS 3CL proteinase (0–10 μM). All separations were in triplicate. Relative standard deviations (RSDs) of the migration times were calculated from a series of three experiments carried out with the same sample within 1 day.
Results and discussion
Determination of binding constants
The binding constants (K b) are the most fundamentally important parameters for biologically active molecules [12], [13], [14]. The principle of this method for the determination of binding constants is to exploit the changes in the migration time of the complexed sample in the background electrolyte containing the complexation agent. After adding SARS 3CL proteinase to background solution (BGS), the octapeptide inhibitor can interact with the proteinase. The equilibrium represented by the following equations assumed that a 1:1 complex is formed:
| (1) |
| (2) |
where OP, 3CLP, and OP-3CLP are the octapeptide, SARS 3CL proteinase, and complex, respectively. K b is the binding constant for Eq. (1). As pointed out by Yang et al. [20], the capacity factor k, which is used as an indicator to describe the relationship between retention time and mass distribution equilibrium in chromatography, can be used here to describe the migration behaviors of the active compounds in CE. Moreover, the addition of SARS 3CL proteinase in the BGS is applied as a pseudo-stationary phase. Based on our previous work [21], [22], the binding constant can be measured according to the following equation:
| (3) |
where ϕ is phase ratio, [OP]s is the concentration of the octapeptide in stationary phase, and [OP]m0 is the initial concentration of the octapeptide without the addition of SARS 3CL proteinase in the mobile phase.
As defined in chromatography, the capacity factor k is equal to (t − t 0)/t 0, where t and t 0 are the migration times of the sample and neutral marker, respectively. With the linear relationship of k versus the concentration of SARS 3CL proteinase ([3CLP]), the binding constant is calculated from the slope (a) and intercept (b):
| (4) |
The weak interactions between several anti-HIV compounds with HIV transactivation-responsive (TAR) RNA and BSA were successfully studied simultaneously and by quickly using this method with the binding constants of 103 to 104 M−1 [21], [22].
To evaluate the pharmacological activity of inhibitor binding to SARS 3CL proteinase in a physiological system or simulated physiological system, the phosphate buffer system of 25 mM at pH 7.4 was used for the binding study. The migration times of the octapeptide (t) and DMSO (t 0) were measured in seven running buffer solutions containing different concentrations of SARS 3CL proteinase (0–10 μM). The experimental data and RSD values are summarized in Table 1 . The reproducibility of the migration times was very well based on the values of RSD less than 0.5%. A set of electropherograms of migration time shift of the octapeptide inhibitor versus SARS 3CL proteinase concentration change is shown in Fig. 1 . A plot of k versus the concentration of SARS 3CL proteinase at two temperatures based on the Table 1 data is shown in Fig. 2 . The binding constants of the inhibitor with the proteinase were determined four times at two temperatures and calculated according to Eq. (4), with the average values being 2.44 × 104 M−1 (RSD = 6.0%, n = 4) at 20 °C and 2.11 × 104 M−1 (RSD = 15%, n = 4) at 37 °C. The RSD value at 37 °C was larger than that at 20 °C due to the instability of the higher capillary temperature.
Table 1.
Experimental data of the migration times and RSDs (n = 3)
| [3CLP] (× 10−6 M) | 20 °C |
37 °C |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| t (min) | RSD (%) | t0 (min) | RSD (%) | k | t (min) | RSD (%) | t0 (min) | RSD (%) | k | |
| 0 | 6.573 | 0.48 | 8.050 | 0.47 | −0.183 | 4.704 | 0.20 | 5.898 | 0.36 | −0.202 |
| 0.953 | 6.715 | 0.10 | 8.328 | 0.10 | −0.194 | 4.791 | 0.22 | 6.089 | 0.12 | −0.213 |
| 1.91 | 6.821 | 0.11 | 8.511 | 0.14 | −0.199 | 4.838 | 0.18 | 6.180 | 0.11 | −0.217 |
| 3.81 | 6.966 | 0.13 | 8.787 | 0.22 | −0.207 | 4.969 | 0.10 | 6.411 | 0.26 | −0.225 |
| 5.72 | 7.137 | 0.12 | 9.108 | 0.18 | −0.216 | 5.083 | 0.11 | 6.603 | 0.34 | −0.230 |
| 7.62 | 7.289 | 0.17 | 9.376 | 0.23 | −0.223 | 5.186 | 0.10 | 6.796 | 0.14 | −0.237 |
| 9.53 | 7.407 | 0.28 | 9.600 | 0.36 | −0.228 | 5.305 | 0.08 | 7.008 | 0.15 | −0.243 |
Note. [3CLP], concentration of SARS 3CL proteinase; t, migration time of the octapeptide; t0, migration time of DMSO; k, capacity factor.
Fig. 1.
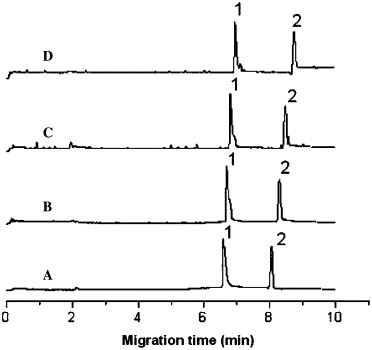
Electropherogram of the migration time shift of the octapeptide versus SARS 3CL proteinase concentration change at 20 °C. Conditions: running buffer, 25 mM phosphate solution (pH 7.4); applied voltage, +15 kV; pressure injection, 10 s at 15 psi; wavelength, 214 nm. Peaks 1 and 2 represent octapeptide and DMSO, respectively, and A, B, C, and D represent SARS 3CL proteinase concentrations of 0, 0.953, 1.91, and 3.81 μM, respectively. The constant concentrations of octapeptide and DMSO in the sample were 1.50 × 10−4 M and 0.01% (v/v), respectively.
Fig. 2.
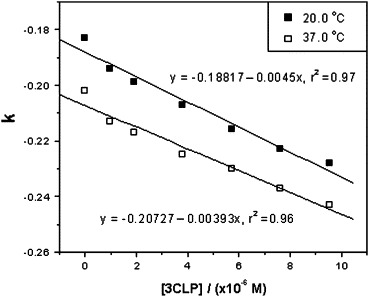
Plot of k versus the concentration of SARS 3CL proteinase at two temperatures.
We demonstrated previously that the dimer of SARS 3CL proteinase should be the biologically functional form and plays a major role in catalysis. In addition, one monomer of SARS 3CL proteinase binds to the other one specifically at the N-terminal interface [19], [23]. To compete with the dimeric interaction of the proteinase, the octapeptide inhibitor was designed according to the amino acid sequence of the N terminus of SARS 3CL proteinase. Because the concentrations of SARS 3CL proteinase were less than 0.2 mg ml−1 in the current experimental conditions, the main form of the proteinase was thought to be monomer. The binding constant of the octapeptide with SARS 3CL proteinase was measured to be 2.44 × 104 M−1 at 20 °C, and the dissociation constant of the dimer of the proteinase was estimated to be 100 μM [19]; thus, the octapeptide inhibitor can bind to the monomer of the proteinase competitively and can prevent the dimerization effectively. Moreover, the interactions between the octapeptide and the two common proteins, BSA and OVA, were studied as a negative control at the same conditions to avoid the influence of nonspecific absorption. Compared with Fig. 2, the plots of BSA were similar (Fig. 3 A), with the binding constants being calculated as 8.01 × 103 M−1 at 20 °C and 8.20 × 103 M−1 at 37 °C, whereas the plots of OVA were random (Fig. 3B). The results showed that the octapeptide could interact with BSA weakly but could not bind to OVA and that there should be a specific binding between the octapeptide inhibitor and SARS 3CL proteinase.
Fig. 3.
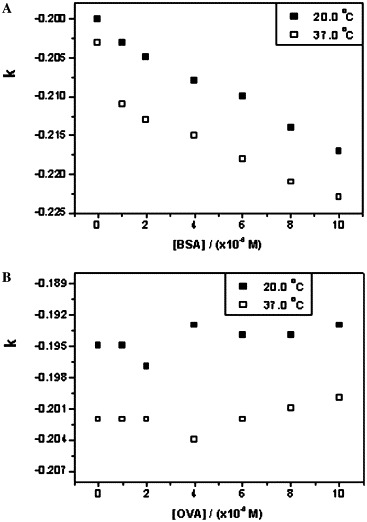
Plots of k versus the concentrations of BSA and OVA. (A) Interaction between octapeptide and BSA at 20 and 37 °C. (B) Interaction between octapeptide and OVA at 20 and 37 °C.
Thermodynamic studies of the interactions
Small molecules bind to macromolecules with four types of interactions: H-bond, van der Waals, electrostatic, and hydrophobic interactions. The thermodynamic parameters, enthalpy change (ΔH) and entropy change (ΔS) of the binding process, are important for confirming binding mode [24], [25]. If ΔH does not vary significantly over the temperature range studied, the enthalpic contribution to the Gibbs free energy (ΔG) can be determined according to the following equation:
| (5) |
where K 1 and K 2 are the binding constants at temperatures T 1 and T2, respectively, and R is the gas constant (8.314 J mol−1 K−1). The free energy change is estimated from the following relationship:
| (6) |
where K is the binding constant at the corresponding temperature. The entropy change can be determined from the following equation:
| (7) |
Based on the values of binding constants, the thermodynamic parameters ΔH, ΔG, and ΔS were measured according to Eqs. (5), (6), (7), respectively. These values are summarized in Table 2 .
Table 2.
Thermodynamic parameters of interactions between SARS 3CL proteinase and the octapeptide
| T (K) | Kb (× 104 M−1) | ΔH (kJ mol−1) | ΔG (kJ mol−1) | ΔS (J mol−1 K−1) |
|---|---|---|---|---|
| 293 | 2.44 | −6.71 | −24.6 | 61.1 |
| 310 | 2.11 | −6.71 | −25.7 | 61.3 |
Note. T, absolute temperature; Kb, binding constant; ΔH, enthalpy change; ΔG, Gibbs free energy; ΔS, entropy change.
As shown in Table 2, ΔH values were negative under the experimental conditions, demonstrating that the binding reaction was an exothermic process. The sign and magnitude of the thermodynamic parameters associated with various kinds of interaction were characterized [24], [25]. From the point of view of water structure, positive entropy was frequently taken as the evidence of hydrophobic interaction, and it was also shown that positive entropy and slightly negative enthalpy might be a manifestation of electrostatic interactions between ionic species in aqueous solution [25]. Based on the experimental data, we conclude that the hydrophobic interaction may play a major role, whereas electrostatic forces also contribute to the binding process of SARS 3CL proteinase and the octapeptide inhibitor, consistent with our previous work.
Conclusion
SARS therapy requires the development of antiviral compounds that effectively prevent or treat this disease. A simple and reliable CE method with very low sample consumption was applied to study the interaction between SARS 3CL proteinase and an octapeptide interface inhibitor. The binding constant was determined by the changes in the migration time of the octapeptide inhibitor at different concentrations of SARS 3CL proteinase in the running buffers. The octapeptide was shown to bind to SARS 3CL proteinase competitively, with binding constant K b values of 2.44 × 104 M−1 at 20 °C and 2.11 × 104 M−1 at 37 °C. Analysis of the thermodynamic parameters, enthalpy change (ΔH) and entropy change (ΔS), indicated that hydrophobic interaction might play a major role, along with electrostatic force, in the binding processes. The ACE method should be widely applicable for large-scale ligand screening against protein targets.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (20375003, 20475006, 10345004, 90103029, 20173001, and 20228306), the 863 High-Technology Project, the National Basic Research Project (2003CB715900), and the Ministry of Education of China.
Footnotes
Abbreviations used: SARS, severe acute respiratory syndrome; SARS–CoV, SARS-associated coronavirus; 3CL, 3C-like; CMK, chloromethyl ketone; TGEV, transmissible gastroenteritis virus; ELISA, enzyme-linked immunosorbent assay; CE, capillary electrophoresis, ACE, affinity capillary electrophoresis; CD, cyclodextrin; BSA, bovine serum albumin; OVA, ovalbumin; DMSO, dimethyl sulfoxide; EOF, electroosmotic flow; RSD, relative standard deviation; BGS, background solution; TAR, transactivation-responsive.
References
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.Drosten C., Gunther S., Preiser W., vander Werf S., Brodt H.R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Peiris J.S.M., Lai S.T., Poon L.L.M., Guan Y., Yam L.Y.C., Lim W., Nicholls J., Yee W.K.S., Yan W.W., Cheung M.T., Cheng V.C.C., Chan K.H., Tsang D.N.C., Yung R.W.H., Ng T.K., Yuen K.Y. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rota P.A., Oberste M.S., Monroe S.S., Nix W.A., Campagnoli R., Icenogle J.P., Penaranda S., Bankamp B., Maher K., Chen M.H., Tong S., Tamin A., Lowe L., Frace M., DeRisi J.L., Chen Q., Wang D., Erdman D.D., Peret T.C., Burns C., Ksiazek T.G., Rollin P.E., Sanchez A., Liffick S., Holloway B., Limor J., McCaustland K., Olsen-Rasmussen M., Fouchier R., Gunther S., Osterhaus A.D., Drosten C., Pallansch M.A., Anderson L.J., Bellini W.J. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 5.Marra M.A., Jones S.J., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 6.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 7.Yang H.T., Yang M.J., Ding Y., Liu Y.W., Lou Z.Y., Zhou Z., Sun L., Mo L.J., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z.H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong B., Gui C.S., Xu X.Y., Luo C., Chen J., Luo H.B., Chen L.L., Li G.W., Sun T., Yu C.Y., Yue L.D., Duan W.H., Shen J.K., Qin L., Shi T.L., Li Y.X., Chen K.X., Luo X.M., Shen X., Shen J.H., Jiang H.L. A 3D model of SARS–CoV 3CL proteinase and its inhibitor design by virtual screening. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 9.Wu C.Y., Jan J.T., Ma S.H., Kuo C.J., Juan H.F., Cheng Y.S.E., Hsu H.H., Huang H.C., Wu D., Brik A., Liang F.S., Liu R.S., Fang J.M., Chen S.T., Liang P.H., Wong C.H. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc. Natl. Acad. Sci. USA. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou K.C., Wei D.Q., Zhong W.Z. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. Biochem. Biophys. Res. Commun. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuo C.J., Chi Y.H., Hsu J.T.A., Liang P.H. Characterization of SARS main protease and inhibitor assay using a fluorogenic substrate. Biochem. Biophys. Res. Commun. 2004;318:862–867. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oravcová J., Böhs B., Lindner W. Drug–protein binding studies: new trends in analytical and experimental methodology. J. Chromatogr. B. 1996;677:1–28. doi: 10.1016/0378-4347(95)00425-4. [DOI] [PubMed] [Google Scholar]
- 13.Østergaard J., Schou C., Larsen C., Heegaard N.H.H. Evaluation of capillary electrophoresis–frontal analysis for the study of low molecular weight drug–human serum albumin interactions. Electrophoresis. 2002;23:2842–2853. doi: 10.1002/1522-2683(200209)23:17<2842::AID-ELPS2842>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka Y., Terabe S. Estimation of binding constants by capillary electrophoresis. J. Chromatogr. B. 2002;768:81–92. doi: 10.1016/s0378-4347(01)00488-1. [DOI] [PubMed] [Google Scholar]
- 15.Little J.N., Hughes D.E., Karger B.L. A powerful screening technology utilizing capillary electrophoresis. Am. Biotechnol. Lab. 1999;8:36. [Google Scholar]
- 16.D.E. Hughes, J.L. Waters, Y.M. Dunayevskiy, Capillary electrophoretic method to detect target-binding ligands and to determine their relative affinities, U.S. patent 6,524,866 (2003)
- 17.Heegaard N.H.H., Robey F.A. Use of capillary zone electrophoresis to evaluate the binding of anionic carbohydrates to synthetic peptides derived from human serum amyloid P component. Anal. Chem. 1992;64:2479–2482. doi: 10.1021/ac00045a004. [DOI] [PubMed] [Google Scholar]
- 18.Fanali S. Controlling enantioselectivity in chiral capillary electrophoresis with inclusion–complexation. J. Chromatogr. A. 1997;792:227–267. doi: 10.1016/s0021-9673(97)00809-1. [DOI] [PubMed] [Google Scholar]
- 19.Fan K.Q., Wei P., Feng Q., Chen S.D., Huang C.K., Ma L., Lai B., Pei J.F., Liu Y., Chen J.G., Lai L.H. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y., Zhang X.X., Korenaga T. Study on the dynamic complexation between protein and PAHs by capillary electrophoresis. Anal. Sci. 2001;17:i1345–i1348. [Google Scholar]
- 21.Ding L., Zhang X.X., Chang W.B., Lin W., Yang M. Study on the interactions between anti-HIV-1 active compounds with trans-activation response RNA by affinity capillary electrophoresis. J. Chromatogr. B. 2005;814:99–104. doi: 10.1016/j.jchromb.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Ding L., Zhang X.X., Chang W.B., Lin W., Yang M. Determination of the binding constants of four anti-HIV-1 active compounds with bovine serum albumin by capillary zone electrophoresis. Chin. J. Chromatogr. 2004;22:624–626. [PubMed] [Google Scholar]
- 23.Liu S.Y., Pei J.F., Chen H., Zhu X.L., Liu Z.M., Ma W.Z., He F.L., Lai L.H. Modeling of the SARS coronavirus main proteinase and conformational flexibility of the active site. Beijing Daxue Xuebao. 2003;35(Suppl.):62–65. [PubMed] [Google Scholar]
- 24.Tian J.N., Liu J.Q., Tian X., Hu Z.D., Chen X.G. Study of the interaction of kaempferol with bovine serum albumin. J. Mol. Struct. 2004;691:197–202. [Google Scholar]
- 25.Ross P.D., Subramanian S. Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry. 1981;20:3096–3102. doi: 10.1021/bi00514a017. [DOI] [PubMed] [Google Scholar]