

Abstract
The ubiquitin proteasome system (UPS) is essential in regulating myriad aspects of protein functions. It is therefore a fundamentally important regulatory mechanism that impacts most if not all aspects of cellular processes. Indeed, malfunction of UPS components is implicated in human diseases such as neurodegenerative and immunological disorders and many cancers. The success of proteasome inhibitors in cancer therapy suggests that modulating enzymes in the ubiquitination cascade would be clinically important for therapeutic benefits. In this review, we summarize advances in developing inhibitors of a variety of UPS components. In particular, we highlight recent work done on the protein engineering of ubiquitin as modulators of the UPS, a novel approach that may shed light on innovative drug discovery in the future.
Keywords: Ubiquitination, Deubiquitinating enzyme (DUB), Small molecule, Ubiquitin variant (Ubv), Protein engineering, Phage display
1. Introduction
Ubiquitination is a dynamic and reversible post‐translational process initially discovered in proteasome mediated protein degradation [1]. Since then, it has become clear that ubiquitination plays a critical role in many key cellular processes, by means of a variety of mechanisms, from changing protein stability and modulating protein activity to promoting or disrupting protein–protein interactions [2, 3]. The central player, ubiquitin (Ub), is a small (8.5 kDa) but evolutionarily conserved protein. Several proteins related to Ub in sequence and three‐dimensional structure have also been identified as Ub‐like proteins (UBLs), such as NEDD8 (neural precursor cell expressed, developmentally downregulated 8), SUMO (small ubiquitin modifier), and ISG15, which are important for a diverse set of signaling pathways including nuclear transport, autophagy, and antiviral pathways [4]. In a highly coordinated multi‐step enzymatic cascade, ubiquitination covalently attaches one or more Ubs to internal lysine (Lys) residues or the N termini (rarely) of target proteins through the sequential actions of ubiquitin‐activating enzymes (E1), ubiquitin‐conjugating enzymes (E2), and ubiquitin ligases (E3) [5, 6, 7, 8] (Fig. 1 A). First, E1 employs ATP to adenylate Ub to form a high‐energy thioester bond between the Ub C‐terminal carboxyl group and the thiol group of the E1 active site cysteine residue (E1∼Ub). The activated ubiquitin is then transferred to the cysteine residue of E2 enzymes through a similar thioester linkage (E2∼Ub). Finally, the E3 ligases recruit charged E2 enzymes and facilitate specific transfer of Ub to protein substrates [9, 10]. Importantly, a family of deubiquitinating enzymes (DUBs) catalyzes the cleavage of Ub from polypeptides to make it a reversible process and also contribute to Ub homeostasis [11].
Figure 1.
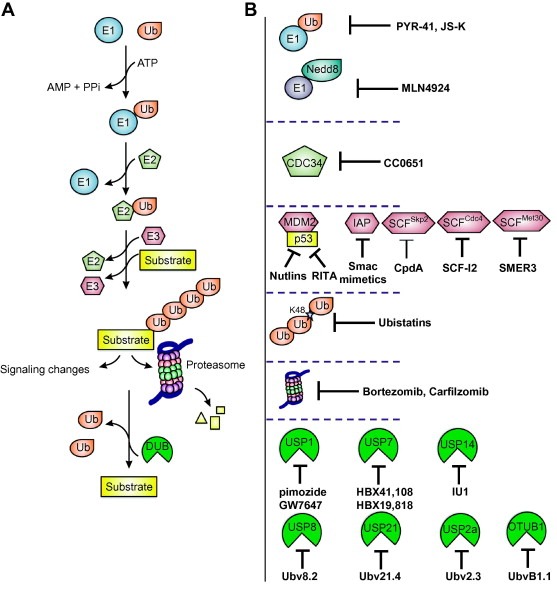
The UPS and its inhibitors. (A) Ubiquitination involves a three‐step enzymatic cascade. First, E1 employs ATP to adenylate Ub to form a high‐energy thioester bond between the Ub C‐terminal carboxyl group and the thiol group of the active site cysteine residue. The activated Ub is then transferred to the cysteine residue of an E2 enzyme through a thioester linkage. Finally, an E3 ligase recruits a charged E2 and helps transfer Ub to specific protein substrates to form mono‐ (not shown) or poly‐Ub chains, which will lead to different cellular responses, including signaling events or protein degradation through the proteasome. Deubiquitinating enzymes (DUBs) catalyze the cleavage of Ub from polypeptides to reverse the ubiquitination process and to maintain Ub homeostasis. (B) Inhibitors that block various steps in the ubiquitination pathway.
E3 ligases can be further divided into the HECT (homologous to E6‐associated protein carboxy terminus) family, the RING (really interesting new gene) family, and the U‐box family [7]. The major difference among them is that the HECT E3 ligases form an intermediate thioester bond with Ub before linkage to substrate. In contrast, the RING and U‐box E3 ligases facilitate Ub transfer from E2 enzymes by an allosteric mechanism [10, 12]. E3 ligases can modify proteins by attachment of a single Ub to one (monoubiquitination) or more Lys residues (multi‐monoubiquitination) in a substrate [13, 14], or by sequential attachment of multiple Ub moieties (assembled through isopeptide bond formation between the C‐terminal Gly carboxylate and internal Lys side chains in Ub) to a Lys residue in the substrate to form Ub chains (polyubiquitination) [10]. Ub has seven Lys residues (K6, K11, K27, K29, K33, K48, and K63), all of which can potentially participate in chain formation, with K48 and K63 being the most common and intensively studied residues involved in polyubiquitination [6, 15]. Ub can also form linear chains by linking to the amine of the N‐terminal methionine (M1) [16].
Given the essential role of the ubiquitin proteasome system (UPS) in regulating protein function, it was not surprising that dysfunction of UPS components is implicated in the occurrence of many human pathological disorders, including numerous cancers, cardiovascular diseases, viral diseases, and neurodegenerative disorders [5]. For example, HUWE1 (encoding a HECT E3 ligase) is highly expressed in lung, breast, and colorectal carcinomas [17, 18], mutational inactivation of E3 ligase BRCA1 causes breast cancer predisposition [19], and overexpression of MDM2 was observed in several types of cancer, as MDM2 promotes the degradation of p53 [20, 21]. Therefore, developing inhibitors to target specific components of the UPS is predicted to have significant therapeutic potential. This has been demonstrated by the clinical success of the proteasome inhibitor Bortezomib (PS341/Velcade, Millennium Pharmaceuticals), the first drug targeting the UPS approved by the US Food and Drug Administration (FDA) in 2003 for treatment of relapsed or refractory multiple myeloma [22, 23]. Bortezomib is a dipeptidyl boronic acid derivative (Fig. 2 A) that is a reversible inhibitor of the chymotrypsin‐like activity of the 20S proteasome core particle [24]. It is also in clinical trials for follicular non‐Hodgkin's lymphoma (Phase III), diffuse large B cell lymphoma (Phase II), and many other cancers [9]. Although one would assume that inhibition of the proteasome blocks a common step of the UPS and is therefore non‐specific, interestingly, Bortezomib showed selective cytotoxicity to cancer cells compared with normal cells, probably because cancer cells have much more proteasome workload as they may generate higher concentrations of aberrant proteins [25, 26]. An irreversible proteasome inhibitor structurally and mechanistically distinct from Bortezomib, Carfilzomib (PR‐171/Kyprolis, Onyx Pharmaceuticals, Fig. 2A), was approved by FDA in 2012 for patients previously treated with Bortezomib and immunomodulatory compounds [27]. Carfilzomib can induce cell cycle arrest and apoptosis in a variety of human cancer cell lines and importantly, it is more potent and more selective than Bortezomib and therefore displays activity against Bortezomib‐resistant cancer cells and its cytotoxicity against RPMI 8226 multiple myeloma cells is higher that that of Bortezomib [27, 28, 29, 30]. Efforts are ongoing to develop second‐generation proteasome inhibitors, and hopefully these may yield drugs displaying better potency and fewer side effects [5, 24, 27, 31]. Detailed review about proteasome inhibitors for their synthesis, pharmacology, and clinical treatment of multiple myeloma and other types of cancer have been published elsewhere and will not be discussed further [27, 29, 32].
Figure 2.
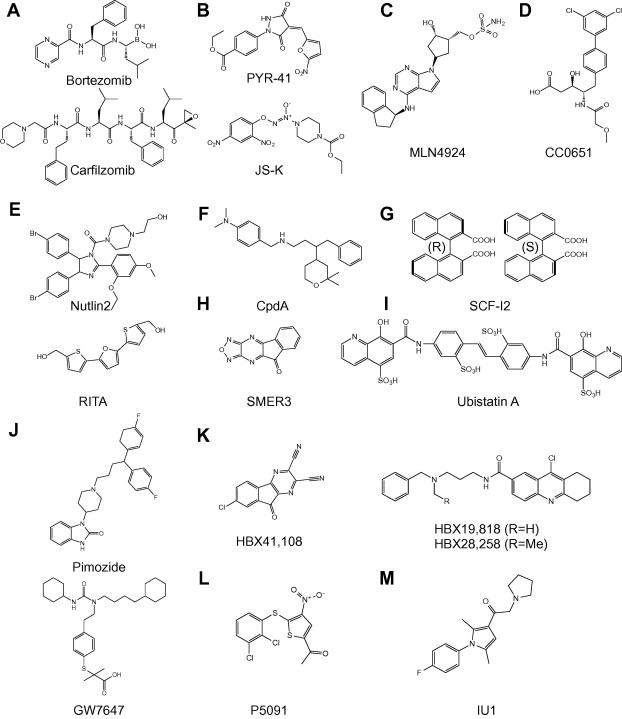
Chemical structures of small molecule inhibitors. (A) Proteasome inhibitors Bortezomib (top) and Carfilzomib (bottom) [27]. (B) E1Ub inhibitors PYR‐41 (top) and JS‐K (bottom) [34, 35]. (C) E1NEDD8 inhibitor MLN4924 [41, 42]. (D) CDC34 inhibitor CC0651 [57]. (E) MDM2–p53 inhibitors Nutlin2 (top) and RITA (bottom) [66, 67]. (F) SCFskp2 inhibitor CpdA [80]. (G) SCFCdc4 inhibitor SCF‐I2 (R‐ and S‐enantiomers) [83]. (H) SCFMet30 inhibitor SMER3 [85]. (I) K48 poly‐Ub chain inhibitor Ubistatin A [100]. (J) USP1/UAF1 inhibitors pimozide (top) and GW7647 (bottom) [113]. (K) USP7 inhibitors HBX41,108 (left) and HBX19,818/28,258 (right) [119, 121]. (L) USP7 inhibitor P5091 [120]. (M) USP14 inhibitor IU1 [122].
With rapidly growing understanding of the fundamental importance of the UPS in cell signaling and disease, it was realized that the system provides a rich source of attractive molecular targets for pharmacological intervention [10]. Many groups and companies are gearing up to search for small molecule inhibitors that selectively block certain steps of the UPS. The success of Bortezomib and Carfilzomib in anti‐cancer therapy suggests that the development of such compounds would be clinically important to target individual pathways and substrate proteins for therapeutic benefits. Such drugs should increase the effectiveness of the treatment and should also have fewer non‐specific side effects compared with general proteasome inhibitors. In this review, we illustrate recent progress in finding specific inhibitors for UPS components (Fig. 1B) that have been implicated in human diseases and therefore represent potential therapeutic targets. We then highlight the invention of Ub variant technology as a rapid means for generating tools to modulate UPS enzymes.
2. Small‐molecule inhibitors targeting steps in the ubiquitination cascade
2.1. Ub activation
E1 Ub activating enzymes (two members in the human genome) catalyze the first step in the ubiquitination pathway in an ATP‐dependent manner [8]. E1 first binds ATP and adenylates the C‐terminal glycine of Ub, and then forms a covalent thioester bond between its catalytic cysteine and Ub. Subsequently, E1 binds a second ATP and Ub to form a ternary complex that is competent to transfer thioester‐bound Ub to a variety of E2 Ub conjugating enzymes [5]. Biochemically, there are three major points where Ub activation can be targeted for inhibition. First, binding of ATP to the E1 enzyme may be blocked, using an ATP‐competitive small‐molecule inhibitor analogous to those used to inhibit kinases [33]. Second, formation of the covalent E1∼Ub complex may be prevented by targeting the active‐site thiol. Third, the interaction between the E1 and E2 enzymes may be blocked [5, 25]. However, inhibition of Ub activation is predicted to be non‐specific in principle, as it is required for ubiquitination of many substrates in cells. Indeed, temperature‐sensitive mutations in E1 arrest the cell cycle in late S phase and G2, indicating that multiple pathways required for cell proliferation in normal cells are affected by mutating E1. Below, we summarize advances in developing inhibitors for Ub and NEDD8 E1 enzymes (Fig. 1B).
2.1.1. E1Ub inhibitor
PYR‐41 (Fig. 2B), an irreversible pyrazone derivative inhibitor was identified in a screen for inhibitors of MDM2‐dependent p53 ubiquitination, and it was found that the nitrogen dioxide group on the furan ring of PYR‐41 likely blocked the catalytic cysteine of E1Ub [34]. Importantly, the compound did not demonstrate inhibitory activity against other thiol‐dependent enzymes, including several E2 enzymes [34]. However, PYR‐41 can partially inhibit HECT E3 ligase activity in vitro, raising specificity concerns. The effects in cells treated with PYR‐41 included inhibition of cytokine‐induced nuclear factor κB (NF‐κB) activation, stabilization of p53 protein levels, induction of p53‐dependent transcription, and preferential killing of transformed cells with wild‐type (wt) p53 [34]. Interestingly, PYR‐41 treatment causes increase of cellular sumoylation but neddylation levels stay the same [34]. The potential therapeutic value of targeting E1Ub is further underscored by the unexpected finding that the nitric oxide (NO)‐producing prodrug JS‐K (Fig. 2B) inhibited E1∼Ub thioester formation through an interaction between NO and the E1Ub active‐site thiol [35]. Consistent with other E1Ub inhibitors, the downstream effects of JS‐K treatment included decreased levels of total ubiquitinated proteins and increased p53 expression.
2.1.2. NEDD8 activating enzyme inhibitor
Interestingly, the UBL protein NEDD8 was found to be required for activation of a large subfamily of more than 100 E3 ligases, the cullin‐RING ligases (CRLs) [36, 37, 38]. The NEDD8 protein shares ∼60% sequence identity with Ub, and it also can be conjugated to proteins through a process initiated by a specific E1, NAE (NEDD8 activating enzyme). Therefore, inhibiting NAE would be more specific than inhibiting E1Ub, and many CRL substrates are important regulators for cancer cell growth and survival, including the cell cycle inhibitor p27 [39]. Inhibition of the NEDD8 pathway was recently demonstrated using a small‐molecule inhibitor, MLN4924 [40, 41]. MLN4924 (Fig. 2C) is an adenosine sulfamate analog that binds to the nucleotide‐binding pocket of the NEDD8 thioester of NAE and forms a covalent adduct that effectively depletes all neddlyation activity through substrate‐assisted inhibition, thereby driving all CRLs into inactive non‐neddylated forms [42]. As shown in mouse cancer models, MLN4924 appears to exert its effect by stabilizing CDT1, a DNA replication licensing factor and a substrate of the SCFskp2 and CRL4Cdt2 ligases, leading to DNA synthesis misregulation and apoptosis and senescence of proliferating cancer cells [43]. Preclinical data show high efficacy of MLN4924 in diffuse large B cell lymphoma and acute myeloid leukemia (AML) [44, 45], and the compound has entered phase I/II clinical trials for the treatment of multiple myeloma and non‐Hodgkins lymphoma. Notably, analogues of MLN4924 form similar adducts with other UBL proteins and inhibit their cognate E1 enzymes, including Ub and SUMO‐activating enzyme, indicating that “substrate‐assisted” inhibition may prove useful for targeting other UBL pathways [42].
2.2. Ub conjugation
The second step of the ubiquitination cascade is controlled by E2 Ub conjugating enzymes (∼30 members in the human genome). Other UBL proteins also have their specific E2 enzymes, such as UBE2I for SUMO and UBE2M/F for NEDD8 (reviewed in [46]). E2 enzymes have been linked to cancer and other diseases, including UBE2Q2 in head and neck carcinoma [47], UBE2T in lung cancer [48] and UBE2C in chromosomal instability and tumor formation [49]. An E2 enzyme accepts the activated Ub from E1 through a thioester bond with its catalytic cysteine and then transfers Ub to substrate proteins with assistance from E3 ligases [46, 50]. E2 enzymes catalyze both Ub chain initiation and elongation and, in some cases, also govern the type and extent of Ub linkage [46, 50, 51]. However, some E2 enzymes only function in a specific step, as demonstrated by UBE2W and UBE2E for chain initiation of BRCA1 substrates and UBE2N‐UBE2V1 and UBE2K for chain elongation [46, 52], and some can mediate both processes, such as yeast Cdc34 [53]. The catalytic properties of E2 enzymes, and the binding of E1, E3 and Ub, are mediated by a highly conserved 150–200 amino acid Ub‐conjugating catalytic domain (UBC) [46, 50, 51]. E2 enzymes can be further classified based on the presence of N‐ or C‐terminal extensions to the catalytic core, which have important functional implications [54]. Each E2 enzyme can interact with multiple E3 ligases, as for example, UBE2R1 recognizes several SCF complexes [55]. This is also further shown in a network interaction study of E2‐RING E3 pairs [56]. Therefore, targeting Ub conjugation for inhibition at E2‐E3 interaction interfaces may provide better specificity than targeting the interfaces of E1–E2 and E2–Ub.
2.2.1. CDC34 inhibitor
CDC34, or UBE2R1, is the dedicated E2 enzyme for CRL E3 ligases in promoting K48‐linked polyubiquitination and degradation of cell cycle regulatory proteins including the cell cycle inhibitor p27, which functions to prevent G1/S progression [39]. In a high‐throughput screen for inhibition of p27 ubiquitination, the Sicheri group found a small molecule named CC0651 [57]. CC0651 (Fig. 2D) turns out to be a specific inhibitor for human CDC34 (Fig. 1B) but not for its homolog UBE2R2 or its yeast counterpart Cdc34. The inhibitor acts allosterically by binding to a pocket that is 19 Å away from the active site and induces a number of translocations of secondary structural elements in the enzyme (Fig. 3 A). CC0651 and its closely related derivatives caused accumulation of p27 and cyclin E in cells and inhibited proliferation of human cancer cells. These findings suggest that it may be feasible to develop highly selective inhibitors for other E2 enzymes in a similar manner.
Figure 3.
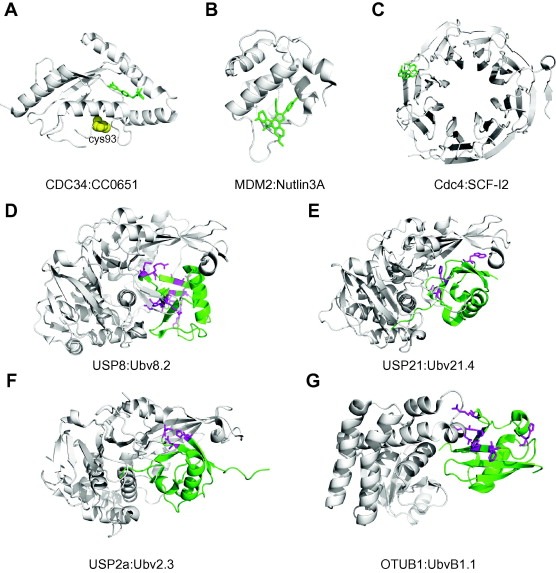
Structures of inhibitors in complex with UPS components. (A) E2 CDC34/UBE2R1 (white) in complex with CC0651 (green). The catalytic cysteine is depicted as spheres (yellow) (PDB entry 3RZ3). (B) E3 MDM2 (white) in complex with Nutlin3A (green) (PDB entry 4HG7). (C) Yeast F‐box protein Cdc4 WD40 domain (white) in complex with SCF‐I2 (green) (PDB entry 3MKS). (D) USP8 (white) in complex with Ubv8.2 (green) (PDB entry 3N3K). (E) USP21 (white) in complex with Ubv21.4 (green) (PDB entry 3MTN). (F) USP2a (white) in complex with Ubv2.3 (green) (PDB entry 3V6E). (G) OTUB1 (white) in complex with UbvB1.1 (green) (PDB entry 4I6L). Differences in Ubvs relative to wt Ub are shown as magenta side chains.
2.3. Ub ligation
The final step of ubiquitination is controlled by E3 Ub ligases (more than 600 members in the human genome), which determine the selectivity of ubiquitination by interacting with both E2∼Ub and the substrates to which Ub is transferred. Inhibitors of E3–E2 or E3–substrate interactions may thus enable specific targeting of a limited number of proteins, which may translate into a better therapeutic ratio and fewer side effects. To this end, structural studies of E3 ligase substrate‐binding surfaces or sites mediating ubiquitination activity and assembly of E3 complexes not only reveal molecular mechanisms, but also facilitate the discovery of small‐molecule inhibitors. As discussed above, E3 ligases function by one of two general mechanisms: they either form covalent Ub adducts on their active sites (HECT) or serve as non‐covalent adaptors that facilitate the transfer of Ub from E2∼Ub to substrate (RING and U‐box). The three classes of E3 ligases have different protein interaction domains (HECT, RING, and U‐box) that bind to E2 enzymes, and other domains recruit substrates [58, 59, 60]. The HECT domain is a ∼350‐residue polypeptide, which adopts a bilobal structure [61]; the N‐terminal lobe contains the E2‐binding site and the C‐terminal lobe harbors the catalytic cysteine. The lobes are separated by a small linker stretch, forming a cleft with the active site [46]. However, these surfaces are quite shallow and are not well adapted for targeting by small molecules. On the other hand, the RING finger domain is formed by conserved patterns of cysteine and histidine residues that hold two Zn2+ ions in a conformation referred to as a “cross‐brace” motif that bind E2 enzymes [62]. Finally, the RING‐like U‐box family adopts a similar structure but without employing Zn2+ coordination. One might think that inhibition of a canonical catalytic site (HECT) would be easier than targeting protein–protein interaction domains (RING or U‐box), but all reported small molecule inhibitors of E3 ligases target RING‐type enzymes (Fig. 1B).
2.3.1. MDM2–p53 inhibitor
Stabilization of the tumor suppressor p53 is critical for cells to respond to stress and limit cancer development. MDM2 is a RING E3 ligase that has a crucial role in regulating the abundance of p53. Many tumors that retain wild type p53 show evidence of MDM2 overexpression [63, 64]. Thus, targeting MDM2 in these cancer cells is an attractive strategy for cancer therapy [65]. Nutlins (imidazoline derivatives developed by Roche) are the first group of small molecules that can interfere with the ability of MDM2 to mediate p53 ubiquitination [66]. Nutlin‐2 (Fig. 2E) disrupts the interaction between MDM2 and p53 by binding directly to the p53‐binding site of MDM2 and thereby stabilize p53. Indeed, Nutlins induce p53‐dependent cell‐cycle arrest, apoptosis, and cellular senescence in cancer cell lines. Importantly, Nutlins have a significant anti‐tumor effect as shown by growth inhibition of human tumor xenografts in mice, with no obvious toxicity to healthy tissues [66]. Nutlin 3A/R7112 (Fig. 3B) has advanced to clinical trials for solid tumors and leukemia given its favorable preclinical characteristics in terms of pharmacological properties and toxicity [9]. RITA (Fig. 2E) is another small molecule that was shown to regulate the interaction between MDM2 and p53 in a chemical screen designed to find compounds that specifically arrested growth of a p53‐positive cancer cell line [67]. Instead of binding MDM2, RITA probably binds the N‐terminus of p53, thereby inducing a conformational change that stabilizes the N‐terminal α‐helix and prevents the recognition of p53 by MDM2 [67]. Interestingly, RITA does not affect the transcriptional activity of p53, but rather, prevents p53 from interacting with other regulatory proteins, such as PARC (p53‐Associated Parkin‐like cytoplasmic protein) and p300, which can promote p53 polyubiquitination by MDM2 [68]. As with Nutlins, RITA induces apoptosis in human tumor cells with little effect on normal cells, and it slows down the growth of tumour xenografts in mice [67]. It will be interesting to test whether RITA and Nutlins can act synergistically to induce apoptosis in cancer cells [25]. Inhibitors of MDM2 E3 ligase activity were also reported to be able to promote p53 stabilization and activation [69, 70]. Further studies are required to determine whether these molecules will be useful in the treatment of human cancer.
2.3.2. IAP inhibitors
Another class of RING E3 ligases actively targeted in the field is the IAPs (inhibitor‐of‐apoptosis proteins), and several antagonists of IAPs (Fig. 1B) have already entered clinical trials [9]. In a variety of cell lines, IAPs effectively suppress the enzymatic activity of caspases and therefore inhibit apoptosis mediated by both intrinsic and extrinsic pathways, including death receptor activation, ionizing radiation and viral infection [71]. The E3 ligase activity of IAPs is a key regulatory component in the apoptotic program since it mediates both autoubiquitination and degradation, and also, that of their substrates [71, 72]. Smac is a protein that interacts with the surface groove of the BIR3 domain of IAPs and prevents IAP inhibition of both initiator and effector caspases [73]. A small molecule mimic of Smac was found to bind to cellular IAPs and potently induce caspase activation and apoptosis in human cancer cells [74]. Further studies of Smac mimetic small molecules revealed that they induce the autoubiquitination and degradation of the IAPs, which then leads to the death of cancer cells by stimulating the tumor necrosis factor α (TNF‐α) pathway [75].
2.3.3. SCF inhibitors
Small molecule inhibitors of several SCF family members of CRL E3 ligases have also been identified. The SCF complex is composed of a modular E3 core containing scaffold CUL1 and RING domain containing protein RBX1, and an ubiquitination substrate specificity module composed of SKP1 and a member of the F‐box family of proteins, many of which are overexpressed in many cancers and correlate with poor prognosis [55, 76, 77]. In the case of SCF complexes, inhibition of either the F‐box protein–substrate interface or the recruitment of the F‐box protein to the SCF core are attractive strategies to selectively inhibit ubiquitination events [78]. SCFskp2 ubiquitinates p27 and targets it for proteasomal destruction, and decreased levels of p27 are a poor prognosis factor in many malignancies [79]. Researchers have identified a small molecule CpdA (Fig. 2F) that prevents the incorporation of Skp2 into the SCFskp2 complex, and thus induces G1/S cell cycle arrest by stabilizing p27 and other substrates. Importantly, CpdA sensitized multiple myeloma cells to Bortezomib and was active against both myeloid and lymphoblastoid leukemia cells derived from patients [80]. In addition, a recent study screened for inhibitors that selectively target the p27‐binding interface formed by Skp2‐Cks1. This approach yielded four compounds that stabilize the expression of p27 in different cancer cell lines and induce cell cycle arrest in G1 [81]. SCFβTrCP1 is a CRL E3 ligase that triggers the degradation of IkBα, the inhibitory component of the transcription factor NF‐kB, and inhibitors of SCFβTrCP1 may therefore have potential to prevent the polyubiquitination and degradation of IkBα [82].
Researchers have also identified a small‐molecule inhibitor of Cdc4, the yeast ortholog of the mammalian F‐box protein Fbw7 (F box and WD repeat domain‐containing 7). Structural studies revealed that the inhibitor named SCF‐I2 (Fig. 2G) inserts between the β‐strands of the WD40 domain of Cdc4, which are remote from the substrate‐binding site (Fig. 3C). Binding of SCF‐I2 induces a series of conformational changes that distort the substrate‐binding pocket and impair binding and ubiquitination of its substrates, phosphorylated Cdk1 inhibitor Sic1 and Far1 [83]. Thus, SCF‐I2 is one of the first allosteric inhibitors of an E3 ligase identified thus far and its discovery raises the possibility of a similar strategy for allosteric modulation of the WD40‐repeat class of proteins [84]. A small molecule that inhibits the SCFMet30, but not the closely related SCFCdc4, was recently identified in a chemical genetics screen in yeast for enhancers of rapamycin [85]. The compound SMER3 (Fig. 2H) bound to the F‐box protein Met30 and diminished its binding to Skp1 in vivo and thereby disrupted the assembly of a functional SCF complex and stabilized substrates such as Met4 [85]. Finally, the drug thalidomide, a sedative that causes birth effects, was identified as an inhibitor for cereblon (CRBN), a component of the CRL4DDB1 complex important for limb outgrowth and the expression of a fibroblast growth factor (FGF8) during embryonic development [86]. Collectively, these proof‐of‐principle studies demonstrate the feasibility of obtaining selective pharmacological inhibitors of SCF ligases, which will offer highly specific therapeutic approaches for a wide variety of malignancies.
2.4. Ub chains
In general, Ub chains are homogenous as the same residue is modified during elongation, although mixed linkage chains have been observed [6, 87]. Mass spectrometry studies revealed that all seven Lys residues and the N‐terminal Met residue can participate in the formation of Ub linkages [88, 89]. Interestingly, the different linkages result in different chain topologies as shown by the structures of di‐Ub molecules [6]. Ub chains adopt either compact conformations, where adjacent moieties interact with each other (e.g., K6, K11 and K48 linkages), or open conformations, where no interfaces are present except for the linkage site (e.g., M1 and K63 linkages) [6, 90, 91, 92]. The differences in the chain topologies likely determine the fate of ubiquitinated proteins [10]. For instance, proteins tagged with K48‐linked poly‐Ub chains are generally labeled for 26S proteasome‐mediated recognition and protein degradation. In contrast, K63‐linked poly‐Ub chains have been implicated in a variety of non‐proteolytic functions, including DNA repair, protein trafficking, and ribosomal protein synthesis [3]. The roles of other types of Ub chains have not been studied in as great detail as K48‐ and K63‐linked chains, but their specific roles are starting to emerge [2]. For example, K11‐linked chains are highly abundant in mitotic cells when the anaphase‐promoting complex cyclosome (APC/C), a key cell cycle regulator, degrades its substrates [93, 94]. K11‐linked chains also play an important role in endoplasmic reticulum‐associated degradation and NF‐κB essential modulator (NEMO)‐dependent activation of NF‐κB [95, 96]. Both K27 and K33 linkages can be assembled by U‐box E3 ligases during the stress response [58], and K29‐linked chains play a role in Ub fusion degradation [97], while K6‐linked chains are not likely to have a proteolytic role [98]. Linear chains are assembled by the linear ubiquitin chain assembly complex and play a crucial role in NF‐κB signaling [99].
2.4.1. Ubistatins
Unexpectedly, a recent large‐scale chemical genetics screen identified small molecules known as ubistatins (Fig. 2I for ubistatin A) that specifically bind to K48‐linked ubiquitin chains [100]. In a reconstituted in vitro system, ubistatins can stabilize both cyclin B and Sic1, substrates of APC/C and SCFCdc4, respectively. NMR and in vitro binding assays showed that ubistatins bind interfaces between K48‐linked Ub molecules and thereby change the conformation of the Ub chains and impair binding and recognition by poly‐Ub receptors of the proteasome [100]. These molecules have strong negative charges and therefore are not cell permeable, but they provide evidence for a new therapeutic approach for blocking protein degradation by modification of poly‐Ub chains [25].
2.5. Deubiquitination
In recent years, reversible ubiquitination has attracted increasing attention as it is a crucial mediator within intracellular signaling such as p53 and NF‐κB pathways [101]. This reversibility is accomplished through deubiquitinating enzymes (DUBs), which can remove Ub moieties from substrates to prevent protein degradation or signaling. DUBs also contribute to Ub homeostasis and can edit the form of modification by trimming Ub chains [101, 102]. Therefore, it is not surprising that misregulation of DUBs is implicated in a growing number of diseases, including neurological disorders, viral infections and cancer. In humans, there are approximately 100 DUBs belonging to five different structural families: approximately 60 ubiquitin specific proteases (USPs), four ubiquitin C‐terminal hydrolases (UCHs), four Josephin domain (Machado–Joseph disease protein domain) proteases, 16 ovarian tumor (OTU) proteases and eight JAB1/MPN+/Mov34 domain (JAMM) metallo‐enzyme proteases [103, 104]. It is well accepted that the DUB family of enzymes are “druggable”, because they demonstrate a high degree of substrate specificity and contain well defined catalytic pockets that makes them amenable to screening with libraries of small molecules [103]. Indeed, Inhibitors of the UCH family of DUBs have been reported with modest selectivity and affinity [105, 106, 107]. More encouragingly, there has been good progress in developing small molecule inhibitors targeting the SARS CoV (severe acute respiratory syndrome coronavirus) DUB PLpro (papain‐like protease) [107, 108], confirming the potential of the DUB family as one of the most druggable families in the UPS. Novartis has patented compounds that inhibit USP2 and UCH‐L3, which are implicated in the MDM2–p53 pathway or neurodegenerative disorders, respectively [9]. Here we summarize recent advances in the development of inhibitors for USPs (Fig. 1B), the most intensively studied sub‐family of DUBs.
2.5.1. USP1 inhibitor
USP1 was recently found to deubiquitinate inhibitor of DNA binding (ID) 1‐3 proteins and, consequently, to promote stem cell characteristics in osteosarcoma [109]. USP1 mRNA and protein levels were also increased in a subset of human osteosarcoma biopsy samples. Finally, USP1 knockdown in osteosarcoma cells causes cell‐cycle arrest and osteogenic differentiation. Interestingly, USP1 also deubiquitinates FANCD2, a central player in the Fanconi Anemia (FA) pathway, and is required for FANCD2 foci formation, release from chromatin, and essential function in DNA repair [110, 111]. USP1 is therefore a promising target for pharmacological intervention, as inhibiting its protease activity in malignant osteosarcoma should lead to reduced proliferative capacity, the potential to reverse its transformed phenotype, and defective DNA repair to sensitize cells to chemotherapeutic agents. It should be noted that USP1 forms a stable complex with UAF1, which stimulates the catalytic activity of USP1 [112]. Recently cell active small‐molecule reversible inhibitors against USP1/UAF1 were identified (pimozide and GW7647, Fig. 2J) and they displayed selectivity against a number of DUBs, deSUMOylase, and cysteine proteases [113]. Further studies of the USP1/UAF1 inhibitors suggested that pimozide and GW7647 inhibit USP1/UAF1 by a non‐competitive mechanism and bind at a site other than the active site of USP1/UAF1 [113]. Importantly, the USP1 inhibitors act synergistically with the chemotherapy drug cisplatin to inhibit the proliferation of cisplatin‐resistant non‐small cell lung cancer (NSCLC) cells [113]. However, further biochemical and genetic studies in osteosarcoma cells treated with USP1 inhibitors will be critical to assess their therapeutic potential [114].
2.5.2. USP7 inhibitor
USP7 is a key regulator of p53, as well as many other substrates that are involved in various cellular pathways [115]. As discussed above, the E3 ligase MDM2 binds and ubiquitinates p53 to regulate its cellular protein level and activity [116]. USP7 is a critical component of this pathway as it deubiquitinates and stabilizes both p53 and MDM2. As shown by the Gu group, partial reduction of USP7 levels in several human cell lines promotes decreased levels of both MDM2 and p53, yet total abolition of USP7 stabilizes p53 levels by decreasing MDM2 [117, 118]. This observation suggests that the absence of USP7 promotes MDM2 downregulation, which in turn eliminates the activity of MDM2 as the Ub ligase for p53. Hybrigenics has developed a cyano‐indenopyrazine derivative small molecule compound (HBX41, 108, Fig. 2K) that can inhibit USP7 activity in the submicromolar range but is not quite selective against a panel of cysteine proteases [119]. Further kinetics data indicate that HBX 41,108 is an uncompetitive reversible inhibitor and it allosterically modulates the catalytic reaction of USP7 [119]. Similar to RNAi‐mediated USP7 silencing in cancer cells, HBX41,108 treatments stabilized p53, activated transcription of a p53 target gene without inducing genotoxic stress, and inhibited cancer cell growth [119]. Very recently, other USP7 inhibitors have been developed and they will likely help characterize the potential for USP7 inhibitors in preclinical settings [120, 121]. For instance, Hybrigenics developed second‐generation irreversible USP7 inhibitors (Fig. 2K), which are capable of regulating USP7 substrates in cancer cells and recapitulate the USP7 knockdown phenotype [121]. Indeed, incubation of exponentially growing HCT116 cells with HBX 19,818 led to an increase in the levels of p53 and its target protein cyclin‐dependent kinase inhibitor p21. Most importantly, HBX19,818 binds selectively to the active site of USP7 and is much more specific than HBX41,108 [121]. Another selective USP7 inhibitor, P5091 (Fig. 2L), was able to induce apoptosis in multiple myeloma cells resistant to conventional and Bortezomib therapies [120].
2.5.3. USP14 inhibitor
As we discussed above, the proteasome is the major regulator of Ub‐mediated protein degradation. USP14 is one of three human DUBs, along with UCH37 and RPN11, which associate with the 19S regulatory particle of the multi‐subunit 26S proteasome [103]. The N‐terminal UBL domain of USP14 is required for its association with the proteasome and this association can stimulate its catalytic activity [122, 123]. USP14 is shown to inhibit the degradation of Ub–protein conjugates both in vitro and in cells by trimming Ub chains on substrates [122]. Recently, a high‐throughput screen identified a selective reversible small‐molecule inhibitor of the deubiquitinating activity of human USP14. The compound IU1 (Fig. 2M) binds specifically to the activated form of USP14 (proteasome bound) and enhances the degradation of several proteasome substrates in cells, including some implicated in neurodegenerative disease (i.e., Tau and ataxin‐3), suggesting a potential strategy to reduce the levels of misfolded and aggregated proteins in cells under proteotoxic stress [5, 122].
3. Ubiquitin variants as modulators of UPS components
3.1. Rationale and strategy
We devised an inhibitor design strategy based on the fact that most enzymes and proteins that interact with Ub recognize a common surface with low affinity but high specificity. For example, although catalytic domains of USPs often share low sequence homology, crystal structures have revealed a common Ub‐binding site [124, 125, 126]. In different USPs, Ub is bound in the same orientation and the isopeptide linkage is aligned in the active site. Interestingly, the interaction surface on each side is very large but binding is generally weak [127]. However, despite the common fold, the Ub‐binding sites of USP family members differ in sequence, and consequently exhibit significant topological variation. Importantly, this paradigm of weak interactions through large, diverse surfaces also extends to other Ub‐binding proteins in the UPS, including E2 enzymes [128], E3 ligases [129, 130] and Ub‐binding domains (UBDs) [131].
Based on these observations, we reasoned that it should be possible to use Ub as a scaffold to engineer high affinity binders to virtually any Ub‐interacting protein. This would be accomplished by introducing mutations in the Ub surface that would enhance binding towards a particular protein. Since Ub‐binding sites are large and variable, this strategy should produce Ub variants (Ubvs) that bind tightly and specifically. Importantly, these Ubvs should act as potent modulators of UPS proteins by blocking the binding of low affinity Ub substrates at natural binding sites.
We displayed Ub on bacteriophage and identified ∼30 residues that make contact with USP surfaces and constitute the USP‐binding site [132] (Fig. 4 A). We constructed a library by simultaneously targeting the entire USP‐binding site for combinatorial mutagenesis, using three mutagenic oligonucleotides (each covering a contiguous region of primary sequence). By using a “soft” randomization strategy, the library was biased in favor of the wt sequence but allowed for significant diversity across the entire USP‐binding surface. We reasoned that this would enable the selection of variants with mutations that improve affinity for a particular USP without drastically altering the binding site. The library was used to perform phage display selections (Fig. 4B) and yielded not only USP inhibitors, but also inhibitors and even activators of other DUBs, Ub ligases and UBDs [132].
Figure 4.
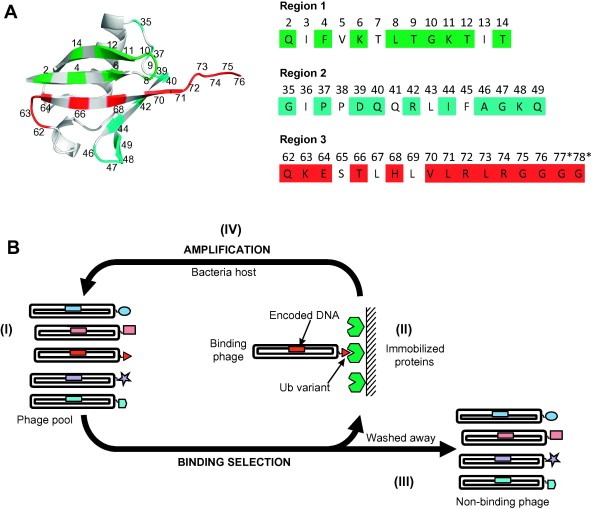
Phage display of Ub variant libraries. (A) Ub variant library design. The Ub structure (PDB entry 1UBQ) is shown (grey) and positions diversified in the library are colored as follows: region 1 (green), region 2 (cyan), and region 3 (red). The sequences of the regions targeted in the library design are shown to the right and are colored as in the structure. (B) Phage display cycle for the selection of Ub variants binding to an immobilized protein. (I) Within the phage pool, each phage particle displays a unique Ubv and encapsulates the encoding DNA. (II) Protein‐binding phage are captured with immobilized proteins. (III) Non‐binding phages are washed away. (IV) Bound phages are amplified by infection of an Escherichia coli host. The amplified pool can be cycled through additional rounds of selection to further enrich for Protein‐binding Ubvs, and individual clones can be subjected to DNA sequencing to decode sequences of displayed Ubvs.
3.2. Ubvs as modulators of DUBs and ligases
3.2.1. DUB inhibitors
USP8 has been implicated in ubiquitin remodeling, clathrin‐mediated internalization, endosomal sorting and regulation of receptor tyrosine kinases, including the epidermal growth factor receptor (EGFR) [133, 134, 135]. We identified a Ubv (Ubv.8.2) that bound specifically to the Ub‐binding site of USP8 (Fig. 3D) and inhibited catalytic activity with potency in the single‐digit nanomolar range [132]. Interestingly, Ubv8.2 binds to USP8 in a non‐canonical fashion and inhibits its function by stabilizing an auto‐inhibited state in which the active site is held in a closed conformation [132]. In cells, Ubv.8.2 interacts with USP8 and causes ubiquitination, lyososomal localization and down‐regulation of activated endogenous EGFR, which is the expected outcome of USP8 inhibition [132, 133, 136].
We also identified Ubvs that targeted the Ub‐binding sites of USP21 (Fig. 3E) and USP2a (Fig. 3F) and others have identified inhibitors of USP7 [137]. Ubiquitinated receptor‐interacting protein 1 (RIP1) is a positive regulator of NF‐κB activation induced by TNF‐α, and in turn, USP21 has been shown to down‐regulate TNF‐α‐induced NF‐κB activation by deubiquitinating RIP1 [138]. We identified a Ubv (Ubv.21.4) that inhibited USP21 with single‐digit nanomolar potency [132]. Ubv.21.4 blocked the deubiquitination of RIP1 by USP21 and restored NF‐κB activation, showing that it acts as an inhibitor of USP21 in cells. Finally, in both USP21 and USP2a complex structures, the Ubv binds in a manner similar to that of wild type Ub and the mutations lead to improved hydrophobic contacts and a rewiring of hydrogen bond networks [132].
Extending beyond USPs, the Ubv library also yielded an inhibitor of OTUB1, a member of the OTU DUB family that is required for the clearance of Ub conjugates after DNA damage [139]. Ubv.B1.1 efficiently inhibited OTUB1 catalyzed cleavage of K48‐linked di‐Ub [132]. OTUB1 also binds to the activated E2‐conjugating enzyme Ub∼UbcH5b, and the binding is enhanced by the presence of Ub [140]. Although Ubv.B1.1 binds to OTUB1 much more tightly than wt Ub, it showed a compromised ability to promote complex formation, suggesting an alteration in the binding mode. Indeed, the structure of OTUB1 in complex with Ubv.B1.1 revealed that the inhibitor binds to the distal Ub binding site of OTUB1 and enables a rationalization of its altered binding and allosteric properties (Fig. 3G).
3.2.2. NEDD4 activator
Although HECT E3 ligases play critical roles in cancer and possess intrinsic catalytic activity, which makes them potential targets for drug development [10, 61, 141], no small molecule inhibitors have yet been identified for any family member. Using the Ubv library we successfully generated specific binders for NEDD4 and ITCH. Surprisingly, a NEDD4 binder (Ubv.N4.02) acted as an activator and increased auto‐ubiquitination in vitro [132]. This activator activity was also observed in cells, where Ubv.N4.02 increased the ubiquitination of the yin‐yang protein 1 (YY1), a known substrate of NEDD4 [142]. These unexpected results underscore the need for ligands that act directly on discrete Ub‐binding sites to confirm or refine effects observed with RNAi approaches that work by reducing levels of the entire protein. Moreover, the results demonstrate that the engineered Ub variants can also be utilized as activators of proteins in the UPS and, together with the already established inhibition of DUBs, represent a general strategy that could be used to develop enzyme modulators to study the UPS on a system‐wide scale.
4. Conclusions and perspectives
Over the past decade, Ub has taken center stage in cell signaling. It is now clear that this humble protein plays a leading role in the control of numerous signaling pathways. Consequently, the enzymes that drive ubiquitination and deubiquitination have attracted considerable interest as potential targets for the treatment of cancer and other diseases. Just as research into protein phosphorylation has driven targeted cancer therapies in the last decade, hopes are high that basic discoveries in Ub biology will yield a new class of therapies in the upcoming decade. Unfortunately, these hopes have thus far been stymied by a paucity of specific inhibitors of the ubiquitination and deubiquitination pathways. Nonetheless, recent developments in both small‐molecule chemistry and protein engineering provide hope that inhibitor design for proteins in the UPS may be entering an accelerated phase.
Following the development of clinically successful proteasome inhibitors [22, 23], significant progress has been made to identify small molecules targeting specific UPS components (Fig. 1B) for the potential treatment of diseases such as cancer and neurodegenerative diseases [5, 143]. Most of these compounds have not yet entered clinical trials, where they will face the ultimate test of therapeutic efficacy in humans. Nevertheless, these proofs‐of‐principle show that it is feasible to selectively inhibit many steps of the ubiquitination pathway. It is noteworthy that, unlike ATP‐competitive inhibitors for kinases [33], no general small‐molecule approaches are available for the inhibition of E2, E3, or DUBs. Therefore, custom designed high‐throughput screening strategies remain crucial for small‐molecule inhibitor discovery.
In parallel with small molecule development, Ubvs derived by protein engineering have been shown to be ideal tools for conventional cell biology experiments to elucidate the molecular mechanisms and biological functions of Ub ligases and DUBs. The recombinant nature of Ubvs enables facile manipulations at the genetic level to adapt the proteins for different assays and applications. Ubvs directly target functional sites and act as potent and specific inhibitors or activators of catalytic function. Consequently, they can be used to assess directly the effects of active site inhibition for validation of UPS enzymes as drug targets for therapy of cancer and other diseases. Moreover, insights from structural studies may be applicable to the design of mechanism‐based therapeutic small molecule inhibitors.
Acknowledgments
We apologize to all groups whose important contributions to ubiquitin biology were not mentioned in this review due to space limitations. W.Z. is supported by a post‐doctoral fellowship from the . Ubiquitin variant research projects in the Sidhu laboratory are funded by the CIHR operating Grant MOP‐111149.
Zhang Wei and Sidhu Sachdev S.(2014), Development of inhibitors in the ubiquitination cascade, FEBS Letters, 588, doi: 10.1016/j.febslet.2013.11.003
References
- 1. Hershko A., Heller H., Elias S., Ciechanover A., Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem., 258, (1983), 8206– 8214. [PubMed] [Google Scholar]
- 2. Husnjak K., Dikic I., Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem., 81, (2012), 291– 322. [DOI] [PubMed] [Google Scholar]
- 3. Al-Hakim A., Escribano-Diaz C., Landry M.C., O'Donnell L., Panier S., Szilard R.K., Durocher D., The ubiquitous role of ubiquitin in the DNA damage response. DNA Repair, 9, (2010), 1229– 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der Veen A.G., Ploegh H.L., Ubiquitin-like proteins. Annu. Rev. Biochem., 81, (2012), 323– 357. [DOI] [PubMed] [Google Scholar]
- 5. Bedford L., Lowe J., Dick L.R., Mayer R.J., Brownell J.E., Ubiquitin-like protein conjugation and the ubiquitin–proteasome system as drug targets. Nat. Rev. Drug Disc., 10, (2011), 29– 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Komander D., Rape M., The ubiquitin code. Annu. Rev. Biochem., 81, (2012), 203– 229. [DOI] [PubMed] [Google Scholar]
- 7. Clague M.J., Liu H., Urbe S., Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell, 23, (2012), 457– 467. [DOI] [PubMed] [Google Scholar]
- 8. Hershko A., Ciechanover A., The ubiquitin system. Annu. Rev. Biochem., 67, (1998), 425– 479. [DOI] [PubMed] [Google Scholar]
- 9. Cohen P., Tcherpakov M., Will the ubiquitin system furnish as many drug targets as protein kinases?. Cell, 143, (2010), 686– 693. [DOI] [PubMed] [Google Scholar]
- 10. Bernassola F., Karin M., Ciechanover A., Melino G., The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell, 14, (2008), 10– 21. [DOI] [PubMed] [Google Scholar]
- 11. Nijman S.M., Luna-Vargas M.P., Velds A., Brummelkamp T.R., Dirac A.M., Sixma T.K., Bernards R., A genomic and functional inventory of deubiquitinating enzymes. Cell, 123, (2005), 773– 786. [DOI] [PubMed] [Google Scholar]
- 12. Ozkan E., Yu H., Deisenhofer J., Mechanistic insight into the allosteric activation of a ubiquitin-conjugating enzyme by RING-type ubiquitin ligases. Proc. Natl. Acad. Sci. USA, 102, (2005), 18890– 18895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hicke L., Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol., 2, (2001), 195– 201. [DOI] [PubMed] [Google Scholar]
- 14. Haglund K., Di Fiore P.P., Dikic I., Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem. Sci., 28, (2003), 598– 603. [DOI] [PubMed] [Google Scholar]
- 15. Haglund K., Dikic I., Ubiquitylation and cell signaling. EMBO J., 24, (2005), 3353– 3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kirisako T., Kamei K., Murata S., Kato M., Fukumoto H., Kanie M., Sano S., Tokunaga F., Tanaka K., Iwai K., A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J., 25, (2006), 4877– 4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adhikary S., Marinoni F., Hock A., Hulleman E., Popov N., Beier R., Bernard S., Quarto M., Capra M., Goettig S., Kogel U., Scheffner M., Helin K., Eilers M., The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell, 123, (2005), 409– 421. [DOI] [PubMed] [Google Scholar]
- 18. Chen D., Brooks C.L., Gu W., ARF-BP1 as a potential therapeutic target. Br. J. Cancer, 94, (2006), 1555– 1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruffner H., Joazeiro C.A., Hemmati D., Hunter T., Verma I.M., Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc. Natl. Acad. Sci. USA, 98, (2001), 5134– 5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li M., Brooks C.L., Wu-Baer F., Chen D., Baer R., Gu W., Mono-versus polyubiquitination: differential control of p53 fate by Mdm2. Science, 302, (2003), 1972– 1975. [DOI] [PubMed] [Google Scholar]
- 21. Kussie P.H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A.J., Pavletich N.P., Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science, 274, (1996), 948– 953. [DOI] [PubMed] [Google Scholar]
- 22. Richardson P.G., Barlogie B., Berenson J., Singhal S., Jagannath S., Irwin D., Rajkumar S.V., Srkalovic G., Alsina M., Alexanian R., Siegel D., Orlowski R.Z., Kuter D., Limentani S.A., Lee S., Hideshima T., Esseltine D.L., Kauffman M., Adams J., Schenkein D.P., Anderson K.C., A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med., 348, (2003), 2609– 2617. [DOI] [PubMed] [Google Scholar]
- 23. Richardson P.G., Sonneveld P., Schuster M.W., Irwin D., Stadtmauer E.A., Facon T., Harousseau J.L., Ben-Yehuda D., Lonial S., Goldschmidt H., Reece D., San-Miguel J.F., Blade J., Boccadoro M., Cavenagh J., Dalton W.S., Boral A.L., Esseltine D.L., Porter J.B., Schenkein D., Anderson K.C., Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med., 352, (2005), 2487– 2498. [DOI] [PubMed] [Google Scholar]
- 24. Petroski M.D., The ubiquitin system, disease, and drug discovery. BMC Biochem., 9, Suppl 1 (2008), S7– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nalepa G., Rolfe M., Harper J.W., Drug discovery in the ubiquitin–proteasome system. Nat. Rev. Drug Discovery, 5, (2006), 596– 613. [DOI] [PubMed] [Google Scholar]
- 26. Bianchi G., Oliva L., Cascio P., Pengo N., Fontana F., Cerruti F., Orsi A., Pasqualetto E., Mezghrani A., Calbi V., Palladini G., Giuliani N., Anderson K.C., Sitia R., Cenci S., The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood, 113, (2009), 3040– 3049. [DOI] [PubMed] [Google Scholar]
- 27. Rentsch A., Landsberg D., Brodmann T., Bulow L., Girbig A.K., Kalesse M., Synthesis and pharmacology of proteasome inhibitors. Angew. Chem., Int. Ed. Engl., 52, (2013), 5450– 5488. [DOI] [PubMed] [Google Scholar]
- 28. Demo S.D., Kirk C.J., Aujay M.A., Buchholz T.J., Dajee M., Ho M.N., Jiang J., Laidig G.J., Lewis E.R., Parlati F., Shenk K.D., Smyth M.S., Sun C.M., Vallone M.K., Woo T.M., Molineaux C.J., Bennett M.K., Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res., 67, (2007), 6383– 6391. [DOI] [PubMed] [Google Scholar]
- 29. Moreau P., Richardson P.G., Cavo M., Orlowski R.Z., San Miguel J.F., Palumbo A., Harousseau J.L., Proteasome inhibitors in multiple myeloma: 10 years later. Blood, 120, (2012), 947– 959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuhn D.J., Chen Q., Voorhees P.M., Strader J.S., Shenk K.D., Sun C.M., Demo S.D., Bennett M.K., van Leeuwen F.W., Chanan-Khan A.A., Orlowski R.Z., Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin–proteasome pathway, against preclinical models of multiple myeloma. Blood, 110, (2007), 3281– 3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kupperman E., Lee E.C., Cao Y., Bannerman B., Fitzgerald M., Berger A., Yu J., Yang Y., Hales P., Bruzzese F., Liu J., Blank J., Garcia K., Tsu C., Dick L., Fleming P., Yu L., Manfredi M., Rolfe M., Bolen J., Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res., 70, (2010), 1970– 1980. [DOI] [PubMed] [Google Scholar]
- 32. Ruschak A.M., Slassi M., Kay L.E., Schimmer A.D., Novel proteasome inhibitors to overcome bortezomib resistance. J. Natl. Cancer Inst., 103, (2011), 1007– 1017. [DOI] [PubMed] [Google Scholar]
- 33. Zhang J., Yang P.L., Gray N.S., Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer, 9, (2009), 28– 39. [DOI] [PubMed] [Google Scholar]
- 34. Yang Y., Kitagaki J., Dai R.M., Tsai Y.C., Lorick K.L., Ludwig R.L., Pierre S.A., Jensen J.P., Davydov I.V., Oberoi P., Li C.C., Kenten J.H., Beutler J.A., Vousden K.H., Weissman A.M., Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res., 67, (2007), 9472– 9481. [DOI] [PubMed] [Google Scholar]
- 35. Kitagaki J., Yang Y., Saavedra J.E., Colburn N.H., Keefer L.K., Perantoni A.O., Nitric oxide prodrug JS-K inhibits ubiquitin E1 and kills tumor cells retaining wild-type p53. Oncogene, 28, (2009), 619– 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chiba T., Tanaka K., Cullin-based ubiquitin ligase and its control by NEDD8-conjugating system. Curr. Protein Pept. Sci., 5, (2004), 177– 184. [DOI] [PubMed] [Google Scholar]
- 37. Petroski M.D., Deshaies R.J., Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol., 6, (2005), 9– 20. [DOI] [PubMed] [Google Scholar]
- 38. Goldenberg S.J., Cascio T.C., Shumway S.D., Garbutt K.C., Liu J., Xiong Y., Zheng N., Structure of the Cand1–Cul1–Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell, 119, (2004), 517– 528. [DOI] [PubMed] [Google Scholar]
- 39. Skaar J.R., Pagano M., Control of cell growth by the SCF and APC/C ubiquitin ligases. Curr. Opin. Cell Biol., 21, (2009), 816– 824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soucy T.A., Smith P.G., Rolfe M., Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin. Cancer Res., 15, (2009), 3912– 3916. [DOI] [PubMed] [Google Scholar]
- 41. Soucy T.A., Smith P.G., Milhollen M.A., Berger A.J., Gavin J.M., Adhikari S., Brownell J.E., Burke K.E., Cardin D.P., Critchley S., Cullis C.A., Doucette A., Garnsey J.J., Gaulin J.L., Gershman R.E., Lublinsky A.R., McDonald A., Mizutani H., Narayanan U., Olhava E.J., Peluso S., Rezaei M., Sintchak M.D., Talreja T., Thomas M.P., Traore T., Vyskocil S., Weatherhead G.S., Yu J., Zhang J., Dick L.R., Claiborne C.F., Rolfe M., Bolen J.B., Langston S.P., An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature, 458, (2009), 732– 736. [DOI] [PubMed] [Google Scholar]
- 42. Brownell J.E., Sintchak M.D., Gavin J.M., Liao H., Bruzzese F.J., Bump N.J., Soucy T.A., Milhollen M.A., Yang X., Burkhardt A.L., Ma J., Loke H.K., Lingaraj T., Wu D., Hamman K.B., Spelman J.J., Cullis C.A., Langston S.P., Vyskocil S., Sells T.B., Mallender W.D., Visiers I., Li P., Claiborne C.F., Rolfe M., Bolen J.B., Dick L.R., Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell, 37, (2010), 102– 111. [DOI] [PubMed] [Google Scholar]
- 43. Lin J.J., Milhollen M.A., Smith P.G., Narayanan U., Dutta A., NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res., 70, (2010), 10310– 10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swords R.T., Kelly K.R., Smith P.G., Garnsey J.J., Mahalingam D., Medina E., Oberheu K., Padmanabhan S., O'Dwyer M., Nawrocki S.T., Giles F.J., Carew J.S., Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood, 115, (2010), 3796– 3800. [DOI] [PubMed] [Google Scholar]
- 45. Milhollen M.A., Traore T., Adams-Duffy J., Thomas M.P., Berger A.J., Dang L., Dick L.R., Garnsey J.J., Koenig E., Langston S.P., Manfredi M., Narayanan U., Rolfe M., Staudt L.M., Soucy T.A., Yu J., Zhang J., Bolen J.B., Smith P.G., MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood, 116, (2010), 1515– 1523. [DOI] [PubMed] [Google Scholar]
- 46. van Wijk S.J., Timmers H.T., The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J., 24, (2010), 981– 993. [DOI] [PubMed] [Google Scholar]
- 47. Maeda H., Miyajima N., Kano S., Tsukiyama T., Okumura F., Fukuda S., Hatakeyama S., Ubiquitin-conjugating enzyme UBE2Q2 suppresses cell proliferation and is down-regulated in recurrent head and neck cancer. Mol. Cancer Res.: MCR, 7, (2009), 1553– 1562. [DOI] [PubMed] [Google Scholar]
- 48. Hao J., Xu A., Xie X., Hao J., Tian T., Gao S., Xiao X., He D., Elevated expression of UBE2T in lung cancer tumors and cell lines. Tumour Biol., 29, (2008), 195– 203. [DOI] [PubMed] [Google Scholar]
- 49. van Ree J.H., Jeganathan K.B., Malureanu L., van Deursen J.M., Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J. Cell Biol., 188, (2010), 83– 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ye Y., Rape M., Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol., 10, (2009), 755– 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Burroughs A.M., Jaffee M., Iyer L.M., Aravind L., Anatomy of the E2 ligase fold: implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J. Struct. Biol., 162, (2008), 205– 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Christensen D.E., Brzovic P.S., Klevit R.E., E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol., 14, (2007), 941– 948. [DOI] [PubMed] [Google Scholar]
- 53. Petroski M.D., Deshaies R.J., Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell, 123, (2005), 1107– 1120. [DOI] [PubMed] [Google Scholar]
- 54. Winn P.J., Religa T.L., Battey J.N., Banerjee A., Wade R.C., Determinants of functionality in the ubiquitin conjugating enzyme family. Structure, 12, (2004), 1563– 1574. [DOI] [PubMed] [Google Scholar]
- 55. Skowyra D., Craig K.L., Tyers M., Elledge S.J., Harper J.W., F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell, 91, (1997), 209– 219. [DOI] [PubMed] [Google Scholar]
- 56. Markson G., Kiel C., Hyde R., Brown S., Charalabous P., Bremm A., Semple J., Woodsmith J., Duley S., Salehi-Ashtiani K., Vidal M., Komander D., Serrano L., Lehner P., Sanderson C.M., Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res., 19, (2009), 1905– 1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ceccarelli D.F., Tang X., Pelletier B., Orlicky S., Xie W., Plantevin V., Neculai D., Chou Y.C., Ogunjimi A., Al-Hakim A., Varelas X., Koszela J., Wasney G.A., Vedadi M., Dhe-Paganon S., Cox S., Xu S., Lopez-Girona A., Mercurio F., Wrana J., Durocher D., Meloche S., Webb D.R., Tyers M., Sicheri F., An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell, 145, (2011), 1075– 1087. [DOI] [PubMed] [Google Scholar]
- 58. Hatakeyama S., Yada M., Matsumoto M., Ishida N., Nakayama K.I., U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem., 276, (2001), 33111– 33120. [DOI] [PubMed] [Google Scholar]
- 59. Zheng N., Schulman B.A., Song L., Miller J.J., Jeffrey P.D., Wang P., Chu C., Koepp D.M., Elledge S.J., Pagano M., Conaway R.C., Conaway J.W., Harper J.W., Pavletich N.P., Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature, 416, (2002), 703– 709. [DOI] [PubMed] [Google Scholar]
- 60. Huang L., Kinnucan E., Wang G., Beaudenon S., Howley P.M., Huibregtse J.M., Pavletich N.P., Structure of an E6AP–UbcH7 complex: insights into ubiquitination by the E2–E3 enzyme cascade. Science, 286, (1999), 1321– 1326. [DOI] [PubMed] [Google Scholar]
- 61. Scheffner M., Kumar S., Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim. Biophys. Acta, (2013), http://10.1016/j.bbamcr.2013.03.024 [DOI] [PubMed] [Google Scholar]
- 62. Metzger M.B., Pruneda J.N., Klevit R.E., Weissman A.M., RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta, (2013), http://10.1016/j.bbamcr.2013.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Momand J., Jung D., Wilczynski S., Niland J., The MDM2 gene amplification database. Nucleic Acids Res., 26, (1998), 3453– 3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Reifenberger G., Liu L., Ichimura K., Schmidt E.E., Collins V.P., Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res., 53, (1993), 2736– 2739. [PubMed] [Google Scholar]
- 65. Hock A.K., Vousden K.H., The role of ubiquitin modification in the regulation of p53. Biochim. Biophys. Acta, (2013), http://10.1016/j.bbamcr.2013.05.022 [DOI] [PubMed] [Google Scholar]
- 66. Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E.A., In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science, 303, (2004), 844– 848. [DOI] [PubMed] [Google Scholar]
- 67. Issaeva N., Bozko P., Enge M., Protopopova M., Verhoef L.G., Masucci M., Pramanik A., Selivanova G., Small molecule RITA binds to p53, blocks p53–HDM-2 interaction and activates p53 function in tumors. Nat. Med., 10, (2004), 1321– 1328. [DOI] [PubMed] [Google Scholar]
- 68. Grossman S.R., Deato M.E., Brignone C., Chan H.M., Kung A.L., Tagami H., Nakatani Y., Livingston D.M., Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science, 300, (2003), 342– 344. [DOI] [PubMed] [Google Scholar]
- 69. Yang Y., Ludwig R.L., Jensen J.P., Pierre S.A., Medaglia M.V., Davydov I.V., Safiran Y.J., Oberoi P., Kenten J.H., Phillips A.C., Weissman A.M., Vousden K.H., Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell, 7, (2005), 547– 559. [DOI] [PubMed] [Google Scholar]
- 70. Roxburgh P., Hock A.K., Dickens M.P., Mezna M., Fischer P.M., Vousden K.H., Small molecules that bind the Mdm2 RING stabilize and activate p53. Carcinogenesis, 33, (2012), 791– 798. [DOI] [PubMed] [Google Scholar]
- 71. Hunter A.M., LaCasse E.C., Korneluk R.G., The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis, 12, (2007), 1543– 1568. [DOI] [PubMed] [Google Scholar]
- 72. Yang Y., Fang S., Jensen J.P., Weissman A.M., Ashwell J.D., Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science, 288, (2000), 874– 877. [DOI] [PubMed] [Google Scholar]
- 73. Du C., Fang M., Li Y., Li L., Wang X., Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell, 102, (2000), 33– 42. [DOI] [PubMed] [Google Scholar]
- 74. Li L., Thomas R.M., Suzuki H., De Brabander J.K., Wang X., Harran P.G., A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science, 305, (2004), 1471– 1474. [DOI] [PubMed] [Google Scholar]
- 75. Wu H., Tschopp J., Lin S.C., Smac mimetics and TNFalpha: a dangerous liaison?. Cell, 131, (2007), 655– 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jin J., Cardozo T., Lovering R.C., Elledge S.J., Pagano M., Harper J.W., Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev., 18, (2004), 2573– 2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cardozo T., Pagano M., The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol., 5, (2004), 739– 751. [DOI] [PubMed] [Google Scholar]
- 78. Bassermann F., Eichner R., Pagano M., The ubiquitin proteasome system – implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta, (2013), http://10.1016/j.bbamcr.2013.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Merlet J., Burger J., Gomes J.E., Pintard L., Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell. Mol. Life Sci.: CMLS, 66, (2009), 1924– 1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen Q., Xie W., Kuhn D.J., Voorhees P.M., Lopez-Girona A., Mendy D., Corral L.G., Krenitsky V.P., Xu W., Moutouh-de Parseval L., Webb D.R., Mercurio F., Nakayama K.I., Nakayama K., Orlowski R.Z., Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood, 111, (2008), 4690– 4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wu L., Grigoryan A.V., Li Y., Hao B., Pagano M., Cardozo T.J., Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem. Biol., 19, (2012), 1515– 1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nakajima H., Fujiwara H., Furuichi Y., Tanaka K., Shimbara N., A novel small-molecule inhibitor of NF-kappaB signaling. Biochem. Biophys. Res. Commun., 368, (2008), 1007– 1013. [DOI] [PubMed] [Google Scholar]
- 83. Orlicky S., Tang X., Neduva V., Elowe N., Brown E.D., Sicheri F., Tyers M., An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat. Biotechnol., 28, (2010), 733– 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lydeard J.R., Harper J.W., Inhibitors for E3 ubiquitin ligases. Nat. Biotechnol., 28, (2010), 682– 684. [DOI] [PubMed] [Google Scholar]
- 85. Aghajan M., Jonai N., Flick K., Fu F., Luo M., Cai X., Ouni I., Pierce N., Tang X., Lomenick B., Damoiseaux R., Hao R., Del Moral P.M., Verma R., Li Y., Li C., Houk K.N., Jung M.E., Zheng N., Huang L., Deshaies R.J., Kaiser P., Huang J., Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat. Biotechnol., 28, (2010), 738– 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H., Identification of a primary target of thalidomide teratogenicity. Science, 327, (2010), 1345– 1350. [DOI] [PubMed] [Google Scholar]
- 87. Bianchi K., Meier P., A tangled web of ubiquitin chains: breaking news in TNF-R1 signaling. Mol. Cell, 36, (2009), 736– 742. [DOI] [PubMed] [Google Scholar]
- 88. Peng J., Schwartz D., Elias J.E., Thoreen C.C., Cheng D., Marsischky G., Roelofs J., Finley D., Gygi S.P., A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol., 21, (2003), 921– 926. [DOI] [PubMed] [Google Scholar]
- 89. Tokunaga F., Sakata S., Saeki Y., Satomi Y., Kirisako T., Kamei K., Nakagawa T., Kato M., Murata S., Yamaoka S., Yamamoto M., Akira S., Takao T., Tanaka K., Iwai K., Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol., 11, (2009), 123– 132. [DOI] [PubMed] [Google Scholar]
- 90. Eddins M.J., Varadan R., Fushman D., Pickart C.M., Wolberger C., Crystal structure and solution NMR studies of Lys48-linked tetraubiquitin at neutral pH. J. Mol. Biol., 367, (2007), 204– 211. [DOI] [PubMed] [Google Scholar]
- 91. Varadan R., Assfalg M., Haririnia A., Raasi S., Pickart C., Fushman D., Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J. Biol. Chem., 279, (2004), 7055– 7063. [DOI] [PubMed] [Google Scholar]
- 92. Komander D., Reyes-Turcu F., Licchesi J.D., Odenwaelder P., Wilkinson K.D., Barford D., Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep., 10, (2009), 466– 473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Matsumoto M.L., Wickliffe K.E., Dong K.C., Yu C., Bosanac I., Bustos D., Phu L., Kirkpatrick D.S., Hymowitz S.G., Rape M., Kelley R.F., Dixit V.M., K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol. Cell, 39, (2010), 477– 484. [DOI] [PubMed] [Google Scholar]
- 94. Jin L., Williamson A., Banerjee S., Philipp I., Rape M., Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell, 133, (2008), 653– 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xu P., Duong D.M., Seyfried N.T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J., Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell, 137, (2009), 133– 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Dynek J.N., Goncharov T., Dueber E.C., Fedorova A.V., Izrael-Tomasevic A., Phu L., Helgason E., Fairbrother W.J., Deshayes K., Kirkpatrick D.S., Vucic D., C-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J., 29, (2010), 4198– 4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Johnson E.S., Ma P.C., Ota I.M., Varshavsky A., A proteolytic pathway that recognizes ubiquitin as a degradation signal. J. Biol. Chem., 270, (1995), 17442– 17456. [DOI] [PubMed] [Google Scholar]
- 98. Kim W., Bennett E.J., Huttlin E.L., Guo A., Li J., Possemato A., Sowa M.E., Rad R., Rush J., Comb M.J., Harper J.W., Gygi S.P., Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell, 44, (2011), 325– 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sato Y., Fujita H., Yoshikawa A., Yamashita M., Yamagata A., Kaiser S.E., Iwai K., Fukai S., Specific recognition of linear ubiquitin chains by the Npl4 zinc finger (NZF) domain of the HOIL-1L subunit of the linear ubiquitin chain assembly complex. Proc. Natl. Acad. Sci. USA, 108, (2011), 20520– 20525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Verma R., Peters N.R., D'Onofrio M., Tochtrop G.P., Sakamoto K.M., Varadan R., Zhang M., Coffino P., Fushman D., Deshaies R.J., King R.W., Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science, 306, (2004), 117– 120. [DOI] [PubMed] [Google Scholar]
- 101. Komander D., Clague M.J., Urbe S., Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol., 10, (2009), 550– 563. [DOI] [PubMed] [Google Scholar]
- 102. Fraile J.M., Quesada V., Rodriguez D., Freije J.M., Lopez-Otin C., Deubiquitinases in cancer: new functions and therapeutic options. Oncogene, 31, (2012), 2373– 2388. [DOI] [PubMed] [Google Scholar]
- 103. Eletr Z.M., Wilkinson K.D., Regulation of proteolysis by human deubiquitinating enzymes. Biochim. Biophys. Acta, (2013), http://10.1016/j.bbamcr.2013.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sacco J.J., Coulson J.M., Clague M.J., Urbe S., Emerging roles of deubiquitinases in cancer-associated pathways. IUBMB Life, 62, (2010), 140– 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liu Y., Lashuel H.A., Choi S., Xing X., Case A., Ni J., Yeh L.A., Cuny G.D., Stein R.L., Lansbury P.T. Jr., Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem. Biol., 10, (2003), 837– 846. [DOI] [PubMed] [Google Scholar]
- 106. Mermerian A.H., Case A., Stein R.L., Cuny G.D., Structure-activity relationship, kinetic mechanism, and selectivity for a new class of ubiquitin C-terminal hydrolase-L1 (UCH-L1) inhibitors. Bioorg. Med. Chem. Lett., 17, (2007), 3729– 3732. [DOI] [PubMed] [Google Scholar]
- 107. Edelmann M.J., Nicholson B., Kessler B.M., Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev. Mol. Med., 13, (2011), e35– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., Baker S.C., Ghosh A.K., Mesecar A.D., A non-covalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA, 105, (2008), 16119– 16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Williams S.A., Maecker H.L., French D.M., Liu J., Gregg A., Silverstein L.B., Cao T.C., Carano R.A., Dixit V.M., USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell, 146, (2011), 918– 930. [DOI] [PubMed] [Google Scholar]
- 110. Kee Y., D'Andrea A.D., Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev., 24, (2010), 1680– 1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kim J.M., Parmar K., Huang M., Weinstock D.M., Ruit C.A., Kutok J.L., D'Andrea A.D., Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev. Cell, 16, (2009), 314– 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cohn M.A., Kowal P., Yang K., Haas W., Huang T.T., Gygi S.P., D'Andrea A.D., A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol. Cell, 28, (2007), 786– 797. [DOI] [PubMed] [Google Scholar]
- 113. Chen J., Dexheimer T.S., Ai Y., Liang Q., Villamil M.A., Inglese J., Maloney D.J., Jadhav A., Simeonov A., Zhuang Z., Selective and cell-active inhibitors of the USP1/UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells. Chem. Biol., 18, (2011), 1390– 1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Jacq X., Kemp M., Martin N.M., Jackson S.P., Deubiquitylating enzymes and DNA damage response pathways. Cell Biochem. Biophys., 67, (2013), 25– 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Nicholson B., Suresh Kumar K.G., The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem. Biophys., 60, (2011), 61– 68. [DOI] [PubMed] [Google Scholar]
- 116. Chene P., Inhibiting the p53–MDM2 interaction: an important target for cancer therapy. Nat. Rev. Cancer, 3, (2003), 102– 109. [DOI] [PubMed] [Google Scholar]
- 117. Li M., Chen D., Shiloh A., Luo J., Nikolaev A.Y., Qin J., Gu W., Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature, 416, (2002), 648– 653. [DOI] [PubMed] [Google Scholar]
- 118. Li M., Brooks C.L., Kon N., Gu W., A dynamic role of HAUSP in the p53–Mdm2 pathway. Mol. Cell, 13, (2004), 879– 886. [DOI] [PubMed] [Google Scholar]
- 119. Colland F., Formstecher E., Jacq X., Reverdy C., Planquette C., Conrath S., Trouplin V., Bianchi J., Aushev V.N., Camonis J., Calabrese A., Borg-Capra C., Sippl W., Collura V., Boissy G., Rain J.C., Guedat P., Delansorne R., Daviet L., Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther., 8, (2009), 2286– 2295. [DOI] [PubMed] [Google Scholar]
- 120. Chauhan D., Tian Z., Nicholson B., Kumar K.G., Zhou B., Carrasco R., McDermott J.L., Leach C.A., Fulcinniti M., Kodrasov M.P., Weinstock J., Kingsbury W.D., Hideshima T., Shah P.K., Minvielle S., Altun M., Kessler B.M., Orlowski R., Richardson P., Munshi N., Anderson K.C., A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell, 22, (2012), 345– 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Reverdy C., Conrath S., Lopez R., Planquette C., Atmanene C., Collura V., Harpon J., Battaglia V., Vivat V., Sippl W., Colland F., Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol., 19, (2012), 467– 477. [DOI] [PubMed] [Google Scholar]
- 122. Lee B.H., Lee M.J., Park S., Oh D.C., Elsasser S., Chen P.C., Gartner C., Dimova N., Hanna J., Gygi S.P., Wilson S.M., King R.W., Finley D., Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature, 467, (2010), 179– 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Leggett D.S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R.T., Walz T., Ploegh H., Finley D., Multiple associated proteins regulate proteasome structure and function. Mol. Cell, 10, (2002), 495– 507. [DOI] [PubMed] [Google Scholar]
- 124. Hu M., Li P., Li M., Li W., Yao T., Wu J.W., Gu W., Cohen R.E., Shi Y., Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell, 111, (2002), 1041– 1054. [DOI] [PubMed] [Google Scholar]
- 125. Hu M., Li P., Song L., Jeffrey P.D., Chenova T.A., Wilkinson K.D., Cohen R.E., Shi Y., Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J., 24, (2005), 3747– 3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Renatus M., Parrado S.G., D'Arcy A., Eidhoff U., Gerhartz B., Hassiepen U., Pierrat B., Riedl R., Vinzenz D., Worpenberg S., Kroemer M., Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure, 14, (2006), 1293– 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hurley J.H., Lee S., Prag G., Ubiquitin-binding domains. Biochem. J., 399, (2006), 361– 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Brzovic P.S., Lissounov A., Christensen D.E., Hoyt D.W., Klevit R.E., A UbcH5/ubiquitin non-covalent complex is required for processive BRCA1-directed ubiquitination. Mol. Cell, 21, (2006), 873– 880. [DOI] [PubMed] [Google Scholar]
- 129. Ogunjimi A.A., Wiesner S., Briant D.J., Varelas X., Sicheri F., Forman-Kay J., Wrana J.L., The ubiquitin binding region of the Smurf HECT domain facilitates polyubiquitylation and binding of ubiquitylated substrates. J. Biol. Chem., 285, (2010), 6308– 6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Maspero E., Mari S., Valentini E., Musacchio A., Fish A., Pasqualato S., Polo S., Structure of the HECT:ubiquitin complex and its role in ubiquitin chain elongation. EMBO Rep., 12, (2011), 342– 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Sgourakis N.G., Patel M.M., Garcia A.E., Makhatadze G.I., McCallum S.A., Conformational dynamics and structural plasticity play critical roles in the ubiquitin recognition of a UIM domain. J. Mol. Biol., 396, (2010), 1128– 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ernst A., Avvakumov G., Tong J., Fan Y., Zhao Y., Alberts P., Persaud A., Walker J.R., Neculai A.M., Neculai D., Vorobyov A., Garg P., Beatty L., Chan P.K., Juang Y.C., Landry M.C., Yeh C., Zeqiraj E., Karamboulas K., Allali-Hassani A., Vedadi M., Tyers M., Moffat J., Sicheri F., Pelletier L., Durocher D., Raught B., Rotin D., Yang J., Moran M.F., Dhe-Paganon S., Sidhu S.S., A strategy for modulation of enzymes in the ubiquitin system. Science, 339, (2013), 590– 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Berlin I., Schwartz H., Nash P.D., Regulation of epidermal growth factor receptor ubiquitination and trafficking by the USP8.STAM complex. J. Biol. Chem., 285, (2010), 34909– 34921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Balut C.M., Loch C.M., Devor D.C., Role of ubiquitylation and USP8-dependent deubiquitylation in the endocytosis and lysosomal targeting of plasma membrane KCa3.1. FASEB J., 25, (2011), 3938– 3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Meijer I.M., van Leeuwen J.E., ERBB2 is a target for USP8-mediated deubiquitination. Cell. Signal., 23, (2011), 458– 467. [DOI] [PubMed] [Google Scholar]
- 136. Niendorf S., Oksche A., Kisser A., Lohler J., Prinz M., Schorle H., Feller S., Lewitzky M., Horak I., Knobeloch K.P., Essential role of ubiquitin-specific protease 8 for receptor tyrosine kinase stability and endocytic trafficking in vivo. Mol. Cell. Biol., 27, (2007), 5029– 5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhang Y., Zhou L., Rouge L., Phillips A.H., Lam C., Liu P., Sandoval W., Helgason E., Murray J.M., Wertz I.E., Corn J.E., Conformational stabilization of ubiquitin yields potent and selective inhibitors of USP7. Nat. Chem. Biol., 9, (2013), 51– 58. [DOI] [PubMed] [Google Scholar]
- 138. Xu G., Tan X., Wang H., Sun W., Shi Y., Burlingame S., Gu X., Cao G., Zhang T., Qin J., Yang J., Ubiquitin-specific peptidase 21 inhibits tumor necrosis factor alpha-induced nuclear factor kappaB activation via binding to and deubiquitinating receptor-interacting protein 1. J. Biol. Chem., 285, (2010), 969– 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Nakada S., Tai I., Panier S., Al-Hakim A., Iemura S., Juang Y.C., O'Donnell L., Kumakubo A., Munro M., Sicheri F., Gingras A.C., Natsume T., Suda T., Durocher D., Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature, 466, (2010), 941– 946. [DOI] [PubMed] [Google Scholar]
- 140. Juang Y.C., Landry M.C., Sanches M., Vittal V., Leung C.C., Ceccarelli D.F., Mateo A.R., Pruneda J.N., Mao D.Y., Szilard R.K., Orlicky S., Munro M., Brzovic P.S., Klevit R.E., Sicheri F., Durocher D., OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Mol. Cell, 45, (2012), 384– 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Rotin D., Kumar S., Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol., 10, (2009), 398– 409. [DOI] [PubMed] [Google Scholar]
- 142. Persaud A., Alberts P., Amsen E.M., Xiong X., Wasmuth J., Saadon Z., Fladd C., Parkinson J., Rotin D., Comparison of substrate specificity of the ubiquitin ligases Nedd4 and Nedd4-2 using proteome arrays. Mol. Syst. Biol., 5, (2009), 333– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Harper J.W., King R.W., Stuck in the middle: drugging the ubiquitin system at the e2 step. Cell, 145, (2011), 1007– 1009. [DOI] [PubMed] [Google Scholar]