

Abstract
Lipid rafts are membrane microdomains enriched in cholesterol, sphingolipids, and glycolipids that have been implicated in many biological processes. Since cholesterol is known to play a key role in the entry of some other viruses, we investigated the role of cholesterol and lipid rafts in the host cell plasma membrane in Newcastle Disease Virus (NDV) entry. We used methyl-β-cyclodextrin (MβCD) to deplete cellular cholesterol and disrupt lipid rafts. Our results show that the removal of cellular cholesterol partially reduces viral binding, fusion and infectivity. MβCD had no effect on the expression of sialic acid containing molecule expression, the NDV receptors in the target cell. All the above-described effects were reversed by restoring cholesterol levels in the target cell membrane. The HN viral attachment protein partially localized to detergent-resistant membrane microdomains (DRMs) at 4 °C and then shifted to detergent-soluble fractions at 37 °C. These results indicate that cellular cholesterol may be required for optimal cell entry in NDV infection cycle.
Keywords: Lipid raft, Cholesterol, Viral entry, Paramyxovirus, NDV
Highlights
► We examine the role of cellular cholesterol on NDV activities. ► Removal of cholesterol partially reduces viral binding, fusion and infectivity. ► This negative effect was reversible. ► The HN viral attachment protein partially localized to cell lipid rafts at 4 °C. ► Cellular cholesterol may be required for optimal cell entry in NDV infection.
1. Introduction
Newcastle Disease Virus (NDV), a prototype of paramyxoviruses, is an avian enveloped RNA-negative strand virus that causes respiratory disease in domestic fowls leading to huge economic losses in the poultry industry. The envelope of NDV contains two associated glycoproteins that mediate viral entry: the hemagglutinin-neuraminidase (HN) and fusion (F) proteins. HN is the receptor-binding protein that recognizes and binds to sialoglycoconjugates at the cell surface [1]. The fusion protein is a metastable protein that undergoes a series of irreversible conformational changes to trigger the fusion of the viral and plasma membrane in a pH-independent manner [2]. Nevertheless, we have previously reported an additional pathway of NDV entry through caveolae-dependent endocytosis [3]. For most paramyxoviruses, including NDV, the triggering of F protein is HN-dependent through its fusion promotion activity. The mechanism by which HN protein activates F in a homotypic manner is not well understood [4], [5].
Lipid rafts are dynamic membrane microdomains preferentially containing cholesterol as a major constituent, together with sphingolipids and specific associated proteins. Although their existence is still controversial, the resistance of these components to cold detergent extraction and mechanical disruption has been considered proof of their existence (for a review, see [6]). Caveolae, a particular membrane raft subset, are cholesterol- and glycosphingolipid-enriched plasma membrane microdomains, identifiable as stable invaginations of the plasma membrane, which are enriched in caveolin, a membrane protein that is tightly bound to cholesterol [7]. Lipids rafts have been implicated in functions such as membrane signaling and trafficking, signal transduction and the regulation of cell adhesion [8].
Moreover, the involvement of lipid rafts and cell membrane cholesterol has been demonstrated in different stages of the viral life cycle, such as viral entry, assembly and budding (reviewed in [9], [10], [11]).
Some enveloped viruses have been shown to enter host cells in a cholesterol-dependent manner, including poliovirus [12], polyomavirus SV40 [13], [14], coronavirus [15], [16], arenavirus [17], togavirus [18], [19], poxvirus, such as vaccinia virus [20] and herpes virus [21], [22]. Lipid rafts have also been implicated in HIV entry [23], [24], and Ebola and Marburg filoviruses also require cholesterol in the plasma cell membrane [25], [26]. For some of these viruses, caveolae have been proposed to be the portal of entry, including SV40 [13], picornavirus Echovirus 1 [27], the amphotropic murine leukemia virus (A-MLV) [28] and the Ebola and Marburg filoviruses [29].
There are few reports describing the role of cell membrane cholesterol in paramyxovirus entry, but with no evidence that lipid rafts might be directly involved in it. It has been shown that the paramyxovirus canine distemper virus, genus Morbillivirus, does not require cholesterol in the plasma membrane but does require it in the viral envelope [30]. The data concerning RSV are controversial. Thus, some authors have reported the independence of RSV entry from cholesterol [31], whereas in other works the entry of RSV through caveolae in dendritic cells has been proposed [32]. Moreover, using small interfering RNA technology [33] showed that RSV use clathrin-mediated endocytosis to productively infect cells. For NDV, it has been shown that lipid rafts are sites of NDV assembly and release [34], [35].
We have previously shown that the caveolin phosphorylating factor PMA inhibits NDV fusion [3] and that cholesterol depletion exerts an inhibitory effect on NDV interaction with the target cell, leading us to propose that a fraction of NDV may penetrate the cell through caveolae-mediated endocytosis [3]. In the present paper, we extend these studies with a view to clarifying the functional role of cholesterol in NDV entry. Depletion of cellular cholesterol by treatment with methyl-β-cyclodextrin (MβCD) inhibited NDV binding, fusion and infectivity. This inhibition was almost completely compensated by replenishing cellular cholesterol levels, suggesting that the effect of MβCD treatment on virus activities would be due to the removal of cholesterol. Analysis of the distribution of viral envelope glycoproteins in cholesterol-rich resistant membranes showed that viral glycoproteins partially associate with lipid rafts during initial virus adsorption and entry. Our data corroborate a role of cell membrane cholesterol during the early stages of NDV infection. A clear understanding of the role of lipid membrane components in viral entry should be useful in the future for designing antiviral agents and vaccines.
2. Materials and methods
2.1. Cell lines and virus
East Lansing Line (ELL-0) avian fibroblasts and HeLa cells were obtained from the American Type Culture Collection and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with L-GlutaMax (580 mg l− 1, Invitrogen), penicillin–streptomycin (100 U ml− 1–100 μg ml− 1) and 10% heat inactivated fetal bovine serum (complete medium). The lentogenic “Clone 30” strain of NDV was obtained from Intervet Laboratories (Salamanca, Spain). The virus was grown and purified mainly as described previously [3].
2.2. Reagents and antibodies
Methyl-β-cyclodextrin (MβCD), cholesterol, lovastatin, Triton-X-100, OptiPrep, FITC-MALI, monoclonal anti-β-tubulin antibody and 3-(4,5-dimethyl-2-thiazoyl)-2,5-diphenyl tetrazolium bromide (MTT) were from Sigma-Aldrich; Octadecylrhodamine B chloride (R18), Hoechst 33258 and Alexa fluor 488 donkey anti-mouse antibody were from Molecular Probes. Polyclonal anti-caveolin-1 (N20) and anti-GAPDH antibodies were from Santa Cruz Biotechnology; FITC-SNA was from Vector Laboratories; polyclonal anti-NDV, monoclonal anti-F (2A6) and HN (7B1) antibodies were generous gifts from Dr. Adolfo García-Sastre (Emerging Pathogens Institute, Mount Sinai School of Medicine, New York, USA).
2.3. Virus titration
Viral titres were calculated in plaque formation assays in Vero cells, as described previously [36], or in monolayers of ELL-0 cells in 24-well plates infected with different dilutions of NDV for 24 h. Cells were fixed for 30 min with PBS containing 2.5% paraformaldehyde, blocked in PBS containing 1% BSA for 1 h, and incubated for 1 h at room temperature with 5 μg ml− 1 2A6 anti-F monoclonal antibody. Cells were then incubated with anti-mouse Alexa Fluor 488-conjugated IgGs (dilution 1:400) for 45 min. After a wash with PBS, cells were visualized under an inverted fluorescence microscope (Olympus IX51) at 10 × magnification. NDV infectivity was calculated from the dilution of NDV that infected all cells of the monolayer and was expressed as p.f.u. ml− 1.
2.4. Fluorescence-activated cell sorting (FACS) analysis
Monolayers of ELL-0 cells in 60 mm plates were infected with 5 mois of NDV for 24 h at 37 °C. Infected cells were removed from the plates by scraping, pelleted by centrifugation and incubated for 1 h at room temperature in the presence of both 5 μg ml− 1 mAb anti-F and mAb anti-HN. After centrifugation, cells were resuspended in PBS and incubated with anti-mouse Alexa Fluor 488-conjugated IgG (dilution 1:100) for 45 min in the dark at room temperature, followed by fixation in FACS lysing solution (Becton-Dickinson) for 10 min. Cells were then pelleted by centrifugation and resuspended in a suitable volume of PBS for analysis in a FACScalibur flow cytometer (Becton Dickinson). At least 5 × 104 cells were analyzed for each sample. Mean fluorescence values and the % of cells with fluorescence higher than the background were combined to quantify viral protein expression and were normalized to the control values. Data were analyzed with the WinMDI 2.9 software.
2.5. Cholesterol depletion and replenishment experiments
2.5.1. Cholesterol depletion
ELL-0 cells seeded on 35 mm plates were left either untreated or treated with increasing concentrations of MβCD and 4 μg ml− 1 lovastatin in OptiMEM (Gibco) for 1 h at 37 °C. Under these conditions, cell viability was not significantly affected, as determined with the trypan blue exclusion method. The viability of untreated or MβCD-treated cells was found to be higher than 90% as analyzed by trypan blue exclusion. Then, the medium was removed and the cells were washed 3 times with OptiMEM to remove MβCD. Following this, the cells were processed as required for each experiment in the presence of 4 μg ml− 1 lovastatin a competitive inhibitor of the rate limiting enzyme HMG-CoA reductase in the cholesterol synthesis pathway, preventing the synthesis of endogenous cholesterol [37].
2.5.2. Replenishment of cholesterol
The replenishment of cellular cholesterol was accomplished as described previously [12], [38]. Briefly, after depletion of cellular cholesterol as described above, ELL-0 cells were treated either with medium alone or with cholesterol-MβCD-containing medium, with soluble cholesterol at final concentrations ranging from 50 to 200 μM for 1 h at 37 °C. Then, cells were washed six times with OptiMEM to remove MβCD.
2.5.3. Determination of cholesterol levels
Cellular cholesterol was extracted according to [12], [20]. Briefly, ELL-0 cells were lysed by three cycles of freeze-thawing followed by ultrasonication and the cholesterol was extracted from the cell lysates by the addition of chloroform/methanol 1:1. The bottom layer (chloroform) was collected and evaporated under vacuum. The residual cholesterol was dissolved in ethanol and assayed with a fluorimetric Amplex Red cholesterol assay kit (Molecular Probes) according to the manufacturer's protocol. Data were referred to the cholesterol levels of the same number of untreated control cells, considered as 100%.
2.6. Virus-binding assays
Plated control, cholesterol-depleted and ELL-0 cells replenished with cholesterol as described above were washed three times with ice-cold OptiMEM, and incubated with 5 mois of NDV for 1 h at 4 °C. Then, the cells were washed with ice-cold PBS to remove unbound virus and were collected by scraping, performing FACS analysis as reported above.
2.7. NDV–cell fusion assays
Purified NDV was labeled with the fluorescent probe octadecylrhodamine (R18) essentially as described previously [39]. Control, cholesterol-depleted and cholesterol-replenished ELL-0 cells plated in 35-mm plates were incubated with 3 μg of R18-NDV per plate in OptiMEM for 1 h at 37 °C. Then, the plates were washed 3 times with PBS and the transfer of the rhodamine probe to cells was observed under an Olympus IX51 inverted fluorescence microscope. R18 transfer was considered to be represented by bright, full cell-shaped events (Supplementary Fig. S1). Nuclei were stained with Hoechst 33258 (10 μg ml− 1). The percentage of fusion was calculated as the number of positive red-stained cells in 6 random fields with respect to the total number of cells in these areas of the well.
2.8. NDV infectivity assays
2.8.1. FACS assay
Monolayers of control and cholesterol-depleted ELL-0 cells were infected with 5 mois of NDV in OptiMEM for 1 h at room temperature. Then, the viral inoculum was removed and complete medium was added. At 24 h post-infection, cells were washed three times with PBS and processed for FACS analysis as described above.
2.8.2. Fluorescent microscopy assays
Monolayers of control, cholesterol-depleted and cholesterol replenished ELL-0 cells were infected with 1 moi of a recombinant NDV (rNDV-F3aa-mRFP [40], that expresses a monomeric red fluorescent protein [41], kindly provided by Dr. Adolfo García-Sastre) for 1 h at room temperature. After 24 h at 37 °C, the cells were observed under an Olympus IX51 inverted fluorescence microscope with a 10 × objective. Quantification of infectivity was accomplished by measuring the area of red fluorescence (infected cells that express the RFP) in pixels, referred to the area of nuclei (stained with Hoechst 33258) of the field in three to seven random fields. Areas were quantified using the analysis tool in ImageJ software.
To analyze the effect of cholesterol removal on the course of viral infection, ELL-0 cells were infected with the recombinant NDV at 1 moi. At different times post-infection, 10 mM of MβCD and 4 μg ml− 1 of lovastatin were added for 1 h at 37 °C, after which the cells were washed and maintained in complete medium in the presence of lovastatin for 24 h at 37 °C. The percentage of infectivity was calculated as the number of red fluorescent cells (infected cells) out of the total number of cells in three random fields.
2.9. Lectin staining
Lectin staining of ELL-0 cells was performed by incubation with FITC-labeled lectins. MβCD-treated or untreated ELL-0 cells were detached from the plates by scraping and fixed with FACS lysing solution for 10 min at room temperature, washed with PBS and incubated for 30 min at 4 °C either with 10 μg/ml FITC-labeled Maackia amurensis lectin (FITC-MALI), which specifically recognizes α2,3-bound sialic acids [42], or with FITC-labeled Sambucus nigra lectin (FITC-SNA), which recognizes α2,6-bound sialic acids [43]. Lectin binding to cells was detected by FACS as above. For confocal visualization of lectin staining, MβCD-treated or untreated ELL-0 cells were washed once with OptiMEM and then fixed with 2.5% paraformaldehyde in PBS for 30 min at 4 °C and then incubated with 10 μg ml− 1 FITC-MALI lectin for 30 min at 4 °C. After a short rinse with PBS, cells were mounted in Prolong Gold antifade reagent with DAPI (Invitrogen) and viewed with a confocal microscope (Leica SP5 equipped with a 488 nm argon laser for FITC and a 405 nm diode laser for DAPI, 63 × objective lens).
2.10. Fractionation of detergent-insoluble and -soluble membranes
Cells were fractionated into detergent-soluble and insoluble fractions mainly as described previously [20], [44], [45]. Monolayers of ELL-0 or HeLa cells grown on 60 mm plates were infected with 25 mois of NDV for 1 h at 4 °C and, where indicated, shifted to 37 °C for an additional hour. Cells were then washed in cold PBS and lysed in ice-cold lysis buffer (10 mM Tris HCl, pH 7.6, 140 mM NaCl, 5 mM EDTA, 1% sodium deoxycholate, 1% TX-100) containing a cocktail of protease inhibitors for 30 min at 4 °C. The lysates were then passed through a 20-gauge needle 20 times, and the nuclei and cellular debris were pelleted by centrifugation at 12,000 rpm for 30 min. The supernatants were layered at the bottom of a 40%–30%–5% discontinuous OptiPrep gradient formed by overlaying 2 ml 40% (containing the lysates), 6.5 ml 30% and 3.5 ml 5% of OptiPrep in buffer lysis. Gradients were centrifuged at 34,000 rpm in a Beckman SW40 Ti rotor for 20 h at 4 °C. A total of 12 fractions of 1 ml were collected from the top of the gradient by pipetting, after which they were analyzed for the presence of viral and cellular proteins by Western blotting.
2.11. Western blot analyses
Samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes (GE Healthcare). Membranes were blocked overnight at 4 °C in TBS (50 mM Tris–HCl, pH 7.6, 150 mM NaCl) blocking buffer containing 5% dry skimmed milk. Then, membranes were incubated with individual primary rabbit polyclonal anti-NDV (1:5000 dilution), rabbit polyclonal anti-caveolin N20 (1:300 dilution), mouse monoclonal anti-β-tubulin (1:2000 dilution), and monoclonal anti-GAPDH (1:2000 dilution) antibodies, for 1 h at room temperature. After extensive washes with blocking buffer, the membranes were incubated for 1 h at room temperature with secondary anti-rabbit or anti-mouse antibodies conjugated with horseradish peroxidase (1:5000 and 1:5000 dilution respectively, GE Healthcare), washed, and developed with ECL Plus Western blotting reagent system (GE Healthcare).
2.12. MTT cell viability assay
Cell viability after lovastatin incubation for 24 h in the infectivity experiments was assessed with a tetrazolium bromide colorimetric assay. Briefly, Ell-0 were seeded in a 96-well plate and incubated for 24 h in the presence of 4 μg ml− 1 of lovastatin. Then, the culture medium was removed and 100 μl of complete medium containing 10 μl MTT (5 mg/ml in PBS) was added. The absorbance of each well was measured at 620 nm on a Microplate Reader (Mutiskan Ex, Thermo Scientific) with pure DMSO as a blank. Non-treated cells were used as a control and relative cell viability (mean % ± SD, n = 3) was expressed as ODlovastatin / ODcontrol × 100%. The data indicate that lovastatin did not elicit a negative effect on cell viability/proliferation, even higher OD values (132.9% +/− 17.6, mean ± SD, n = 3) being obtained.
3. Results
To determine whether the removal of cholesterol from the target membrane affected NDV entry into cells, ELL-0 cells were treated with increasing concentrations of MβCD. MβCD treatment of cells resulted in a dose-dependent reduction in the cellular cholesterol content (Fig. 1a). Approximately, 85–90% of cellular cholesterol was removed at a concentration of 10 mM MβCD. The fusion of NDV with MβCD-treated ELL-0 cells was significantly reduced in a dose-dependent manner, as determined by an R18 transfer assay (Fig. 1b and Table 1 ). At a concentration of 5 mM MβCD (60% of cholesterol reduction, Fig. 1a), viral fusion was reduced by about 25% (Fig. 1b and Table 1), whereas the highest concentration of MβCD assayed, (10 mM, which led to 80% of cholesterol reduction (Fig. 1a)) elicited about 40% of NDV fusion inhibition (Fig. 1b and Table 1).
Fig. 1.

Effect of cellular cholesterol removal on NDV fusion. (a) Uninfected avian ELL-0 cell monolayers were incubated in the presence of increasing concentrations of MβCD for 1 h at 37 °C. Cholesterol was extracted and quantified as described in Materials and methods. Data are means ± standard deviations of three independent experiments. (b) R18-labeled NDV was bound to MβCD-treated cells and untreated cells (control) for 1 h at 4 °C, after which they were allowed to fuse for 1 h at 37 °C. Fusion was assessed by transfer of the R18 red dye to the cell membrane. As a control of non-specific dye transfer, NDV was inactivated by incubation at 100 °C for 2 min (NDV-R18 inact). In these samples, non-significant dye transfer was observed in both untreated and MβCD-treated cells.
Table 1.
Quantification of the effect of cholesterol removal on NDV activities.a
| Concentration of MβCD |
|||
|---|---|---|---|
| 5 mM | 10 mM | 15 mM | |
| Fusionb | 74.5 ± 8.02 | 63.0 ± 8.18 | ND |
| Bindingb | 85.0 ± 7.0 | 43.7 ± 12.05 | ND |
| Infectivityc | 98.0 | 61.0 | 47.0 |
ND: not determined.
Percentage of activity from control untreated cells.
Data are means ± SD of three or four independent experiments.
Data from those as shown in Fig. 3a are means of two independent experiments.
The effect of cholesterol depletion on NDV attachment was analyzed by flow cytometric assays. The binding of NDV to cholesterol-depleted cells was reduced by about 50% as compared with the controls in 10 mM MβCD-treated cells (Fig. 2a and Table 1). As a negative control, the virus was inactivated by heating at 100 °C for 2 min. No differences with samples in the absence of virus were detected, as shown in Supplementary Fig. S2. To analyze whether cholesterol depletion was affecting the expression of viral receptors at the cell surface, after the MβCD treatment, cells were labeled with two fluorescent-labeled lectins, FITC-MALI and FITC-SNA. The binding of FITC-MALI to cells preincubated in the presence of sialidase from Vibrio cholerae was reduced as assessed by fluorescence microscopy (Supplementary Fig. S3) revealing the specificity of lectin binding to sialoglycoconjugates. The degree of binding of both lectins to treated cells was similar to that of control cells (Fig. 2b), indicating that after cholesterol removal the receptor concentration was not modified at the cell surface. Nevertheless, when we analyzed the lectin-labeled cells by confocal microscopy, the FITC-MALI lectin-staining pattern was altered (Fig. 2c and Supplementary Fig. S4): upon MβCD treatment, the cell surface displayed patches enriched in sialic acid compound (Fig. 2c), in agreement with previous reports [34], [46].
Fig. 2.

Effect of cellular cholesterol depletion on NDV binding and receptor expression. (a) NDV at a moi of 5 was allowed to bind to MβCD-treated and -untreated cells for 1 h at 4 °C. Then, the binding of NDV to target cells was analyzed by a FACS-based immunoassay (see Materials and methods). The negative control corresponds to cells in the absence of virus without MβCD (red) or with MβCD (black). (b) 10 mM MβCD-treated or -untreated ELL-0 cells were incubated either with 10 μg ml− 1 of FITC-MALI lectin or with FITC-SNA lectin, and analyzed by FACSs. Negative control: cells without lectin nor MβCD. (c) Untreated control cells, lovastatin-treated and MβCD-incubated cells were stained with FITC-MALI lectin and analyzed under confocal microscopy as detailed in Materials and methods.
Fig. S2.

Negative control for the FACS binding assay. NDV at a moi of 5 was inactivated by incubation at 100 °C for 2 min and then was allow to bind to MβCD-treated (magenta) and -untreated cells (green) for 1 h at 4 °C. Then, the binding of NDV to target cells was analyzed by a FACS-based immunoassay as detailed in Materials and methods. Additional control corresponds to cells in the absence of virus without MβCD (red) or with MβCD (blue).
Fig. S3.
Lectin binding to sialidase-treated cells. Untreated (control) and V. cholerae- (25 mU/ml) sialidase-treated Ell-0 cells were stained with FITC-labeled lectin from Maackia amurensis (FITC-MALI), which recognizes α2,3 sialic acids. The binding of FITC-MALI to cells preincubated in the presence of sialidase from Vibrio cholerae was reduced revealing the specificity of lectin binding to sialoglycoconjugates.
Fig. S4.

Untreated control cells, lovastatin-treated and MβCD-incubated cells were stained with FITC-MALI lectin and analyzed under confocal microscopy as detailed in Materials and methods.
The infectivity of NDV was probed by FACS-based assays after 24 h of infection (Fig. 3a). Pre-treatment of ELL-0 cells with increasing concentrations of MβCD prior to exposure to 5 mois of NDV resulted in a reduction in viral infectivity, being about 50% less than that of untreated control cells at the highest MβCD concentration assayed (15 mM) (Fig. 3a and Table 1). In negative controls, the virus was inactivated by heating at 100 °C for 2 min. No differences with samples in the absence of virus were detected, as shown in Supplementary Fig. S5. Additionally, cellular cholesterol was depleted at several time points post-infection by treatment with 10 mM MβCD (Fig. 3b). ELL-0 cells were infected with rNDV-F3aa-mRFP recombinant NDV. Viral infectivity was monitored at 24 h post-infection, as detailed in Materials and methods. The results showed that the addition of MβCD at 1 h post-infection did not exert any inhibitory effect on NDV. Moreover, treatment with MβCD at 20 min post-infection only had a moderate effect (Fig. 3b). These results suggest that cholesterol is required during the entry of NDV into the host cell during the first stages of viral infectivity.
Fig. 3.
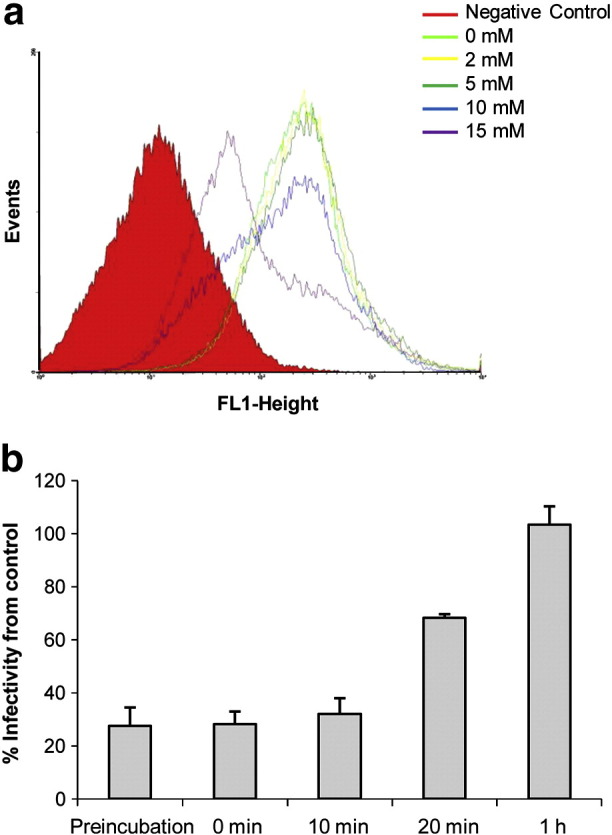
Effect of cholesterol removal from cells on NDV infectivity. (a) NDV at 5 mois was allowed to bind to MβCD-treated and -untreated cells for 1 h at 4 °C, and was then transferred to 37 °C. At 24 h post-infection, viral infectivity was analyzed by a FACS-based immunoassay, probing with anti-F and anti-HN primary antibodies (see Materials and methods). The negative control corresponds to cells in the absence of virus with or without MβCD. (b) Effect of cholesterol depletion on the course of NDV entry. Monolayers of ELL-0 cells were infected with a 10− 4 dilution of the recombinant rNDV-F3aa-mRFP NDV, meaning a moi of 1. At different times post-infection, 10 mM of MβCD and lovastatin were added for 1 h at 37 °C. After 24 h at 37 °C, infectivity was quantified as the number of red fluorescent cells out of the total number of cells in a fluorescent microscopy assay, and is referred to control untreated cells. Preincubation data refer to cells incubated with 10 mM of MβCD for 1 h at 37 °C before virus infection. Data are means ± SD of three independent experiments.
Fig. S5.

Negative control for the FACS infectivity assay. NDV at 5 mois was inactivated by incubation at 100 °C for 2 min and then was allowed to bind to MβCD-treated (green) and -untreated (black) cells for 1 h at 4 °C, and was then transferred to 37 °C. At 24 h post-infection, viral infectivity was analyzed by a FACS-based immunoassay, probing with anti-F and anti-HN primary antibodies (see Materials and methods). Additional control corresponds to cells in the absence of virus without MβCD (red) or with MβCD (blue).
To further check that the inhibitory effects of MβCD treatment on NDV activities described above were due to the removal of cellular cholesterol, the effect of cholesterol replenishment was analyzed (Fig. 4 and Table 2 ). After the depletion of cellular cholesterol by pre-treatment with 10 mM MβCD, cholesterol-depleted cells were incubated in the presence of MβCD previously complexed to cholesterol, acting as a cholesterol donor to cells [12]. The addition of 200 μM of exogenous cholesterol almost completely restored cellular cholesterol levels to those of the controls (Fig. 4a). Our data revealed a correlation between the degree of cholesterol replacement (Fig. 4a) and the degree of recovery of NDV activities (Fig. 4 and Table 2). Viral fusion and infectivity were recovered by almost 100% in comparison with the control (Fig. 4b, d and Table 2); viral binding was recovered up to 70% (Fig. 4c). In sum, the recovery of viral activities following cholesterol replenishment strongly supports the notion that the inhibitory effects of MβCD treatment on NDV activities would be due specifically to cholesterol depletion per se, and that this effect would be reversible.
Fig. 4.

Effect of cellular cholesterol replenishment on NDV activities. (a) Cholesterol-depleted cells were incubated with increasing concentrations of cholesterol for 1 h at 37 °C. Cholesterol was extracted and quantified as described in Materials and methods. Data are expressed as percentages of total cholesterol with respect to untreated control cells. Data are means ± standard deviations of three independent experiments. (b) The fusion activity of R18-labeled NDV with replenished-cholesterol cells was analyzed by fluorescence microscopy, as detailed in the legend to Fig. 1b. (c) After cellular cholesterol replenishment, NDV at a moi of 5 was allowed to bind cells for 1 h at 4 °C. Then, the binding of NDV to target cells was analyzed by a FACS-based immunoassay as detailed in Materials and methods. The negative control corresponds to cells in the absence of virus. (d) Viral infectivity was analyzed by infecting control, cholesterol-depleted and cholesterol replenished ELL-0 cells with 1 moi of the recombinant rNDV-F3aa-mRFP for 1 h at room temperature. After 24 h at 37 °C, the cells were observed under an Olympus IX51 inverted fluorescence microscope with a 10 × objective. Quantification of infectivity was accomplished by measuring the area of red fluorescence (infected cells that express the RFP) in pixels referred to the area of nuclei (blue fluorescence) of the field in three to seven random fields. Areas were quantified using the analysis tool in ImageJ software.
Table 2.
Quantification of the effect of cholesterol replenishment on NDV activities.a
| MβCD concentration + cholesterol concentration |
|||
|---|---|---|---|
| 10 mM | 10 mM + 100 μM | 10 mM + 200 μM | |
| Fusionb | 59.9 ± 8.78 | 90.0 ± 10.45 | 96.1 ± 7.96 |
| Bindingb | 47.9 ± 10.27 | 62.1 ± 12.35 | 69.9 ± 14.41 |
| Infectivityc | 46.6 | 101.6 | 112.7 |
Data are percentages of activity from control untreated cells.
Means ± SD of three or four independent experiments.
Data from experiments as those shown in Fig. 4d, means of two independent experiments.
The effect of MβCD treatment on the synthesis of viral proteins in NDV-infected cells was also analyzed (Fig. 5 ). In these experiments, viral protein synthesis was assayed by immunoblot analysis using a rabbit polyclonal anti-NDV antibody. ELL-0 cells were infected with NDV at 5 mois. At 24 h post-infection, cell extracts were analyzed by Western blot as detailed in Materials and methods. As can be seen, in 10 mM MβCD-treated cells the synthesis of viral F and NP proteins was reduced. The addition of cholesterol to MβCD-treated cells prior to virus infection led to an increase in the synthesis of viral proteins as compared to MβCD-treated ELL-0 cells. The levels of cellular GAPDH were unaffected by MβCD treatment or virus infection. These results indicated that the synthesis of viral proteins was reduced in cholesterol-depleted cells, in support of the infectivity inhibition data shown in Fig. 3. We observed a certain stimulatory effect of protein expression in the presence of 200 μM exogenous cholesterol, which correlates with some enhancement of viral infectivity (Table 2). This enhancement might be related to the increase of cholesterol in the cell membrane above normal values (Fig. 4a), although further research is needed to clarify the significance of this enhancement.
Fig. 5.

Effect of depletion and replenishment of cellular cholesterol on viral protein expression in NDV-infected ELL-0 cells. Cells were either untreated or treated with different concentrations of MβCD for 1 h at 37 °C, after which they were incubated or not in the presence of exogenous cholesterol for an additional hour at 37 °C and infected with NDV at 5 mois. Total cell protein extracts were separated by SDS-PAGE and analyzed by Western blot, using polyclonal anti-NDV antibodies. The viral protein band belonging to F and NP is shown. As a control, the expression of cellular GAPDH was analyzed.
Since it is reported that MβCD-driven removal of cholesterol led to a disruption of the cholesterol-rich DRMs of lipid rafts, we next wished to determine whether NDV might localize to these membrane microdomains during entry. We analyzed the association of viral proteins with isolated DRMs from ELL-0 cells by treatment with TX100 at low temperatures, followed by discontinuous OptiPrep flotation gradients, as detailed in Materials and methods. As a control, caveolin, a protein raft marker, was mainly present in the detergent-resistant fractions 5 and 6, whereas β-tubulin was mainly found in the detergent-soluble membrane fractions 11 and 12 (Fig. 6a). For some samples, NDV was bound to cells for 1 h at 4 °C (Fig. 6a, HN 4 °C); for other samples, after virus binding for 1 h at 4 °C, cells were shifted to 37 °C for an additional hour (Fig. 6a, HN 4 °C/37 °C). Then, the cells were lysed and layered at the bottom of a 40%–30%–5% OptiPrep density gradient. Fractions were collected from the top of the gradient and analyzed by Western blotting, probing with a polyclonal anti-NDV antibody. We found that after virus binding at 4 °C, HN was distributed in both the low density Triton-insoluble fractions 5 and 6 as well as in the detergent-soluble fractions. Nevertheless, after virus–cell samples shifting to 37 °C, the association of HN to raft fractions was limited to fraction 6. These results were also corroborated in HeLa cells, where HN protein was distributed in both the raft and non-raft fractions (Fig. 6b, HN 4 °C/37 °C). After MβCD incubation of cells (Fig. 6b, HN 10 mM MβCD), the viral protein concentration was reduced and was mainly associated with the non-raft fractions. After the replenishment of cholesterol (Fig. 6b, HN 10 mM MβCD + 200 μM Cho), the distribution of HN in the flotation gradient fractions was as observed in the case of the control cells (Fig. 6b HN 4 °C/37 °C).
Fig. 6.

Distribution of NDV HN protein in DRMs. Membrane microdomains were isolated from ELL-0 (a) or HeLa (b) cells by OptiPrep density gradient centrifugation as detailed in Materials and methods. Caveolin and β-tubulin were used as positive and negative controls respectively for lipid raft association. (a) NDV alone, distribution of HN protein of 5 mois of purified NDV; HN (4 °C) is from cells incubated with 25 mois of NDV for 1 h at 4 °C; in HN (4 °C/37 °C), cells were incubated NDV for 1 h at 4 °C and then shifted to 37 °C for 1 h. (b) In HN (4 °C/37 °C), cells were incubated with NDV for 1 h at 4 °C and then shifted to 37 °C for 1 h; in HN (10 mM MβCD), prior to viral infection the cells were treated with 10 mM MβCD; in HN (10 mM MβCD + 200 μM cholesterol), cells were incubated with 10 mM MβCD and then with 200 μM cholesterol prior to viral infection. HN was detected by Western blot probing with an anti-NDV polyclonal antibody.
4. Discussion
Cholesterol and lipid raft domains have been implicated in several stages of the life cycle of different animal virus families (reviewed in [9], [11]). The involvement of lipid rafts in viral assembly and budding has been demonstrated mainly for enveloped viruses, including HIV and influenza [9], [47]. Additionally, cholesterol and lipid rafts have been implicated in viral entry [9], [10], a physical association between viral glycoproteins and cellular membrane lipid rafts having been reported [21], [48]. For viruses that use caveolar/lipid raft endocytic pathways (for a review of viral entry by endocytosis, see [49]), the dependence of the viruses on cholesterol has been related to the integrity of their portal of entry, i.e., lipid rafts.
In the present study, we have demonstrated that NDV requires cell membrane cholesterol for optimal infection of ELL-0 cells. This is supported by several lines of evidence: i) MβCD treatment of cells partially inhibited NDV binding, fusion and infectivity; ii) the effect of cholesterol removal was reversible; iii) the effect of MβCD treatment following NDV absorption was not significant, suggesting that cholesterol must be essential during the initial virus entry stages; iv) cholesterol depletion led to a reduction in viral protein synthesis in infected cells; and v) the partial association of the HN NDV attachment protein to lipid DRMs.
Among their different functions, membrane rafts exert a concentrating effect on proteins. Receptors and coreceptors of several viruses have been reported to be associated with lipid rafts on cell membranes such, as CD4 for HIV-1 [50]. During viral attachment, protein recruitment would facilitate protein interactions, leading to viral entry either through a concentration of viral receptors in lipid rafts and/or through a concentration of viral envelope proteins facilitating the formation of the viral protein fusion scaffold [48]. The dependence of the binding of HIV-1 envelope gp120 glycoprotein to lipid rafts on cholesterol strongly supports the idea that raft-colocalized receptors would be directly involved in virus entry [50], [51], [52]. Similarly, for NDV it could be speculated that rafts might act as a platform for local concentrations of receptors and coreceptors, such as glycosphingolipids and/or glycoproteins. During the very early binding steps, a percentage of HN would colocalize to lipid rafts (Fig. 6a, HN 4 °C) shifting to non-raft fractions as the viral–cell interaction progresses (Fig. 6a, HN 4 °C/37 °C). This could explain the negative effect of cholesterol removal on NDV binding. The partial colocalization of viral proteins to DRMs during binding suggests a weak or partial interaction with rafts, as proposed for other viruses [21], [22]. The different patterns of lectin staining in control and cholesterol-depleted cells suggest that a pool of sialic acid glycoconjugates recognized by NDV might be associated with lipid rafts and that could be modified after cholesterol removal, whereas sialic acid concentrations at the cell surface would not be modified (Fig. 2). Surprisingly, the data presented here differ from our previous data, which showed that most HN was located in the DRM fractions after viral binding to COS-7 cells [45]. These differences might be cell-dependent, ELL-0 avian fibroblasts being the natural viral host. Additionally, the formation and enlargement of a fusion pore requires the concerted action of several viral proteins [53]. Therefore, the disruption of lipid rafts might hinder the assembly of the viral envelope glycoproteins needed for the formation of the fusion scaffold [54]. In this sense, it has been proposed that the increase in the antiviral potency of cholesterol-tagged-peptides derived from the C-terminal heptad repeat regions of viral fusion proteins, including those of paramyxoviruses, would be related to the preconcentration or targeting of these peptides to lipid rafts [55], [56].
Cholesterol removal and/or lipid raft disruption negatively affect the caveolae- and lipid raft-mediated endocytosis that is used as pathway of entry by several viruses [57], [58], [59]. Cholesterol has also been implicated as a regulatory factor in the entry of viruses that use clathrin and non-clathrin non caveolar/lipid raft endocytic pathways [49]. Recently, Melikyan's team [60] have demonstrated that HIV productively infects cells via clathrin-mediated endocytosis, which helps to explain the dependence of HIV on target membrane cholesterol [61]. Previously, we have reported the colocalization of NDV with caveolin and with the early endosome marker EEA1, leading us to propose that a certain percentage of the virus manages to penetrate the cell through caveolin-dependent endocytic pathways [3]. In this context, it is possible that removal of the cholesterol could negatively affect NDV entry by altering endocytic pathways. The absence of a total blockade of virus–cell fusion and infectivity after MβCD treatment (Table 1), as well as the partial association of HN with DRM fractions, may suggest that NDV entry would take place in both raft and non-raft environments. Nevertheless, further research into how the association of NDV with lipid rafts facilitates viral entry is required. Our data indicate a stronger effect of cholesterol removal on NDV binding than on fusion (Table 1). Moreover, after cholesterol replenishment, fusion and infectivity were almost completely recovered, whereas binding was not (Table 2). Therefore, it could be argued that some non-productive binding sites were permanently removed after MβCD treatment, whereas productive binding sites were recovered after cholesterol replenishment, allowing viral fusion and infectivity to occur efficiently.
NDV entry might also depend on the presence of cholesterol not related to lipid rafts, but also on membrane fluidity; the stabilization the local bilayer bending that takes place during membrane fusion [62]; or on some specific requirement for viral protein activities [11]. In addition, raft association might be required for virus-induced signal pathways leading to virus entry and replication [63], [64]. In Sendai virus, another paramyxovirus, the role of AKT1 and Raf/MEK/ERK cascades during viral fusion has been recently reported [65]. Moreover, it has been reported that cholesterol removal alters the actin cytoskeleton [66], which might negatively affect viral interaction with cells since cortical actin has been implicated in virus entry [49].
For some viruses, including paramyxoviruses [30], the dependence on cholesterol did not appear to correlate with the interaction with lipid rafts in the target membranes but with the cholesterol content of the viral envelope [11]. Additionally, some viruses require cholesterol in both viral and cell membranes [21]. Cholesterol is the major neutral lipid component of the NDV envelope present at a high cholesterol/phospholipid ratio [67]. Nevertheless, Morrison's team [35] have reported that viral envelope cholesterol is not essential for NDV infectivity, whereas cellular lipid rafts have been implicated in viral assembly and release.
In sum, our data show that cholesterol in the target membrane is required for optimal NDV fusion and infectivity, suggesting that the association of NDV proteins with lipid rafts might enhance viral entry, although further studies are needed to understand the mechanism(s) involved in the cholesterol dependence of NDV entry. A clear understanding of the role of lipid membrane components in viral entry will be useful for designing antiviral agents and vaccines.
The following are the supplementary materials related to this article.
R18 transfer from virus to target cell after virus–cell fusion. R18-labeled NDV was bound to MβCD-treated cells and untreated cells (control) for 1 h at 4 °C, after which they were allowed to fuse for 1 h at 37 °C. Nuclei were stained with Hoechst 33258 (10 μg ml− 1). Photographs were taken under an Olympus IX51 inverted fluorescence microscope using the 40 × objective.
Acknowledgments
This work was partially supported by grants from Junta de Castilla y León (SA009A08) to I.M.B. and from the Fondo de Investigaciones Sanitarias (FIS) (PI08/1813) cofinanced by FEDER funds from the EU to E.V. We thank Dr. Adolfo García-Sastre for providing recombinant rNDV-F3aa-mRFP NDV, and anti-HN and polyclonal anti-NDV antibodies. Thanks are also due to N. Skinner for language corrections.
References
- 1.Villar E., Barroso I.M. Role of sialic acid-containing molecules in paramyxovirus entry into the host cell: a minireview. Glycoconj. J. 2006;23:5–17. doi: 10.1007/s10719-006-5433-0. [DOI] [PubMed] [Google Scholar]
- 2.Lamb R.A., Jardetzky T.S. Structural basis of viral invasion: lessons from paramyxovirus F. Curr. Opin. Struct. Biol. 2007;17:427–436. doi: 10.1016/j.sbi.2007.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cantin C., Holguera J., Ferreira L., Villar E., Muñoz-Barroso I. Newcastle disease virus may enter cells by caveolae-mediated endocytosis. J. Gen. Virol. 2007;88:559–569. doi: 10.1099/vir.0.82150-0. [DOI] [PubMed] [Google Scholar]
- 4.Lamb R.A., Parks G.D. Paramyxoviridae: The Viruses and Their Replication. In: Knipe D.M., Howley P.M., editors. 1[5th] Wolters Kluwer/Lippincott Williams & Wilkins; 2007. pp. 1449–1496. [Google Scholar]
- 5.Ayllon J., Villar E., Muñoz-Barroso I. Mutations in the ectodomain of Newcastle disease virus fusion protein confer a hemagglutinin-neuraminidase-independent phenotype. J. Virol. 2010;84:1066–1075. doi: 10.1128/JVI.01473-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lingwood D., Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 7.Parton R.G., Simons K. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 2007;8:185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 8.Simons K., Gerl M.J. Revitalizing membrane rafts: new tools and insights. Nat. Rev. Mol. Cell Biol. 2010;11:688–699. doi: 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- 9.Chazal N., Gerlier D. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 2003;67:226–237. doi: 10.1128/MMBR.67.2.226-237.2003. (table) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rawat S.S., Viard M., Gallo S.A., Rein A., Blumenthal R., Puri A. Modulation of entry of enveloped viruses by cholesterol and sphingolipids (Review) Mol. Membr. Biol. 2003;20:243–254. doi: 10.1080/0968768031000104944. [DOI] [PubMed] [Google Scholar]
- 11.Teissier E., Pecheur E.I. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur. Biophys. J. 2007;36:887–899. doi: 10.1007/s00249-007-0201-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danthi P., Chow M. Cholesterol removal by methyl-beta-cyclodextrin inhibits poliovirus entry. J. Virol. 2004;78:33–41. doi: 10.1128/JVI.78.1.33-41.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson H.A., Chen Y., Norkin L.C. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell. 1996;7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pelkmans L., Kartenbeck J., Helenius A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001;3:473–483. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 15.Choi K.S., Aizaki H., Lai M.M. Murine coronavirus requires lipid rafts for virus entry and cell–cell fusion but not for virus release. J. Virol. 2005;79:9862–9871. doi: 10.1128/JVI.79.15.9862-9871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li G.M., Li Y.G., Yamate M., Li S.M., Ikuta K. Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microbes. Infect. 2007;9:96–102. doi: 10.1016/j.micinf.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vela E.M., Zhang L., Colpitts T.M., Davey R.A., Aronson J.F. Arenavirus entry occurs through a cholesterol-dependent, non-caveolar, clathrin-mediated endocytic mechanism. Virology. 2007;369:1–11. doi: 10.1016/j.virol.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bron R., Wahlberg J.M., Garoff H., Wilschut J. Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J. 1993;12:693–701. doi: 10.1002/j.1460-2075.1993.tb05703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Y.E., Cassese T., Kielian M. The cholesterol requirement for sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J. Virol. 1999;73:4272–4278. doi: 10.1128/jvi.73.5.4272-4278.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung C.S., Huang C.Y., Chang W. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 2005;79:1623–1634. doi: 10.1128/JVI.79.3.1623-1634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bender F.C., Whitbeck J.C., Ponce d.L., Lou H., Eisenberg R.J., Cohen G.H. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 2003;77:9542–9552. doi: 10.1128/JVI.77.17.9542-9552.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desplanques A.S., Nauwynck H.J., Vercauteren D., Geens T., Favoreel H.W. Plasma membrane cholesterol is required for efficient pseudorabies virus entry. Virology. 2008;376:339–345. doi: 10.1016/j.virol.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liao Z., Cimakasky L.M., Hampton R., Nguyen D.H., Hildreth J.E. Lipid rafts and HIV pathogenesis: host membrane cholesterol is required for infection by HIV type 1. AIDS Res. Hum. Retroviruses. 2001;17:1009–1019. doi: 10.1089/088922201300343690. [DOI] [PubMed] [Google Scholar]
- 24.Yi L., Fang J., Isik N., Chim J., Jin T. HIV gp120-induced interaction between CD4 and CCR5 requires cholesterol-rich microenvironments revealed by live cell fluorescence resonance energy transfer imaging. J. Biol. Chem. 2006;281:35446–35453. doi: 10.1074/jbc.M607302200. [DOI] [PubMed] [Google Scholar]
- 25.Bavari S., Bosio C.M., Wiegand E., Ruthel G., Will A.B., Geisbert T.W., Hevey M., Schmaljohn C., Schmaljohn A., Aman M.J. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J. Exp. Med. 2002;195:593–602. doi: 10.1084/jem.20011500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu X., Xiong Y., Silver J. Asymmetric requirement for cholesterol in receptor-bearing but not envelope-bearing membranes for fusion mediated by ecotropic murine leukemia virus. J. Virol. 2002;76:6701–6709. doi: 10.1128/JVI.76.13.6701-6709.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pietiainen V., Marjomaki V., Upla P., Pelkmans L., Helenius A., Hyypia T. Echovirus 1 endocytosis into caveosomes requires lipid rafts, dynamin II, and signaling events. Mol. Biol. Cell. 2004;15:4911–4925. doi: 10.1091/mbc.E04-01-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beer C., Andersen D.S., Rojek A., Pedersen L. Caveola-dependent endocytic entry of amphotropic murine leukemia virus. J. Virol. 2005;79:10776–10787. doi: 10.1128/JVI.79.16.10776-10787.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Empig C.J., Goldsmith M.A. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 2002;76:5266–5270. doi: 10.1128/JVI.76.10.5266-5270.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imhoff H., von Messling V., Herrler G., Haas L. Canine distemper virus infection requires cholesterol in the viral envelope. J. Virol. 2007;81:4158–4165. doi: 10.1128/JVI.02647-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gutierrez-Ortega A., Sanchez-Hernandez C., Gomez-Garcia B. Respiratory syncytial virus glycoproteins uptake occurs through clathrin-mediated endocytosis in a human epithelial cell line. Virol. J. 2008;5:127. doi: 10.1186/1743-422X-5-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Werling D., Hope J.C., Chaplin P., Collins R.A., Taylor G., Howard C.J. Involvement of caveolae in the uptake of respiratory syncytial virus antigen by dendritic cells. J. Leukoc. Biol. 1999;66:50–58. doi: 10.1002/jlb.66.1.50. [DOI] [PubMed] [Google Scholar]
- 33.Kolokoltsov A.A., Deniger D., Fleming E.H., Roberts N.J., Jr., Karpilow J.M., Davey R.A. Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosome formation for infection by respiratory syncytial virus. J. Virol. 2007;81:7786–7800. doi: 10.1128/JVI.02780-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laliberte J.P., McGinnes L.W., Peeples M.E., Morrison T.G. Integrity of membrane lipid rafts is necessary for the ordered assembly and release of infectious Newcastle disease virus particles. J. Virol. 2006;80:10652–10662. doi: 10.1128/JVI.01183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laliberte J.P., McGinnes L.W., Morrison T.G. Incorporation of functional HN-F glycoprotein-containing complexes into Newcastle disease virus is dependent on cholesterol and membrane lipid raft integrity. J. Virol. 2007;81:10636–10648. doi: 10.1128/JVI.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.San Roman K., Villar E., Muñoz-Barroso I. Mode of action of two inhibitory peptides from heptad repeat domains of the fusion protein of Newcastle disease virus. Int. J. Biochem. Cell Biol. 2002;34:1207–1220. doi: 10.1016/s1357-2725(02)00045-6. [DOI] [PubMed] [Google Scholar]
- 37.Keller P., Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J. Cell Biol. 1998;140:1357–1367. doi: 10.1083/jcb.140.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang H., Li Y., Sadaoka T., Tang H., Yamamoto T., Yamanishi K., Mori Y. Human herpesvirus 6 envelope cholesterol is required for virus entry. J. Gen. Virol. 2006;87:277–285. doi: 10.1099/vir.0.81551-0. [DOI] [PubMed] [Google Scholar]
- 39.Connolly S.A., Lamb R.A. Paramyxovirus fusion: real-time measurement of parainfluenza virus 5 virus–cell fusion. Virology. 2006;355:203–212. doi: 10.1016/j.virol.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 40.Mibayashi M., Martinez-Sobrido L., Loo Y.M., Cardenas W.B., Gale M., Jr., Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 2007;81:514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell R.E., Tour O., Palmer A.E., Steinbach P.A., Baird G.S., Zacharias D.A., Tsien R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. U. S. A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Imberty A., Gautier C., Lescar J., Perez S., Wyns L., Loris R. An unusual carbohydrate binding site revealed by the structures of two Maackia amurensis lectins complexed with sialic acid-containing oligosaccharides. J. Biol. Chem. 2000;275:17541–17548. doi: 10.1074/jbc.M000560200. [DOI] [PubMed] [Google Scholar]
- 43.Nokhbeh M.R., Hazra S., Alexander D.A., Khan A., McAllister M., Suuronen E.J., Griffith M., Dimock K. Enterovirus 70 binds to different glycoconjugates containing alpha2,3-linked sialic acid on different cell lines. J. Virol. 2005;79:7087–7094. doi: 10.1128/JVI.79.11.7087-7094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thorp E.B., Gallagher T.M. Requirements for CEACAMs and cholesterol during murine coronavirus cell entry. J. Virol. 2004;78:2682–2692. doi: 10.1128/JVI.78.6.2682-2692.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anastasia L., Holguera J., Bianchi A., D'Avila F., Papini N., Tringali C., Monti E., Villar E., Venerando B., Muñoz-Barroso I., Tettamanti G. Over-expression of mammalian sialidase NEU3 reduces Newcastle disease virus entry and propagation in COS7 cells. Biochim. Biophys. Acta. 2008;1780:504–512. doi: 10.1016/j.bbagen.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 46.Hao M., Mukherjee S., Maxfield F.R. Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13072–13077. doi: 10.1073/pnas.231377398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ono A., Freed E.O. Role of lipid rafts in virus replication. Adv. Virus Res. 2005;64:311–358. doi: 10.1016/S0065-3527(05)64010-9. [DOI] [PubMed] [Google Scholar]
- 48.Umashankar M., Sanchez-San Martin C., Liao M., Reilly B., Guo A., Taylor G., Kielian M. Differential cholesterol binding by class II fusion proteins determines membrane fusion properties. J. Virol. 2008;82:9245–9253. doi: 10.1128/JVI.00975-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mercer J., Schelhaas M., Helenius A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010;79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 50.Popik W., Alce T.M., Au W.C. Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J. Virol. 2002;76:4709–4722. doi: 10.1128/JVI.76.10.4709-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manes S., del Real G., Lacalle R.A., Lucas P., Gomez-Mouton C., Sanchez-Palomino S., Delgado R., Alcami J., Mira E., Martinez A. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 2000;1:190–196. doi: 10.1093/embo-reports/kvd025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen D.H., Giri B., Collins G., Taub D.D. Dynamic reorganization of chemokine receptors, cholesterol, lipid rafts, and adhesion molecules to sites of CD4 engagement. Exp. Cell Res. 2005;304:559–569. doi: 10.1016/j.yexcr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 53.Melikyan G.B. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology. 2008;5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blumenthal R., Sarkar D.P., Durell S., Howard D.E., Morris S.J. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell–cell fusion events. J. Cell Biol. 1996;135:63–71. doi: 10.1083/jcb.135.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ingallinella P., Bianchi E., Ladwa N.A., Wang Y.J., Hrin R., Veneziano M., Bonelli F., Ketas T.J., Moore J.P., Miller M.D., Pessi A. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. U. S. A. 2009;106:5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Porotto M., Yokoyama C.C., Palermo L.M., Mungall B., Aljofan M., Cortese R., Pessi A., Moscona A. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 2010;84:6760–6768. doi: 10.1128/JVI.00135-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parton R.G., Richards A.A. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic. 2003;4:724–738. doi: 10.1034/j.1600-0854.2003.00128.x. [DOI] [PubMed] [Google Scholar]
- 58.Pelkmans L., Helenius A. Insider information: what viruses tell us about endocytosis. Curr. Opin. Cell Biol. 2003;15:414–422. doi: 10.1016/s0955-0674(03)00081-4. [DOI] [PubMed] [Google Scholar]
- 59.Marsh M., Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miyauchi K., Kim Y., Latinovic O., Morozov V., Melikyan G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137:433–444. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viard M., Parolini I., Sargiacomo M., Fecchi K., Ramoni C., Ablan S., Ruscetti F.W., Wang J.M., Blumenthal R. Role of cholesterol in human immunodeficiency virus type 1 envelope protein-mediated fusion with host cells. J. Virol. 2002;76:11584–11595. doi: 10.1128/JVI.76.22.11584-11595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chernomordik L. Non-bilayer lipids and biological fusion intermediates. Chem. Phys. Lipids. 1996;81:203–213. doi: 10.1016/0009-3084(96)02583-2. [DOI] [PubMed] [Google Scholar]
- 63.Raghu H., Sharma-Walia N., Veettil M.V., Sadagopan S., Caballero A., Sivakumar R., Varga L., Bottero V., Chandran B. Lipid rafts of primary endothelial cells are essential for Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8-induced phosphatidylinositol 3-kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J. Virol. 2007;81:7941–7959. doi: 10.1128/JVI.02848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Das S., Chakraborty S., Basu A. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3′ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells. J. Neurochem. 2010;115:537–549. doi: 10.1111/j.1471-4159.2010.06951.x. [DOI] [PubMed] [Google Scholar]
- 65.Sharma N.R., Mani P., Nandwani N., Mishra R., Rana A., Sarkar D.P. Reciprocal regulation of AKT and MAP kinase dictates virus–host cell fusion. J. Virol. 2010;84:4366–4382. doi: 10.1128/JVI.01940-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kwik J., Boyle S., Fooksman D., Margolis L., Sheetz M.P., Edidin M. Membrane cholesterol, lateral mobility, and the phosphatidylinositol 4,5-bisphosphate-dependent organization of cell actin. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13964–13969. doi: 10.1073/pnas.2336102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muñoz-Barroso I., Cobaleda C., Zhadan G., Shnyrov V., Villar E. Dynamic properties of Newcastle Disease Virus envelope and their relations with viral hemagglutinin-neuraminidase membrane glycoprotein. Biochim. Biophys. Acta. 1997;1327:17–31. doi: 10.1016/s0005-2736(97)00040-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
R18 transfer from virus to target cell after virus–cell fusion. R18-labeled NDV was bound to MβCD-treated cells and untreated cells (control) for 1 h at 4 °C, after which they were allowed to fuse for 1 h at 37 °C. Nuclei were stained with Hoechst 33258 (10 μg ml− 1). Photographs were taken under an Olympus IX51 inverted fluorescence microscope using the 40 × objective.