

Abstract
Study objectives
The inflammatory responses and associated clinical severity of COPD exacerbations are greatly variable, and the determinants of these factors are poorly understood. We examined the hypothesis that bacteria and viruses may modulate this heterogeneity and that interactions between bacterial and viral infection may affect changes in airway bacterial load and the clinical features and inflammatory responses of exacerbations in patients with COPD.
Design
Prospective cohort study.
Setting
Outpatient Department, London Chest Hospital, London, UK.
Patients
Thirty-nine patients with COPD.
Measurements
We prospectively studied 56 COPD exacerbations, obtaining clinical data and paired sputum and serum samples at baseline and exacerbation. Qualitative and quantitative microbiology, polymerase chain reaction detection for rhinovirus, and estimation of cytokine levels by enzyme-linked immunosorbent assay were performed.
Results
A total of 69.6% of exacerbations were associated with a bacterial pathogen, most commonly Haemophilus influenzae. Rhinovirus was identified in 19.6% of exacerbations. The rise in bacterial load at exacerbation correlated with the rise in sputum interleukin (IL)-8 (r = 0.37, p = 0.022) and fall in FEV1 (r = 0.35, p = 0.048). Exacerbations with both rhinovirus and H influenzae had higher bacterial loads (108.56 cfu/mL vs 108.05cfu/mL, p = 0.018) and serum IL-6 (13.75 pg/mL vs 6.29 pg/mL, p = 0.028) than exacerbations without both pathogens. In exacerbations with both cold symptoms (a marker of putative viral infection) and a bacterial pathogen, the FEV1 fall was greater (20.3% vs 3.6%, p = 0.026) and symptom count was higher (p = 0.019) than those with a bacterial pathogen alone.
Conclusions
The clinical severity and inflammatory responses in COPD exacerbations are modulated by the nature of the infecting organism: bacterial and viral pathogens interact to cause additional rises in inflammatory markers and greater exacerbation severity.
Keywords: bacteria, COPD, exacerbations, viruses
Abbreviations: CI, confidence interval; HRV, human rhinovirus; IL, interleukin; IQR, interquartile range; PCR, polymerase chain reaction; PPM, potentially pathogenic microorganism
Exacerbations of COPD are characterized by increased airway1 2 and systemic inflammation.3 However, there is marked variability in the nature of the inflammatory response at exacerbation and thus the symptoms, clinical severity, and time course of these events.4 Individual factors such as respiratory viruses5 6 7 and particular bacterial pathogens8 are associated with indexes of more severe exacerbations. However, interactions between individual pathogens and the mechanisms that underlie the heterogeneity of exacerbations are poorly understood.
Bacterial pathogens are commonly identified in the lower airway of COPD patients in the stable state.1 9 10 Airway inflammation is directly related to the number of bacteria in the lower airway in stable COPD, and greater airway bacterial load is itself a stimulus to faster disease progression.11 While it is known that airway bacterial load rises at exacerbation,9 it is not known to what extent these rises modulate changes in airway inflammation and exacerbation severity, or what factors determine changes in bacterial load.
Respiratory viruses have been implicated as important infective triggers of exacerbations,5 6 7 12 with human rhinovirus (HRV) being the most commonly identified viral pathogen.5 Virus-associated exacerbations are longer and thus more severe than nonviral exacerbations,5 but whether this is due to the direct effects of viral infection on the airway or a mechanism involving changes in lower airway bacteria is not known. We examined the hypothesis that the heterogeneity of inflammatory, symptomatic, and physiologic responses at COPD exacerbation is modulated by airway bacterial and viral infection and that a combination of these pathogens would result in greater airway and systemic inflammation and hence clinical and physiologic indexes of exacerbation severity.
Materials and Methods
Patient Selection
Patients with COPD were recruited from the outpatient department of the London Chest Hospital into the East London COPD cohort. The inclusion criteria for this study have previously been published2 3 4 5 6 11 13 14 and include a postbronchodilator FEV1 < 70% of predicted for age and height, β2-agonist reversibility < 15% of baseline and/or < 200 mL, and a FEV1/FVC ratio < 70%. Patients were assessed clinically and with chest radiography at recruitment to ensure the absence of other significant respiratory disease. Patients were observed for this study from April 2001 to July 2002. Ethics approval for the study was obtained from the East London and City Health Authority Research Ethics committee; all patients gave written informed consent. This patient cohort has been the subject of previous articles2 3 4 5 6 11 on various aspects of COPD exacerbation.
Diary Card Monitoring and Follow-up
At recruitment, patients were taught how to record on diary cards each morning postbronchodilator peak expiratory flow (Mini-Wright; Clement Clark International; Harlow, UK). Patients recorded a change in their symptoms using a letter-annotated system. When well or stable, the patients were instructed not to record any of the symptom letters in the diary. However, when they perceived an increase over their normal, stable condition in symptoms (major and minor, see below), they noted the corresponding symptom letter on their diary card. Therefore, the patients recorded symptom letters if a symptom was perceived as worse, eg, dyspnea, or of new onset, eg, a sore throat (as the latter is not usually present).
Stable State
Patients were reviewed at recruitment and with their diary cards every 3 months in the study clinic to monitor compliance with data collection and to record changes in medication and baseline lung function. A review of diary cards was utilized to ensure that stable sampling was performed when subjects had been clear of exacerbation symptoms and had completed any exacerbation treatment for at least 6 weeks.
Exacerbations
Patients were encouraged to report symptom changes to the study team; they were assessed within 24 to 48 h in the study clinic by a respiratory physician prior to initiation of therapy for the exacerbation. The diagnosis of an exacerbation was based on symptomatic criteria previously validated by our group.2 3 4 5 6 An exacerbation was defined as the presence for at least 2 consecutive days of increase in any two major symptoms (dyspnea, sputum purulence, sputum amount) or increase in one major and one minor symptom (wheeze, sore throat, cough, symptoms of a common cold). Exacerbation symptoms were binary coded as present or absent, and the sum of these at exacerbation onset was termed the symptom count, which has been validated as a marker of clinical severity.4 Lung function measurement and sputum and blood sampling were performed on patients prior to the initiation of exacerbation treatment.
Measurement of Lung function
Lung function was measured with a rolling seal spirometer (SensorMedics; Yorba Linda, CA). Lung function measurements were obtained between 9:30 am and 11:30 am, 1 h after the patient's usual bronchodilator medication. At least three spirometry readings were obtained at each visit, and the best performance was recorded.
Sputum and Blood Sampling
Sputum was sampled if the subject met criteria for the stable state at the 3-month review and also at presentation of exacerbation. Immediately following lung function measurement, the patients were asked to spontaneously expectorate sputum into a sterile container. Patients unable to produce a sample of sputum spontaneously underwent sputum induction.13
Once a sample was obtained, sputum plugs were separated from saliva using sterile forceps, and one third of the sputum was taken and analyzed for quantitative bacterial culture.15 The remainder was homogenized and centrifuged, and aliquots of supernatant were stored at − 70°C for later cytokine analysis.2 11 13 Sputum samples containing < 25 squamous epithelial cells per low-power field and > 25 leukocytes per high-power field were accepted for analysis. An aliquot of phosphate-buffered saline solution-processed sputum was frozen at – 80°C for subsequent RNA extraction and polymerase chain reaction (PCR). The remainder was analyzed for inflammatory cytokines.2 11 13 Sputum interleukin (IL)-6 and IL-8 levels were measured using an enzyme-linked immunosorbent assay (R&D Systems; Abingdon, UK).2 Contemporaneous blood samples were obtained, centrifuged at 4°C, and serum decanted and stored at − 80°C for subsequent analysis of IL-6 levels using an enzyme-linked immunosorbent assay (R&D Systems).
Quantitative Bacterial Analysis
Samples were processed homogenized. Tenfold serial dilutions of the homogenized sample were made in brain heart infusion broth, and 100-μL aliquots were plated out onto the surface of a range of different media, including blood agar, chocolate agar, MacConkey agar, and cysteine lactose electrolyte-deficient agar. These were incubated for 18 to 36 h at 37°C in an atmosphere of air + 5% carbon dioxide. After incubation, bacterial colonies were counted and subcultured for identification by standard methods.15 Haemophilus influenzae and Haemophilus parainfluenzae were identified and differentiated by their growth patterns on peptone agar on which discs containing nicotinamide adenosine dinucleotide or haemin were placed (Oxoid Unipath; Basingstoke, UK). The number of colony forming units per milliliter of sputum was calculated from the number of colonies obtained and the dilution of the sputum. Bacteriology data are expressed as the total bacterial count in log base 10 U. Potentially pathogenic microorganisms (PPMs) are bacteria known to be common pathogens of the respiratory tract in subjects with COPD (Streptococcus pneumoniae, H influenzae, H Moraxella catarrhalis, Staphylococcus aureus, Pseudomonas aeruginosa, and other Gram-negative enteric bacteria).
RNA Extraction, Reverse Transcription, and Picornavirus PCR
RNA extraction from the sputum was performed using a standard extraction kit (Qiagen; Southampton, UK). Reverse transcription was performed using random hexamers, and picornavirus PCR was performed as previously described.6 This PCR technique has been validated in the detection of rhinovirus in these samples using confirmatory nucleic acid sequencing.6
Statistical Analysis
Normally distributed data are reported by means and SDs and skewed data are presented as medians and interquartile range (IQR). Correlations were assessed using the Pearson or Spearman correlation coefficient (two tailed), as appropriate. Continuous variables with normal distributions were compared by t test, whereas those with nonnormal distributions were compared by the Mann-Whitney U or Wilcoxon signed-ranks test. Changes in parameters from the stable state to exacerbation were assessed using a paired analysis of the stable sample data taken preceding the exacerbation studied; p values ≤ 0.05 were regarded as significant. In the analysis of changes in parameters between stable state and exacerbation, the prior stable sampling point closest to the subjects corresponding exacerbation was used creating a data set of paired baseline and exacerbation samples for each exacerbation. The analysis of group data was initially adjusted for repeated measures by selecting the first exacerbation sampled per patient (n = 39) to assess the changes in measured indexes from baseline to exacerbation. These observed changes were comparable to the larger data set of 56 exacerbations (in 39 patients), which was therefore used to compare individual exacerbation characteristics and etiologies, as in previous studies.5 Multivariate analysis was performed using a multiple linear regression analysis. Data analysis was performed using statistical software (SPSS version 10.0; SPSS; Chicago, IL).
Results
Patient Characteristics
Table 1 shows the baseline characteristics of the 39 patients in the East London COPD Cohort sampled during the study. Fifty-six paired stable and exacerbation samples were obtained from 39 patients for this analysis. Of these 39 patients, 15 were receiving long-term oxygen therapy, all patients were receiving long-term inhaled corticosteroids (median, 500 μg/d; IQR, 400 to 1,500 μg/d of beclomethasone equivalents), no patients were receiving long-term oral corticosteroids, and all patients received regular inhaled bronchodilators. The remainder of the patients did not have an exacerbation during the sampling period (n = 26), did not report an exacerbation to the study team, received antibiotic treatment before sampling, or were unable to provide an adequate sputum sample (n = 14). The sampled patients did not differ significantly in terms of baseline characteristics from those who were not sampled (Table 1).
Table 1.
Patient Baseline Characteristics (n = 39)*
| Characteristics | Data |
|---|---|
| Age, yr | 68.8 (6.9) |
| FEV1% predicted | 40.6 (15.6) |
| Pao2, kPa | 8.86 (0.89) |
| Paco2, kPa | 5.67 (0.74) |
| Smoking history, pack-yr | 41.0 (30–52) |
| Male gender, % | 60 |
| Active smokers, % | 23 |
| Chronic sputum producers, % | 43 |
Data are presented as mean (SD) or median (IQR) unless otherwise indicated.
Changes in Lung Function and Inflammatory Markers at Exacerbation
Table 2 shows the stable and exacerbation FEV1, airway bacterial load, sputum IL-6 and IL-8, and blood IL-6 levels for all the 56 sampled exacerbations and on a per-patient basis (n = 39). In both analyses, the mean FEV1 fell at exacerbation and the mean airway bacterial load rose significantly. Exacerbations were associated with increased airway inflammation in terms of sputum IL-8. The rises in levels of sputum and serum IL-6 did not reach statistical significance.
Table 2.
FEV1, Infective and Inflammatory Changes for Baseline (Stable State) and Exacerbation Sample Points and on a Per-Patient Basis (n = 39) and for All 56 Sampled Exacerbations (n = 56)*
| Variables | Baseline | Exacerbation | p Value† |
|---|---|---|---|
| Per patient (n = 39) | |||
| FEV1, L | 0.95 (0.36) | 0.87 (0.30) | 0.012 |
| Bacterial load, log cfu/mL | 7.47 (0.73) | 8.16 (0.76) | 0.001 |
| Sputum IL-8, pg/mL | 3,647 (2,930–4,466) | 4,409 (3,983–4,787) | 0.002 |
| Sputum IL-6, pg/mL | 146.0 (20.4–246.9) | 187.6 (49.9–269.1) | 0.322 |
| Serum IL-6, pg/mL | 4.73 (3.34–7.07) | 6.0 (4.25–13.18) | 0.228 |
| All patients (n = 56) | |||
| FEV1, L | 0.96 (0.37) | 0.89 (0.32) | 0.015 |
| Bacterial load, log cfu/mL | 7.50 (0.74) | 8.09 (0.76) | 0.001 |
| Sputum IL-8, pg/mL | 3,604 (2,913–4,390) | 4,288 (3,991–4,765) | 0.005 |
| Sputum IL-6, pg/mL | 146.7 (29.4–233.0) | 185.0 (50.0–280.0) | 0.477 |
| Serum IL-6, pg/mL | 4.6 (3.1–7.1) | 6.6 (4.0–11.7) | 0.136 |
Data are presented as mean (SD) or median (IQR).
Statistical comparison of baseline and exacerbation.
Airway Bacteriology
Airway bacterial load rose in all samples (n = 56) from 107.50 (0.74) log cfu/mL in the stable state to 108.09(0.76) log cfu/mL at exacerbation, and also rose significantly in data adjusted for repeated measures (n = 39) [107.47 (0.73) to 108.16 (0.76); p = 0.001]. The prevalence of PPMs rose from 48.2% at baseline to 69.6% at exacerbation (n = 56), with the remainder of samples demonstrating nonspecific bacterial growth. The most frequently isolated organism was H influenzae in 14.3% of stable and 37.5% of exacerbation samples, with S pneumoniae in 8.9% and 14.3%, M catarrhalis in 7.1% and 14.3%, H parainfluenzae in 10.7% and 0%, S aureus in 3.6% and 0%, P aeruginosa in 1.8% and 1.8%, and Gram-negative enteric bacteria in 1.8% and 1.8%, respectively; the remainder demonstrated nonspecific bacterial growth.
Relationships Between Bacterial Load, Airway Inflammation, and Lung Function Changes at Exacerbation
Changes in airway bacterial load (n = 39) were related to exacerbation severity in terms of changes in lung function and airway inflammation. The rise in airway bacterial load from baseline to exacerbation was related to the percentage fall in FEV1 (r = 0.35, p = 0.048). The magnitude of the rise in airway IL-8 at exacerbation was related to the rise in airway bacterial load (ρ = 0.37, p = 0.022).
The changes in airway and serum IL-6 observed from stable state to exacerbation were not related to changes in bacterial load (ρ = − 0.76, p = 0.649 and ρ = 0.144, p = 0.482, respectively). However, the rise in systemic inflammation was related to that of airway inflammation; the change in sputum IL-6 correlated with the change in serum IL-6 (ρ = 0.435, p = 0.023, n = 39).
Changes in airway and systemic markers of inflammation at exacerbation were modulated by existing disease severity. The observed change in sputum IL-8 at exacerbation per patient was inversely related to the baseline FEV1 (percentage of predicted) [ρ = − 0.298, p = 0.05], as was the change in sputum IL-6 (ρ = − 0.358, p = 0.02) and the change in serum IL-6 (ρ = − 0.392, p = 0.03). Thus, patients with more severe COPD exhibited greater rises in inflammation at exacerbation compared with those with more mild disease.
Effects of Individual Pathogens and Their Interactions
H influenzae
H influenzae-related exacerbations were associated with higher airway bacterial load (n = 56; 108.52 [0.39] log cfu/mL) compared to cases in which it was not isolated (107.85 (0.81) log cfu/mL; p = 0.001). There was a trend toward more severe drops in FEV1 (expressed as a percentage of baseline): − 11.91% (SD, 15.32%) with H influenzae present, vs − 1.20 without H influenzae (SD, 15.09%) [p = 0.057]. Where de novo H influenzae infection occurred (ie, H influenzae present at exacerbation but not present in stable sample), there was again a greater exacerbation bacterial load (108.56 [0.40] log cfu/mL) compared to when H influenzae was not isolated (107.81 [0.84] log cfu/mL) [p = 0.001], and the percentage fall in FEV1 was significantly worse in this group (− 14.60% [SD, 12.39%]) compared to the non-H influenzae exacerbations (− 1.17% [SD, 15.99%]) [p = 0.027].
HRV and Colds
HRV PCR findings were positive in 11 of 56 exacerbations (19.6%); cold symptoms, a measure of putative viral infections, were present in 6 of 18 cases (32.1%). The presence of cold symptoms and HRV-positive PCR sputum were related (continuity adjusted χ2, 4.11; p = 0.04). Exacerbations associated with colds were associated with a greater percentage fall in FEV1 (− 14.03%; SD, 13.91%) than those without colds (− 3.01%; SD, 16.38%) [p = 0.043].
Effect of Viral and Bacterial Infections on All Sampled Exacerbations
The observed FEV1 fall associated with colds at exacerbation was more marked in the presence of a lower airway bacterial pathogen: − 20.3% (SD, 14.81%) with both colds and a bacterial pathogen, compared to − 3.63% (SD, 5.57%) with a cold alone (p = 0.026) or − 3.13% (SD, 14.88%) with a bacterial pathogen alone (p = 0.001) [Fig 1 ]. The specific effect of the interaction between colds and bacterial pathogens was assessed with a multivariate regression analysis with the percentage of FEV1 fall at exacerbation as the dependent variable; the effect of the interaction was additional to the independent effects of each individual factor (95% confidence interval [CI], − 13.13 to − 2.09; p = 0.009).
Figure 1.
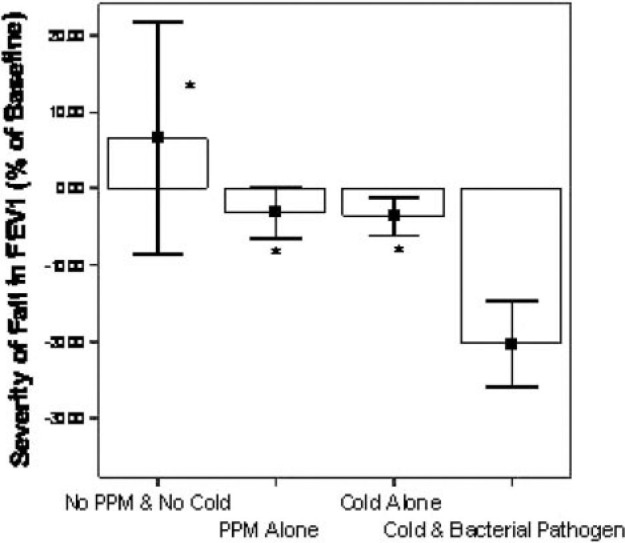
Effect of airway pathogens and pathogen combinations on percentage fall in FEV1 at exacerbation. Columns represent mean values with error bars as SEM *Significant (p < 0.05) difference between this category and cold and bacterial pathogen category (n = 56).
Similarly, exacerbation symptoms were more severe (higher symptom count at exacerbation onset) in those exacerbations associated with a PPM in the presence of cold symptoms (4.0; IQR, 3.0 to 4.5) compared to those with a PPM alone (3.0; IQR, 2.0 to 3.0) [p = 0.019] or with neither a PPM nor cold symptoms (3.0; IQR, 2.0 to 3.0) [p = 0.029; Fig 2 ].
Figure 2.
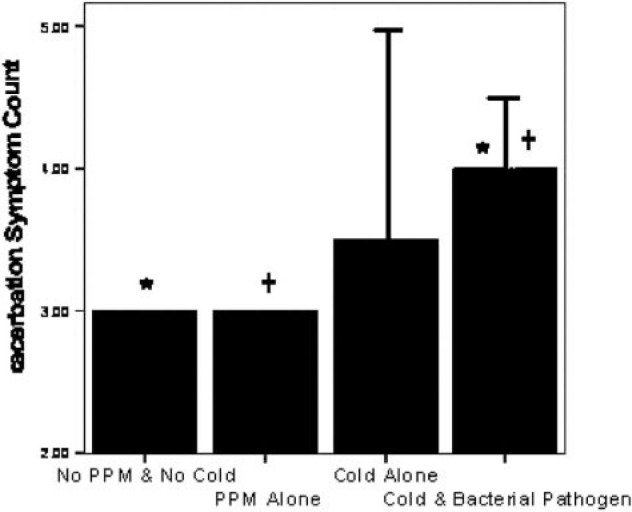
Effect of airway pathogens and pathogen combinations on symptom severity (median symptom count at exacerbation onset). Columns represent median values, bars indicate IQR, and * and + denote statistically significant (p < 0.05) differences between corresponding labeled categories (n = 56; *p = 0.029; + p = 0.019).
Exacerbations associated with both H influenzae and HRV exhibited a greater bacterial load (108.56 [0.31] log cfu/mL vs 108.05 [0.77] log cfu/mL, p = 0.018) and serum IL-6 (13.75 pg/mL; IQR, 10.53 to 16.91 pg/mL; vs 6.29 pg/mL; IQR, 3.31 to 9.75 pg/mL, p = 0.028) than those without both pathogens. The exacerbation of airway bacterial load associated with H influenzae and HRV compared to other PPMs is illustrated in Figure 3 .
Figure 3.
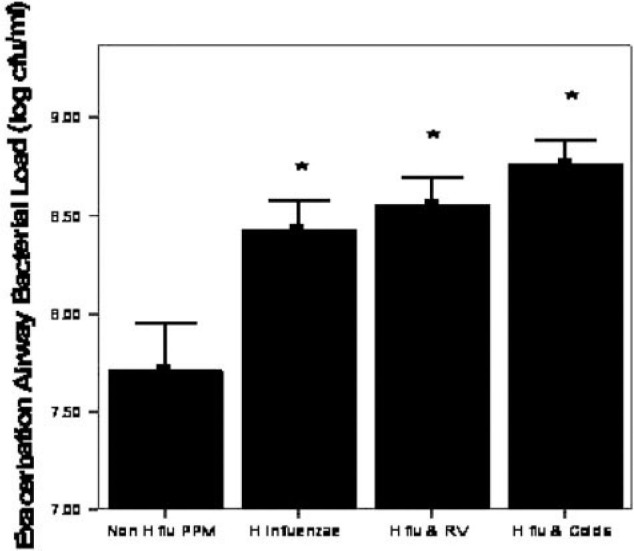
Airway bacterial load at exacerbation for different categories of isolated pathogen(n = 56). Columns represent mean values, with error bars as SEM. RV = rhinovirus; H flu = H influenzae. *Significantly different from all PPM categories.
Discussion
The results of this study show for the first time a synergistic effect of viral and bacterial infections in modulating the severity of symptoms, lung function changes, and inflammation at exacerbations of COPD. The findings demonstrate that changes in lower airway bacterial load are associated with the variability in inflammation and lung function seen at exacerbation in patients with moderate-to-severe COPD, effects that were more pronounced in proven rhinoviral and putative viral infections. These data also suggest that pathogens associated with more severe exacerbations, such as H influenzae, may act at least in part via a greater stimulus to inflammation, associated with higher airway bacterial loads. Patients with more severe disease in this study demonstrated greater rises in airway and systemic inflammation than those with milder disease. This suggests that the heterogeneous nature of exacerbation severity is dependent not only on the nature of infective triggers but also on the baseline severity of disease.
This study has been performed using the well-validated technique of daily diary card symptom recording and analysis to confirm both the diagnosis of exacerbations and also the stable state.2 3 4 5 6 11 13 14 The study design has allowed us to sample the same patients in both clinical states and to describe not only cross-sectional analyses at exacerbation but also changes from baseline, and furthermore how these changes in exacerbation parameters were modulated by the corresponding infectious agents.
We have found that that the severity of the fall in lung function and the rise in inflammation seen at exacerbation are related to the extent of the rise in airway bacterial load. A relationship between airway inflammation and airway bacterial load has previously been described in the stable state,1 16 17 18 19 20 21 22 with higher loads associated with greater falls in FEV1 over a 1-year study.11 A number of previous studies8 18 19 have identified that bacterial pathogens are commonly found in the lower airway at exacerbation with higher loads than in the stable state.9 However, the effect of rising numbers of bacteria on the nature of exacerbations has not been investigated. These findings suggest that changes in bacterial load may play a role in the heightened levels of airway inflammation characteristic of exacerbations. However, evidence for an association between changes in bacterial load and indexes of exacerbation severity does not prove causality; it is possible that changes in airway bacterial load may simply be a secondary phenomenon to other causes of inflammation. Indeed, the findings of this study show that the key changes in symptoms and lung function at exacerbation were observed when the synergistic effects of viral and bacterial infection were found. In vitro and intervention studies are required to differentiate the exact contribution of a particular pathogen or pathogens to the inflammatory and pathophysiologic changes at exacerbation.
H influenzae was found in this study, as in previous studies,8 9 10 to be the most important bacterial pathogen identified both in terms of prevalence in the stable state and at exacerbation, and in determining the airway bacterial load. H influenzae, unlike a number of other bacterial pathogens, may colonize not only the airway but the respiratory epithelium itself. H influenzae colonization has been shown to be a greater stimulus to airway inflammation than other commonly isolated pathogens.8 23 This is in agreement with the findings of our study that demonstrate that H influenzae was present in greater numbers than the other PPMs identified, and that its presence at exacerbation was associated with more severe drops in FEV1. The role of less prevalent bacterial pathogens at exacerbation, in particular, their interactions with respiratory viruses requires further study.
The stimulus of newly acquired H influenzae at exacerbation provided a greater deleterious effect on FEV1 than H influenzae-associated exacerbations in patients already having colonization with this pathogen. These findings compliment previous work19 of the role that strain changes of particular bacterial species play in the etiology of exacerbations. This has identified the role that a new antigenic stimulus to the airway immune system plays in the pathogenesis of an exacerbation. It is feasible that acquisition of a new bacterial strain or type may not only provide a direct antigenic stimulus but also overcome the established host/pathogen balance allowing bacterial proliferation, and thus a further inflammatory stimulus due to greater bacterial numbers. To date, studies to determine the possible interactions between viral infection and bacterial strain changes have not been performed; these may provide information on the complex mechanisms that result in triggering exacerbations.
Respiratory viral infection is an important trigger to the airway immune system. In our cohort of influenza-vaccinated patients, we have previously demonstrated that HRV is the most commonly isolated virus at exacerbation.5 Rhinovirus can be isolated from lower airway samples6 and is associated with greater levels of inflammation than nonviral infections.6 Similarly, we have shown that colds, a marker of putative viral infections,6 are associated with more severe exacerbations. In this study, systemic inflammation (serum IL-6), exacerbation symptoms, and lung function changes were all more severe when evidence for both bacterial and viral infection was present. It is possible that this effect may have been due to the separate additional inflammatory stimuli of two separate pathogens in the airway; however, this explanation is not supported by the multivariate analysis that indicated a synergistic effect on lung function in addition to the individual effects of each pathogen type. Furthermore, these exacerbations were associated with higher bacterial loads than when both pathogens were not present, which may suggest a synergistic interactive effect of viral infection which allows greater proliferation of airway bacteria. Viral infection therefore may impact on exacerbation severity indirectly by increasing bacterial load in addition to the direct effects of viral infection itself, eg, heightened inflammation or airway hyperresponsiveness, independent of other pathogens. While HRV is the most common virus identified at exacerbation5 and hence the target of investigation in this study, a number of other respiratory viruses have been identified in the airway during these events, for example coronavirus.5 7 The role of these other viral pathogens and atypical bacteria at exacerbation remains uncertain24 25 and requires investigation.
The mechanisms by which viral infection may facilitate airway bacterial growth are likely to be complex. However, any disruption of the innate defenses of the respiratory epithelium in a lower airway colonized with bacteria may unsettle a fine balance between host immunity and bacterial numbers. Rhinoviral infection is known to increase mucous production and neutrophilic inflammation.26 Direct evidence that rhinoviral infection increases susceptibility to bacterial adherence to airway epithelial cells, a key process in bacterial infection, is available from in vitro studies.27 Indeed, the key cell surface binding site for HRV infection, intracellular adhesion molecule-1, is itself up-regulated by HRV infection28 and by bacterial colonization29; this increase may play a key role in neutrophil elastase-mediated inflammation.30 Hence, by a number of mechanisms, viral infection may alter the immune environment that may allow either proliferation of colonizing airway bacteria or a new pathogen to infect the lower airway.
This study was performed in patients with moderate-to-severe COPD. The role of bacterial infection and therefore potential bacterial-viral interaction is likely to vary with disease severity and therefore prevalence of bacterial colonization.10 18 Indeed, we have shown that the degree of airway and systemic inflammatory response at exacerbation was related to baseline disease severity. This suggests that the severity of inflammatory response may progress with disease severity, which is in agreement with the findings of a longitudinal analysis31 of exacerbations. Further studies are required to determine if these findings can be extrapolated to COPD patients with milder disease. Indeed, the observed heterogeneity of exacerbations is likely to be further modulated by the relative frequency of particular pathogens and hence may show seasonality; this may explain differences in associated cytokine responses found in studies2 of comparable sample size. Similarly, differences in the technique of sampling, spontaneous or induced sputum, may affect the observed results. However, we have previously demonstrated the two techniques are comparable in assessing lower airway inflammation.13 Therapy must also be considered important when considering factors modulating inflammatory responses. The patients sampled for this study were all receiving inhaled steroids both at baseline and when sampled at exacerbation. It is possible that the inflammatory responses observed at exacerbation were modified by effects of this treatment.32 A statistical analysis of this effect was not feasible due to the ubiquity of inhaled steroid use in this patient group. Therefore, the modulating influences on the nature of exacerbations are numerous. It is probable that any individual factor plays a contributing rather than a definitive role in determining the nature and severity of a particular exacerbation, and furthermore that potential interactions between these factors further modulate the characteristics of these events.
The findings of this study suggest that changes in airway bacterial load, the nature of the individual infective pathogens, and interactions between multiple pathogens and the airway modulate exacerbation severity. Further studies are required to improve understanding of the pathogen/host interactions at exacerbation and indeed also in the stable state. Manipulation of this complex relationship with appropriate anti-infective and anti-inflammatory therapies may benefit COPD patients by reducing both exacerbation severity and slowing progression of this highly prevalent disease.
ACKNOWLEDGMENT
The authors thank Angela Whiley for assistance with the quantitative bacteriology and Professor Sebastian Johnston and his department for advice and tuition in PCR techniques.
Footnotes
Reproduction of this article is prohibited without written permission from the American College of Chest Physicians (www.chestjournal.org/misc/reprints.shtml).
Funding was provided by the Joint Research Board, St. Bartholomew's Hospital Special Trustees, The British Lung Foundation.
References
- 1.Hill A, Campbell EJ, Hill SL. Association between bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med. 2000;109:288–295. doi: 10.1016/s0002-9343(00)00507-6. [DOI] [PubMed] [Google Scholar]
- 2.Bhowmik A, Seemungal TAR, Sapsford RJ. Relation of sputum inflammatory markers to symptoms and physiological changes at COPD exacerbations. Thorax. 2000;55:114–120. doi: 10.1136/thorax.55.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wedzicha JA, Seemungal TAR, MacCallum PK. Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost. 2000;84:210–215. [PubMed] [Google Scholar]
- 4.Seemungal TAR, Donaldson GC, Bhowmik A. Time course and recovery of exacerbations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:1608–1613. doi: 10.1164/ajrccm.161.5.9908022. [DOI] [PubMed] [Google Scholar]
- 5.Seemungal TAR, Harper-Owen R, Bhowmik A. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1618–1623. doi: 10.1164/ajrccm.164.9.2105011. [DOI] [PubMed] [Google Scholar]
- 6.Seemungal TAR, Harper-Owen R, Bhowmik A. Detection of rhinovirus in induced sputum at exacerbation of chronic obstructive pulmonary disease. Eur Respir J. 2000;16:677–683. doi: 10.1034/j.1399-3003.2000.16d19.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohde G, Wiethege A, Borg I. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax. 2003;58:37–42. doi: 10.1136/thorax.58.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sethi S, Muscarella K, Evans N. Airway inflammation and etiology of acute exacerbations of chronic bronchitis. Chest. 2000;118:1557–1565. doi: 10.1378/chest.118.6.1557. [DOI] [PubMed] [Google Scholar]
- 9.Monso E, Ruiz J, Rosell A. Bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1995;152:1316–1320. doi: 10.1164/ajrccm.152.4.7551388. [DOI] [PubMed] [Google Scholar]
- 10.Zalacain R, Sobradillo V, Amilibia J. Predisposing factors to bacterial colonization in chronic obstructive pulmonary disease. Eur Respir J. 1999;13:343–348. doi: 10.1034/j.1399-3003.1999.13b21.x. [DOI] [PubMed] [Google Scholar]
- 11.Wilkinson TMA, Patel IS, Wilks M. Airway bacterial load and FEV1decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167:1090–1095. doi: 10.1164/rccm.200210-1179OC. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg SB, Allen M, Wilson J. Respiratory viral infections in adults with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162:167–173. doi: 10.1164/ajrccm.162.1.9911019. [DOI] [PubMed] [Google Scholar]
- 13.Bhowmik A, Seemungal TAR, Sapsford RJ. Comparison of spontaneous and induced sputum for investigation of airway inflammation in chronic obstructive pulmonary disease. Thorax. 1998;53:953–956. doi: 10.1136/thx.53.11.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilkinson TMA, Donaldson GC, Hurst JR. Early therapy improves outcomes of exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;169:1298–1303. doi: 10.1164/rccm.200310-1443OC. [DOI] [PubMed] [Google Scholar]
- 15.Barrow GI, Feltham RK. Cambridge University Press; Cambridge, UK: 1993. (Cowan and Steel's manual for the identification of medical bacteria). 3rd ed. [Google Scholar]
- 16.Patel IS, Seemungal TAR, Wilks M. Relationship between bacterial colonisation and the frequency character and severity of COPD exacerbations. Thorax. 2002;57:759–764. doi: 10.1136/thorax.57.9.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banerjee D, Khair OA, Honeybourne D. Impact of sputum bacteria on airway inflammation and health status in clinical stable COPD. Eur Respir J. 2004;23:685–691. doi: 10.1183/09031936.04.00056804. [DOI] [PubMed] [Google Scholar]
- 18.Miravitlles M, Espinosa C, Fernandez-Laso E. Relationship between bacterial flora in sputum and functional impairment in patients with acute exacerbations of COPD. Chest. 1999;116:40–46. doi: 10.1378/chest.116.1.40. [DOI] [PubMed] [Google Scholar]
- 19.Sethi S, Evans N, Brydon JB. New strains of bacteria and exacerbations of chronic obstructive pulmonary disease. N Engl J Med. 2002;347:465–471. doi: 10.1056/NEJMoa012561. [DOI] [PubMed] [Google Scholar]
- 20.Wilson R. The pathogenesis and management of bronchial infections: the vicious circle of respiratory decline. Rev Contemp Pharmacother. 1992;3:103–112. [Google Scholar]
- 21.Sethi S, Murphy TF. Bacterial infection in chronic obstructive pulmonary disease in 2000: a state of the art review. Clin Microbiol Rev. 2001;14:336–363. doi: 10.1128/CMR.14.2.336-363.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill A, Gompertz S, Stockley RA. Factors influencing airway inflammation in chronic obstructive pulmonary disease. Thorax. 2000;55:970–977. doi: 10.1136/thorax.55.11.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bresser P, Out TA, van Alphen L. Airway inflammation in non obstructive and obstructive chronic bronchitis with chronicHaemophilus influenzaeairway infection. Am J Respir Crit Care Med. 2000;162:947–952. doi: 10.1164/ajrccm.162.3.9908103. [DOI] [PubMed] [Google Scholar]
- 24.Seemungal TAR, Wedzicha JA, MacCallum PK. Chlamydia pneumoniaeand COPD exacerbation. Thorax. 2002;57:1087–1089. doi: 10.1136/thorax.57.12.1087-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blasi F, Damato S, Cosentini R. Chlamydia pneumoniaeand chronic bronchitis: association with severity and bacterial clearance following treatment. Thorax. 2002;57:672–676. doi: 10.1136/thorax.57.8.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White AJ, Gompertz S, Stockley RA. Chronic obstructive pulmonary disease: 6. The aetiology of exacerbations of chronic obstructive pulmonary disease. Thorax. 2003;58:73–80. doi: 10.1136/thorax.58.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishizuka S, Yamaya M, Suzuki T. Rhinovirus infection and Pneumococcus adhesion. J Infect Dis. 2003;188:1929–1940. doi: 10.1086/379833. [DOI] [PubMed] [Google Scholar]
- 28.Papi A, Johnston SL. Rhinovirus infection induces expression of its own receptor ICAM-1 via increased NF-κB mediated transcription. J Biol Chem. 1999;274:9707–9720. doi: 10.1074/jbc.274.14.9707. [DOI] [PubMed] [Google Scholar]
- 29.Patel IS, Roberts NJ, Lloyd-Owen SJ. Airway epithelial inflammatory responses and clinical parameters in COPD. Eur Respir J. 2003;22:94–99. doi: 10.1183/09031936.03.00093703. [DOI] [PubMed] [Google Scholar]
- 30.Nadel JA. Role of neutrophil elastase in hypersecretion during COPD exacerbations, and proposed therapies. Chest. 2000;117:386–389S. doi: 10.1378/chest.117.5_suppl_2.386s. [DOI] [PubMed] [Google Scholar]
- 31.Donaldson GC, Seemungal TAR, Patel IS. Longitudinal changes in the nature, severity and frequency of COPD exacerbations. Eur Respir J. 2003;22:931–936. doi: 10.1183/09031936.03.00038303. [DOI] [PubMed] [Google Scholar]
- 32.Patel IS, Roberts NJ, Lloyd-Owen SJ. Airway epithelial responses and clinical parameters in COPD. Eur Respir J. 2003;22:94–99. doi: 10.1183/09031936.03.00093703. [DOI] [PubMed] [Google Scholar]