

Abstract
Hurler's syndrome (HS), the most severe form of mucopolysaccharidosis type-I, causes progressive deterioration of the central nervous system and death in childhood. Allogeneic stem cell transplantation (SCT) before the age of 2 years halts disease progression. Graft failure limits the success of SCT. We analyzed data on HS patients transplanted in Europe to identify the risk factors for graft failure. We compared outcomes in 146 HS patients transplanted with various conditioning regimens and grafts. Patients were transplanted between 1994 and 2004 and registered to the European Blood and Marrow Transplantation database. Risk factor analysis was performed using logistic regression. ‘Survival’ and ‘alive and engrafted’-rate after first SCT was 85 and 56%, respectively. In multivariable analysis, T-cell depletion (odds ratio (OR) 0.18; 95% confidence interval (CI) 0.04–0.71; P=0.02) and reduced-intensity conditioning (OR 0.08; 95% CI 0.02–0.39; P=0.002) were the risk factors for graft failure. Busulfan targeting protected against graft failure (OR 5.76; 95% CI 1.20–27.54; P=0.028). No difference was noted between cell sources used (bone marrow, peripheral blood stem cells or cord blood (CB)); however, significantly more patients who received CB transplants had full-donor chimerism (OR 9.31; 95% CI 1.06–82.03; P=0.044). These outcomes may impact the safety/efficacy of SCT for ‘inborn-errors of metabolism’ at large. CB increased the likelihood of sustained engraftment associated with normal enzyme levels and could therefore be considered as a preferential cell source in SCT for ‘inborn errors of metabolism’.
Keywords: cord blood, inherited disorders, Hurler's syndrome
Introduction
Mucopolysaccharidosis type I (MPS-1) is an autosomal recessive metabolic storage disease caused by the deficiency of α-L-iduronidase, resulting in accumulation of heparan and dermatan sulfate substrates (glycosaminoglycans) in various tissues.1 Hurler's syndrome (HS) is the most severe phenotype, with symptoms appearing shortly after birth, characterized by upper airway obstruction and recurrent chest infections, hepatosplenomegaly, corneal clouding, cardiac dysfunction, skeletal abnormalities, progressive deterioration of the central nervous system and death in early childhood.1, 2, 3, 4, 5, 6, 7 Hematopoietic stem cell transplantation (SCT) can prevent the progression of HS and provides maximal benefit when performed early in life.6, 8 Enzyme replacement therapy (ERT; Aldurazyme) became recently available for the treatment of MPS-1. However, intravenously administered enzyme will not cross the blood–brain barrier; therefore, SCT remains the treatment of choice in children with HS.
The estimated number of HS patients who underwent SCT worldwide, since the early 1980s, is more than 400 (based on European Blood and Marrow Transplantation (EBMT)- and Center for International Blood and Marrow Transplant Research databases). Donor engraftment after SCT leads to a rapid reduction of obstructive airway symptoms, hepatosplenomegaly and corneal clouding. Hydrocephalus is either prevented or stabilized and hearing impairment improves in many children.9, 10, 11, 12, 13 Additionally, successful SCT averts death from cardiac dysfunction2, 5 improves growth and psychomotor development, and prolongs survival.4, 6, 7, 11, 12, 14 Significant and often progressive orthopedic anomalies persist however, despite successful SCT, often needing additional interventions.6, 11, 12
While the quality of life of successfully transplanted children seems encouraging, the relatively high rates of graft failure (15–75%)9, 10, 11, 12 and ‘transplant-related morbidity’ limit its success.9, 10, 15 Optimizing transplantation techniques resulting in less graft failure and less ‘transplant-related morbidity/mortality’ could improve the outcome of HS patients.
We now report data on 146 patients with HS transplanted in Europe between 1994 and 2004. We studied different conditioning regimens, donor types and cell sources to identify factors associated with graft failure and morbidity after SCT.
Methods
Collection of data
All HS patients reported to the EBMT-registry between January 1994 and September 2004 were included. Since the follow-up of these patients was incomplete, an additional ‘simple questionnaire’ was prepared and sent to the various centers that transplanted these patients to complete the data set on (1) basic patient characteristics, (2) basic donor information (cell source, donor relationship, human leukocyte antigen (HLA)-matching and, in the case of cord blood (CB), cell count of the product, pre-transplant ERT), (3) conditioning regimen used, (4) survival and engraftment including chimerism and enzyme activity and (5) transplant-related morbidity and mortality with particular reference to graft-versus-host disease (GvHD), veno-occlusive disease (VOD), pulmonary complications (infection, idiopathic pneumonia syndrome/diffuse alveolar hemorrhage (IPS/DAH)).
Patients
From a total of 154 patients entered in the EBMT registry, questionnaires were completed in 146 patients (82 male/64 female). These were included in the analyses of outcomes. The diagnosis of HS was confirmed on the basis of the α-L-iduronidase activity in peripheral blood leukocytes, increased excretion of glycosaminoglycans in the urine and the clinical phenotype. The median age of diagnosis was 10.5 (range 0–55) months, while the median age at transplantation was 18 (range 1–96) months. The follow-up after first SCT was 44 (median; range 6–120) months. Patients were transplanted in 16 centers: Manchester (39), Lyon (20), Dublin (20), London (15), Paris (12), Utrecht (8), Monza (6), Hannover (5), Prague (5), Vienna (4), Zurich (4), Ghent (4), Padua (1), Jena (1), Leiden (1) and Nancy (1).
Groups to be analyzed: grafts and conditioning
HLA matching was based on high-resolution (HR) typing for class I and class II (10 antigens) for bone marrow (BM) and peripheral blood stem cell (PBSC) donors, and for CB donors lower resolution criteria were used (loci A and B by serology and DRB1 by HR typing). A DPB1 mismatch was not taken into account. Mainly due to retrospective HR typing in some centers, the number of patients transplanted with ⩾2 mismatches is relatively high. For the analyses, patients were simply divided into matched or mismatched group. CB grafts identical according to the lower resolution criteria mentioned above were regarded as matched. Other factors analyzed included (a) cell source (BM, PBSC, CB), (b) donor relationship and (c) T-cell depletion (TCD).
Conditioning regimens were grouped as follows: busulfan–cyclophosphamide 200 mg/kg, busulfan with high-dose cyclophosphamide (240–260 mg/kg), busulfan-targeting, fludarabine-based myeloablative (Flud-MA) and reduced-intensity conditioning (RIC). The various subgroups within the main groups are described (in the legend of Table 2).
Table 2.
Univariate predictors of survival and being alive and engrafted
| A&E | Alive | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| n | % | OR | 95% CI | P-value | % | OR | 95% CI | P-value | |
| Overall | 146 | 56 | 85 | ||||||
| Age | 146 | 0.98 | 0.96–1.01 | 0.23 | 1.02 | 0.99–1.05 | 0.23 | ||
| Gender | 146 | 1.15 | 0.60–2.21 | 0.67 | 0.87 | 0.45–1.72 | 0.70 | ||
| Heterozygote donora | 146 | 1.46 | 0.41–5.2 | 0.90 | 0.49 | 0.055–4.32 | 0.52 | ||
| HLA disparity b | |||||||||
| Matched | 96 | 64 | 1 | 85 | 1 | ||||
| Mismatched | 50 | 42 | 0.41 | 0.21–0.84 | 0.014 | 84 | 0.78 | 0.31–1.95 | 0.59 |
| Conditioning c | |||||||||
| Bu/Cy | 68 | 53 | 1 | 87 | 1 | ||||
| Bu/Cyhi | 30 | 67 | 1.78 | 0.72–4.36 | 0.21 | 80 | 0.55 | 0.57–5.79 | 0.32 |
| Bu target | 15 | 87 | 5.76 | 1.20–27.54 | 0.028 | 87 | 0.89 | 0.17–4.73 | 0.90 |
| Flud-MA | 17 | 70 | 2.13 | 0.67–6.71 | 0.19 | 88 | 0.97 | 0.19–5.04 | 0.97 |
| RIC | 18 | 11 | 0.11 | 0.02–0.52 | 0.005 | 78 | 0.48 | 0.13–1.83 | 0.29 |
| TCD d | |||||||||
| No | 118 | 64 | 1 | 84 | 1 | ||||
| Yes | 28 | 25 | 0.19 | 0.08–0.49 | 0.001 | 89 | 1.15 | 0.36–1.95 | 0.81 |
| Donor | |||||||||
| Family | 52 | 58 | 1 | 90 | 1 | ||||
| Unrelatede | 94 | 55 | 0.91 | 0.46–1.80 | 0.78 | 82 | 2.08 | 0.72–6.01 | 0.18 |
| Source | |||||||||
| BM | 103 | 55 | 1 | 84 | 1 | ||||
| PBSC | 20 | 50 | 0.81 | 0.31–2.11 | 0.67 | 95 | 4.02 | 0.51–32.02 | 0.19 |
| CBf | 23 | 65 | 1.51 | 0.59–3.88 | 0.39 | 83 | 1.00 | 0.31–3.31 | 0.99 |
| ERT g | |||||||||
| No | 127 | 55.9 | 1 | 82.7 | 1 | ||||
| Yes | 19 | 57.9 | 1.09 | 0.41–2.88 | 0.87 | 94.7 | 3.77 | 0.48–29.75 | 0.21 |
Abbreviations: A&E=alive and engrafted; BM=bone marrow; CB=cord blood; CI=confidence interval; ERT, enzyme replacement therapy; Flud-MA=fludarabine-based myeloablative; HLA=human leukocyte antigen; OR=odds ratios; RIC=reduced-intensity conditioning; TCD=T-cell depletion.
aUnrelated donors were regarded as not carrying the α-L-iduronidase mutation.
bFrom the matched donors, 46 were family members and 50 were unrelated. From the mismatched donors, 6 were family members and 44 unrelated: 32 had 1 mismatch, 6 had 2 mismatches and 11 more than 2 mismatches. From one patient, the mismatch grade is unclear.
cBusulfan was given in the regular myeloablative doses (16 or 20 mg/kg) p.o., unless otherwise indicated. Groups were subdivided: Bu/Cy=busulfan+cyclophosphamide 200 mg/kg including one patient receiving+10 mg/kg thiothepa, Bu/Cyhi=busulfan+cyclophosphamide 240 or 260 mg/kg, Bu target=Bu/Cy (4) or Bu/Cyhi (9) or Bu/Cy+fludarabine 150 mg/m2 (2), Flud-based myeloablation (MA)=Bu/Cy+fludarabine 150 mg/m2 (9) or busulfan+fludarabine 180 mg/m2 (4) or busulfan+melphalan 4 mg/kg+fludarabine 150 mg/m2 (4) and reduced-intensity conditioning (RIC)=melphalan 140 mg/m2+fludarabine 150 mg/m2 (8) or melphalan 140 mg/m2+TLI 2 Gy+fludarabine 150 mg/m2 (2) or busulfan 10 mg/kg+fludarabine 150 mg/m2 (2) and treosulfan 36 or 42 g/m2+fludarabine 150 mg/m2 (6). Busulfan target: either steady state 600–900 ng/ml (n=10) or daily areas under the curves (AUCs) of 17 500–25 000 μg/l × h (n=5). Twelve received an adjusted dose on the second day. Busulfan was given either p.o. or i.v. No VOD was seen in this group.
dCD3+ ranging from <5 × 104/kg to 107/kg.
eFor unrelated donors, serotherapy was given: either ATG or Campath-1H depending on institutional protocols.
f Median cell dose of the CBs used was as follows: in NC/kg 7.8 (range 2.7–20.0) × 107 and in CD34+/kg (n=13) 2.5 (1.1–10.0) × 105. Three of the 23 patients received a CB from an HLA-identical sibling donor and the rest was unrelated.
gERT=enzyme replacement therapy, pre-SCT.
P-values <0.1 were selected for multivariate analysis. Bold and italic indicates the P-value, bold alone for the OR.
Donor chimerism was determined by various standard procedures (cytogenetic/molecular/X–Y FISH), depending on the center and grouped as follows: >95, 75–95, 50–75, 25–50 and 11–25%. A donor chimerism of >95% was regarded as having full donor chimerism (>10 and <95% as mixed chimerism).
Enzyme levels in leukocytes were measured (locally) by various standard procedures and grouped as follows: normal, high heterozygote for heterozygote individuals (15–25 nmol/h/mg) and low heterozygote for heterozygote individuals (4.5–15 nmol/h/mg).
Prophylaxis against GvHD consisted of cyclosporin (aiming at 100–200μg/l) for all patients, ±addition of methylprednisolone (1–2 mg/kg/day) in the case of a CB donors and plus methotrexate (MTX) in the case of a unrelated donor (UD). In the case of a sibling donor, the use of MTX was based on institutional protocols. When in the case of TCD (by any method: for example, using antibodies, CD34+ selection), the number of cells was reduced <50 000 CD3+/kg, no GvHD prophylaxis was given.
End points
Primary end points were the ‘alive and engrafted’ rate and ‘survival’ after the first SCT at the latest follow-up time point (at least >6 months). ‘Engraftment’ was defined as a donor chimerism of >10% and an α-L-iduronidase level of more than the lower limit of normal for the heterozygote individuals (>4.5 nmol/h/mg). Patients receiving successful ‘donor lymphocyte infusions (DLI)’ (defined as subsequent increase of donor chimerism) because of mixed chimerism were regarded as successful transplants.
Secondary end points were transplantation-associated morbidity defined as acute GvHD or chronic GvHD, VOD and IPS/DAH. Acute GvHD was diagnosed and graded according to Glucksberg et al.16 Severity of chronic GvHD was graded according to Shulman et al.17 The diagnosis of VOD was made according to the Seattle or Baltimore criteria.18 Other secondary end points were as follows: the effect of DLI given on mixed chimerism, ‘alive and engrafted’ and ‘survival’ after second and third SCTs, the effect of cell source (CB vs the combined group BM+PBSC) on mixed chimerism and the influence of period (1994–1998 vs 1999–2004) of transplantation on ‘alive and engrafted’ and ‘survival’ rate.
Statistical analysis
The associations between age, sex, donor relationship, heterozygote donor, stem cell source, HLA disparity, conditioning regimen used and TCD and the primary and secondary end points were analyzed in univariable and multivariable logistic regression analyses. Dichotomous outcomes (for example, alive and engrafted: yes/no) were used as dependent and predictors as independent variables. Univariable predictors of outcome that were statistically significant (P<0.10) were selected for multivariable logistic regression analysis. Results are expressed as odds ratios (OR) and corresponding 95 percent confidence intervals (95% CI). CI not including 1 (P-values<0.05) were considered statistically significant. The time to event (graft failure and dead) in association to the primary end point ‘alive and engrafted’ was expressed in Kaplan–Meier curves. Statistical analysis was performed using SPSS 12.1 (SPSS Inc., Chicago, IL, USA).
Results
Survival and ‘alive and engrafted’ rate
The ‘survival’ and ‘alive and engrafted’ rate after first transplantation was 124/146 (85%) and 82/146 (56%), respectively. The overall ‘alive and engrafted’ rate after one to three transplantations was 111/146 (76%) with a survival rate of 118/146 (81%). At the time of analysis, some patients were awaiting a second graft. Twenty-two patients (15%) died after initial transplant, 19 from transplantation-related causes (Table 1). Infection, mainly viral, was the most prevalent cause of death.
Table 1.
Mortality (overall) and morbidity (after first SCT)
| n | % | |
|---|---|---|
| Causes of death (after 1–3 SCTs) | ||
| Infection | 15 | 10.0 |
| Viral | ||
| Adeno, EBV and CMV | 8 | |
| RSV | 1 | |
| Bacterial | 5 | |
| Fungal | 1 | |
| GvHD | 3 | 2.0 |
| Cardial/respiratory ECI | 2 | 1.4 |
| VOD (+parainfluenza III/enterocolitis) | 2 | 1.4 |
| DAH | 1 | 0.7 |
| Multiorgan failure ECI | 1 | 0.7 |
| Hurler (disease progression) | 4 | 2.7 |
| Morbidity after first SCT | ||
| Acute GvHD (n=146) | ||
| Grade I | 26 | 17.8 |
| Grade II | 15 | 10.3 |
| Grade III | 3 | 2.1 |
| Grade IV | 5 | 3.5 |
| Chronic GvHD (n=114a) | ||
| Limited | 6 | 4.1 |
| Extensive | 2 | 1.4 |
| VOD (n=134) | ||
| Pulmonary complications (ventilated) (n=132b) | 12 | 8.3 |
| IPS/DAH | 4 | 2.8 |
| Infection/ARDS | 7 | 4.8 |
| Pulmonary hypertension | 1 | 0.7 |
Abbreviations: ARDS=acute respiratory distress syndrome; CMV=cytomegalovirus; DAH=diffuse alveolar hemorrhage; ECI=e causa ignota (= of unknown origin); GvHD=graft-versus-host disease; IPS=idiopathic pneumonia syndrome; RSV=respiratory syncytial virus; SCT=stem cell transplantation; VOD=veno-occlusive disease.
aPatients at risk.
bFor 14 patients, data are missing.
Values with no significance are shown in bold type.
The association between the various variables and the primary end points are shown in Table 2. Data for this analysis were complete for all variables. RIC (P=0.005), TCD (P=0.001) and HLA disparity (P=0.014) were found to be predictors for lower rates of being ‘alive and engrafted’. Within the CB group, HLA disparity was not found to be a predictor for a lower ‘alive and engrafted’ rate (OR 0.88; 95% CI 0.16–4.87; P=0.88): after first CB transplant, 8/12 (6/6 match), 5/8 (5/6 match) and 2/3 (4/6 match) were successful. Busulfan targeting was a predictor for a higher rate of being ‘alive and engrafted’ (P=0.028). None of the characteristics clearly predicted survival and, therefore, predictors of the primary end point ‘alive and engrafted’ may be taken to be the predominant predictors of graft failure. In addition, Kaplan–Meier curves, showing the influence of conditioning, TCD and cell source on the end point ‘alive and engrafted’ over time are shown in Figure 1.
Figure 1.
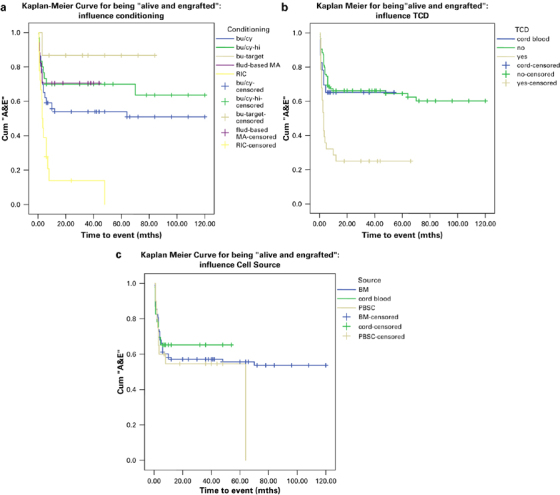
Kaplan–Meier curves for being ‘alive and engrafted’. Influence of (a) conditioning, (b) TCD and (c) cell source are shown. A&E, alive and engrafted; BM, bone marrow; Flud-MA=fludarabine-based myeloablative; PBSC, peripheral blood stem cell; RIC=reduced-intensity conditioning; TCD, T-cell depletion.
The multivariable analyses on the primary end point ‘alive and engrafted’ showed similar results to those of the univariable analysis (Figure 2), except that the Flud-MA regimens were found to protect against graft failure in this analysis (univariable analysis P=0.19). After deleting the three patients who received successful DLI, Flud-MA regimen was no longer a predictor for a higher ‘alive and engrafted’ rate.
Figure 2.
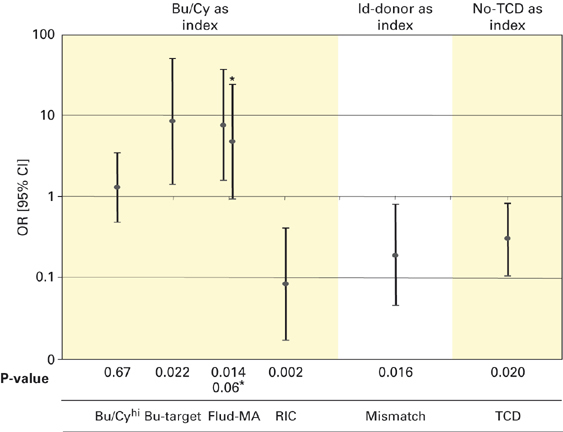
Multivariate predictors of being ‘alive and engrafted’ after first SCT. In the Flud-MA group: Bu/Cy/Flud was successful in 4/9 cases, Bu/Flud in 4/4 (of whom three patients received successful DLI because of progressive mixed chimerism) and Bu/Me/Cy in 4/4 cases. Without the three patients patients receiving successful DLI Flud-MA is no longer a predictor for a higher rate of being ‘alive and engrafted’. Bu/Cyhi=busulfan/cyclophosphamide-high dose (either 240 or 260 mg/kg); Bu-target=doses adjusted busulfan; Flud-MA=fludarabine-based myeloablative conditioning; matching=matched donor vs mismatched donor (Id-donor denotes identical donor); OR=odds ratio; TCR=T-cell depletion; 95% CI=95% confidence interval.
There was no difference in outcome between the periods 1994–1998 and 1999–2004, although almost all TCD and RIC transplantations were performed in the second period (Table 3). In the earlier period, unrelated donor and mismatched donors were risk factors, but not in the later period.
Table 3.
Univariate predictors of survival and being alive and engrafted for the period 1994–1999 and 1999–2004
| (A&E): 1994–1998 | (A&E): 1999–2004 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | (%) | OR | 95% CI | P-value | n | (%) | OR | 95% CI | P-value | |
| Overall | 51 | 57 | 95 | 56 | ||||||
| HLA disparity | ||||||||||
| Matched | 36 | 67 | 1 | 60 | 62 | 1 | ||||
| Mismatched | 15 | 33 | 0.25 | 0.07–0.90 | 0.033 | 35 | 46 | 0.53 | 0.22–1.22 | 0.13 |
| Conditioning a | ||||||||||
| Bu/Cy | 30 | 54 | 1 | 36 | 53 | 1 | ||||
| Bu/Cyhi | 17 | 59 | 1.25 | 0.38–4.16 | 0.72 | 13 | 77 | 2.98 | 0.70–12.67 | 0.14 |
| Bu target | 4 | 75 | 2.63 | 0.24–28.20 | 0.43 | 11 | 91 | 8.95 | 1.04–77.37 | 0.046 |
| Flud-based MA | — | — | — | — | — | 17 | 77 | 2.15 | 0.63–7.36 | 0.22 |
| RIC | — | — | — | — | — | 18 | 11 | 0.11 | 0.02–0.56 | 0.008 |
| TCD b | ||||||||||
| No | 47 | 62 | — | — | — | 61 | 65 | 1 | ||
| Yes | 4 | 0 | — | — | — | 24 | 29 | 0.22 | 0.08–0.61 | 0.004 |
| Donor | ||||||||||
| Family | 21 | 70 | 1 | 29 | 48 | 1 | ||||
| Unrelated | 30 | 46 | 0.38 | 0.12–1.21 | 0.10 | 66 | 59 | 1.55 | 0.65–3.73 | 0.33 |
| Source | ||||||||||
| BM | 47 | 62 | 1 | 56 | 50 | 1 | ||||
| PBSC | 1 | 0 | — | — | — | 19 | 52 | 1.11 | 0.39–3.15 | 0.84 |
| CB | 3 | 0 | — | — | — | 20 | 75 | 3.00 | 0.96–9.37 | 0.059 c |
Abbreviations: A&E=alive and engrafted; BM=bone marrow; CB=cord blood; CI=confidence interval; ERT=enzyme replacement therapy; Flud-MA=fludarabine-based myeloablative; HLA=human leucocyte antigen; OR=odds ratios; PBSC=peripheral blood stem cell; RIC=reduced-intensity conditioning; TCD=T-cell depletion.
Multivariate analysis resulted in a similar outcome (predictors of being A&E) to that found in the overall group.
aBusulfan was given in the regular myeloablative doses (16 or 20 mg/kg) p.o., unless otherwise indicated. Groups were subdivided as described in Table 1.
bCD3+ ranging from <5 × 104/kg to 107/kg.
cIn the multivariate analysis P=0.20.
P-values <0.1 were selected for multivariate analysis. Bold and italic indicates the P-value, bold alone for the OR.
Morbidity associated with SCT
Acute GvHD (⩾grade 2) was noted in 16% of patients (Table 1) and was found to be independent of sex, age at transplantation, matching, conditioning, cell source and donor relationship. A similar analysis for VOD showed, for every month older in age, 12% less chance of developing VOD (OR 0.88; 95% CI 0.81–0.97; P=0.009), while Bu/Cyhi (OR 5.25; 95% CI 1.22–22.69; P=0.026) was a predictor for VOD. The median age of patients with a VOD was 9 (range 4–18) months. The multivariable analyses suggested only older age (9% less chance for every month older in age) to be a protector (OR 0.91; 95% CI 0.83–1.01; P=0.063) against VOD. Additionally, there was a nonsignificant trend to more pulmonary complications (n=11) in the mismatched group (OR 2.96; 95% CI 0.89–9.87; P=0.077). Morbidity rates associated with second SCT were similar to those after first SCT.
Chimerism and enzyme activity
Donor chimerism and enzyme activity after first SCT and overall are shown in Table 4. After first SCT, 58/82 (71%) patients achieved full donor chimerism. When subdivided by source, 44/67 (66%) for the combined BM/PBSC group and 14/15 (93%; 1 patient with 91% donor chimerism) of the patients receiving CB achieved full donor chimerism. The univariable analysis showed CB to be a predictor for developing full donor chimerism (OR 7.14; 95% CI 0.91–58.82; P=0.062). In the multivariable analyses (source and conditioning used: unadjusted P<0.10), this association became significant (OR 9.31; 95% CI 1.06–82.03; P=0.044). The enzyme level in the CB group was normal in all (n=15; 100%) patients while only 72% (40/55; 12 missing of whom 6 were with mixed chimerism) had normal enzyme levels in the combined BM/PBSC group.
Table 4.
Donor chimerism and enzyme activity in leucocytes (α-iduronidase in nmol/h/mg) in leukocytes
| After first n=82 | Overall n=111 | |
|---|---|---|
| Donor chimerism a | n (%) | n (%) |
| >95% | 58 (70.7) | 91 (81.9) |
| 75–95% | 14 (17.1) | 11 (9.9) |
| 50–75% | 7 (8.5) | 6 (5.4) |
| >10–50% | 3 (3.7) | 3 (2.7) |
| Enzyme activity b | ||
| Normal | 55 (67.1) | 74 (66.7) |
| High heterozygote (15–25) | 10 (12.2) | 13 (11.7) |
| Low heterozygote (5–15) | 5 (6.1) | 6 (5.4) |
| Missing/not measured | 12 (14.6) | 17 (15.3) |
aThe median level of mixed chimerism was 75 (15–91)%.
bAfter first SCT (most recent measurement): from those who are having a full donor chimerism, 5 of the 58 patients measured had a heterozygote enzyme-activity. From those with a mixed chimerism, 10 of the 18 patients had a heterozygote enzyme activity.
Effect of DLI and outcome of second (and third) SCT
Because of progressive mixed chimerism (progressive donor signal decrease in at least two chimerism measurements), 10 patients received DLI. The number of DLI given per patient varied from 1 to 17. The amount of CD3+ cells in the DLI varied from 2.5 × 104 to 107/kg. In three patients, a conversion in the mixed chimerism was seen. All other patients, with the exception of one who had a stable low level (16%) of donor chimerism, were re-transplanted. No toxicity was seen in any of the patients who received DLI.
Thirty-three patients were re-transplanted, three of whom received a third transplant. The ‘alive and engrafted’ rate for the second SCT was 79% (median follow-up 12; range 7–50 months): 16/21 (76.2%) using the same donor, 10/12 (83.3%) using a different donor, 16/19 after myeloablative conditioning and 10/14 after RIC. Two of three patients who received a third-SCT (20 and 28 months of follow-up) are alive with a functioning graft.
Discussion
In this European retrospective study on the outcome of SCT, HS, TCD and RIC were found to be risk factors for graft failure. Busulfan targeting influenced engraftment positively, while cell source used (BM, PBSC or CB) did not influence the end point. CB; however, was found to be a predictor for achieving full donor chimerism associated with normal enzyme levels.
Since the first published report of SCT for HS,3 no large series has been published analyzing the risk factors for graft failure. The ‘alive and engrafted’ rates in this paper are similar to published data10, 11 as others have noted,10, 19 TCD was a risk factor for graft failure. Figures may even be overestimated since three patients receiving successful DLI, because of early mixed chimerism, were regarded as successful. Data in relation to HLA disparity and graft failure in HS are conflicting: Peters et al.10 noted inferior outcome with HLA mismatch while Souillet et al.11 did not. In the study by Souillet et al.,11 16/27 patients received a mismatched graft (1–4 mismatches) with no documented higher incidence of graft failure or GvHD. In this study, the largest series, including some (n=20) of the patients described by Souillet et al.,11 HLA disparity was found to be a risk factor in the 1994–1998 period but not thereafter. Improved supportive care (for example, firmer GvHD prophylaxis) is probably an important factor in this observation. Additionally, the use of (mismatch) CB (mainly used after 1998) might have influenced the observation that mismatching was not associated with a higher incidence of graft failure after 1998, since mismatches in CB vs BM/PBSC are not biologically equivalent. Similar experience has been observed elsewhere.12, 20, 21
Cell source (PBSC, BM or CB) was not found to influence the end point ‘alive and engrafted’. An interesting observation, however, is that after first SCT, significantly more patients receiving CB achieved full donor chimerism in comparison with patients receiving PBSC/BM. Similar results have been observed in the above-mentioned studies and mixed chimerism was not seen.12, 21 Mixed chimerism was associated with lower enzyme levels in this study as in previous studies.10, 11 Although no large-scale studies have examined the impact of enzyme levels after SCT on long-term outcome, recent data appear to suggest that low levels may negatively influence neurocognitive outcome.22 Future studies are eagerly awaited.
The observation of less mixed chimerism in patients receiving CB is intriguing. It might be that the higher degree of HLA mismatch23 exerts a stronger graft-versus-marrow (GvM) effect. The fact that this GvM effect is associated with subclinical GvHD might be due to the fact that CB cells have a more naive phenotype. This might make the marrow more sensitive to GvM in comparison to the known target organs of GvHD. Another explanation might be the increased pluripotential capability of the CB stem cell, relative to adult stem cell, with higher proliferative potentials in comparison to BM.24, 25, 26 We are aware from animal studies that the addition of mesenchymal stem cells to the SCT product results in less graft rejection, due to the immunomodulating potential of these cells.27, 28 Other advantages of CB include (1) lower rates for acute GvHD and extensive chronic GvHD in comparison to unrelated donors,29, 30, 31, 32 (2) immediate availability,23 reducing the period between diagnosis and SCT (in this study 7.5 months), which might improve the long-term outcome, (3) reduced likelihood of transmitting infection (viral) and (4) the suggestion that a more primitive stem cell population might have a greater capacity for trans-differentiation.24, 25, 26 This latter capability might be particularly important in SCT for inborn errors of metabolism, by theoretically improving delivery of enzyme to bone, cartilage and brain tissue. More research is warranted to study this hypothesis.
Inevitably, in the absence of international standardized conditioning protocols, various regimens have been challenged to improve the engraftment in SCT for HS: Souillet et al.,11 as well as the Dublin group (unpublished data), have used higher doses of cyclophosphamide (240–260 mg/kg) with the aim of increasing myeloablation and immunosuppression/ablation, which failed to influence engraftment. Other groups have increased immunosuppression by adding fludarabine to a myelo-ablative regimen (Flud-MA) which, in the multivariable analysis, was found to be a predictor for a higher ‘alive and engrafted’ rate, in contrast to the results from the univariable analysis (P=0.19). Any improvement may have been partly offset due to the fact that this group contained relatively more patients receiving a TCD and HLA mismatch graft (three of whom received successful DLI), both being identified as risk factors for graft failure. Additionally, the Flud-MA group (n=17) consisted of three different regimens, including four patients receiving busulfan+melphalan. This combination is more myeloablative than busulfan alone.
Targeting the busulfan dose by pharmacokinetic monitoring has been shown to optimize myeloablation, reduce toxicity and increase engraftment rates, because of the known narrow therapeutic range and wide variability in busulfan exposure in children receiving oral busulfan.33, 34, 35 In this study, busulfan targeting was associated with better engraftment. The optimized myeloablation might be required to overcome the effect of abundance of ‘glycosaminoglycans’ in the BM matrix, which has been suggested to negatively influence stem cell homing.36 In line with this, we found RIC to be a risk factor for graft failure. Although for malignant and some nonmalignant diseases RIC is feasible37, 38 for genetic diseases (for example; HS, Thalassemia-major) RIC is associated with graft failure.39
The incidence of GvHD and ‘IPS/DAH’ using BM/PBSCs reported by Peters et al.9, 10 was considerably higher than that observed in this series. While no obvious explanation is apparent, alternative conditioning and GVHD prophylaxis regimens may well be a factor.
Survival after second transplant was better in this study than reported by others (80 vs 50%),40 and two of three patients achieved sustained engraftment following a third graft. There was no difference observed between patients receiving either a myeloablative or RIC second SCT and whether the same or an alternative donor was chosen. These data indicate that after an unsuccessful first SCT, a second SCT is a feasible option.
In conclusion, TCD and RIC were associated, in this series, with increased incidence of graft failure, while busulfan targeting (therapeutic drug monitoring) increased the likelihood of sustained engraftment. The same criteria may apply in relation to other inborn errors of metabolism and hemoglobinopathies. CB increased the likelihood of sustained engraftment associated with normal enzyme levels and could therefore be considered as a preferential stem cell source. A further study of long-term outcome on successfully transplanted patients is now proposed.
Acknowledgements
We thank our collaborators for sharing/providing patient information: Drs Pierre Bordigoni and Alexandra Salmon (Nancy, France), Mary Coussons (Data manager Manchester, UK), Anne Gahan (Data manager, Dublin, Ireland), Isabelle Hirsch (Data manager, Necker Hospital, Paris), Dr Claudia Haase (Jena, Germany), Susanne Matthes-Martin (Vienna, Austia), Tayfun Guengoer (Zurich, Switserland), Robbert Bredius (Leiden, the Netherlands), Stefania Varotto (Padova, Italy) and Vicky Borbon (Ghent, Belgium).
Footnotes
M Cavazzana-Calvo: For the Working Party Inborn Errors of the European Blood and Marrow Transplantation (EBMT) group.
References
- 1.Neufeld EF, Muenzer J. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill: New York; 2001. The mucopolysaccharidoses; pp. 3421–3452. [Google Scholar]
- 2.Braunlin EA, Rose AG, Hopwood JJ, Candel RD, Krivit W. Coronary artery patency following long-term successful engraftment 14 years after bone marrow transplantation in the Hurler syndrome. Am J Cardiol. 2001;88:1075–1088. doi: 10.1016/S0002-9149(01)01999-3. [DOI] [PubMed] [Google Scholar]
- 3.Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981;2:709–712. doi: 10.1016/S0140-6736(81)91046-1. [DOI] [PubMed] [Google Scholar]
- 4.Summers CG, Purple RL, Krivit W, Pineda R, Copland GT, Ramsay NKC. Ocular changes in the mucopolysaccharidoses after bone-marrow transplantation. Ophthalmology. 1989;96:977–984. doi: 10.1016/S0161-6420(89)32795-3. [DOI] [PubMed] [Google Scholar]
- 5.Vinallonga X, Sanz N, Balaguer A, Miro L, Ortega JJ, Casaldaliga J. Hypertrophic cardiomyopathy in mucopolysaccharidoses – regression after bone-marrow transplantation. Pediatr Cardiol. 1992;13:107–109. doi: 10.1007/BF00798216. [DOI] [PubMed] [Google Scholar]
- 6.Krivit W, Henslee-Downey J, Klemperer M. Survival in Hurler's disease following bone marrow transplantation in 84 patients. Bone Marrow Transplant. 1995;15:S182–S185. [Google Scholar]
- 7.Shapiro EG, Lockman LA, Balthazor M, Krivit W. Neuropsychological outcomes of several storage diseases with and without bone-marrow transplantation. J Inherited Metab Dis. 1995;18:413–429. doi: 10.1007/BF00710053. [DOI] [PubMed] [Google Scholar]
- 8.Vellodi A, Young EP, Cooper A, Wraith JE, Winchester B, Meaney C. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Childhood. 1997;76:92–99. doi: 10.1136/adc.76.2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peters C, Balthazor M, Shapiro EG, King RJ, Kollman C, Hegland JD. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood. 1996;87:4894–4902. [PubMed] [Google Scholar]
- 10.Peters C, Shapiro EG, Anderson J, Henslee-Downey PJ, Klemperer MR, Cowan MJ. Hurler syndrome: II. Outcome of HLA genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. Blood. 1998;91:2601–2608. [PubMed] [Google Scholar]
- 11.Souillet G, Guffon N, Maire I, Pujol M, Taylor P, Sevin F. Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003;31:1105–1117. doi: 10.1038/sj.bmt.1704105. [DOI] [PubMed] [Google Scholar]
- 12.Staba SL, Escolar ML, Poe M, Kim Y, Martin PL, Szabolcs P. Cord-blood transplants from unrelated donors in patients with Hurler's syndrome. N Engl J Med. 2004;350:1960–1969. doi: 10.1056/NEJMoa032613. [DOI] [PubMed] [Google Scholar]
- 13.Kurtzberg J, Krivit W . Cord blood transplantation for lysosomal storage diseases demonstrates the potential of cord blood cells for future cellular therapies. Blood (ASH meeting) 2004; 3602 (abstract).
- 14.Peters C, Shapiro EG, Krivit W. Hurler syndrome: past, present, and future. J Pediatr. 1998;133:7–9. doi: 10.1016/S0022-3476(98)70170-2. [DOI] [PubMed] [Google Scholar]
- 15.Orchard PJ, Grewal S, Milla C, Braunlin EA, Defor T, Panoskaltsis-Mortari A. Pulmonary risk factors in allogeneic transplantation for Hurler syndrome. Blood. 2004;104:592A. [Google Scholar]
- 16.Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18:295–304. doi: 10.1097/00007890-197410000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Shulman HM, Sullivan KM, Weiden PL, McDonald GB, Striker GE, Sale GE. Chronic graft-versus-host syndrome in man: a long-term clinicopathologic study of 20 Seattle patients. Am J Med. 1980;69:204–217. doi: 10.1016/0002-9343(80)90380-0. [DOI] [PubMed] [Google Scholar]
- 18.Reiss U, Cowan M, McMillan A, Horn B. Hepatic venoocclusive disease in blood and bone marrow transplantation in children and young adults: incidence, risk factors, and outcome in a cohort of 241 patients. J Pediatr Hematol Oncol. 2002;24:746–750. doi: 10.1097/00043426-200212000-00013. [DOI] [PubMed] [Google Scholar]
- 19.Martin P, Carter S, Kernan N, Sahdev I, Wall D, Pietryga D. Results of the Cord Blood Transplantation Study (COBLT): outcomes of unrelated donor umbilical cord blood transplantation in pediatric patients with lysosomal and peroxisomal storage diseases. Biol Blood Marrow Transplant. 2006;12:184–194. doi: 10.1016/j.bbmt.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 20.Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- 21.Guffon N, Fouilhoux A, Bertrand Y, Thomalla M . Longitudinal neurocognitive follow-up of 26 MPSI patients with human stem cell transplantation or with enzyme replacement therapy. Ninth International Symposium on Mucopolysaccharide and Related Diseases, Venice, 2006, p 166.
- 22.Rubinstein P, Dobrila L, Rosenfield RE. Processing and cryopreservation of placental/umbilical cord blood for unrelated bone marrow reconstitution. Proc Natl Acad Sci USA. 1995;92:10119–10122. doi: 10.1073/pnas.92.22.10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kogler G, Sensken S, Airey JA, Trapp T, Muschen M, Feldhahn N. A new human somatic stem cell from placental cord blood with intrinsic pluripotent differentiation potential. J Exp Med. 2004;200:123–135. doi: 10.1084/jem.20040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen N, Hudson JE, Walczak P, Misiuta I, Garbuzova-Davis S, Jiang L. Human umbilical cord blood progenitors: the potential of these hematopoietic cells to become neural. Stem Cells. 2005;23:1560–1570. doi: 10.1634/stemcells.2004-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meier C, Middelanis J, Wasielewski B, Neuhoff S, Roth-Haerer A, Gantert M. Spastic paresis after perinatal brain damage in rats is reduced by human cord blood mononuclear cells. Pediatr Res. 2006;59:244–249. doi: 10.1203/01.pdr.0000197309.08852.f5. [DOI] [PubMed] [Google Scholar]
- 26.Le Blanc K, Ringden O. Immunobiology of human mesenchymal stem cells and future use in hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11:321–334. doi: 10.1016/j.bbmt.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Ryan JM, Barry FP, Murphy JM, Mahon BP. Mesenchymal stem cells avoid allogeneic rejection. J Inflamm (London) 2005;2:8. doi: 10.1186/1476-9255-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ballen KK. New trends in umbilical cord blood transplantation. Blood. 2005;105:3786–3792. doi: 10.1182/blood-2004-10-4125. [DOI] [PubMed] [Google Scholar]
- 29.Gluckman E, Rocha V, Arcese W, Michel G, Sanz G, Chan KW. Factors associated with outcomes of unrelated cord blood transplant: guidelines for donor choice. Exp Hematol. 2004;32:397–407. doi: 10.1016/j.exphem.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 30.Wagner JE, Barker JN, Defor TE, Baker KS, Blazar BR, Eide C. Transplantation of unrelated donor umbilical cord blood in 102 patients with malignant and nonmalignant diseases: influence of CD34 cell dose and HLA disparity on treatment-related mortality and survival. Blood. 2002;100:1611–1618. doi: 10.1182/blood-2002-01-0294. [DOI] [PubMed] [Google Scholar]
- 31.Chao N. J. Stem Cell Transplantation (Cord Blood Transplants) Hematology. 2004;2004(1):354–371. doi: 10.1182/asheducation-2004.1.354. [DOI] [PubMed] [Google Scholar]
- 32.Bolinger AM, Zangwill AB, Slattery JT, Glidden D, DeSantes K, Heyn L. An evaluation of engraftment, toxicity and busulfan concentration in children receiving bone marrow transplantation for leukemia or genetic disease. Bone Marrow Transplant. 2000;25:925–930. doi: 10.1038/sj.bmt.1702371. [DOI] [PubMed] [Google Scholar]
- 33.Zwaveling J, Bredius RGM, Cremers SCLM, Ball LM, Lankester AC, Teepe-Twiss IM. Intravenous busulfan in children prior to stem cell transplantation: study of pharmacokinetics in association with early clinical outcome and toxicity. Bone Marrow Transplant. 2005;35:17–23. doi: 10.1038/sj.bmt.1704707. [DOI] [PubMed] [Google Scholar]
- 34.Vassal G, Koscielny S, Challine D, ValteauCouanet D, Boland I, Deroussent A. Busulfan disposition and hepatic veno-occlusive disease in children undergoing bone marrow transplantation. Cancer Chemother Pharmacol. 1996;37:247–253. doi: 10.1007/BF00688324. [DOI] [PubMed] [Google Scholar]
- 35.Bolinger AM, Zangwill AB, Slattery JT, DeSantes K, Heyn L, Risler LJ. Target dose adjustment of busulfan using pharmacokinetic parameters in pediatric patients undergoing bone marrow transplantation for malignancy or inborn errors. Blood. 1997;90:1665. [Google Scholar]
- 36.Baxter MA, Wynn RF, Schyma L, Holmes DK, Wraith JE, Fairbairn LJ. Marrow stromal cells from patients affected by MPS I differentially support haematopoietic progenitor cell development. J Inherited Metab Dis. 2005;28:1045–1053. doi: 10.1007/s10545-005-0136-4. [DOI] [PubMed] [Google Scholar]
- 37.Resnick IB, Shapira MY, Slavin S. Nonmyeloablative stem cell transplantation and cell therapy for malignant and non-malignant diseases. Transplant Immunol. 2005;14:207–219. doi: 10.1016/j.trim.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 38.Slavin S, Aker M, Shapira MY, Resnick I, Bitan M, Or R. Reduced-intensity conditioning for the treatment of malignant and life-threatening non-malignant disorders. Clin Transplant. 2003;26:275–282. [PubMed] [Google Scholar]
- 39.Jacobsohn DA, Duerst R, Tse W, Kletzel M. Reduced intensity haemopoietic stem-cell transplantation for treatment of non-malignant diseases in children. Lancet. 2004;364:156–162. doi: 10.1016/S0140-6736(04)16628-2. [DOI] [PubMed] [Google Scholar]
- 40.Grewal SS, Krivit W, Defor TE, Shapiro EG, Orchard PJ, Abel SL. Outcome of second hematopoietic cell transplantation in Hurler syndrome. Bone Marrow Transplant. 2002;29:491–496. doi: 10.1038/sj.bmt.1703395. [DOI] [PubMed] [Google Scholar]