

Abstract
The SARS coronavirus main proteinase (Mpro) is a key enzyme in the processing of the viral polyproteins and thus an attractive target for the discovery of drugs directed against SARS. The enzyme has been shown by X-ray crystallography to undergo significant pH-dependent conformational changes. Here, we assess the conformational flexibility of the Mpro by analysis of multiple crystal structures (including two new crystal forms) and by molecular dynamics (MD) calculations. The MD simulations take into account the different protonation states of two histidine residues in the substrate-binding site and explain the pH-activity profile of the enzyme. The low enzymatic activity of the Mpro monomer and the need for dimerization are also discussed.
Keywords: SARS-CoV Mpro, molecular dynamics simulation, new crystal forms, multiple X-ray structures, conformational flexibility
Abbreviations used: CoV, coronavirus; MD, molecular dynamics; Mpro, main proteinase; RMS, root-mean-square; RMSD, root-mean-square deviation; RMSF, root-mean-square fluctuation; SARS, severe acute respiratory syndrome
Introduction
Severe acute respiratory syndrome (SARS) became a global threat due to its rapid transmission and high fatality rate.1 Up to now, neither an efficacious therapy nor a reliable preventive treatment has become available, although several approaches towards these goals are being tested at the moment.
Soon after the initial outbreak, a new coronavirus, SARS-CoV, was discovered as the etiological agent of SARS.1, 2, 3 Coronaviruses are positive-strand RNA viruses featuring the largest viral RNA genomes (about 30 kb) found to date. It has been shown that all the protein functions required for SARS coronavirus replication are encoded by the replicase gene.4, 5 This gene encodes two overlapping polyproteins (pp1a and pp1ab), from which the functional proteins are released by extensive proteolytic processing. This is primarily achieved by the 33 kDa main proteinase (Mpro), which is frequently also called 3C-like proteinase (3CLpro) to indicate a similarity in substrate specificity with the 3C proteinase of picornaviruses. The 3C proteinase of rhinovirus has been selected as a target for the development of drugs against the common cold.6, 7 Similarly, the functional importance of the SARS-CoV Mpro in the viral life-cycle makes it a preferred target for discovering anti-SARS drugs.8, 9, 10, 11
Sequence alignment of the SARS-CoV Mpro with orthologues from other coronaviruses indicates that the enzyme is highly conserved, with 40% and 44% sequence identity, respectively, to human CoV (HCoV) 229E Mpro and porcine transmissible gastroenteritis virus (TGEV) Mpro, the crystal structures of which have been reported.8, 12 Homology modeling for the SARS-CoV Mpro has been performed by several groups.8, 9 More recently, the crystal structure of the SARS-CoV Mpro has been determined.13, 14 This structure, like those of other CoV Mpros, comprises three domains (Figure 1 (a)). Domains I (residues 8–101) and II (residues 102–184) are β-barrels and together resemble the structure of chymotrypsin, whereas domain III (residues 201–306) consists mainly of α-helices. Domains II and III are connected by a long loop (residues 185–200). Located in a cleft between domains I and II, the active site of the Mpro comprises a catalytic dyad consisting of the conserved residues Cys145 and His41. A water molecule is hydrogen-bonded to His41 and can be considered the third component of a catalytic triad. In addition, in vitro experiments demonstrated that truncations of domain III reduced significantly or abolished completely the proteolytic activities of the main proteinases of HCoV 229E15 and TGEV.12 It was suggested that domain III of Mpro is required for keeping the proteolytic activity by holding domain II and the long loop (residues 185–200) in a catalytically competent orientation,12 and/or that its main role is orienting the N-terminal residues that play an important role for the catalytic activity of the enzyme (see below and Yang et al.13).
Figure 1.
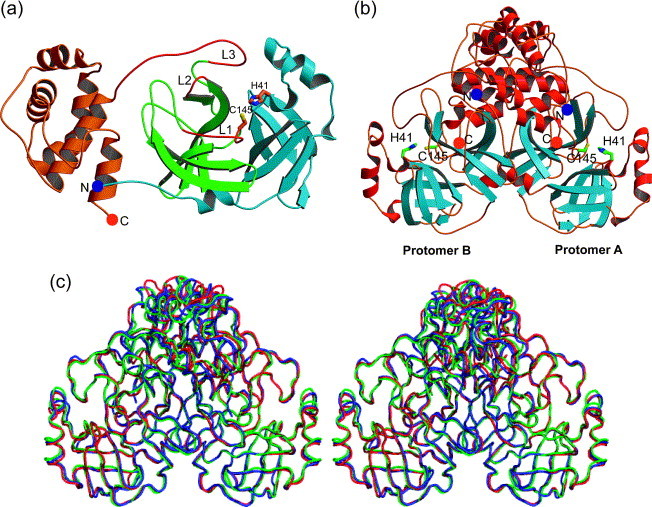
Structure of the (a) monomer and (b) dimer of SARS-CoV Mpro. (a) Domains I (light blue) and II (green) each contain a six-stranded β-barrel and domain III (orange) is composed mainly of α-helices. The amino and the carboxy terminus are marked by a blue and an orange sphere, respectively. The flexible loops L1, L2, and L3 (red) comprise residues 138–145 (the oxyanion-binding loop), 165–172, and 185–200, respectively. (b) α-Helices are red and β-strands are light blue. The amino and the carboxy termini are marked by blue and orange spheres, respectively. Dimerization is mainly due to interactions between the helical domains III of each monomer (top). (c) Superimposition (in stereo) of the Cα backbone as determined in three different crystal forms. Blue, monoclinic form; red, tetragonal form; green, orthorhombic form. (a) and (b) were prepared by MOLSCRIPT,40 (c) was prepared by PyMOL.41
In all known crystal structures of coronavirus main proteinases, the enzyme exists as a dimer (Figure 1(b)),8, 12, 13, 14 and dimerization is also observed in solution at slightly elevated concentrations.12, 16, 17, 18 It is believed that the dimer is the enzymatically active species, since the specific activity increases linearly with increasing enzyme concentration.18 A special feature first discovered for the SARS-CoV Mpro (but most probably present in all coronavirus main proteinases) is that in the monoclinic crystals grown at pH 6.0, the two monomers have different conformations around the S1 substrate-binding site, since the loop 138–145, in particular Phe140, as well as Glu166 undergo dramatic conformational rearrangements. As a result, one protomer exists in an active and the other in an inactive conformation.13 In the latter, the S1 substrate-binding pocket has virtually collapsed as the consequence of the reorientation of Glu166, and the oxyanion hole no longer exists due to the conformational change of residues 138–145 (see Figure 6(a) below). On the other hand, when the crystals are equilibrated at pH 7.6 or pH 8.0, both monomers are in an active conformation.13
Figure 6.
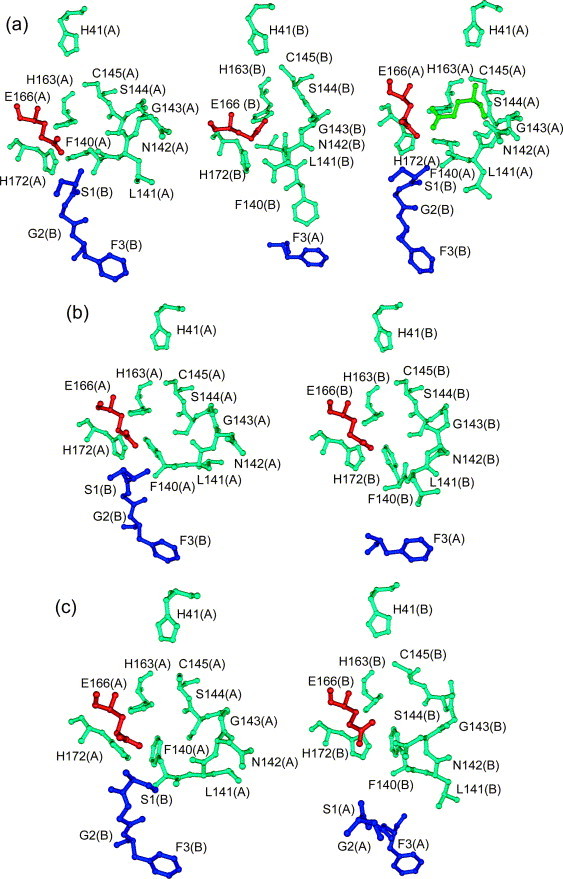
The S1 binding pocket and the oxyanion loop (residues 138–145, first two residues not shown) as revealed by X-ray crystallography (Yang et al.13 and this work, monoclinic crystal form). Left panel: monomer A; right panel: monomer B. Residues of the parent monomer are shown in light blue, with the exception of Glu166, which is red. The N-terminal residues of the other monomer in the dimer are shown in dark blue. (a), (b) and (c) The crystal structures at pH 6.0, pH 7.6 and pH 8.0, respectively.13 To visualize substrate binding to the enzyme, an additional panel is included in (a) (far right) showing the P1 Gln residue (green) of a substrate-analogous inhibitor as bound in the S1 specificity pocket.13 Note the interaction with His163 at the distal end of the P1 glutamine.
Thus, the coronavirus Mpro is a very flexible protein, the conformational state of which appears to depend on the pH value of the medium. This is probably of biological significance, because the viral polyproteins (of which the Mpro is a domain before self-release by autocleavage) assemble on the late endosome where local pH tends to be acidic. Here, we analyze the conformational flexibility of the SARS-CoV Mpro in detail, and take the pH of the medium into account. We compare the results of molecular dynamics simulations with multiple crystal structures of the free enzyme, including two new crystal forms. In addition to the previously described monoclinic crystal form (space group P21), we obtained a tetragonal and an orthorhombic crystal form for the SARS-CoV Mpro (space groups P43212 and P21212). The corresponding refined structures provide valuable information on the flexibility of the enzyme when compared to one another and to the original structure in the monoclinic form. In addition, we describe several independent determinations of the structure in the monoclinic crystal form. The motivation for this is that unless protein crystals diffract to Bragg spacings of 1.5 Å or better, it is normally difficult to identify minor, i.e. less frequently occupied, conformations of flexible parts of a protein from one crystal. However, if different crystals that are grown at slightly deviating conditions are analyzed, one conformation of a flexible surface loop or amino acid side-chain may be selected as the predominant one in one crystal, and another conformation in the other. Thus, independent structural analyses of multiple crystals grown at almost but, in practice, never absolutely identical conditions, will provide useful information on the flexibility of the macromolecule under study. In the work presented here, minor differences in crystallization conditions were introduced by the presence of different weak inhibitors of the SARS-CoV Mpro in the crystallization set-ups. However, none of these compounds was found to have bound to the enzyme, as evidenced by the complete absence of even spurious difference density in the substrate-binding site. In our analysis, we focus on the pH-dependent rearrangements observed in the substrate-binding site and investigate the conformational changes that lead to activation/deactivation of the enzyme. We also address the question of why the monomeric proteinase shows little enzymatic activity, whereas the dimer is fully active.
Results and Discussion
X-ray structures
A total of seven X-ray structures of the free SARS-CoV main proteinase have been determined as part of this study. One is derived from newly obtained tetragonal crystals grown at pH 5.9 (space group P43212, resolution 2.0 Å, R factor 17.5%), the second from orthorhombic crystals obtained at pH 6.6 (space group P21212, resolution 2.8 Å, R factor 20.8%). The crystallization conditions for the tetragonal form were identical to the previously established ones of the monoclinic form, and in fact, crystals of both forms often appeared simultaneously in the same crystallization droplet. Lower pH and slightly higher concentrations of the precipitant polyethylene glycol (PEG) 6000 (up to 20%) tended to favor the tetragonal form. The orthorhombic crystals were grown using 0.7 M sodium malonate (pH 6.6) as a precipitant. Details of diffraction data collection and structure solution as well as refinement of the new structures are provided in Table 1 . The other five structures were determined from crystals of the original monoclinic form that had been grown at pH 6.0. The resolutions of the latter structural models varied between 2.14Å and 2.8 Å, and the crystallographic R factors were between 21.0% and 23.8%. The structures derived from monoclinic crystals confirm the findings reported with the original structure of the enzyme:13 monomer A is in an active conformation, with the S1 substrate-binding subsite accessible and the oxyanion loop, residues 138–145, in the catalytically competent conformation, whereas both features have collapsed in monomer B of the dimer, which therefore is considered inactive. In contrast to the monoclinic crystals, which feature an Mpro dimer in the asymmetric unit, there is only a monomer in the asymmetric unit in the tetragonal (P43212) and orthorhombic (P21212) crystal forms, with the dimer being created through crystallographic symmetry. In both of the new crystal forms, which had been obtained at pH 5.9 and 6.6, respectively, the monomers are in the inactive conformation, leading to dimers with both subunits being inactive.
Table 1.
Crystallographic data for the three crystal forms of SARS-CoV Mpro examined in this study
| Crystal form | Monoclinic | Tetragonal | Orthorhombic |
|---|---|---|---|
| Space groups | P21 | P43212 | P21212 |
| pH of crystallization | 6.0 | 5.9 | 6.6 |
| Wavelength (Å) | 0.8046 | 0.8031 | 0.8125 |
| Resolution limits (Å) | 2.14 | 2.0 | 2.8 |
| a (Å) | 52.02 | 69.68 | 108.23 |
| b (Å) | 96.21 | 69.68 | 44.56 |
| c (Å) | 67.53 | 100.19 | 54.20 |
| β (°) | 102.84 | 90.00 | 90.00 |
| Total no. of reflections | 190,084 | 243,889 | 47,275 |
| Unique reflections | 33,801 | 15,515 | 6962 |
| Completeness (%) | 98.46 | 99.7 | 98.1 |
| Redundancy | 3.7 | 14.7 | 6.9 |
| Rmerge (%) | 10.7 | 13.9 | 7.2 |
| I/σ(I) | 10.4 | 20.3 | 19.6 |
Apart from the differences in detail in the substrate-binding site, the SARS-CoV Mpro dimers as seen in the new structures are very similar to the dimer in the original monoclinic crystals grown at pH 6.0 (Figure 1(c)). From the latter structure, the monomers in the new crystal forms display overall root-mean-square (RMS) deviations for Cα atoms (monomers A and B, respectively) of 0.95/0.76 Å (tetragonal form) and 1.10/0.78 Å (orthorhombic form). It is reassuring that the monomers in the new crystal forms, which are in the inactive conformation, are more similar to the inactive monomer B of the dimer in the monoclinic form (second number), than to the active monomer A (first number).
The overall RMS deviation (for Cα atoms) of each of the five repetitive monoclinic structures from the original structure of Yang et al. 13 is between 0.3 Å and 0.5 Å (Figure 3(e) and (f)). The crystal structures provide information on the flexibility of the polypeptide chains. The regions exhibiting the largest deviations in both monomers include residues 43–49, 117–122, and 140–144 in domains I and II. Segment 43–49 shows the largest deviations among the A monomers, with RMS values up to 3.5 Å, whereas the most flexible region in monomer B appears to be the segment 140–144, with one structure having a peak of 5.5 Å at residue Phe140. Domain III also shows some flexibility, but this is less focused on a limited number of short segments of the polypeptide chain, with the exception of the C-terminal three or four residues, which have in fact large deviations. The RMS deviations of the tetragonal and orthorhombic structures from the original monoclinic structure (molecule B) follow the same pattern (see Figure 3(f)). These data on polypeptide-chain flexibility are compared below to those obtained from the molecular dynamics (MD) calculations and from atomic temperature factors of the original crystal structure of the SARS-CoV Mpro 13 and the new crystal forms.
Figure 3.
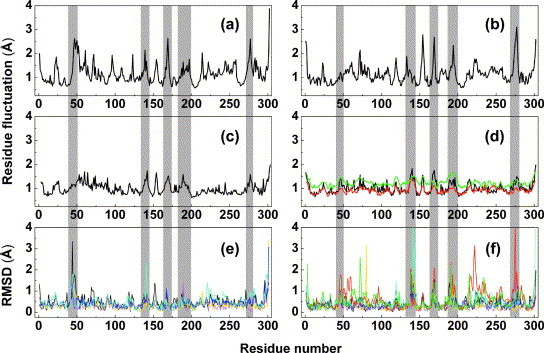
Residue fluctuations for the SARS-CoV Mpro. (a) and (b) Atomic fluctuations of (a) chain A and (b) chain B over the 10 ns equilibrium simulation at pH 6.0. (c) and (d) Mean atomic deviations (〈r〉 values) computed from the experimentally derived B factors using the equation , for (c) chain A and (d) chain B in the SARS-CoV Mpro monoclinic crystal structure (1UJ1.pdb); the corresponding values derived from the new crystal structures are shown in (d) for the tetragonal form (red) and the orthorhombic form (green). (e) and (f) RMSD for (e) monomer A and (f) monomer B for five independent monoclinic crystal structures of SARS-CoV Mpro at pH 6.0, fitted to 1UJ1.pdb, and colored black, yellow, cyan, blue and magenta, respectively. The values for the new crystal structures are shown in (f) for the tetragonal form (red) and the orthorhombic form (green).
Quality of the MD simulations
We have carried out 10 ns MD simulations of the SARS-CoV Mpro dimer at four different protonation states of two histidine residues (His163 and His172) involved in the substrate-binding site (Table 2 ). These protonation states were correlated with the pH of the system, and were assumed on the basis of the hydrogen-bonding pattern derived from the crystal structures and chosen to represent all relevant possible combinations (see Materials and Methods for charge assignment). In the cases of the simulations at pH 6.0, 7.6, and 8.0, the starting structure was the (energy-minimized) X-ray structure derived from monoclinic crystals grown at the respective pH.13 For the state in which His163 is protonated in both monomers of the dimer (here called pH 5.0), no crystal structure was available at the time of the simulation; therefore, the starting model for this was the monoclinic crystal structure at pH 6.0, but with His163 in both protomers positively charged. Since this simulation was carried out, we have obtained the new crystal forms, which presumably have His163 protonated in both monomers. Thus, the new structures provide an experimental check of the predictions derived from the MD calculations. In fact, a comparison of the 10 ns snapshot of the MD simulation at pH 5.0 and the new crystal structures shows that, while there are significant deviations on a residue-by-residue basis in domain III, where some loops have a rather large amplitude of motion during the simulation, the region around the substrate-binding site, including the oxyanion loop 138–145, agrees quite well. The new X-ray structures thus confirm the relevance of our MD simulations, which were apparently able to transform one conformation of the SARS-CoV Mpro into the other. The nature of these pH-dependent rearrangements will be discussed below.
Table 2.
Protonation states of His163 and His172 in the MD simulation models
| H163(A) | H163(B) | H172(A) | H172(B) | |
|---|---|---|---|---|
| pH 5.0 | + | + | + | + |
| pH 6.0 | − | + | + | + |
| pH 7.6 | − | − | + | + |
| pH 8.0 | − | − | − | − |
The system temperature and the total energy of each simulation model were monitored during the MD simulation and found to converge to stable values. The system temperature was coupled at 300 K and the total energy fluctuated within less than 0.2%. The root-mean-square deviation (RMSD) from the starting structure is an important criterion for the convergence of the system. The RMSDs of the Cα atoms are shown in Figure 2 , indicating that the whole simulation system appears to have been stable after 2 ns of equilibration.
Figure 2.
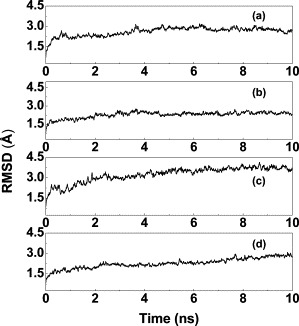
Time dependence of the RMSDs from the starting structure of the SARS-CoV Mpro dimer for Cα atoms during the 10 ns MD simulation. (a), (b), (c), and (d) correspond to pH 6.0, pH 7.6, pH 8.0, and pH 5.0, respectively, shown as 10 ps averages.
Flexibility of the polypeptide chains
To identify the most flexible parts of the proteinase, we calculated RMS fluctuations (RMSFs) for individual residues, and compared them with the crystallographic results. Figure 3 (a) and (b) show the atomic fluctuations averaged over residues for Mpro protomers A and B, derived from the 10 ns MD trajectory at pH 6.0. There are two ways of extracting information on residue flexibilities from the crystallographic data. The first is to derive the corresponding RMSF values from the atomic temperature factors (B factors). The experimental B factors are transformed to the RMSF using the formula , where is a coordinate vector of the ith atom. This is shown in Figure 3(c) and (d) for the monoclinic crystal structure at pH 6.0 (molecules A and B). Also, the RMSF values derived from the tetragonal and orthorhombic crystal structures are given in Figure 3(d) (red and green, respectively). It is obvious that the fluctuations are larger in the orthorhombic crystal form, which correlates well with the high Wilson B factor (76 Å2) of these crystals and the relatively low resolution of 2.8 Å.
The second method for obtaining dynamic information from crystal structures is careful analysis of multiple determinations of the same structure, either from crystals in different space groups, or from different crystals in one and the same space group, but grown under slightly different conditions. Figure 3(e) and (f) display the RMS values for both protomer A and B obtained from the multiple X-ray structure determinations carried out with monoclinic crystals grown at pH 6.0. Also, the corresponding RMS values for the tetragonal and orthorhombic crystal structures are included in Figure 3(f). These comparative data show a reasonably good agreement between the residue fluctuations derived from the MD simulations (Figure 3(a) and (b)) and the two sets of values obtained from the X-ray determination (Figure 3(c)–(f)). The RMS profiles reveal the most flexible parts of the proteinase, which are the loops L1 (residues 138–145), L2 (residues 165–172) and L3 (residues 185–200) around the substrate-binding pocket (as shown in Figure 1(a)), as well as surface residues 153–155 and 274–277 in each of the two protomers (shaded areas in Figure 3). As will be discussed below, all these loops except the last are associated with the flexibility of the substrate-binding pocket. This is also true for the 310-helical segment 45–49, where an insertion is found in SARS-CoV Mpro (compared to other coronavirus main proteinases) and which was difficult to model in the X-ray structures due to its flexibility. In addition, most residues involved in the monomer–monomer interface of the dimer, such as Ala7, Val125, and Glu290, show reduced fluctuations, while Ser139, Phe140, and Glu166, although contributing to the same dimerization interface, exhibit very high fluctuations, indicating that these residues play a special role (see below). The dimer appears not to be very stable, given the fact that at concentrations below 1 mg/ml, the coronavirus main proteinase exists predominantly as a monomer.12 The K D for dissociation of the SARS-CoV dimer has been reported as about 100 μM18 or 227 μM,17 although a much lower value has also been determined.42
Dynamic behavior of the catalytic site
Residues Cys145 (Cys144 in TGEV and HCoV 229E Mpro) and His41 constitute the catalytic dyad of coronavirus main proteinases.8, 12, 13 There is a long-standing debate over the ionization state of the Cys⋯His active-site residue pair in cysteine proteinases. Polgar19 has shown that it exists as a thiolate⋯imidazolium pair in papain-like proteinases, where the thiolate ion can be stabilized by the positive end of the permanent dipole of a long (24 residue) α-helix. However, coronavirus main proteinases (domains I/II) and picornavirus proteinases have a fold that is completely different from papain; instead, it resembles that of the serine proteinase, chymotrypsin, but with an active-site serine-to-cysteine substitution. As a consequence, there is no long α-helix available that could stabilize the thiolate anion, and indeed, Polgar20 has proposed that rhinovirus 2A proteinase contains a hydrogen-bonded thiol⋯imidazole pair.
Huang et al.21 have replaced the catalytic Cys residue by Ser in SARS-CoV Mpro and were able to detect residual proteolytic activity (at pH 7.0) for the mutated enzyme. This observation appears to support the idea that the nucleophile in this proteinase is the uncharged thiol, with His41 acting as a general base. Therefore, Cys145 and His41 of the catalytic dyad were left uncharged in all MD simulation runs in this work. In the various crystal structures of SARS-CoV Mpro, the cysteine–histidine distance is between 3.6 Å and 3.9 Å. The cysteine–histidine distance of SARS-CoV Mpro as a function of simulation time was monitored during the MD calculation and found to fluctuate in the range of 3.5–4.5 Å. In a recent MD simulation of a homology model for the SARS-CoV Mpro monomer built on the basis of the TGEV Mpro crystal structure, Pang22 obtained distances of 1.8–3.3 Å between the Sγ atom of Cys145 and the Nε2 of His41. The lower value is definitely by far too short and would actually correspond to a covalent S–N bond between the two residues. The values obtained in our simulations certainly agree much better with the experimental observations made by X-ray crystallography.
It was shown that coronavirus main proteinases do not possess a third catalytic residue at the canonical position,8, 12, 13 at variance with most other proteinases, including picornavirus 2A and 3C proteinases, chymotrypsin, and papain, which have Asp, Glu or Asn as the third component of the catalytic triad. In the crystal structures of the coronavirus Mpros, a buried water molecule is found in the position that would normally be occupied by the side-chain of the third member of the catalytic triad.8, 12, 13 The water molecule forms three hydrogen bonds with His41, His163/164 (or Gln163, in HCoV 229E Mpro), and Asp186/187 (Figure 4 ; where two residue numbers are given, the first refers to TGEV and HCoV 229E, and the second to SARS-CoV). This hydrogen-bonding system remains intact almost over the entire simulation time, with little fluctuation. At no time during our simulations did residue Asp187 change its conformation to replace the water molecule and form a direct interaction with His41; instead, during most of the simulation time, the aspartate residue remained engaged in a salt-bridge with Arg40 (see Figure 4), thereby bridging domains I and II of the proteinase. However, in his recent MD simulation on a homology model of the SARS-CoV Mpro monomer in complex with a peptide substrate, Pang22 observed that Asp187 did change its conformation and interacted with His41, thereby replacing the water molecule and forming a catalytic triad. He proposed that the presence of a substrate with a P1′ residue would shield the catalytic triad from solvent so that the conformational change could take place. Since we do not have a substrate in our simulations of the Mpro dimer, we cannot comment on this observation; unfortunately, Pang's report neither mentions the charge of His41 in the simulation nor the fate of Arg40, which would lose its interaction partner as a consequence of the reorientation of Asp187.
Figure 4.
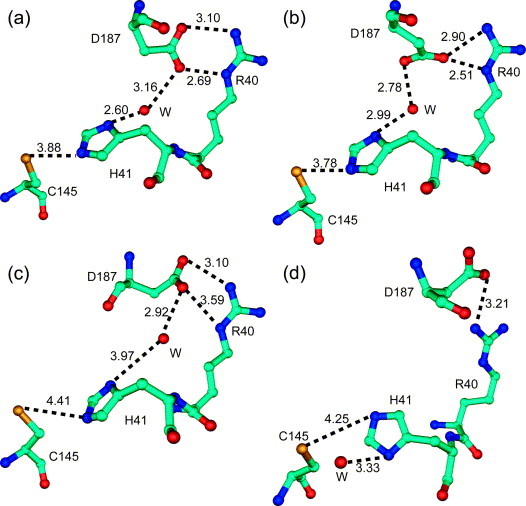
Conformations of the catalytic site at pH 6.0. (a) and (b) Monoclinic X-ray structure, protomers A and B, respectively; (c) and (d) 10 ns snapshot of the MD simulation; protomers A and B, respectively. Water molecules are indicated by W. Some key distances (not necessarily hydrogen bonds) are indicated by broken lines and values given in Å. Note the salt-bridge between Asp187 and Arg40 (see the text).
pH-dependent conformational changes
A major goal of the present study was the investigation of the pH-dependent conformational changes in the substrate-binding site of the SARS-CoV Mpro that had been observed in the crystal structures.13 To this end, we determined the pH-activity curve of our SARS-CoV Mpro preparation, using an HPLC-based peptide cleavage assay that made use of differences in the fluorescence of substrate and products. The substrate was the pentadecapeptide SWTSAVLQSGFRKWA. This peptide corresponds to the N-terminal autocleavage site of the SARS-CoV Mpro, with the exception of the P7 Ile, which had been replaced by Trp, and the P6′ Met, which had also been replaced by Trp. The fluorescence emission of the two tryptophan residues at 353 nm was used to quantify substrate and products. The k cat/K M of our enzyme preparation with this substrate was 61.9(±1.5) mM−1min−1 and thus about sixfold higher than what was reported by Fan et al.,18 albeit for a shorter peptide substrate. We found the pH-activity curve to be bell-shaped, with the maximum enzymatic activity at pH 7.0 (see Figure 5 ). This finding agrees reasonably well with that by Huang et al.,21 who reported an activity maximum at pH 7.4 using a colorimetric cleavage assay.
Figure 5.
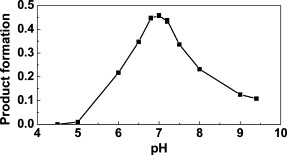
pH-activity curve for SARS-CoV Mpro with a pentadecapeptide substrate (see the text for details).
The four different MD simulations of the SARS-CoV Mpro dimer described here correspond to the various pH-dependent protonation states of residues His163 and His172 in the substrate-binding site (see Table 2). His163, an absolutely conserved residue at the bottom of the S1 specificity pocket, has been shown to form a hydrogen bond between its Nε2 atom and the side-chain carbonyl oxygen of the P1 glutamine residue of the substrate (Figure 6 (a), right image).8, 13 It is important for maintaining the absolute specificity of the Mpro for glutamine in the P1 position of the substrate that His163 is not protonated; a positive charge of this histidine in an otherwise largely hydrophobic environment would probably enable glutamate (in addition to glutamine) to enter the S1 pocket. Near neutral pH and above, the protonation of His163 is prevented by a hydrophobic interaction with the aromatic ring of Phe140, and also by a strong hydrogen bond between the hydroxyl group of Tyr161 (hydrogen donor) and His163 Nδ1. Yang et al.13 had proposed that the conformational changes of the SARS-CoV Mpro observed in one monomer (B) but not the other (monomer A) in the dimer crystallized at pH 6.0 could be caused by protonation of His163 at this pH, which is presumably close to the pK of this residue. The main elements of the observed conformational transition (see Figure 6(a), left and middle images) from monomer A (His163 unprotonated) to monomer B (His163 protonated) and their interpretation by Yang et al.13 were as follows.
-
(1)
The side-chain of Phe140 retracts itself from the interaction with His163, presumably due to the protonation of the latter.
-
(2)
Since Phe140 is part of the oxyanion loop, this segment (residues 138–145) changes its structure significantly. In the resulting conformation, the loop is no longer able to stabilize the tetrahedral transition state of the proteolytic reaction.
-
(3)
In order to neutralize the positive charge on His163, Glu166, which along with His172 forms a wall of the S1 pocket in the conformation observed at pH 7.6 as well as in molecule A of the pH 6.0 dimer, moves “inward” towards His163, thereby breaking its interactions with His172 and the amino terminus (residue Ser1) of the other monomer (A) in the dimer.
-
(4)
When the amino terminus of molecule A loses its interaction with Glu166 of molecule B, the hydrogen bonds between Ser1 and the main chain of Phe140 are also lost. This contributes to the conformational change of the oxyanion loop, residues 138–145.
As a consequence of these concerted movements, the proteinase is presumably inactivated, owing to the oxyanion loop no longer having a competent conformation, and to Glu166 blocking the S1 substrate-binding pocket. This alternative, inactive conformation appears to be quite stable, as it even persisted upon addition of a substrate-analogous inhibitor that was observed to covalently bind to the enzyme in its inactive conformation but was not able to induce the active form and penetrate the S1 pocket.13 The fact that the inactivating conformational changes occur only in one of the two molecules in the monoclinic crystal structure of the SARS-CoV Mpro dimer is in agreement with the proteolytic activity of the enzyme being about 50% at pH 6.0 (see Figure 5). This pH value is presumably close to the pK value of His163, a notion that is confirmed by the fact that in our new tetragonal crystal form of the SARS-CoV Mpro, which was obtained at a pH value slightly below pH 6.0, both monomers adopt the inactive conformation, with the amino terminus of the other protomer in the dimer not interacting with Phe140 and Glu166. This is also the case for the other new crystal form, which belongs to the orthorhombic system and was obtained at pH 6.6, i.e. again near the presumable pK of His163. However, it should be noted that both these new crystal forms contain a SARS-CoV Mpro dimer that is created by crystallographic symmetry from identical monomers; thus, there is no option for the dimer to adopt different conformations in its monomers. Interestingly, the pH-dependent conformational switch can apparently also occur in the orthorhombic form, since an isomorphous crystal structure recently deposited at the Protein Data Bank (PDB) (code 2BW6) but not yet published has both monomers in the active conformation, even though it was obtained at pH 6.5 (J. Lescar, personal communication).
The other histidine residue involved in the S1 specificity pocket, His172, is also subject to protonation/deprotonation depending on pH. As it is engaged in a salt-bridge with Glu166 at the outer wall of the site (Figure 6(a)), its pK value should be elevated by about two units, compared to isolated histidine residues.23 Yang et al. 13 had also determined the X-ray structure of the SARS-CoV Mpro after equilibration of the crystals at pH 8.0 and described it as being very similar to the structure at pH 7.6, i.e. both monomers in the dimer were in the active conformation. However, when we closely inspected their pH 8.0 structure, we noticed that there was no salt-bridge any longer between His172 and Glu166 in either of the two monomers, while the interaction between Glu166 of monomer A and the amino terminus of the other monomer (B) in the dimer still existed (see Figure 6(b) and (c)). As a consequence, Glu166 should have more freedom to move at pH 8.0 than at pH 7.6, and one motivation for our MD simulations was to find out whether it makes use of this potential.
Any interpretation of crystal structures making use of protonation states has to be seen in light of the inability of X-ray crystallography (except at resolutions better than 1 Å) to directly determine the positions of hydrogen atoms because of their low scattering power for X-rays. Therefore, one purpose of the MD study presented here is to examine whether protonation/deprotonation of His163 and His172 would really lead to the conformational rearrangements seen in the crystal structures.
Our MD simulations at the different pH values (Figure 7 ) started with the crystallographic coordinates of the structure determined at that particular pH, with the protonation state of His163 and His172 as indicated in Table 2. Several key distances indicative of conformational changes were monitored throughout the simulation: (1) Glu166–His163; (2) Glu166–His172; (3) Glu166–Ser1 amide nitrogen of the other monomer; and (4) Phe140 (center of mass of phenyl ring)–His163 (center of mass of imidazole ring) (Figure 8 ). At the outset of the pH 6.0 MD simulation (Figure 7(a)), Glu166 does not orient towards the non-protonated His163 in protomer A, but is stably oriented towards the charged His163 in protomer B. The interaction between Glu166 and His172 is a stable ion pair in the A chain during the first three nanoseconds. Thereafter, the distance between these two residues fluctuates up to about 8 Å and then back to form a hydrogen bond at about 8.5 ns. We analyzed the intermediate structure and found that sometimes Glu166 can form hydrogen bonds with the amides of Gly143, Ser144 or Cys145, i.e. the hydrogen-bonding donors of the oxyanion hole. These hydrogen bonds are not stable throughout the simulation, but form and break occasionally. In contrast, in protomer B, Glu166 adopts a relatively stable intermediate position where it interacts simultaneously with His163 and His172 (both of which are protonated). His163 of protomer A makes a weak hydrophobic interaction with Phe140 for most of the simulation time. The amino terminus of the other protomer (B) in the dimer forms a salt-bridge with Glu166 of protomer A and hydrogen bonds with the main chain of Phe140, although this is clearly getting weaker after about 3 ns into the simulation. In molecule B, in contrast, Glu166 does not interact with Ser1A and is instead oriented towards the now protonated His163, thereby blocking the S1 pocket. Furthermore, Phe140B stays away from His163B (as in the X-ray structure), and the oxyanion hole collapses. Thus, the MD simulation at pH 6.0 nicely confirms what has been seen in the crystal structures13 and supports the interpretation made on this basis (compare Figure 6, Figure 7).
Figure 7.
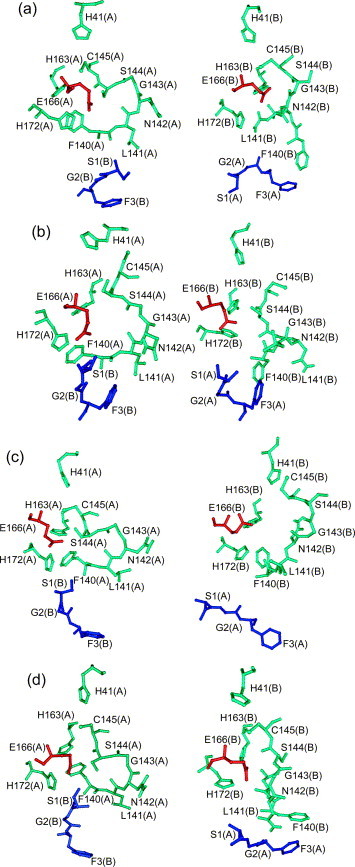
The S1-binding pocket and the oxyanion loop (residues 138–145, first two residues not shown) in the MD simulations (snapshots after 10 ns). (a), (b), (c) and (d) pH 6.0, pH 7.6, pH 8.0 and pH 5.0, respectively. Note that interaction of Glu166 (red) with His172 (to the left) constitutes the active conformation of the SARS-CoV Mpro, whereas its interaction with His163 (to the right) blocks the S1 pocket (compare also Figure 6(a), middle panel) and therefore leads to inactivation of the enzyme.
Figure 8.
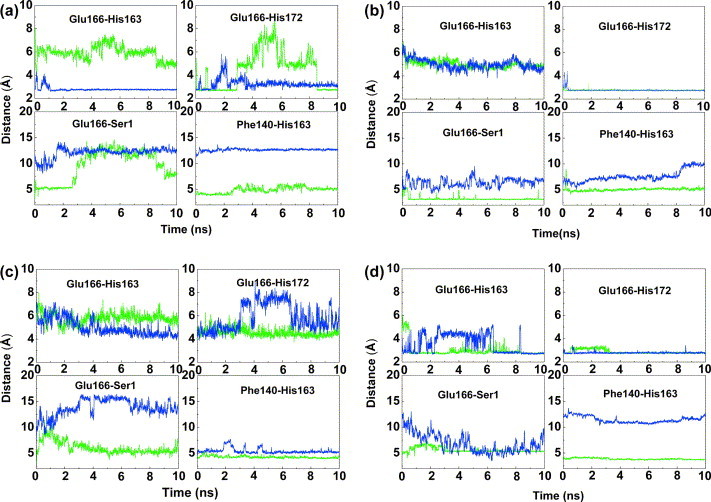
Some characteristic distances in the MD simulations of the SARS-CoV Mpro dimer at (a) pH 6.0, (b) pH 7.6, (c) pH 8.0, and (d) pH 5.0. For each simulation, the distance between Glu166 and His163, Glu166 and His172, Glu166 and Ser1(N) of the other monomer in the dimer, and Phe140 (center of mass of phenyl ring) and His163 (center of mass of imidazole ring) are shown. The shorter of the two distances to the carboxylate oxygen atoms of Glu166, Oε1 and Oε2, is displayed. Green, monomer A; blue, monomer B.
Similar analyses were carried out for the pH 7.6 and pH 8.0 MD simulations (Figure 7, Figure 8). At pH 7.6, Glu166 is more stably oriented towards (protonated) His172 in both monomers, while the distance between the phenyl ring of Phe140 and the imidazole ring of (non-protonated) His163 in protomer B is relatively large compared to the X-ray structure (Figure 7, Figure 8). It is found that this is due to the fact that in the initial structure, these two rings are not stacked, so their interaction may be weaker than the stack in monomer A at pH 6.0. At pH 8.0, Glu166 is more flexible with no hydrogen bonds with either His163 or His172 (Figure 7, Figure 8). This is likely due to the fact that His172 as well as His163 are non-protonated at this pH. For a significant part of the simulation time, Glu166 blocks the entry of substrate to the S1 pocket, thus explaining why the enzymatic activity is decreased at this pH (see Figure 5).
At the outset of the pH 5.0 MD simulation (Figure 7, Figure 8), protomer A is in the active conformation as in the starting model, the pH 6.0 X-ray structure, so that at first, Glu166 is oriented towards His172, and relatively far from His163. After about 1 ns, Glu166 reorients to the protonated His163. During most of the simulation time, it is stable in this orientation. As for protomer B, the distance between Glu166 and His163 fluctuates during the first 6 ns, and then tends to be a stable interaction. From the distance between Glu166 and Ser1 of the other chain, it can be seen that this has a very similar tendency in the two chains. Thus, after 10 ns, the two chains have a similar conformation, which is the inactive conformation (Figure 7(d)).
Our molecular dynamics simulations have confirmed that the protonation states of His163 and His172 are important, since they can affect the conformation of the binding pocket, and thereby control the enzymatic activity. In conclusion, then, the two flanks of the bell-shaped pH-activity curve (Figure 5) are governed by the protonation of His163 (low-pH side) and the deprotonation of His172 (high-pH side).
Intriguingly, a comparison of the key distances after 10 ns of simulation time to the corresponding X-ray structures shows very good qualitative agreement (Table 3 ; also compare Figure 6, Figure 7). Most importantly, where there is a hydrogen-bonding or ion-pair interaction in the crystal, it is almost invariably also found in the 10 ns snapshot, and where there is not, it is absent from the simulation as well. This is important, because formation and disruption of the hydrogen bonds/ion pairs involving His163 and His172 depend on the protonation state of these side-chains. Thus, the very similar interaction pattern seen by X-ray crystallography (which cannot directly determine hydrogen positions) and MD simulations (which make full account of hydrogen atoms bound to polar atoms) lends strong support to the conclusions drawn from the crystal structures of SARS-CoV Mpro by Yang et al.13
Table 3.
Distances between E166(Oε1/Oε2) and H163(Nε2), and E166(Oε1/Oε2) and H172(Nε2)
| pH 5.0 | pH 6.0 | pH 7.6 | pH 8.0 | |
|---|---|---|---|---|
| E166-H163(A) | 2.63/2.68a | 6.29/5.36 | 6.55/4.79 | 7.14/5.33 |
| E166-H163(B) | 2.63/2.73 | 2.44/2.75 | 6.23/4.47 | 7.79/5.58 |
| E166-H172(A) | 3.76/2.93 | 3.25/2.75 | 3.73/2.57 | 5.17/4.76 |
| E166-H172(B) | 3.76/2.75 | 3.70/3.50 | 3.46/2.60 | 5.78/5.99 |
First number, X-ray structure; second number, MD simulation after 10 ns. pH 5.0, distances for the crystal structure are from the tetragonal form grown at pH 5.9. Other pH values, distances for the crystal structures are derived from the monoclinic structures determined by Yang et al.13 Distances corresponding to hydrogen-bonding or ion-pair interactions are printed in bold. Note that interactions of Glu166 with His172 lead to activation of the enzyme, whereas those with His163 lead to inactivation. Other key distances are displayed in Figure 8.
In order to further investigate the influence of the conformational differences between the active and the inactive conformation on the shape of the substrate-binding side, we monitored the volume of the binding pocket every 50 ps during the simulations. Figure 9 shows the results of this analysis for the simulation at pH 6.0. During the first 2 ns, the volume of the pocket in protomer A shrunk significantly. Afterwards, the system appeared to have reached equilibrium and the volume of the pocket fluctuated around 800 Å3. In contrast, the corresponding volume in protomer B fluctuated around 600 Å3 from the beginning. The significantly smaller value indicates that substrate binding is not favored in this conformation.
Figure 9.
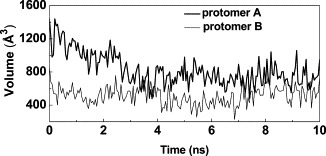
Volume of the binding pocket of protomers A and B at pH 6.0 during the simulation time.
Why is the SARS-CoV Mpro monomer inactive?
Fan et al.18 have proposed that the dimer is the enzymatically active species of the SARS-CoV Mpro, since the specific activity increased linearly with enzyme concentration. Dimerization of the proteinase occurs mainly through the interactions between the helical domain III of each monomer, and through hydrogen bonding between the amino-terminal residues of one monomer and residues near the S1 subsite of the other monomer, in particular Phe140 and Glu166.8, 12, 13 The N-terminal residue (Ser1 in SARS-CoV) has been proposed to keep both the oxyanion hole and Glu166 of the other monomer in a catalytically competent conformation; Yang et al. 13 coined the term of an “N-finger” that switches on the activity of the other monomer. In agreement with this, Anand et al. 12 observed in the TGEV Mpro system that deletion of the first five residues from the N terminus abolished peptidolytic activity of the enzyme almost entirely. This is not due to a destruction of the dimer, since the SARS-CoV Mpro still forms a dimer when the first seven residues from the amino terminus are missing.17 Thus, the amino terminus, as such, is essential for enzymatic activity of the dimer. Accordingly, we carried out preliminary 10 ns MD simulations of the monomer, starting from the active conformation as found in monomer A of the crystal structure determined at pH 6.0. Intriguingly, these resulted in a collapse of the oxyanion hole because of the missing hydrogen bond between the N terminus of the other chain and the main chain of Phe140. Also, not held in position by the same N terminus, Glu166 tended to move towards His163 and block the entry of the S1 pocket, at least during part of the simulation time. These observations clarify the role of the N terminus of the other monomer in the dimer: it is needed to maintain the proper shape of the substrate-binding site, including the oxyanion hole, of the “parent” monomer.
Conclusions
The SARS coronavirus main proteinase (Mpro) shows pH-dependent conformational flexibility, in particular around the substrate-binding site. We have assessed this flexibility by multiple X-ray structures (in the original crystal form as well as in two new crystal forms) and by MD simulations. The latter take into account different protonation states of two histidine residues (His163 and His172) in the substrate-binding site. Overall, we find good agreement between the flexibility data derived from the multiple crystal structures, from atomic temperature factors, and from the MD calculations. The catalytic dyad of Cys145 and His41 is confirmed to exist throughout the MD simulation; at no time is it complemented by a third residue such as Asp187. The conformation and the volume of the S1 binding pocket are controlled by the protonation states of the two histidine residues that are part of the pocket and we propose that the flanks of the bell-shaped pH-activity curve are governed by the protonation of His163 (on the low-pH side) and the deprotonation of His172 (on the high-pH side). Intriguingly, in our MD simulation of the SARS-CoV Mpro monomer, the S1 pocket spontaneously collapses and adopts the inactive conformation that is also seen upon protonation of His163 in the dimeric structure. We conclude that the amino terminus of the other monomer in the dimer plays an important role for the enzyme's activity by maintaining the shape of the oxyanion loop and of the S1 pocket through hydrogen bonds.
Materials and Methods
Crystallization and X-ray structure determination
SARS-CoV Mpro was produced in large quantities as described.8, 13 Crystals belonging to monoclinic space group P21 were grown at pH 6.0 using the published conditions.13 Under the same conditions, especially at slightly lower pH (5.9) and higher (up to ∼20%) concentrations of polyethylene glycol 6000, a new tetragonal crystal form was observed, often in the same droplets where the monoclinic crystals appeared. An additional screen for improving the crystallization conditions was carried out using the “Anion Suite” of Nextal Biotechnologies (Montreal, Canada) and condition no. 36 (1.2 M sodium malonate in 0.1 M Mes, pH 6.5) yielded orthorhombic crystals belonging to space group P21212. These were optimized at 0.7 M sodium malonate (pH 6.6). This crystal form is similar, if not identical, to the one recently described by Hsu et al.14 Since coordinates of the structure described by these authors are not yet available, we solved the structure of the orthorhombic crystal form as well. To some crystallization setups, inhibitors were added that had been identified in an in silico screening effort9 and subsequently synthesized or purchased. In other cases, crystals of the free enzyme were soaked in solutions containing the inhibitor in DMSO. For several of these cases, X-ray structure analysis showed that the inhibitors had not bound to the proteinase. Therefore, the resulting multiple structures of the free enzyme were used to analyze the flexibility of the Mpro. Diffraction data were collected at 100 K at the Joint University of Hamburg/IMB Jena/EMBL synchrotron beamline X13 at DESY, Hamburg, Germany, at a wavelength of ∼0.80 Å. Intensities were measured by a MarCCD detector and processed using the HKL suite.24 Crystal structures were determined by molecular replacement and difference Fourier methods, using the original X-ray structure for the SARS-CoV Mpro crystallized at pH 6.0 (PDB code 1UJ113) as the initial model. CNS25 and, in later stages of refinement, REFMAC26 were employed for X-ray structure refinement. Electron density interpretation and model building were performed using the computer graphics programs O27 and XtalView.28
Molecular dynamics simulations of the Mpro dimer
The starting structures at the different pH values were taken from the Protein Data Bank (PDB entry code 1UJ1 at pH 6.0, 1UK3 at pH 7.6, and 1UK2 at pH 8.0).13 In addition to simulations of the structural dynamics of the Mpro at these pH values, calculations were also carried out at more acidic pH (pH 5.0). Since a crystal structure at a pH below 6.0 was not available at the time when these simulations were carried out, we used 1UJ1.pdb (pH 6.0) as the initial structural model, but with all histidine residues in the substrate-binding site (except His41) fully protonated. Prior to the simulations at pH 5.0, pH 6.0, and pH 7.6, residues SerA1 and GlyA2, which had not been visible in the electron density maps,13 were modeled using the molecular modeling software Sybyl 6.8 (Tripos Associates, St Louis, MO, 2000). The modeled conformation at the beginning of each simulation was similar to that of residues SerB1 and GlyB2 of the other polypeptide chain in the dimer, which did exhibit well-defined electron density. The program package MacroDox version 3.2.229 was used to assign the titratable residues in the protein. The resulting charges of ionizable groups were appropriate for all four pH values studied, with all Arg and Lys residues protonated, and all Asp and Glu deprotonated. The protonation state of histidine residues was adopted from the MacroDox results, with the exception of His41 (see below) and of His163 and His172, which are associated with the S1 substrate-binding subsite and are at the focus of the present study. Both the His163 residues of the A and the B chain of the Mpro dimer were protonated in the simulation at pH 5.0, and unprotonated at pH 7.6 and 8.0. At pH 6.0, His163 was deprotonated in the A chain of the dimer, and protonated in the B chain. His172 of both chains was protonated at pH 5.0, pH 6.0 and pH 7.6, but deprotonated in both chains at pH 8.0 (Table 2). Cys145 and His41 of the catalytic dyad were uncharged in all simulations, in agreement with the recent findings by Huang et al. 21
All simulations were performed using the GROMACS (version 3.1.4) package.30, 31 The GROMOS87 force field32 was used with modifications as suggested by van Buuren et al. 33 and explicit hydrogen atoms in aromatic rings.34 Electrostatic interactions between charged groups at a distance less than 9 Å were calculated explicitly, long-range electrostatic interactions were calculated using the particle-mesh Ewald method35 with a grid width of 1.2 Å and a fourth-order spline interpolation. A cutoff distance of 9 Å was applied for Lennard-Jones interactions. To maintain the system at a constant temperature of 300 K, a Berendsen thermostat36 was applied using a coupling time of 0.1 ps. The pressure was held at 1 bar, with a coupling time of 1.0 ps. The isothermal compressibility was 4.5×10−5 bar−1 for water simulations. The time step was set as 2 fs. All bond lengths including hydrogen atoms were constrained by the LINCS algorithm.37
The simulation cell was a rectangular periodic box with a minimum distance of 10 Å between the protein and the box walls, so that the protein would not interact directly with its own periodic image, given the cutoff. The protein was hydrated in a box containing simple point charge (SPC) water molecules.38 A 100 mM NaCl solution was used, alongside a number of counterions to neutralize the total charge of the system. At the end, each system contained about 75,000 atoms.
All systems were minimized using the steepest descent method until the convergence value of 10 kJ mol−1 Å−1 was reached. When restraints were required on, e.g. protein atoms, a harmonic potential with a force constant of 0.1 kJ mol−1 Å−2 was applied. The simulation was initiated by dynamic equilibration of solvent molecules at 300 K for 100 ps, with velocity reassignment from a Maxwellian distribution of 0.2 ps intervals and the protein structure fixed. This was followed by fixing the protein main chain, and then the Cα atoms, in restraint dynamics equilibration runs of 20 ps each. Afterwards, dynamic equilibration of the entire system (solute and solvent) was performed at 300 K for 20 ps. Following the equilibrations, four 10 ns MD simulation runs were performed, with all trajectories sampled every 1.0 ps.
Molecular dynamics simulations of the Mpro monomer
Since protomer A of the Mpro dimer as revealed by X-ray crystallography (pH 6.0; PDB code 1UJ1) was assumed to be in the active conformation,13 it was selected as the reference structure in the MD simulation of the SARS-CoV Mpro monomer. The MD simulations were carried out as described above for the Mpro dimer. The simulation cell was a rectangular periodic box with a minimum distance of more than 7.5 Å between the protein and the box walls. To neutralize the system, 65 water molecules were replaced by 31 Cl and 34 Na ions. In total, the simulation system contained 2989 solute atoms and 11,665 solvent molecules embedded in a 59 Å×99 Å×68 Å box. Initially, all water molecules and ions, with the whole protein fixed, were energy-minimized by steepest descent until the convergence value of 10 kJ mol−1 Å−1 was reached. Afterwards, minimizations were performed continuously on the protein by first fixing the main chain and subsequently the Cα atoms. After the restraint dynamics equilibration, a 10 ns MD simulation was performed.
Calculation of binding site volume
The volume of the substrate-binding pocket was calculated using SURFNET.39 This program first selects all atoms within 7 Å around a ligand (in this case, the peptidyl chloromethylketone inhibitor used by Yang et al.13) and then introduces spheres with a diameter ranging from 4 Å to 10 Å into the space between any two non-hydrogen atoms of the protein. The volume occupied by such spheres is then estimated.
Determination of enzymatic activity
The pentadecapeptide SWTSAVLQSGFRKWA was used as a substrate in an HPLC-based cleavage assay. This peptide corresponds to the N-terminal autocleavage site of the SARS-CoV Mpro, with the exception of the P7 Ile, which had been replaced by Trp, and the P6′ Met, which had also been replaced by Trp. The concentrations of SARS-CoV Mpro and substrate were 0.83 μM and 0.55 mM, respectively. The reactions were carried out at different pH values in 36 mM buffer solution. The buffer was sodium acetate for pH values between 4.5 and 5.6, bis-Tris for pH 5.8–7.0, bis-Tris-propane for pH 7.0–9.4. The enzymatic activity was found to be reduced by 20% in bis-Tris-propane buffer and this was corrected for. Reactions were run for 15 min and then stopped by addition of 0.1% trifluoric acid. After 5 min cooling on ice, samples were centrifuged and 90 μl of the supernatant were mixed with 60 μl of HPLC sample buffer. Separation of products and substrate was carried out using a reverse-phase (RP) HPLC column (Jupiter 4μ Proteo 90A; Phenomenex) and a linear gradient (1%–90%) of acetonitrile in 0.1% trifluoroacetic acid. Detection of products and substrate was achieved using a fluorescence detector (RF1002, Gynkotec) at 353 nm.
For determination of the enzyme kinetics, substrate concentration was varied between 0.1 mM and 1.75 mM, at a constant enzyme concentration of 0.75 μM. Substrate and enzyme were incubated in 25 mM bis–Tris–HCl buffer (pH 7.0) at 25 °C. Reaction aliquots were removed at different times up to 12 min, and analyzed by RP-HPLC as described above.
Protein Data Bank accession codes
The coordinates and structure factors, respectively, are available from the RCSB Protein Data Bank under PDB codes 2BX3 and r2bx3sf (tetragonal form), and 2BX4 and r2bx4sf (orthorhombic form).
Acknowledgements
We are grateful to Walter Verheyen for expert technical assistance. This work was supported by the Sino-European Project on SARS Diagnostics and Antivirals (SEPSDA) of the European Commission (contract no. SP22-CT-2004-003831), the Sino-German Center for the Promotion of Science (Beijing, grant no. GZ 233 - 202/6), the Deutsche Forschungsgemeinschaft (Hi 611/4-1), the Shanghai Basic Research Project from the Shanghai Science and Technology Commission (grant 02DJ14070, 03DZ19212), the National Natural Science Foundation of China (grants 20372069, 29725203 and 20072042), the State Key Program of Basic Research of China (grants 2003CB514125, 2003CB514124, 2002CB512807 and 2002CB512802), the 863 Hi-Tech Program (grants 2001AA235051, 2001AA235071 and 2002AA3301), and the special programs on fighting SARS of the Ministry of Science and Technology, Chinese Academy of Sciences, National Natural Science Foundation of China and Shanghai Science and Technology Commission. M.Y. was supported by a stipend from the University of Lübeck. R.H. thanks the Fonds der Chemischen Industrie for continuous support.
Edited by R. Huber
References
- 1.Peiris J.S., Chu C.M., Cheng V.C., Chan K.S., Hung I.F., Poon L.L. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/S0140-6736(03)13412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Groneberg D.A., Hilgenfeld R., Zabel P. Molecular mechanisms of severe acute respiratory syndrome (SARS) Respir. Res. 2005;6:8–23. doi: 10.1186/1465-9921-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 6.Dragovich P.S., Prins T.J., Zhou R., Brown E.L., Maldonado F.C., Fuhrman S.A. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 6. Structure-activity studies of orally bioavailable, 2-pyridone-containing peptidomimetics. J. Med. Chem. 2002;45:1607–1623. doi: 10.1021/jm010469k. [DOI] [PubMed] [Google Scholar]
- 7.Matthews D.A., Smith W.W., Ferre R.A., Condon B., Budahazi G., Sisson W. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell. 1994;77:761–771. doi: 10.1016/0092-8674(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 8.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 9.Xiong B., Gui C.-S., Xu X.-Y., Luo C., Chen J., Luo H.-B. A 3D model of SARS_CoV 3CL proteinase and its inhibitors design by virtual screening. Acta Pharmacol. Sin. 2003;24:497–504. [PubMed] [Google Scholar]
- 10.Anand K., Yang H., Bartlam M., Rao Z., Hilgenfeld R. Coronavirus main proteinase: target for antiviral drug therapy. In: Schmidt A., Wolff M.H., Weber O., editors. Coronaviruses with Special Emphasis on First Insights Concerning SARS. Birkhäuser; Basel: 2005. pp. 173–199. [Google Scholar]
- 11.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W. Design of wide-spectrum inhibitors targeting coronavirus main proteinases. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030324. e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an α-extra helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl Acad. Sci. USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu M.-F., Kuo C.-J., Chang K.-T., Chang H.-C., Chou C.-C., Ko T.-P. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ziebuhr J., Heusipp G., Siddell S.G. Biosynthesis, purification, and characterization of the human coronavirus 229E 3C-like proteinase. J. Virol. 1997;71:3992–3997. doi: 10.1128/jvi.71.5.3992-3997.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi J., Wei Z., Song J. Dissection study on the severe acute respiratory syndrome 3C-like protease reveals the critical role of the extra domain in dimerization of the enzyme: defining the extra domain as a new target for design of highly specific protease inhibitors. J. Biol. Chem. 2004;279:24765–24773. doi: 10.1074/jbc.M311744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen S., Chen L., Tan J., Chen J., Du L., Sun T. Severe acute respiratory syndrome coronavirus 3C-like proteinase N terminus is indispensable for proteolytic activity but not for enzyme dimerization: biochemical and thermodynamic investigation in conjunction with molecular dynamics simulations. J. Biol. Chem. 2005;280:164–173. doi: 10.1074/jbc.M408211200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004;279:1637–1642. doi: 10.1074/jbc.M310875200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polgar L. Mercaptide-imidazolium ion-pair: the reactive nucleophile in papain catalysis. FEBS Letters. 1974;47:15–18. doi: 10.1016/0014-5793(74)80415-1. [DOI] [PubMed] [Google Scholar]
- 20.Polgar L. Characterization of the active site thiol group of rhinovirus 2A proteinase. FEBS Letters. 2000;481:289–292. doi: 10.1016/s0014-5793(00)02024-x. [DOI] [PubMed] [Google Scholar]
- 21.Huang C., Wei P., Fan K., Liu Y., Lai L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general-base mechanism. Biochemistry. 2004;43:4568–4574. doi: 10.1021/bi036022q. [DOI] [PubMed] [Google Scholar]
- 22.Pang Y.P. Three-dimensional model of a substrate-bound SARS chymotrypsin-like cysteine proteinase predicted by multiple molecular dynamics simulations: catalytic efficiency regulated by substrate binding. Proteins: Struct. Funct. Bioinformat. 2004;57:747–757. doi: 10.1002/prot.20249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J., Yu M., Jan Y.N., Jan L.Y. Stabilization of ion selectivity filter by pore loop ion pairs in an inwardly rectifying potassium channel. Proc. Natl Acad. Sci. USA. 1997;94:1568–1572. doi: 10.1073/pnas.94.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.Brunger A.T., Adams P.D., Clore G.M., DeLano W.L., Gros P., Grosse-Kunstleve R.W. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallog. sect. D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 26.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallog. sect. D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 27.Jones T.A., Zou J.Y., Kjeldgaard M., Cowan S.W. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog. sect. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 28.McRee D.E. XtalView/Xfit–A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 29.Northrup S.H., Laughner T., Stevenson G. Tennessee Technological University, Department of Chemistry; Cookeville, TN: 1999. MacroDox Macromolecular Simulation Program. [Google Scholar]
- 30.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 31.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 32.van Gunsteren, W. F. & Berendsen, H. J. C. (1987). Gromos-87 Manual. Biomos BV, Nijenborgh 4, Groningen, The Netherlands.
- 33.Van Buuren A.R., Marrink S.J., Berendsen H.J.C. A molecular dynamics study of the decane/water interface. J. Phys. Chem. 1993;97:9206–9212. [Google Scholar]
- 34.van Gunsteren, W. F., Billeter, S., Eising, A. A., Hünenberger, P., Kruger, P. & Mark, A. E. et al. (1996). Biomolecular Simulations: GROMOS96 Manual and User Guide. Biomos BV, Zürich, Groningen.
- 35.Darden T., York D., Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 36.Berendsen H.J.C., Postma J.P.M., Gunsteren W.F. DiNola A, Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 37.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 38.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces. D. Reidel Publishing Company; Dordrecht, The Netherlands: 1981. pp. 331–342. [Google Scholar]
- 39.Laskowski R.A. SURFNET: A program for visualizing molecular surfaces, cavities and intermolecular interactions. J. Mol. Graph. 1995;13:323–330. doi: 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- 40.Kraulis P.J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallog. 1991;24:946–950. [Google Scholar]
- 41.DeLano W.L. DeLano Scientific LLC; San Carlos, CA, USA: 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- 42.Chou C.-Y., Chang H.-C., Hsu W.-C., Lin T.-Z., Lin C.-H., Chang G.-G. Quaternary structure of the severe acute respiratory syndrome (SARS) coronavirus main protease. Biochemistry. 2004;43:14958–14970. doi: 10.1021/bi0490237. [DOI] [PubMed] [Google Scholar]