

Abstract
Viruses have been known to subvert the anti-apoptotic pathways of the host cell in order to delay apoptosis. However, the mechanisms utilized by enterovirus 71 (EV71) to mediate anti-apoptotic activity remained undetermined. We observed that EV71 infection induced an early activation of both phosphatidylinositol 3-kinase (PI3K)/Akt and MAPK/ERK signaling pathways. The activity of GSK3β, a downstream target of these pathways, was negatively regulated by the activation of both MAPK/ERK and PI3K/Akt. The phosphorylation of GSK3 could be inhibited by treatment with the specific inhibitors of MAPK/ERK and PI3K/Akt. Other Akt downstream targets, BAD, caspase-9 and the Forkhead transcription factor (FKHR), were not phosphorylated during the course of infection by EV71. We further demonstrated that infection by UV-irradiated, inactivated virus triggered early Akt activation but was insufficient to trigger late Akt activation. These data suggest that with the phosphorylation of MAPK/ERK and PI3K/Akt the subsequent inactivation of GSK3β is utilized by EV71 as a potential mechanism to delay host cell apoptosis.
Keywords: Enterovirus 71, Apoptosis, Akt, MAPK, UV-inactivation
Introduction
Enterovirus 71 (EV71) is a positive-stranded RNA virus in the genus Enterovirus of the family Picornaviridae. EV71 infection can cause hand, foot, and mouth disease (HFMD) and herpangina. However, EV71 has also been associated with fatal pulmonary edema as well as severe neurological complications, including encephalitis, meningitis, and a poliomyelitis-like syndrome (Gilbert et al., 1988, Alexander et al., 1994, Lum et al., 1998, Chang et al., 1999, Ho et al., 1999, Chen et al., 2001).
In 1998, a large EV71 outbreak infecting more than 120,000 children resulted in 78 fatalities (Ho, 2000). Postmortem studies using immunofluorescence and molecular assays during this outbreak clearly showed that EV71 infected the central nervous system (Hsueh et al., 2000). Furthermore, isolates were extracted from the medulla oblongata and spinal cord (Chang et al., 1998, Shih et al., 2000). Due to the consequences of this and other EV71 outbreaks worldwide (McMinn, 2002), further studies of the mechanisms associated with EV71 infection are essential. Furthermore, a greater understanding of key pathways that are necessary for viral entry and replication may lead to novel therapies which may impact on the health of children worldwide.
An increasing number of viruses have been found to induce apoptosis at the late stages of infection. This process may be important in facilitating the spread of progeny virus to neighboring cells and for providing protection for the progeny virus against host enzymes and antibodies (Teodoro and Branton, 1997). However, viruses have also evolved diverse strategies to evade or delay the early onset of apoptosis. This affords a selective advantage to the virus, allowing it to replicate, spread, and maintain persistent infections (O'Brien, 1998, Roulston et al., 1999, Tortorella et al., 2000, Everett and McFadden, 2001). To increase cell survival and proliferation, the viruses target various intracellular signaling pathways (Teodoro and Branton, 1997), including MAPK/ERK and PI3K/Akt. For example, infection with coxsackievirus, an enterovirus related to EV71, results in activation of both of these survival pathways (Huttunen et al., 1998, Luo et al., 2002, Zhang et al., 2003). Furthermore, coxsackievirus B3 (CVB3) triggers bimodal MAPK/ERK activation, in that initial binding of the virus causes first phase activation while the subsequent phase was elicited by viral replication (Luo et al., 2002). In addition, the PI3K/Akt pathway is also modulated during CVB3 infection (Zhang et al., 2003). Although the activation of these important pathways has been described for other enteroviruses, little is known regarding the action of EV71 on cellular signaling and survival.
Several well-characterized physiological substrates for Akt have been identified to date, including glycogen synthase kinase-3 (GSK-3) (Cross et al., 1995). GSK-3, a ubiquitously expressed protein–serine/threonine kinase, is inhibited by Akt phosphorylation in response to growth factor stimulation. In addition to glycogen synthase, GSK-3 phosphorylates a broad range of substrates, including several transcription factors and the translation initiation factor. These studies suggest that GSK-3 is involved in multiple cellular processes, including metabolism, proliferation, and differentiation (Woodgett, 2001). Additional targets of Akt that have been implicated in control of cell survival include apoptosis signal-regulating kinase 1 (Kim et al., 2001), caspase-9 (Cardone et al., 1998), BAD (Franke and Cantley, 1997), Forkhead transcription factor (FHKR) (Brunet et al., 1999), and NF-κB (Madrid et al., 2000).
Here we report that EV71 induces an anti-apoptotic cellular response via the activation of both the MAPK/ERK and PI3K/Akt pathways that, in turn, inhibit the activity of GSK-3. We found that the phosphorylation status of caspase-9, BAD, and FHKR remained unaffected. UV-inactivated EV71 only stimulated early- but not late-phase Akt phosphorylation, strongly suggesting that signaling is initiated directly by the virus binding to the host receptors.
Materials and methods
Viruses and cell cultures
The enterovirus 71 (isolate 71-2231-TW) was isolated in the 1998 outbreak and supplied by Dr. Shin-Ru Shih of the Clinical Virology Laboratory of Chang Gung Memorial Hospital, Taiwan. Virus stocks were propagated in RD cells using Dulbecco's modified Eagle's medium (DMEM) supplemented with 2% fetal bovine serum (FBS). Cell lines including Vero, RD, and MRC-5 were maintained in DMEM supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml) in an incubator at 37 °C containing 5% CO2.
Antibodies and chemicals
Polyclonal antibodies recognizing phosphorylated Akt (Ser473), phosphorylated FKHR, and phosphorylated GSK3β (Ser9) were obtained from New England Biolabs. A monoclonal antibody against phosphorylated ERK1/2 (Thr202/Tyr204, clone E10) was obtained from New England Biolabs. Polyclonal antibodies recognizing ERK2, Akt, β-actin, BAD, and phosphorylated BAD (Ser136) were purchased from Santa Cruz Biotechnology, Inc. A polyclonal antibody against phosphorylated caspase-9 was obtained from Biocarta (San Diego, CA). Wortmannin and PD98059 were supplied by Sigma.
Virus purification
EV71 virus was adsorbed to confluent RD monolayer cells in T175 culture flasks for 2 h at 37 °C with occasional shaking. Cells were then incubated in DMEM supplemented with 2% FBS until a 90% cytopathic effect was reached. After initial centrifugation at 2000 ×g for 5 min, the cell pellet was subjected to three cycles of freezing and thawing followed by centrifugation to release the intracellular virus particles. Viral aliquots were stored at − 80 °C.
Virus plaque assay
RD cells (4 × 105 cells/well) were plated onto a 6-well plate, incubated overnight, and then infected with serially diluted virus suspension. After adsorption for 1 h, the virus suspension was replaced with DMEM containing 2% FBS and 0.3% agarose. The medium was removed at 96 h post infection. The cells were fixed with 10% formaldehyde and subsequently stained with 1% crystal violet. The titer of the virus was expressed as PFU (plaque forming unit) per milliliter.
Virus infection
Cells were grown to 70%–80% confluence in complete medium, whereupon the cells were serum starved by incubation in serum-free DMEM for 24 h. For viral infection, growth-arrested cells were infected at a multiplicity of infection (MOI) of 5 with EV71 or were sham treated (the same growth medium but without virus) for 1 h. Cells were washed with phosphate-buffered saline (PBS) and then cultured in fresh medium for various lengths of time as indicated. For inhibitor experiments, cells were incubated with the PI3K inhibitor, wortmannin, or the MEK inhibitor, PD98059, for 1 h. The 1000× stock of inhibitors was made in DMSO. Cells were then infected for 1 h, washed with PBS, and placed in serum-free media containing fresh doses of the appropriate inhibitor unless otherwise specified.
Solubilization of cells, electrophoresis, and Western blotting
After the EV71 infection, culture medium was removed and cells were resuspended in 170 μl of lysis buffer per 6-cm culture dish (1% Triton X-100, 50 mM sodium chloride, 1 mM EDTA, 1 mM EGTA, 20 mM sodium fluoride, 20 mM sodium pyrophosphate, 1 mM phenylmethylsulfonyl fluoride, 0.5 μg/ml leupeptin, 1 mM benzamidine, and 1 mM sodium orthovanadate in 20 mM Tris–HCl, pH 8.0). After lysis at 4 °C, the samples were sonicated and pelleted at 12,000 rpm for 15 min in a microfuge. The supernatants were normalized for equal protein content (as measured in a BCA assay, Pierce). Samples (30 μg protein per lane) were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (12% polyacrylamide) (SDS-PAGE) under reducing conditions, using a Mini-Protean III system (Bio-Rad). Proteins were transferred to nitrocellulose membrane and the membrane was incubated successively at room temperature with 5% (w / v) non-fat dried milk in PBST (Na2HPO4 80 mM, NaH2PO4 50 mM, NaCl 100 mM, and 0.05% (w / v) Tween 20, pH 7.5) for 1 h. Membranes were incubated overnight at 4 °C with primary antibodies in PBST. Membranes were washed with PBST four times for 5 min each and incubated with a 1 : 2500 dilution of anti-goat or anti-mouse horseradish peroxidase Ab for 1h. Following each incubation, the membrane was washed extensively with PBST. The immunoreactive bands detected by ECL reagents (GE Healthcare) were developed by Super RX film (Fujifilm).
Immunofluorescence
Growth-arrested Vero cells were infected with EV71 for 1 h at MOI of 5 and the cells were harvested at 0.5 h post infection (pi). The cells were washed five times in PBS and fixed with 2% BSA/1% gelatin/0.02% saponin in PBS for 1 h. Cells were incubated with Akt antibodies (Santa Cruz) for 1.5 h and with Rodamine-conjugated secondary antibodies for 1 h. Coverslides were mounted and cells were visualized using a Zeiss Axiovert 200M immunofluorescence microscope.
UV-irradiated EV-71
Two-milliliter aliquots of virus were dispersed in a 6-cm tissue culture dish, followed by UV-irradiation to inactivate the virus. This was accomplished by placing a compact UV lamp (UVP, Upland, CA) directly on the top of the dish for 1 h on ice. The remaining titer of the inactivated virus was determined using the plaque assay.
Data analysis
The results were quantitated by densitometric analysis (ImageQuant, Molecular Dynamics and PDSI, GE Healthcare). The ratio of phosphoprotein to its respective internal control was normalized to the control level at 0.5 h, arbitrarily set to 1. Data were expressed as the means ± S.E.M. and analyzed with two-tailed Student's t-test at a P < 0.05 level of significance.
Results
EV71 infection leads to activation of Akt
To determine the host cell intracellular pathways involved in EV71 infection, we first examined the kinetics of Akt activation. Growth-arrested Vero cells were infected with EV71 at an MOI of 5 for 1 h and cells were harvested at 0.5, 3 and 6 h pi. Akt activity was determined using an antibody to phosphorylated Akt. As shown in Fig. 1A and B, exposure of Vero cells to EV71 stimulated Akt activity, peaking as early as 0.5 h and sustaining at a high level until at least 6 h. The level of phosphorylation was clearly obvious at 10 min pi but peaked at 0.5 h pi (see Fig. 4A). Therefore, we chose 0.5 h pi as the early infection point for the following experiments. Upon EV71 infection at 0.5 h pi, a significant portion of Akt was concentrated locally on the plasma membrane as indicated by the arrow (Fig. 1C). The membrane translocation of Akt is crucial for phosphorylation and activation by phosphoinositide-dependent kinase, which is usually constitutively active in various cell types (reviewed in Chan et al. (1999)). We then determined whether this activation was a cell-specific phenomenon. The activation of Akt was examined in further two cell lines, MRC-5 and RD, infected with EV71 at the same MOI. The MRC-5 cells, a human embryonic lung cell line, showed the same activation kinetic as observed in the Vero cells (Fig. 1D and E). However, the RD cells, a human rhabdomyosarcoma cell line, did not respond to the EV71 infection even at 6 h pi (data not shown). This demonstrates cell-type-specific differences in the ability of EV71 to induce Akt phosphorylation. Furthermore, we investigated the possibility that PI3K was an upstream target in the activation of Akt by EV71, via the use of wortmannin, a potent and specific inhibitor of PI3K. Wortmannin blocked Akt phosphorylation effectively (Fig. 2B and C). The mock infection with the vehicle, DMSO, did not affect Akt phosphorylation (data not shown). Activation of PI3K and its downstream target, Akt, has been widely implicated in transmitting survival signals in response to a wide variety of stimuli in many different cell types. To determine whether PI3K/Akt activity was influenced by other proteins present in the media during virus preparation, we treated growth-arrested Vero cells with RD cell extract processed in an identical freeze–thaw protocol to that used during virus propagation, but in the absence of virus. We did not observe significant modulation of Akt phosphorylation compared to that of the control (data not shown). Therefore, we believe that the activation of Akt is due to the direct interaction between virus and host cells. EV71 can activate the PI3K/Akt survival pathway in the early stage of infection. This may allow the virus to defend against the premature death of the host cell, maximizing virus progeny from a lytic infection and/or facilitating a persistent infection.
Fig. 1.
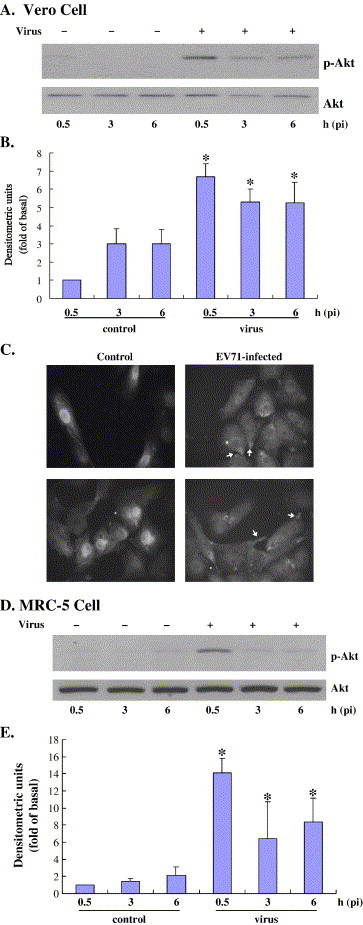
Time course for EV-71 stimulation and Akt phosphorylation. Growth-arrested Vero (A–C) and MRC-5 (D and E) cells were incubated with EV-71 at an MOI of 5 for 1 h and then the cells were washed with PBS twice and replenished with serum-free medium. For immunoblot analysis, cell lysates were collected at the indicated times following EV-71 infection and equal amounts of protein were subjected to SDS-12% PAGE. Proteins were transferred onto nitrocellulose membranes and subjected to Western blotting. Akt activity was analyzed based on phosphorylated Akt (P-Akt). Unphosphorylated Akt was used as the loading control. The control samples contained the same serum content as the virus group. In C, Vero cells were subjected to immunofluorescent staining at 0.5 h pi. Akt redistribution was indicated by arrows. The immunoblot results were quantitated by densitometric analysis and normalized to control 0.5 h levels arbitrarily set to 1 (B and E). Values are means ± S.E.M. from three independent experiments for B and E. *P < 0.05 compared with respective control.
Fig. 4.
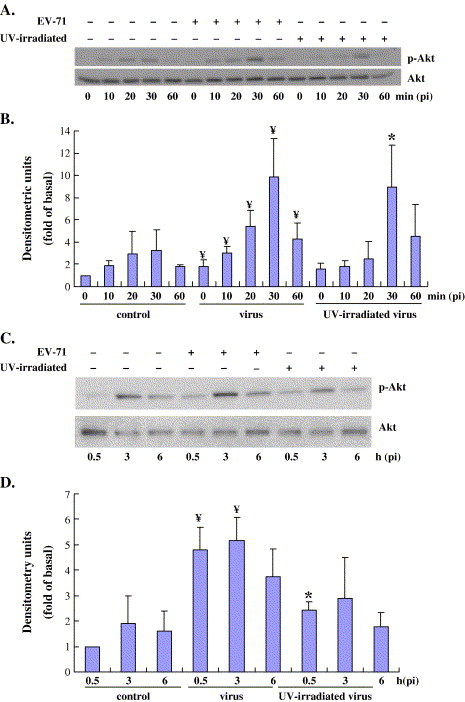
Early- but not late-phase Akt phosphorylation caused by UV-inactivated EV71. The virus was UV-irradiated for 1 h and the remaining titer of the inactivated virus was determined by the plaque assay. The UV-irradiated virus lost infection activity completely after treatment (data not shown). Vero cells were infected with either wild-type virus or UV-inactivated virus. Following 0, 10, 20, 30, and 60 min (A and B) or 0.5, 3 and 6 h (C and D) after infection, cell lysates were harvested and Western blotting was performed to determine Akt activation. Unphosphorylated Akt was used as the control. Values are means ± S.E.M. from four independent experiments. * and ¥, P <0.05 compared with respective control.
Fig. 2.
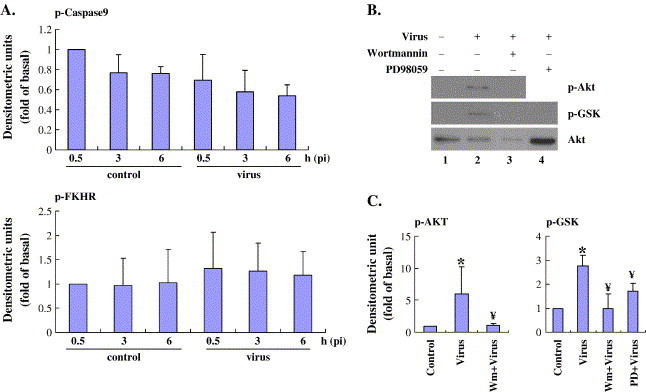
EV71 strongly promotes expression of phospho-Akt and phosphor-GSK-3 but not phospho-caspase 9 and phospho-FKHR. (A) Vero cells were serum-starved overnight, induced with EV71 and harvested at the indicated time points. The phosphorylation of caspase 9 and FKHR was detected by Western blot using respective antibodies. (B and C) GSK phosphorylation was monitored with polyclonal antibodies against phosphorylated GSK (Ser9). Vero cells were pre-treated with or without wortmannin (0.1 μM, Lane 3) for 3 h and PD98059 (PD, 50 μM, Lane 4) for 1 h, followed by infection with EV71 (MOI5) to induce phosphorylation of Akt and GSK3β. Unphosphorylated Akt was used as the control. The results were also quantitated and normalized as in Fig. 1. Values are means ± S.E.M. from three independent experiments. *P < 0.05 compared with control; ¥P < 0.05 compared with virus infection alone.
GSK3β is targeted by EV71 via the activation of the PI3K/Akt and MAPK/ERK pathways
Downstream targets of Akt that have been implicated in control of cell survival include the transcription factor Forkhead (Brunet et al., 1999), human caspase-9 (Cardone et al., 1998), BAD (Franke and Cantley, 1997), and GSK3β (Cross et al., 1995). To determine whether these target proteins are phosphorylated by Akt kinase after EV71 infection, we performed Western blot assays using antibodies against the phosphorylated forms of GSK3β, FKHR, caspase-9, and BAD. No apparent induction of caspase-9 or FKHR (Fig. 2A) or BAD (data not shown) phosphorylation was demonstrated as compared to the non-infected control cells. This negative result was interpreted by reference to a positive control such as 20% FBS-stimulated FKHR pathway (data not shown). Contrastingly, the phosphorylation of GSK-3 was induced approximately 3-fold by activated Akt at 0.5 h pi (Fig. 2B-C). Furthermore, GSK-3 phosphorylation was significantly inhibited by adding wortmannin to the cell culture, as shown in Lane 3 of Fig. 2B. This suggests that EV71 induces cell survival through the PI3K/Akt/GSK3 signaling pathway and that this action is independent of the Akt target proteins, FKHR caspase-9, and BAD.
EV71 infection also induced specific MAPK/ERK phosphorylation
The mitogen-activated protein kinase (MAPK) pathway is another major cell survival mechanism. Therefore, we investigated whether EV71 induces intracellular ERK1/2 phosphorylation events during the course of infection. Vero and MRC-5 cells were either infected with EV71 or mock infected for time intervals ranging from 0.5 to 6 h pi. Immunoblotting was conducted using antibodies specific for phosphorylated ERK1/2. The data demonstrates that in Vero cells, EV71 infection induced modest ERK phosphorylation at 0.5 h pi, reaching a peak at 3 h pi and declining afterwards (Fig. 3A and B). By contrast, in MRC-5 cells, expression of phosphorylated ERK was strong at 0.5 h pi and then rapidly decreasing to basal levels as shown in Fig. 3C and D. β-actin was used as control for loading of total proteins. In addition, the EV71-induced ERK phosphorylation was inhibited by a MAPK-specific inhibitor PD98059 (data not shown). In summary, infection with EV71 induced both PI3K/Akt and MAPK/ERK phosphorylation in a cell-type- and temporal-specific manner.
Fig. 3.
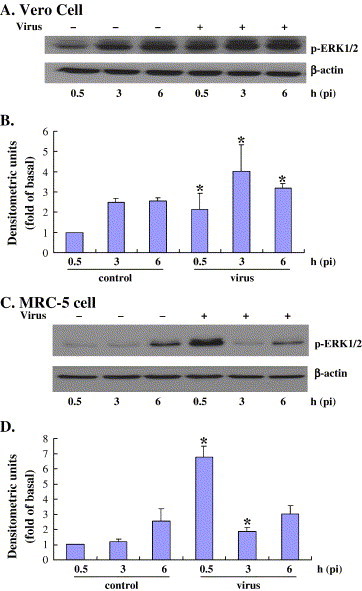
Time course for EV-71 stimulation of ERK1/2 phosphorylation. Growth-arrested Vero (A and B) and MRC-5 (C and D) cells were incubated with EV71 at an MOI of 5 for 1 h. The cells were then washed with PBS twice and replenished with serum-free medium. Cell lysates were collected at the indicated times following EV71 infection and were processed as in Fig. 1. ERK1/2 activity was determined based on phosphorylated ERK1/2 (P-ERK1/2). In the lower panels, the results were quantitated and normalized as in Fig. 1. Values are means ± S.E.M. from three independent experiments. *P < 0.05 compared with control.
In addition to the PI3K/Akt pathway, other studies suggest that activation of ERK1/2 can also phosphorylate GSK3β and promote cell survival (Stambolic and Woodgett, 1994, Eldar-Finkelman et al., 1995). To examine this hypothesis in relation to EV71 infection, Vero cells were pre-incubated with or without 50 μM of PD98059 for 1 h followed by virus infection. The phosphorylation of GSK3β was measured by immunoblot using anti-phospho GSK3β antibodies. Treatment of cells with PD98059 resulted in considerable inhibition of EV71-induced GSK3β phosphorylation (Lane 4 of Fig. 2). This indicated that GSK3 was not only activated by PI3K/Akt but was also regulated by the MAPK/ERK pathway after EV71 infection. Due to the profound inhibition of Akt produced by wortmannin, we could not test whether there was an accumulative effect from the addition of both inhibitors. Furthermore, since Akt protein was reported to be cleaved by caspases during apoptosis (Widmann et al., 1998, Rokudai et al., 2000), we next examined the integrity of total cellular Akt during EV71 infection via Western blot. There was no apparent reduction of the Akt protein levels up to 6 h post infection (data not shown).
UV-irradiated EV71 stimulates early- but not late-phase phosphorylation of Akt
To further elucidate the Akt activation kinetics of EV71, we investigated the response of Vero cells to UV-irradiated EV71 virus. UV-inactivated enterovirus fails to express viral proteins due to the presence of thymidine dimers, which prevent the transcription of viral genes and render the virus incapable of replication. Interestingly, this does not interfere with the virus' capacity for receptor binding nor does it affect endocytosis into host cells (Beck et al., 1990). We hypothesized that the direct binding of the virus to host cells could trigger the survival pathway by phosphorylating Akt because the specific ligand–receptor interactions could result in rapid and transient activation of ERK1/2, as described by Marshall (1995). We investigated the effect of inactive virus on the phosphorylation of Akt. The virus was UV-irradiated for 1 h and the remaining titer of the irradiated virus was completely abolished as determined by the plaque assay (data not shown). The co-culture of Vero cells and UV-irradiated EV71 leads to an early Akt phosphorylation event occurring apparently at 20 min pi (Fig. 4A and B). This phosphorylation peaked at 30 min pi, without significant loss of activity compared to the induction by an active virus. However, the sustained levels of Akt phosphorylation at 6 h pi, as displayed by the active virus, were completely ablated (Fig. 4C and D). This suggests that receptor interaction with EV71 is responsible for the early Akt activation, while viral protein production appears necessary for late-phase Akt activation following EV71 infection.
Discussion
This study has provided new insights into our understanding of the interplay between EV71 and the host signaling pathways that are induced upon infection. We present data demonstrating that EV71 infection activates the PI3K/Akt and MAPK/ERK pathways, which may induce cell survival. Many viruses have been reported to induce either PI3K/Akt and/or MAPK/ERK upon infection resulting in activation of the host cell's anti-apoptotic mechanisms. For example, an interaction with Akt has been observed in a number of other viruses including human papillomavirus type 16 (Nair et al., 2003), Epstein–Barr virus (Dawson et al., 2003), human cytomegalovirus (Johnson et al., 2001), and hepatitis B and C viruses (Lee et al., 2001, He et al., 2002). Such widespread involvement of these pathways suggests the presence of a global virus strategy to prevent host cell death. Furthermore, our results also confirm and expand aspects of previous reports that PI3K/Akt and MAPK/ERK are activated during the process of enterovirus infection (Luo et al., 2002, Huber et al., 1999, Opavsky et al., 2002). Moreover, the phosphorylation of Akt appears to be cell-type specific, in that Vero and MRC5 but not RD cells phosphorylate Akt (Fig. 1). This suggests that not all cell types are conducive to infection or it is possible that this pathway is not inducible in RD cells. Although Akt is not phosphorylated in RD cells, we observed phosphorylation of MAPK/ERK after EV71 infection in RD cells (data not shown). In the present study we have shown that EV71 subverts the cell survival signaling pathways which may enhance its own chance of survival.
Activation of the Akt pathway results in phosphorylation of numerous Akt target proteins, which mediate multiple cellular functions. Targets of Akt implicated in the regulation of cell survival include apoptosis signal-regulating kinase 1 (Kim et al., 2001), caspase-9 (Cardone et al., 1998), the transcription factors Forkhead (Brunet et al., 1999), BAD (Franke and Cantley, 1997), and NF-κB (Madrid et al., 2000). Generally, activation of the Akt pathway should activate a number of its downstream substrates. However, we found that P-Akt specifically phosphorylated only GSK-3β. There are many precedents of such events. Mizutani et al. (2004) have previously investigated the phosphorylation of downstream targets of Akt in SARS CoV-infected Vero E6 cells and reported that the level of phosphorylation of GSK-3β was slightly increased, whereas phosphorylated BAD and FKHR were not detected. In cardiomyocytes, survival signals stimulated by insulin-like growth factor-I do not induce phosphorylation of BAD but suppress activation of caspase-3 (Wu et al., 2000).
The anti-apoptotic activity of Akt can be modulated by caspases, which cleave Akt and thus accelerate apoptotic cell death (Widmann et al., 1998, Rokudai et al., 2000). In EV71-infected cells, we show no evidence for phosphorylation induction of the Akt targets, caspase-9 and Forkhead (Fig. 2). Thus we examined the integrity of total cellular Akt during EV71 infection via Western blot. There is no apparent reduction of the Akt protein levels up to 6 h post infection (data not shown), indicating that at this time point the caspases may not have been activated, although it should be noted that this might not have been a sufficient length of time as there was no evidence of any cell death at the 6 h time point.
Another target of PI3K/Akt signaling, the proapoptotic protein GSK3, is also phosphorylated and inactivated by ERK1/2 signaling (Hetman et al., 2002). The coxsackievirus CVB3, an enterovirus related to EV71, also induces phosphorylation of MAPK/ERK and PI3K/Akt and its downstream target GSK3 (Zhang et al., 2003) which is then followed by a delayed activation of caspase-9 and caspase-3. We demonstrate here that GSK3 is regulated both by MAPK/ERK and PI3K/Akt in EV71-infected cells. Thus, it is proposed that ERK1/2 and Akt signaling can cooperate to reduce the apoptotic threshold in cells.
Immediately following infection with either EV71 or UV-irradiated EV71, Akt and ERK1/2 were significantly phosphorylated. Toward the final stages of virus infection, persistent activation was observed only when the virus was not UV-inactivated. There are two mechanisms an UV-irradiated virus can employ to activate Akt and ERK. These pathways may be induced by direct virus–receptor binding, such as that seen in the HIV activation of ERK (Popik et al., 1998), or by exposure to a viral protein, for example the hepatitis C virus core protein (Fukuda et al., 2001) and the HIV Tat protein (Rusnati et al., 2001). The very early activation peak observed immediately following EV71 infection strongly suggests that signaling is initiated directly by the virus binding to the host receptors. In support of this data, UV-inactivated EV71 only stimulates early-phase phosphorylation of Akt (Fig. 4). UV-irradiated, inactivated EV71 does indeed activate early but not late signaling (at 3 and 6 h pi, Fig. 4). Thus we show that UV-irradiated virus, incapable of replication, does not trigger late Akt activation. This finding suggests that the observed high level of late-phase activation is dependent on viral gene expression as has been observed previously in CVB3 infection (Luo et al., 2002, Carthy et al., 1998).
Taken together, these results indicate that GSK-3 is a key target of PI3K/Akt and MAPK/ERK signaling, which leads to prevention of apoptosis in an early-stage infection of EV71. These findings implicate GSK-3 as a central element in the PI3K/Akt survival pathway, with phosphorylation of one or more targets of GSK-3 presumably serving to activate apoptotic cell death. Identification of the GSK-3 targets that regulate apoptosis at early stage of virus infection thus poses a critical next step to understanding the signaling pathways by which viral protein(s) control cell survival. An investigation to elucidate a more complete picture of the molecular mechanisms involved in cell survival during infection by EV71 is currently underway.
Acknowledgements
We thank Dr. Shin-Ru Shih for the EV71 and Dr. Tzu-Chien Wang for comments. This work was supported in part by grant from the Chang Gung Medical Research Project (CMRP No. 1188), Kweishan, Taoyuan, Taiwan and National Science Council (92-2311-B-182-001), Taiwan.
References
- Alexander J.P., Jr., Baden L., Pallansch M.A., Anderson L.J. Enterovirus 71 infections and neurologic disease—United States, 1977–1991. Journal of Infectious Diseases. 1994;169:905–908. doi: 10.1093/infdis/169.4.905. [DOI] [PubMed] [Google Scholar]
- Beck M., Chapman N., McManus B., Mullican J., Tracy S. Secondary enterovirus infection in the murine model of myocarditis. Pathologic and immunologic aspects. American Journal of Pathology. 1990;136:669–681. [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M.J., Lin M.Z., Juo P., Hu L.S., Anderson M.J., Arden K.C., Blenis J., Greenberg M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Cardone M.H., Roy N., Stennicke H.R., Salvesen G.S., Franke T.F., Stanbridge E., Frisch S., Reed J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- Carthy C.M., Granville D.J., Watson K.A., Anderson D.R., Wilson J.E., Yang D., Hunt D.W., McManus B.M. Caspase activation and specific cleavage of substrates after coxsackievirus B3-induced cytopathic effect in HeLa cells. Journal of Virology. 1998;72:7669–7675. doi: 10.1128/jvi.72.9.7669-7675.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan T.O., Rittenhouse S.E., Tsichlis P.N. AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annual Review of Biochemistry. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- Chang L.Y., Huang Y.C., Lin T.Y. Fulminant neurogenic pulmonary oedema with hand, foot, and mouth disease. Lancet. 1998;352:367–368. doi: 10.1016/S0140-6736(98)24031-1. [DOI] [PubMed] [Google Scholar]
- Chang L.Y., Lin T.Y., Hsu K.H., Huang Y.C., Lin K.L., Hsueh C., Shih S.R., Ning H.C., Hwang M.S., Wang H.S., Lee C.Y. Clinical features and risk factors of pulmonary oedema after enterovirus-71-related hand, foot, and mouth disease. Lancet. 1999;354:1682–1686. doi: 10.1016/S0140-6736(99)04434-7. [DOI] [PubMed] [Google Scholar]
- Chen C.Y., Chang Y.C., Huang C.C., Lui C.C., Lee K.W., Huang S.C. Acute flaccid paralysis in infants and young children with enterovirus 71 infection: MR imaging findings and clinical correlates. AJNR, American Journal of Neuroradiology. 2001;22:200–205. [PMC free article] [PubMed] [Google Scholar]
- Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Dawson C.W., Tramountanis G., Eliopoulos A.G., Young L.S. Epstein–Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. Journal of Biological Chemistry. 2003;278:3694–3704. doi: 10.1074/jbc.M209840200. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H., Seger R., Vandenheede J.R., Krebs E.G. Inactivation of glycogen synthase kinase-3 by epidermal growth factor is mediated by mitogen-activated protein kinase/p90 ribosomal protein s6 kinase signaling pathway in NIH/3T3 cells. Journal of Biological Chemistry. 1995;270:987–990. doi: 10.1074/jbc.270.3.987. [DOI] [PubMed] [Google Scholar]
- Everett H., McFadden G. Viruses and apoptosis: meddling with mitochondria. Virology. 2001;288:1–7. doi: 10.1006/viro.2001.1081. [DOI] [PubMed] [Google Scholar]
- Franke T.F., Cantley L.C. Apoptosis. A bad kinase makes good. Nature. 1997;390:116–117. doi: 10.1038/36442. [DOI] [PubMed] [Google Scholar]
- Fukuda K., Tsuchihara K., Hijikata M., Nishiguchi S., Kuroki T., Shimotohno K. Hepatitis C virus core protein enhances the activation of the transcription factor, Elk1, in response to mitogenic stimuli. Hepatology. 2001;33:159–165. doi: 10.1053/jhep.2001.20794. [DOI] [PubMed] [Google Scholar]
- Gilbert G.L., Dickson K.E., Waters M.J., Kennett M.L., Land S.A., Sneddon M. Outbreak of enterovirus 71 infection in Victoria, Australia, with a high incidence of neurologic involvement. Pediatric Infectious Disease Journal. 1988;7:484–488. doi: 10.1097/00006454-198807000-00007. [DOI] [PubMed] [Google Scholar]
- He Y., Nakao H., Tan S.L., Polyak S.J., Neddermann P., Vijaysri S., Jacobs B.L., Katze M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. Journal of Virology. 2002;76:9207–9217. doi: 10.1128/JVI.76.18.9207-9217.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M., Hsuan S.L., Habas A., Higgins M.J., Xia Z. ERK1/2 antagonizes glycogen synthase kinase-3beta-induced apoptosis in cortical neurons. Journal of Biological Chemistry. 2002;277:49577–49584. doi: 10.1074/jbc.M111227200. [DOI] [PubMed] [Google Scholar]
- Ho M. Enterovirus 71: the virus, its infections and outbreaks. Journal of Microbiology, Immunology, and Infection. 2000;33:205–216. [PubMed] [Google Scholar]
- Ho M., Chen E.R., Hsu K.H., Twu S.J., Chen K.T., Tsai S.F., Wang J.R., Shih S.R. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. New England Journal of Medicine. 1999;341:929–935. doi: 10.1056/NEJM199909233411301. [DOI] [PubMed] [Google Scholar]
- Hsueh C., Jung S.M., Shih S.R., Kuo T.T., Shieh W.J., Zaki S., Lin T.Y., Chang L.Y., Ning H.C., Yen D.C. Acute encephalomyelitis during an outbreak of enterovirus type 71 infection in Taiwan: report of an autopsy case with pathologic, immunofluorescence, and molecular studies. Modern Pathology. 2000;13:1200–1205. doi: 10.1038/modpathol.3880222. [DOI] [PubMed] [Google Scholar]
- Huber M., Watson K.A., Selinka H.C., Carthy C.M., Klingel K., McManus B.M., Kandolf R. Cleavage of RasGAP and phosphorylation of mitogen-activated protein kinase in the course of coxsackievirus B3 replication. Journal of Virology. 1999;73:3587–3594. doi: 10.1128/jvi.73.5.3587-3594.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen P., Hyypia T., Vihinen P., Nissinen L., Heino J. Echovirus 1 infection induces both stress- and growth-activated mitogen-activated protein kinase pathways and regulates the transcription of cellular immediate–early genes. Virology. 1998;250:85–93. doi: 10.1006/viro.1998.9343. [DOI] [PubMed] [Google Scholar]
- Johnson R.A., Wang X., Ma X.L., Huong S.M., Huang E.S. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. Journal of Virology. 2001;75:6022–6032. doi: 10.1128/JVI.75.13.6022-6032.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A.H., Khursigara G., Sun X., Franke T.F., Chao M.V. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Molecular and Cellular Biology. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.I., Kang-Park S., Do S.I. The hepatitis B virus-X protein activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. Journal of Biological Chemistry. 2001;276:16969–16977. doi: 10.1074/jbc.M011263200. [DOI] [PubMed] [Google Scholar]
- Lum L.C., Wong K.T., Lam S.K., Chua K.B., Goh A.Y. Neurogenic pulmonary oedema and enterovirus 71 encephalomyelitis. Lancet. 1998;352:1391. doi: 10.1016/s0140-6736(05)60789-1. [DOI] [PubMed] [Google Scholar]
- Luo H., Yanagawa B., Zhang J., Luo Z., Zhang M., Esfandiarei M., Carthy C., Wilson J.E., Yang D., McManus B.M. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. Journal of Virology. 2002;76:3365–3373. doi: 10.1128/JVI.76.7.3365-3373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid L.V., Wang C.Y., Guttridge D.C., Schottelius A.J., Baldwin A.S., Jr., Mayo M.W. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Molecular and Cellular Biology. 2000;20:1626–1638. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall C.J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- McMinn P.C. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiology Reviews. 2002;26:91–107. doi: 10.1111/j.1574-6976.2002.tb00601.x. [DOI] [PubMed] [Google Scholar]
- Mizutani T., Fukushi S., Saijo M., Kurane I., Morikawa S. Importance of Akt signaling pathway for apoptosis in SARS-CoV-infected Vero E6 cells. Virology. 2004;327:169–174. doi: 10.1016/j.virol.2004.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair P., Somasundaram K., Krishna S. Activated Notch1 inhibits p53-induced apoptosis and sustains transformation by human papillomavirus type 16 E6 and E7 oncogenes through a PI3K–PKB/Akt-dependent pathway. Journal of Virology. 2003;77:7106–7112. doi: 10.1128/JVI.77.12.7106-7112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien V. Viruses and apoptosis. Journal of General Virology. 1998;79(Pt 8):1833–1845. doi: 10.1099/0022-1317-79-8-1833. [DOI] [PubMed] [Google Scholar]
- Opavsky M.A., Martino T., Rabinovitch M., Penninger J., Richardson C., Petric M., Trinidad C., Butcher L., Chan J., Liu P.P. Enhanced ERK-1/2 activation in mice susceptible to coxsackievirus-induced myocarditis. Journal of Clinical Investigation. 2002;109:1561–1569. doi: 10.1172/JCI13971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popik W., Hesselgesser J.E., Pitha P.M. Binding of human immunodeficiency virus type 1 to CD4 and CXCR4 receptors differentially regulates expression of inflammatory genes and activates the MEK/ERK signaling pathway. Journal of Virology. 1998;72:6406–6413. doi: 10.1128/jvi.72.8.6406-6413.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokudai S., Fujita N., Hashimoto Y., Tsuruo T. Cleavage and inactivation of antiapoptotic Akt/PKB by caspases during apoptosis. Journal of Cellular Physiology. 2000;182:290–296. doi: 10.1002/(SICI)1097-4652(200002)182:2<290::AID-JCP18>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Roulston A., Marcellus R.C., Branton P.E. Viruses and apoptosis. Annual Review of Microbiology. 1999;53:577–628. doi: 10.1146/annurev.micro.53.1.577. [DOI] [PubMed] [Google Scholar]
- Rusnati M., Urbinati C., Musulin B., Ribatti D., Albini A., Noonan D., Marchisone C., Waltenberger J., Presta M. Activation of endothelial cell mitogen activated protein kinase ERK(1/2) by extracellular HIV-1 Tat protein. Endothelium. 2001;8:65–74. doi: 10.3109/10623320109063158. [DOI] [PubMed] [Google Scholar]
- Shih S.R., Ho M.S., Lin K.H., Wu S.L., Chen Y.T., Wu C.N., Lin T.Y., Chang L.Y., Tsao K.C., Ning H.C., Chang P.Y., Jung S.M., Hsueh C., Chang K.S. Genetic analysis of enterovirus 71 isolated from fatal and non-fatal cases of hand, foot and mouth disease during an epidemic in Taiwan, 1998. Virus Research. 2000;68:127–136. doi: 10.1016/s0168-1702(00)00162-3. [DOI] [PubMed] [Google Scholar]
- Stambolic V., Woodgett J.R. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochemical Journal. 1994;303:701–704. doi: 10.1042/bj3030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodoro J.G., Branton P.E. Regulation of apoptosis by viral gene products. Journal of Virology. 1997;71:1739–1746. doi: 10.1128/jvi.71.3.1739-1746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella D., Gewurz B.E., Furman M.H., Schust D.J., Ploegh H.L. Viral subversion of the immune system. Annual Review of Immunology. 2000;18:861–926. doi: 10.1146/annurev.immunol.18.1.861. [DOI] [PubMed] [Google Scholar]
- Widmann C., Gibson S., Johnson G.L. Caspase-dependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti-apoptotic signals. Journal of Biological Chemistry. 1998;273:7141–7147. doi: 10.1074/jbc.273.12.7141. [DOI] [PubMed] [Google Scholar]
- Woodgett J.R. Judging a protein by more than its name: GSK-3. Science's STKE. 2001;2001:RE12. doi: 10.1126/stke.2001.100.re12. [DOI] [PubMed] [Google Scholar]
- Wu W., Lee W.L., Wu Y.Y., Chen D., Liu T.J., Jang A., Sharma P.M., Wang P.H. Expression of constitutively active phosphatidylinositol 3-kinase inhibits activation of caspase 3 and apoptosis of cardiac muscle cells. Journal of Biological Chemistry. 2000;275:40113–40119. doi: 10.1074/jbc.M004108200. [DOI] [PubMed] [Google Scholar]
- Zhang H.M., Yuan J., Cheung P., Luo H., Yanagawa B., Chau D., Tozy N.S., Wong B., Zhang J., Wilson J.E., McManus B.M., Yang D. Over-expression of interferon-gamma-inducible GTPase inhibits coxsackievirus B3-induced apoptosis through the activation of the PI3-K/Akt pathway and inhibition of viral replication. Journal of Biological Chemistry. 2003;278:33011–33019. doi: 10.1074/jbc.M305352200. [DOI] [PubMed] [Google Scholar]