

Abstract
A preparative high-speed counter-current chromatography (HSCCC) with a two-phase solvent system composed of ethyl acetate–n-butanol–water (2:7:9, v/v/v) was successfully performed to isolate and separate clemastanin B and indigoticoside A from the plant of Radix Isatidis, a traditional Chinese medicine. A total of 59.2 mg clemastanin B and 66.1 mg indigoticoside A with purities of 94.6% and 99.0% determined by high performance liquid chromatography (HPLC) were obtained in one-step elution from 250 mg crude extract, which contained clemastanin B 24.8% and indigoticoside A 28.4%, and the recoveries of clemastanin B and indigoticoside A were 90.3% and 92.2%, respectively. The chemical structure was identified by IR, MS, 1H NMR and 13C NMR.
Keywords: Lignan, Clemastanin B, Indigoticoside A, Preparative chromatography, Counter-current chromatography, Radix Isatidis
1. Introduction
Radix Isatidis (Banlangen in Chinese), the root of the plant Isatidis indigotica Fort., family Cruciferae, or Baphicacanthus cusia (Nees) Berm, family Acanthaceae, is mainly distributed in Hebei, Jiangsu, Zhejiang, Fujian, Henan and Guangxi province in China. It has been used as a medicinal plant for more than 2000 years from ShenNongBenCaoJing, a famous ancient Chinese medicinal literary and is similar to Folium Isatidis, but lesser bitter in flavor and cold in nature, usually used for seasonal febrile diseases, pestilence, mumps, eruptive diseases, inflammatory diseases with redness of skin and sorethroat [1]. Now pharmacological research indicated that Banlangen has a widely useful activities including anti-virus, anti-bacterial, anti-endotoxic, anti-tumor, anti-inflammatory, immune regulatory effect [2], [3], [4]. Recently, Banlangen has been found to be highly active inhibition replication of the severe acute respiratory syndrome (SARS)-associated virus [5].
Now chemical research indicated that the major constituents are alkaloids [6], [7], [8], [9], [10], [11] and lignans [12], [13] in the plant of Banlangen. Clemastanin B and indigoticoside A (structures shown in Fig. 1 ), two major lignans in Banlangen, have been isolated by some conventional methods including silica gel, sephadex and preparative high-performance liquid chromatography, which are tedious, time consuming and needing multiple steps. So, an effective separation and isolation technique was required. High-speed counter-current chromatography, a support free liquid–liquid partition chromatographic technique, eliminates irreversible adsorption of the sample onto solid support [14], has an excellent sample recovery. The method permits directly introduction of crude samples into the column without more preparation, so it has been successfully applied to isolate and purify a number of natural products [15], [16], [17], [18], [19]. But, there have no scientific literature reports of using HSCCC to isolate and purify clemastanin B and indigoticoside A from the plant of Banlangen.
Fig. 1.
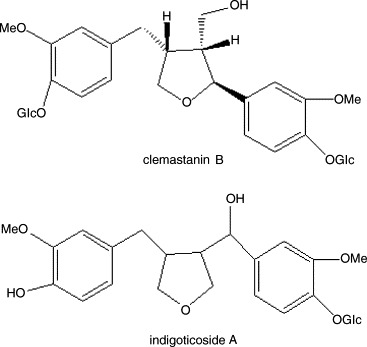
The chemical structures of clemastanin B and indigoticoside A.
The aim of the present paper, therefore, was to develop an efficient method for the isolation and purification of clemastanin B and indigoticoside A with high purities from Banlangen by high-speed counter-current chromatography.
2. Experimental
2.1. Apparatus
Preparative HSCCC was carried out with a model TBE-300A high-speed counter-current chromatography (Shenzhen, Tauto Biotech, China). The apparatus equipped with a polytetrafluoroethylene three preparative coils (diameter of tube, 2.6 mm, total volume, 300 ml) and a 20 ml sample loop. The revolution radius or the distance between the holder axis and central axis of the centrifuge (R) was 5 cm, and the β value varied from 0.5 at the internal terminal to 0.8 at the external terminal (β = r/R where r is the distance from the coil to the holder shaft). The HSCCC system was equipped with a model S constant-flow pump, a model UV-II detector operating at 254 nm, and a model N2010 workstation (Zhejiang University, Hangzhou, China). The experimental temperature was adjusted by HX 1050 constant temperature circulating implement (Beijing Boyikang Lab Implement, Beijing, China).
The analytical HPLC system used throughout this study consisted of LC–10AT pump (Shimadzu, Japan), a SPD–10A UV–vis detector (Shimadzu, Japan), and a model N2000 workstation (Zhejiang University, Hangzhou, China). D101 macroporous resin was purchased from Shanghai Institute of Pharmaceutical Industry (Shanghai, China).
2.2. Reagents
Ethanol, n-butanol, acetic acid were analytical grade and purchased from WuLian Chemical Factory, Shanghai, China. Acetonitrile and methanol were HPLC grade (Merck, Germany). Reverse osmosis Milli-Q water (18 MΩ) (Millipore, USA) was used for all solutions and dilutions.
The Banlangen was purchased from a local drug store and identified by Doctor Luping Qin (Department of Pharmacognosy, College of Pharmacy, the Second Military Medical University, Shanghai, China).
2.3. Preparation of the crude extract
The Banlangen was ground into powder, 300 g of the powder was added to a bottle and extracted by reflux with 1500 ml volume of 50% aqueous ethanol in a haven for 2 h. The mixture was filtered, and the filtrate was collected. The extract was then concentrated to dryness by rotary vaporization at 60 °C under reduced pressure and redissolved in water. The water soluble was added into a glass column (5.0 cm × 75 cm contained 500 g D101 macroporous resin) with 40% aqueous ethanol to yield the crude sample for HSCCC separation.
2.4. Measurement of partition coefficient and separation factor
Approximately 2 mg of the crude extract was weighted in a 10 ml test tube to which 4.0 ml of each phase of the equilibrated two-phase solvent system was added. The tube was shaken vigorously for 2 min to equilibrate the sample thoroughly with the two phases. Then the two-phase was separated and evaporated to dryness under reduced pressure. The residue was diluted with the mobile phase used in HPLC analysis and then analyzed by HPLC. The partition coefficient (K) value was expressed as the peak area of target components in the upper phase divided by that in the lower phase.
2.5. Preparation of two-phase solvent system and sample solution
Two-phase solvent systems were used in the present study, ethyl acetate–n-butanol–water (2:7:9, v/v/v) was prepared. The solvent mixture was thoroughly equilibrated in a separated funnel at room temperature and the two phases were separated shortly before use. The sample solution was prepared by dissolving the sample in the 15 ml lower phase of solvent system for isolation and purification.
2.6. HSCCC separation procedure
In the crude sample isolation and separation, the coil column was first entirely filled with the upper phase of the solvent system. Then the apparatus was rotated at 800 rpm, while the lower phase was pumped into the column at a flow rate of 2.0 ml/min. After the mobile phase front emerged and hydrodynamic equilibrium was established in the column, 15 ml sample solution containing 250 mg of the crude extract was injected through the injection value. The effluent of the column was continuously monitored with a UV–vis detector at 254 nm. Peak fractions were collected according to the elution profile.
2.7. HPLC analysis and identification of CCC peak fractions
The crude sample and the peak fraction obtained by HSCCC were analyzed by high-performance liquid chromatography. The column used was a Lichrospher C18 (6.0 mm × 150 mm i.d. 5 μm) (Hanbang Science, Jiang-Su province, China) with a pre-column equipped with the same stationary phase, the mobile phase was acetonitrile–water–acetic acid (25:75:1, v/v/v). The flow rate was 1.0 ml/min, and the effluent was monitored at 254 nm.
Identification of the CCC peak fractions was carried out by IR (Hitachi 275-50), MS (Finnigan MAT 711), 1H NMR and 13C NMR spectra (Varian Unity Inova-500).
3. Results and discussion
3.1. Optimization of HPLC method
The crude extract and the fractions obtained by HSCCC were determined by HPLC. So, in the first place, a good HPLC separation conditions should be developed for all analyses. Different mobile phase (methanol–water, acetonitrile–water) with different concentrations of acetic acid, flow rate, column temperature and detection wavelength were all tested. The result indicated that acetonitrile–water–acetic acid at a volume ratio of 25:75:1 (v/v/v) was isocratically eluted at a flow rate of 1.0 ml/min, the column temperature and detection wavelength were set at 25 °C and 254 nm. No complex gradient of mobile phase and no buffer were necessary. The HPLC chromatogram of the crude extract is given in Fig. 1A. Peaks 1 and 2 correspond to clemastanin B and indigoticoside A, respectively, which presented at the content of 24.8% and 28.4%.
3.2. Selection of two-phase solvent system and other conditions of HSCCC
In HSCCC separation, a suitable two-phase solvent system was critical for a successful isolation and separation. In our experiment, several kinds of solvent systems were evaluated and the partition coefficients were measured and listed in Table 1 . According to the K values, the result indicated that the solvent systems composed of ethyl acetate–n-butanol–water at the volume ratios of 2:1:3, 4:1:5, 3:2:5 and 2:3:5 (v/v/v) had small K values, which attributed to poor resolution, while n-butanol–water at the volume ratios of 1:1 (v/v) had a large K values and more time will be required to elute the target compounds. So, the solvent system composed of ethyl acetate–n-butanol–water at the volume ratios of 2:7:9 (v/v/v) was most suitable to our experiment.
Table 1.
The partition coefficients (K) and separation factors (α) of clemastanin B and indigoticoside A in different solvent systems
| Solvent system | Clemastanin B, K1 | Separation factor (α) | Indigoticoside A, K2 | |
|---|---|---|---|---|
| Ethyl acetate–n-butanol–water | 2:1:3 | 0.11 | 2.64 | 0.29 |
| Ethyl acetate–n-butanol–water | 4:1:5 | 0.099 | 2.83 | 0.28 |
| Ethyl acetate–n-butanol–water | 2:7:9 | 0.78 | 2.77 | 2.16 |
| Ethyl acetate–n-butanol–water | 3:2:5 | 0.13 | 2.69 | 0.35 |
| Ethyl acetate–n-butanol–water | 2:3:5 | 0.23 | 2.13 | 0.49 |
| n-Butanol–water | 1:1 | 1.12 | 2.80 | 3.14 |
The influence of flow rate of mobile phase, the separation temperature and the revolution speed were also investigated. The result indicated that slow flow speed can produce a good separation, but more time and more mobile phase will be needed, and the chromatogram peak was extended. Considering these aspects, the flow rate was selected 2.0 ml/min in the present study. The temperature has significant effect on K values, the retention of stationary phase and the mutual solvency of the two phases. After tested at 15, 20, 25, 30, 35 and 40 °C, it can be seen that good result can be obtained when the separation temperature was controlled at 25 °C. The revolution speed has a great influence to the retention of stationary phase, high rotary speed can increased the retention of the stationary phase. In our experiment, the revolution speed was set at 800 rpm.
Under the optimized conditions, two fractions were obtained in one-step elution and less than 3 h (HSCCC chromatogram is shown in Fig. 3), and the retention of the stationary phase was 38.6%. The obtained fractions produced 59.2 mg clemastanin B and 66.1 mg indigoticoside A with purities of 94.6% and 99.0% determined by HPLC (shown in Fig. 2B and C). As expected, the HPLC analysis of each fraction revealed that the components eluted in the order of peaks 1 (clemastanin B) and 2 (indigoticoside A). The recoveries of clemastanin B and indigoticoside A were 90.3% and 92.2%, respectively (Fig. 3 ).
Fig. 3.
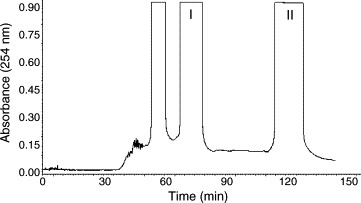
Chromatogram of the crude extract by preparative HSCCC. Solvent system: ethyl acetate–n-butanol–water (2:7:9, v/v/v); stationary phase: upper phase; mobile phase: lower phase; flow rate: 2.0 ml/min; revolution speed: 800 rpm; separation temperature: 25 °C; sample size: 250 mg; sample loop: 20 ml; detection wavelength: 254 nm; retention of the stationary phase: 38.6%.
Fig. 2.
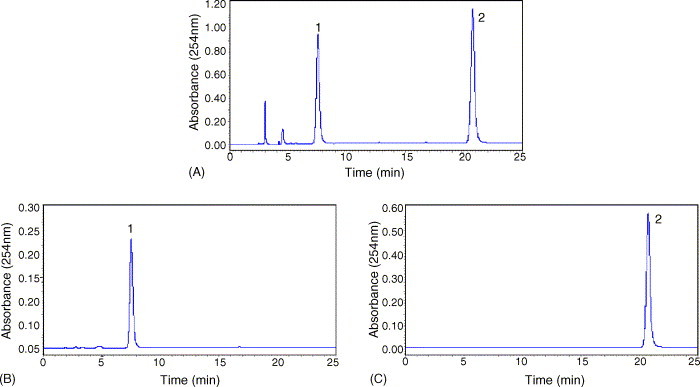
HPLC chromatograms of the crude extract and the fractions obtained by HSCCC. Column: reversed-phase Lichrospher C18 (6.0 mm × 150 mm i.d. 5 μm); mobile phase: CH3CN–H2O–HAC (25:75:1, v/v/v); flow rate: 1.0 ml/min; UV wavelength: 254 nm; (A) crude extract; (B) fraction “I” obtained by HSCCC; (C) fraction “II” obtained by HSCCC; peak 1: clemastanin B; peak 2: indigoticoside A.
3.3. The structural identification
The identification of the obtained materials was carried out by IR, MS, 1H NMR and 13C NMR spectra as follows. Clemastanin B: : 3400 (OH), 2920, 1592, 1510, 1261, 1074. FAB-MS m/z: 707 [M++ + Na], 637, 522, 360, 343, 316, 284, 237, 219. HR-MS: 684.1256. It showed the molecular was 684, which was in agreement with the molecular formula C32H44O16. Indigoticoside A: : 3419 (OH), 1599, 1514, 1276, 1035. FAB-MS m/z: 522 [M+], 545 [M+ + Na], 561[M+ + K]. HR-MS: 522.1947. It showed the molecular was 522, which was in agreement with the molecular formula C26H34O11. Comparing with the reported data, the IR, MS, 1H NMR and 13C NMR data are in agreement with those of clemastanin B and indigoticoside A in literatures [12], [20].
4. Conclusion
Our study demonstrates that HSCCC is a powerful method in separation and isolation bioactive compounds from natural sources. Two lignans including clemastanin B and indigoticoside A were separated from Banlangen by HSCCC with a two-phase solvent system composed of ethyl acetate–n-butanol–water (2:7:9, v/v/v). 59.2 mg clemastanin B and 66.1 mg indigoticoside A with high purities were obtained from 250 mg crude extract in one single isolation. The method is simple, fast and without complex solvent system or gradient elution.
Acknowledgement
Financial support from the Shanghai Key Laboratory for Pharmaceutical Metabolites Research is grateful acknowledged.
References
- 1.Gao X.Z. Res. Trad. Chin. Med. 2001;17:57. [Google Scholar]
- 2.Li S., Chen W.S., Qiao C.Z., Zheng S.Q. J. Second Mil. Med. Univ. 2000;21:204. [Google Scholar]
- 3.Wang Y., Qiao C.Z., Liu S., Zhang H.M. China J. Chin. Mater. Med. 2000;25:327. [PubMed] [Google Scholar]
- 4.Xu H., Fang J.G., Liu Y.H. Chin. Tradit. Herbal Drugs. 2003;34:10. [Google Scholar]
- 5.Shang H.C., Gao X.M., Chen J. Tianjin J. Tradit. Chin. Med. 2003;20:97. [Google Scholar]
- 6.Liu Y.H., Qin G.W., Ding S.P. Chin. Tardit. Herbal Drugs. 2002;33:97. [Google Scholar]
- 7.Chen W.S., Li B., Zhang W.D. Chin. Chem. Lett. 2001;12:501. [Google Scholar]
- 8.Li B., Chen W.S., Zheng S.Q. Acta Pharm. Sinica. 2000;35:508. [Google Scholar]
- 9.Wu X.Y., Liu Y.H., Sheng W.Y. Tetrahedron. 1997;53:13323. [Google Scholar]
- 10.Deng K.M., Wu X.Y., Yang G.J. Chin. Chem. Lett. 1997;8:237. [Google Scholar]
- 11.Wu X.Y., Liu Y.H., Sheng W.Y. Planta Med. 1997;63:55. doi: 10.1055/s-2006-957604. [DOI] [PubMed] [Google Scholar]
- 12.Liu H.L., Wu L.J., Wu B. Chin. J. Magn. Reson. 2002;19:315. [Google Scholar]
- 13.Liu H.L., Wu L.J., Li H. J. Shenyang Pharm. Univ. 2002;19:93. [Google Scholar]
- 14.Ito Y. CRC. Rev Crit. Anal. Chem. 1986;17:65. [Google Scholar]
- 15.Wang X., Wang Y.Q., Geng Y.L., Li F.W., Zheng C.C. J. Chromatogr. A. 2004;1036:171. doi: 10.1016/j.chroma.2004.02.073. [DOI] [PubMed] [Google Scholar]
- 16.Jiang Y., Lu H.T., Chen F. J. Chromatogr. A. 2004;1033:183. doi: 10.1016/j.chroma.2004.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Y., Zhang T.Y., Xu G.Q., Ito Y. J. Chromatogr. A. 2001;929:169. doi: 10.1016/s0021-9673(01)01177-3. [DOI] [PubMed] [Google Scholar]
- 18.Lei L., Yang F.Q., Zhang T.Y., Tu P.F., Wu L.J., Ito Y. J. Chromatogr. A. 2001;912:181. doi: 10.1016/s0021-9673(01)00583-0. [DOI] [PubMed] [Google Scholar]
- 19.Chen L., Han Y.S., Yang F.Q., Zhang T.Y. J. Chromatogr. A. 2001;907:343. doi: 10.1016/s0021-9673(00)00960-2. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y.W., Yu M.Q., Cheng Y.W., Li K.M., Cheng J.M. China J. Chin. Mater. Med. 2005;30:395. [Google Scholar]