

Abstract
We considered the fully overlapping triplets of nucleotide bases and proposed a 2D graphical representation of protein sequences consisting of 20 amino acids and a stop code. Based on this 2D graphical representation, we outlined a new approach to analyze the phylogenetic relationships of coronaviruses by constructing a covariance matrix. The evolutionary distances are obtained through measuring the differences among the two-dimensional curves.
1. Introduction
Compilation of DNA primary sequence data continues unabated and tends to overwhelm us with voluminous outputs that increase daily. Comparison of primary sequences of different DNA strands remains one of the important aspect of the analysis of DNA data banks. Mathematical analysis of the large volume genomic DNA sequence data is one of the challenges for bio-scientists. There are three class methods for the analysis of DNA sequences: (i) Alignment [1], [2]. (ii) Matrices: (1) matrices in which an individual entry corresponds to an individual pair of bases [3], [6], [7] and (2) matrices in which entries summarize information of different X–Y pairs of bases [4], [5], [7]. (iii) Graphical representation: Graphical representation of DNA sequence provides a simple way of viewing, sorting and comparing various gene structures. Graphical techniques have emerged as a very powerful tool for the visualization and analysis of long DNA sequences. These techniques provide useful insights into local and global characteristics and the occurrences, variations and repetition of the nucleotides along a sequence which are not as easily obtainable by other methods. In recent years several authors outlined different graphical representation of DNA sequences based on 2D, 3D or 4D [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20]. Based on these graphical representation, several authors outlined some approaches to make comparison of DNA sequences [21], [22], [23], [24], [25].
All this methods are based on the (four letter alphabet, A, C, G, and T standing for nucleotide bases adenine, cytosine, guanine, and thymine, respectively). We will change to consider the fully overlapping triplets of nucleotide bases. Consideration of triplets of nucleotide bases instead of individual nucleotide bases has several reasons and advantages. There are three of them: (i) The genetic code consists of triplets (codons) of DNA (or RNA in some virus) nucleotides. (ii) The second advantage is that one can easily find the open reading frame as the longest sequence of triplets that contains no stop codons when read in a single reading frame. (iii) The computation will become more simple.
In this Letter, we proposed a 2D graphical representation of the protein sequences consisting of 20 amino acids and a stop code. Based on this 2D graphical representation, we outlined a new approach to analyze the phylogenetic relationships of coronaviruses. The evolutionary distances are obtained through measuring the differences among the two-dimensional curves. Unlike most existing phylogeny construction methods [26], [27], [28], [29], [30], [31], the proposed method does not require multiple alignment.
2. 2D graphical representation of protein sequences and properties
As is known, all of the 64 triplets of nucleotide bases correspond 20 amino acids and a stop code. There are three reading frame start at position 1, 2 and 3, respectively. Using the translate tool, we can obtain three protein sequences consisting of 20 amino acids and a stop code. The 20 amino acids found in proteins can be grouped according to the chemistry of their R groups as in [32]: amino acids A,V,F,P,M,I,L belong to the hydrophobic chemical group; amino acids D,E,K,R belong to charged chemical group; amino acids S,T,Y,H,C,N,Q,W belong to polar chemical group; amino acid belong to glycine chemical group. Then for any DNA sequence, we will transform it into three new sequences defined over alphabet . The rule is as follows:
As shown in Fig. 1 , we construct a pyrimidine–purine graph on two quadrants of the cartesian coordinate system, with pyrimidines ( and ) in the first quadrant and purines ( and ) in the fourth quadrant. The unit vectors representing four alphabets and are as follows:
where m is a real number and , n is a positive real number but not a perfect square number. So that we will reduce a DNA sequence into a series of nodes P 0,P 1,P 2, … ,P ⌊N/3⌋, whose coordinates x i, y i (i = 0,1, 2, … , ⌊N/3⌋, where N is the length of the DNA sequence being studied) satisfy
| (1) |
and satisfy
| (2) |
Fig. 1.
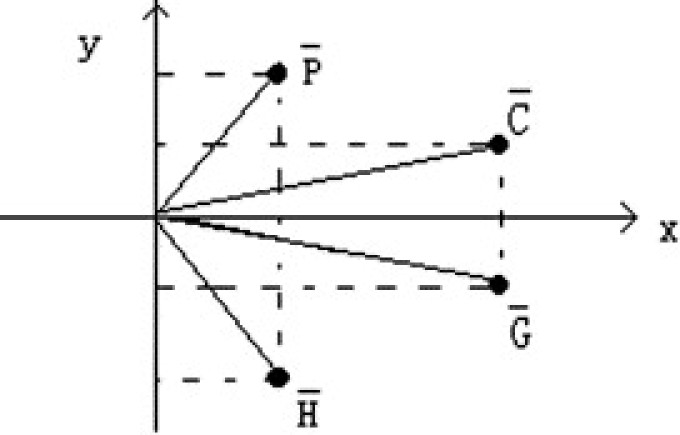
Pyrimidine–purine graph.
where A i,V i,F i,P i,M i,I i,L i,D i,E i,K i,R i,S i,T i,Y i,H i,C i, N i Q i W i,G i,Ω i; are the cumulative occurrence numbers of A, V,F,P,M,I,L,D,E,K,R,S,T,Y,H,C,N,Q,W,G and −(or stop code), respectively, in the subsequence from the 1st base to the ith base in the sequence. And s k,k = 1, … ,17 are positive real number but not perfect square number, s i ≠ s j,i,j = 1, … ,17, and . We define A 0 = V 0 = F 0 = P 0 = M 0 = I 0 = L 0 = D 0 = E 0 = K 0 = R 0 = S 0 = T 0 = Y 0 = H 0 = C 0 = N 0 = Q 0 = W 0 = G 0 = Ω 0 = 0.
We called the corresponding plot set be characteristic plot set. The curve connected all plots of the characteristic plot set in turn is called characteristic curve, which is determined by m, n, that satisfy above mentioned condition. In Fig. 2, Fig. 3, Fig. 4 , we show the SARS corresponding curves with different parameters n and m, where s 1 = 2/3;s 2 = 3/4; s 3 = 4/5;s 4 = 5/6;s 5 = 6/7;s 6 = 7/8;s 7 = 8/9;s 8 = 9/10;s 9 = 10/11; s 10 = 11/12;s 11 = 12/13;s 12 = 13/14;s 13 = 14/15;s 14 = 15/16;s 15 = 16/17;s 16 = 17/18;s 17 = 18/19. Observing Fig. 2, Fig. 3, Fig. 4, we find SARS have similar curves despite with different parameters n and m.
Property 1
For a given DNA sequence there are three 2D representations corresponding to it.
Proof
Using the translate tool, one can obtain three protein sequences consisting of 20 amino acids and a stop code corresponding three reading frame start at position 1, 2 and 3. In a single reading frame, let (x i, y i) be the coordinates of the ith amino acid of protein sequence, then we have
i.e.,
(3) □
Obviously, x i and y i are irrational numbers of form , where s and k are integers. We suppose
then we have
| (4) |
So, for given x-projection and y-projection of any point P = (x, y) on the sequence, after uniquely determining s x,k x,s y,k y from x and y, the number A p,V p,F p,P p,M p,I p,L p,D p,E p,K p,R p,S p,T p,Y p,H p,C p,N p,Q p,W p,G p,Ω p of A,V,F,P,M,I,L,D,E,K,R,S,T,Y,H,C,N,Q,W,G and −(or stop code) from the beginning of the sequence to the point P can be found by solving linear system (2), (4).
Fig. 2.
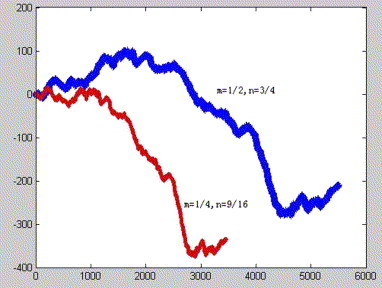
SARS corresponding curve with different parameters n and m based on the first reading frame.
Fig. 3.
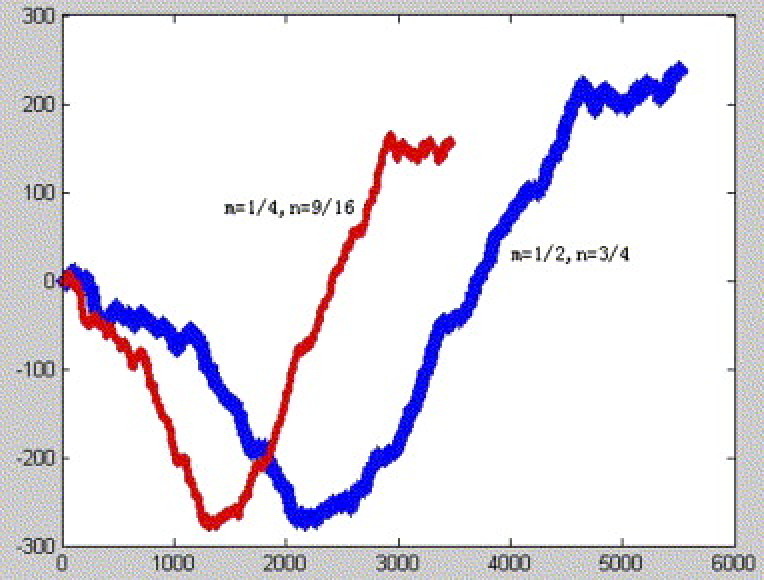
SARS corresponding curve with different parameters n and m based on the second reading frame.
Fig. 4.
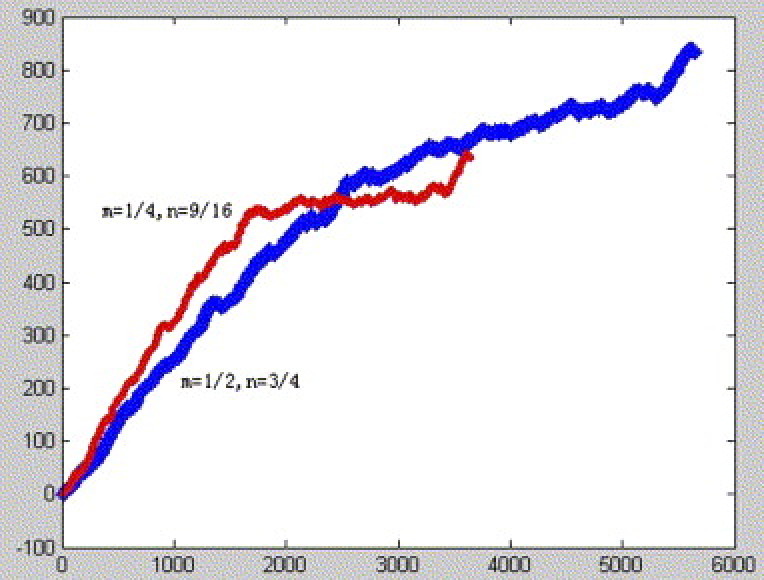
SARS corresponding curve with different parameters n and m based on the third reading frame.
The vector pointing to the point P i from the origin O is denoted by r i. The component of r i, i.e. x i and y i are calculated by Eqs. (1), (2). Let Δr i = r i − r i − 1, then we have Property 2.
Property 2
For any i = 1, 2, … , N′, where N′is the length of protein sequence corresponding the studied DNA sequence, the vector Δr i has only twenty one possible direction. Furthermore, the length of Δr i, i.e.,∣Δ r i∣, is always equal to s k(m 2 + n), for any i = 1, 2, … , N, k = 0,1, … ,17,s 0 = 1.
Proof
Actually, the components of Δr i, i.e., Δx i and Δy i can be calculated for each possible residue (A, V,F,P,M,I,L,D,E,K,R,S,T,Y,H,C,N,Q,W,G and −) at the ith position of the protein sequence by using Eqs. (1), (2). For example, when the ith residue is A, we find Δx i = m and . This result is independent of the conformation state of the (i − 1)th residue. The two numbers are called the direction of Δr i. The direction number and the length of Δr i for each possible residue type at the ith position are summarized. □
Property 3
There is no circuit or degeneracy in our two-dimensional graphical representation.
Proof
We assume that: (1) the number of amino acid forming a circuit is l; (2) the number of A,V,F,P,M,I,L,D,E,K,R,S, T,Y,H,C,N,Q,W,G and −(or stop code) in a circuit is a′,v′,f′,p′,m′,i′,l′,d′,e′,k′,r′,s′,t′,y′,h′,c′,n′,q′,w′,g′ and δ′, respectively. So a′ + v′ + f′ + p′ + m′ + i′ + l′ + d′ + e′ + k′ + r′ + s′ + t′ + y′ + h′ + c′ + n′ + q′ + w′ + g′ + δ′ = l. Because a′A,v′V,f′F,p′P,m′M,i′I,l′L,d′D,e′E,k′K,r′R,s′S,t′T,y′Y,h′H,c′C,n′N,q′Q,w′W,g′G and δ′ −(or stop code) form a circuit, the following equation holds:
(5)
i.e.,
(6) Clearly Eqs. (5), (6) hold if, and only if a′ = v′ = f′ = p′ = m′ = i′ = l′ = d′ = e′ = k′ = r′ = s′ = t′ = y′ = h′ = c′ = n′ = q′ = w′ = g′ = δ′ = 0. Therefore, l = 0, which means no circuit exists in this graphical representation. □
Property 4
The 2D representation possesses the reflection symmetry.
Proof
usually the sequence is expressed in the order from 5′ to 3′. Suppose that the 2D representation for protein sequence is described by (x i, y i),i = 0,1, 2, … , N. Suppose again that the 2D representation for the reverse sequence, i.e, the same sequence but from 3′ to 5′ is described by , we find
(7) □
3. Phylogenetic tree of coronaviruses
For any DNA sequence, we have three translating protein sequences. For any protein sequence, we have a set of points (x i, y i),i = 1,2,3, … ,N, where N is the length of the sequence. The coordinates of the geometrical center of the points, denoted by x 0 and y 0, may be calculated as follows:
| (8) |
The element of covariance matrix CM of the points are defined:
| (9) |
(See Table 1 )The above four numbers give a quantitative description of a set of point (x i, y i),i = 1, 2, … , N, scattering in a two-dimensional space. Obviously, the matrix is a real symmetric 2 × 2 one. There is a leading eigenvalue for a matrix CM. So that there are three geometrical centers and three leading eigenvalue corresponding a DNA sequence. In Table 2 , we list the geometrical centers and leading eigenvalues belonging to 24 species with parameter (See Table 3 ).
Table 1.
The accession number, abbreviation, name and length for the 24 coronavirus geneomes
| No. | Accession | Abbreviation | Genome | Length (nt) |
|---|---|---|---|---|
| l | NC_002645 | HCoV_229E | Human coronavirus 229E | 27 317 |
| 2 | NC_002306 | TGEV | Transmissible gastroenteritis virus | 28 586 |
| 3 | NC_003436 | PEDV | Porcine epidemic diarrhea virus | 28 033 |
| 4 | U00735 | BCoVM | Bovine coronavirus strain Mebus | 31 032 |
| 5 | AF391542 | BCoVL | Bovine coronavirus isolate BCoV-LUN | 31 028 |
| 6 | AF220295 | BCoVQ | Bovine coronavirus Quebec | 31 100 |
| 7 | NC_003045 | BCoV | Bovine coronavirus | 31 028 |
| 8 | AF208067 | MHVM | Murine hepatitis virus strain ML-10 | 31 233 |
| 9 | AF101929 | MHV2 | Murine hepatitis virus strain 2 | 31 276 |
| 10 | AF208066 | MHVP | Murine hepatitis virus strain Penn 97-1 | 31 112 |
| 11 | NC_001846 | MHV | Murine hepatitis virus | 31 357 |
| 12 | NC_001451 | IBV | Avian infectious bronchitis virus | 27 608 |
| 13 | AY278488 | BJ01 | SARS coronavirus BJ01 | 29 725 |
| 14 | AY278741 | Urbani | SARS coronavirus Urbani | 29 727 |
| 15 | AY278491 | HKU-39849 | SARS coronavirus HKU-39849 | 29 742 |
| 16 | AY278554 | CUHK-W1 | SARS coronavirus CUHK-W1 | 29 736 |
| 17 | AY282752 | CUHK-Su10 | SARS coronavirus CUHK-SulO | 29,736 |
| 18 | AY283794 | SIN2500 | SARS coronavirus Sin2500 | 29 711 |
| 19 | AY283795 | SIN2677 | SARS coronavirus Sin2677 | 29 705 |
| 20 | AY283796 | SIN2679 | SARS coronavirus Sin2679 | 29 711 |
| 21 | AY283797 | SIN2748 | SARS coronavirus Sin2748 | 29 706 |
| 22 | AY283798 | SIN2774 | SARS coronavirus Sin2774 | 29 711 |
| 23 | AY291451 | TW1 | SARS coronavirus TW1 | 29 729 |
| 24 | NC_004718 | TOR2 | SARS coronavirus | 29 751 |
Table 2.
Twenty one possible direction
| Δxn | Δyn | ∣Δrn∣ | |
|---|---|---|---|
| A | m | m2 + n | |
| D | m | m2 + n | |
| S | −m | m2 + n | |
| G | m | m2 + n | |
| V | s1(m2 + n) | ||
| F | s2(m2 + n) | ||
| P | s3(m2 + n) | ||
| M | s4(m2 + n) | ||
| I | s5(m2 + n) | ||
| L | s6(m2 + n) | ||
| E | s7(m2 + n) | ||
| K | s8(m2 + n) | ||
| R | s9(m2 + n) | ||
| T | s10(m2 + n) | ||
| Y | s11(m2 + n) | ||
| H | s12(m2 + n) | ||
| C | s13(m2 + n) | ||
| N | s14(m2 + n) | ||
| Q | s15(m2 + n) | ||
| w | s16(m2 + n) | ||
| – | s17(m2 + n) |
Table 3.
The geometric centers and three leading eigenvalues for each of the 24 coronavirus genomes
| i | λ1 | λ2 | λ3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.5692e + 003 | −159.0439 | 2.5566e + 003 | −342.5873 | 2.6794e + 003 | 389.8249 | 2.1520 | 2.2321 | 2.3707 |
| 2 | 2.8619e + 003 | −230.4309 | 2.8245e + 003 | −723.2605 | 2.9971e + 003 | 128.9913 | 2.6999 | 2.8393 | 2.9157 |
| 3 | 2.8626e + 003 | −233.0932 | 2.8231e + 003 | −724.5553 | 2.9976e + 003 | 130.5104 | 2.7034 | 2.8386 | 2.9178 |
| 4 | 2.8602e + 003 | −245.6989 | 2.8245e + 003 | −743.4898 | 2.9985e + 003 | 133.2708 | 2.7056 | 2.8453 | 2.9209 |
| 5 | 2.8688e + 003 | −294.6379 | 2.8364e + 003 | −709.6245 | 3.0012e + 003 | 146.3851 | 2.7519 | 2.8561 | 2.9158 |
| 6 | 2.6263e + 003 | 415.1362 | 2.5204e + 003 | −204.5027 | 2.4666e + 003 | −516.9428 | 2.2817 | 2.0813 | 2.1269 |
| 7 | 2.8773e + 003 | −476.9658 | 2.8773e + 003 | −476.9658 | 2.9006e + 003 | −252.7994 | 2.8910 | 2.8910 | 2.7932 |
| 8 | 2.8902e + 003 | −446.8927 | 2.8902e + 003 | −446.8927 | 2.9139e + 003 | −227.7537 | 2.9004 | 2.9004 | 2.8179 |
| 9 | 2.8853e + 003 | −459.6862 | 3.0344e + 003 | 82.5446 | 2.8912e + 003 | −273.7115 | 2.9146 | 2.9739 | 2.7829 |
| 10 | 2.8582e + 003 | −528.7428 | 3.0320e + 003 | 34.9426 | 2.8807e + 003 | −253.2886 | 2.8697 | 2.9882 | 2.7408 |
| 11 | 2.5137e + 003 | −415.8854 | 2.6893e + 003 | 244.2464 | 2.5817e + 003 | −222.8666 | 2.2271 | 2.3287 | 2.1831 |
| 12 | 2.7670e + 003 | −48.3996 | 2.7276e + 003 | −34.7759 | 2.8570e + 003 | 524.7574 | 2.4705 | 2.5740 | 2.6849 |
| 13 | 2.7255e + 003 | −35.7080 | 2.8550e + 003 | 526.4976 | 2.7646e + 003 | −43.8066 | 2.5698 | 2.6804 | 2.4654 |
| 14 | 2.7656e + 003 | −45.9837 | 2.7262e + 003 | −35.1151 | 2.8557e + 003 | 528.0186 | 2.4675 | 2.5711 | 2.6821 |
| 15 | 2.7659e + 003 | −45.2775 | 2.7260e + 003 | −36.4889 | 2.8558e + 003 | 530.0127 | 2.4680 | 2.5710 | 2.6828 |
| 16 | 2.7656e + 003 | −47.8004 | 2.7267e + 003 | −33.6628 | 2.8560e + 003 | 527.4290 | 2.4680 | 2.5725 | 2.6838 |
| 17 | 2.7239e + 003 | −35.1426 | 2.8535e + 003 | 527.3351 | 2.7632e + 003 | −45.2702 | 2.5669 | 2.6777 | 2.4630 |
| 18 | 2.7233e + 003 | −36.1921 | 2.8529e + 003 | 527.2583 | 2.7627e + 003 | −45.4289 | 2.5657 | 2.6766 | 2.4620 |
| 19 | 2.7239e + 003 | −34.4434 | 2.8535e + 003 | 527.8162 | 2.7633e + 003 | −45.2775 | 2.5667 | 2.6780 | 2.4630 |
| 20 | 2.7239e + 003 | −35.6707 | 2.8525e + 003 | 525.5247 | 2.7621e + 003 | −43.2715 | 2.5678 | 2.6737 | 2.4587 |
| 21 | 2.7241e + 003 | −35.5425 | 2.8535e + 003 | 527.2287 | 2.7634e + 003 | −45.5734 | 2.5675 | 2.6777 | 2.4636 |
| 22 | 2.7647e + 003 | −48.0684 | 2.7258e + 003 | −35.7184 | 2.8553e + 003 | 523.7099 | 2.4661 | 2.5700 | 2.6815 |
| 23 | 2.7647e + 003 | −47.8421 | 2.7252e + 003 | −35.8263 | 2.8547e + 003 | 524.8910 | 2.4661 | 2.5692 | 2.6808 |
| 24 | 2.6110e + 003 | −251.1068 | 2.7585e + 003 | 459.3175 | 2.6727e + 003 | −97.0235 | 2.3573 | 2.4587 | 2.3322 |
In order to facilitate the quantitative comparison of different species in terms of their collective parameters, we introduce a distance scale as defined below. Suppose that there are two species i and j, the parameters are , respectively, where are the three leading eigenvalues of matrix CM i corresponding to species i. The distance d ij between the two points is
| (10) |
where d ij denotes the distance between the geometric centers of the ith and the jth genomes, and M is the total number of all genomes (M = 24, here). Then we obtain a real M × M symmetric matrix whose elements are d ij.
Accordingly, a real symmetric M × M matrix D ij is obtained and used to reflect the evolutionary distance between the species i and j. The clustering tree is constructed using the UPGMA method in Phylip package (http://evolution.genetics.washington.edu/phylip.html). The final phylogenetic tree is drawn using the Drawgram program in the Phylip package. In Fig. 5 , we present the phylogenetic tree belonging to 24 species.
Fig. 5.
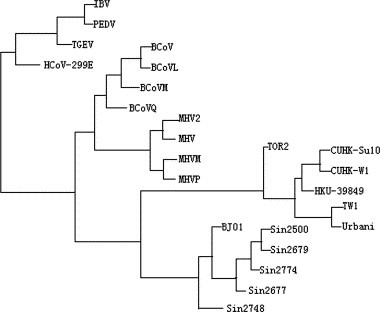
Phylogenetic tree.
4. Conclusion
We made a analysis of DNA sequences by considering the fully overlapping triplets of nucleotide bases. The presented graphical representation can be recaptured mathematically without loss of textual information. And our representation provides a direct plotting method to denote DNA sequences without degeneracy.
Most existing approaches for phylogenetic inference use multiple alignment of sequences and assume some sort of an evolutionary model. The multiple alignment strategy does not work for all types of data, e.g., whole genome phylogeny, and the evolutionary models may not always be correct. The current two-dimensional graphical representation of DNA sequences provides different approach for constructing phylogenetic tree. Unlike most existing phylogeny construction methods, the proposed method does not require multiple alignment. Also, both computational scientists and molecular biologists can use it to analysis protein sequences efficiently. We can obtain some graphical representation of protein sequence based on 2D, 3D and 4D using the following transform: . and satisfy Eq. (2). a i,c i,g i and t i are the cumulative occurrence numbers of A, C, G and T, respectively, in the subsequence from the 1st base to the ith base in the sequence.
Acknowledgments
This work is supported in part by the China Postdoctoral Science Foundation and the National Natural Science Foundation of Hunan University.
References
- 1.Pearson W.R., Lipman D.J. Proc. Natl. Acad. Sci. USA. 1988;85:2444. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sankofff D., Kruskal J.B., editors. String Edits, and Macromolecules: The Theory and Practice of Sequence Comparison. Addison-Wesley Publ. Co.; Reading, MA: 1983. p. 1. [Google Scholar]
- 3.Randic M., Vracko M., Nandy A., Basak S.C. J. Chem. Inf. Comput. Sci. 2000;40:1235. doi: 10.1021/ci000034q. [DOI] [PubMed] [Google Scholar]
- 4.Randic M. J. Chem. Inf. Comput. Sci. 2000;40:50. doi: 10.1021/ci990084z. [DOI] [PubMed] [Google Scholar]
- 5.Randic M. Chem. Phys. Lett. 2000;317:29. [Google Scholar]
- 6.Randic M., Vracko M. J. Chem. Inf. Comput. Sci. 2000;40:599. doi: 10.1021/ci9901082. [DOI] [PubMed] [Google Scholar]
- 7.Randic M., Basak S.C. J. Chem. Inf. Comput. Sci. 2001;41:561. doi: 10.1021/ci0000981. [DOI] [PubMed] [Google Scholar]
- 8.Bo Liao. Chem. Phys. Lett. 2005;401:196. doi: 10.1016/j.cplett.2004.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chunxin Yuan, Bo Liao, Tianming Wang. Chem. Phy. Lett. 2003;379:412. [Google Scholar]
- 10.Bo Liao, Tianming Wang. J. Comput. Chem. 2004;25(11):1364. doi: 10.1002/jcc.20060. [DOI] [PubMed] [Google Scholar]
- 11.Bo Liao, Tianming Wang. J. Mol. Struct. THEOCHEM. 2004;681:209. [Google Scholar]
- 12.Stephn S.-T. Yan, JiaSong Wang, Air Niknejad, Chaoxiao Lu, Ning Jin, Yee-kin Ho. Nucl. Acid Res. 2003;31(12):3078. [Google Scholar]
- 13.Randic M., Vracko M., Nandy A., Basak S.C. J. Chem. Inf. Comput. Sci. 2000;40:1235. doi: 10.1021/ci000034q. [DOI] [PubMed] [Google Scholar]
- 14.Milan Randic, Majan Vracko, Nella Lers, Dejan Plavsic. Chem. Phys. Lett. 2003;368:1. [Google Scholar]
- 15.Hamori E., Ruskin J. J. Biol. Chem. 1983;258:1318. [PubMed] [Google Scholar]
- 16.Hamori E. Nature. 1985;314:585. doi: 10.1038/314585a0. [DOI] [PubMed] [Google Scholar]
- 17.Gates M.A. Nature. 1985;316:219. doi: 10.1038/316219a0. [DOI] [PubMed] [Google Scholar]
- 18.Nandy A. Curr. Sci. 1994;66:309. [Google Scholar]
- 19.Nandy A. Comput. Appl. Biosci. 1996;12:55. doi: 10.1093/bioinformatics/12.1.55. [DOI] [PubMed] [Google Scholar]
- 20.Bo Liao, Mingshu Tan, Kequan Ding. Chem. Phy. Lett. 2005;402:380. [Google Scholar]
- 21.Bo Liao, Tianming Wang. Chem. Phys. Lett. 2004;388:195. [Google Scholar]
- 22.Bo Liao, Yusen Zhang, Kequan Ding, Tianming Wang. J. Mol. Struct.: THEOCHEM. 2005;717:199. [Google Scholar]
- 23.Randic M., Vracko M., Lers N., Plavsic D. Chem. Phys. Lett. 2003;371:202. [Google Scholar]
- 24.Bo Liaoa, Mingshu Tan, Kequan Ding. Chem. Phys. Lett. 2005;414:296. [Google Scholar]
- 25.Bo Liaoa, Kequan Ding. J. Comput. Chem. 2005;14(26):1519. doi: 10.1002/jcc.20287. [DOI] [PubMed] [Google Scholar]
- 26.Jukes T.H., Cantor C.R. Academic Press; New York: 1969. Mammalian Protein Metabolism. 21-132. [Google Scholar]
- 27.Kimura M. J. Mol. Evol. 1980;16:111. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 28.Barry D., Hartigan J.A. Stat. Sci. 1987;2:191. [Google Scholar]
- 29.Kishino H., Hasegawa M. J. Mol. Evol. 1989;29:170. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- 30.Lake J.A. Proc. Natl Acad. Sci. USA. 1994;91:1455. doi: 10.1073/pnas.91.4.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masatoshi Nei, Sudhir Kumar. Oxford University Press; 2000. Molecular Evolution Phylogeny. [Google Scholar]
- 32.Mount D.W. Cold Spring Harbor Laboratory Press; 2001. Bioinformatics: Sequence and Genome Analysis. [Google Scholar]