

Abstract
Translational stalling of ribosome bound to endoplasmic reticulum (ER) membrane requires an accurate clearance of the associated polypeptides, which is not completely understood in mammals. We characterized in mammalian cells the model of ribosomal stalling at the STOP-codon based on proteins tagged at the C-terminus with the picornavirus 2A peptide followed by a termination codon instead of the Proline (2A*). We exploited the 2A* stalling model to characterize the pathway of degradation of ER-targeted polypeptides. We report that the ER chaperone BiP/GRP78 is a new main factor involved. Moreover, degradation of the ER-stalled polypeptides required the activities of the AAA-ATPase VCP/p97, its associated deubiquitinylase YOD1, the ribosome-associated ubiquitin ligase Listerin and the proteasome. In human proteome, we found two human C-terminal amino acid sequences that cause similar stalling at the STOP-codon. Our data suggest that translational stalling at the ER membrane activates protein degradation at the interface of ribosomal- and ER-associated quality control systems.
Abbreviations: ER, endoplasmic reticulum; RET, ribosomal exit tunnel; ERAD, ER-associated degradation; eRF, eukaryotic release factor; FMDV, foot and mouth disease virus; scFv, single-chain variable fragment; MHC-Iα, major histocompatibility complex class iα; CQ, Chloroquine; StrAv, streptavidin; BiP/GRP78, binding immunoglobulin protein/glucose-regulated protein 78 kDa; CD99L2, CD99 molecule-like protein 2; POTEI and POTEE, prostate ovary testis-expressed protein family member I and E; AMD1, adenosylmethionine decarboxylase 1
Keywords: ribosomal stalling, ER-associated degradation, BiP, 2A peptide, STOP-codon
Graphical Abstract
Highlights
-
•
Ribosomal stalling at the STOP-codon causes degradation of the translated protein.
-
•
Picornavirus 2A peptide and related sequences cause ribosome stalling at STOP-codon.
-
•
BiP/GRP78 recognizes polypeptides produced by membrane-bound stalled ribosomes.
-
•
ER-stalled polypeptides are disposed of through the ERAD pathway.
-
•
BIP/GRP78 is a novel key player for ERAD targeting of stalled ribosomal peptides.
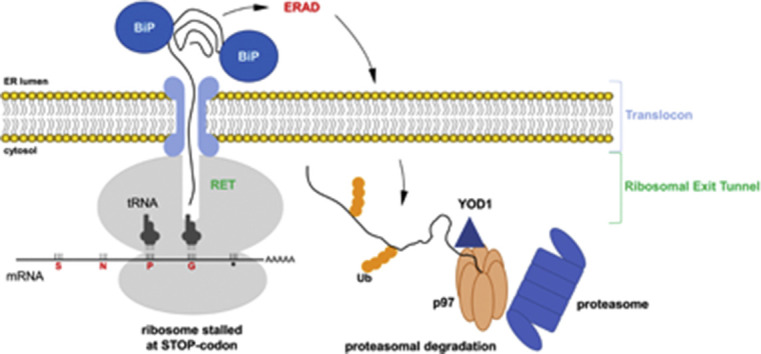
Legend.

BiP/GRP78 recognises proteins derived from ribosomes stalled at the STOP-codon (in red, the stalling peptide), causing their degradation through the ERAD pathway.
Introduction
Proteome homeostasis is crucial for cell survival and involves several levels of quality control mechanisms to avoid production of aberrant protein products [1]. The first checkpoint during biogenesis of polypeptides is co-translational and mediated by ribosomes, which are considered a dynamic center for quality control of both mRNA and nascent proteins. Damaged mRNAs or nascent peptides forming stable interactions with the ribosomal exit tunnel (RET) and the peptidyl-transferase center can cause translational stalling of the ribosomes [2], [3]. Ribosomal stalling is potentially dangerous for the cell survival, therefore different mechanisms have evolved to resolve such events. Translation of erroneous mRNAs owing to a premature termination codon, the lack of termination codon or a broad range of potential stalling sequences often cause degradation of such mRNAs by nonsense-mediated decay (NMD), nonstop decay (NSD) and no-go decay (NGD) pathways, respectively [4], [5], [6]. These pathways trigger either ribosomes dissociation and recycling or their degradation together with associated tRNAs and nascent polypeptides [7]. Impairments of these protein quality control pathways are involved in several pathological conditions, such as the Huntington disease [8] and other neurodegenerative diseases [9], [10].
Nascent polypeptides derived from stalled ribosomes are extracted from the RET and targeted to degradation through the proteasome pathway, which involves the E3 ubiquitin ligase Ltn1/Listerin [11], [12] and the AAA ATPase Cdc48/p97 [13], [14], [15], identified in yeast and mammalian cells. Other players described as part of the ribosome-associated quality control pathway are the integral ribosomal protein Rack1 [16], the surveillance complex formed by Pelota–HBS1–ABCE1 [4], [17] and the ribosome quality control proteins 1 and 2/NEMF [13], [18], [19]. Beyond the poly-Lys sequence translated from poly(A) tails, diverse peptide sequences have been described to cause ribosomal stalling [2], [20]. Some of them cause stalling at the STOP-codon and have been described in proteins of mammalian viruses, bacteria, fungi and plants [21], [22], [23], [24].
In eukaryotic cells, the release from the ribosome of the terminated polypeptide occurs through recognition of the STOP-codon by the eukaryotic release factor (eRF) 1 and is mediated by eRF3 with recycling of the ribosomal subunits [25]. Some strains of mammalian picornaviruses, however, encode for a peptide called 2A, which can trigger the release of the newly synthesized protein independently on the presence of a STOP-codon [26]. This “non-conventional” translation termination is the consequence of the 2A activity that mediates processing at its C-terminus altering ribosome activity to promote hydrolysis of the ester bond between the nascent peptide and the tRNA, rather than the formation of the peptide linkage with the aminoacyl-tRNA located on the downstream codon. The result is a premature release of the upstream protein without ribosome dissociation.
2A is a peptide 18 amino acids (aa) long that strictly requires for its activity the Asn-Pro-Gly sequence at its C-terminus and a Pro immediately downstream (Fig. 1a). As 2A allows co-translation of separate proteins encoded in a single open reading frame (ORF), it has been largely used for several biotechnological applications [27], [28], [29], [30]. Mutation of the last Pro into a STOP-codon was reported to cause stalling of translation in in vitro systems (wheat germ and rabbit reticulocyte lysates) [31], [32], making it an interesting model to study ribosome stalling in living human cells.
Fig. 1.

C-terminal 2A* compromises the expression of ER targeted proteins. (a) Schematic representation of the non-conventional translation termination caused by FMDV peptide 2A, resulting in the release of the upstream protein terminating in Gly (G) and the translation of the downstream protein initiating with Pro (P). (b) Left panel: SV5-tagged secretory reporter constructs (pr1) fused to the C-terminal peptide 2A followed by a STOP-codon (2A*) or including a P-codon before the STOP-codon (2A-P*). Right panel: Western blot of cell culture supernatants and extracts of HEK293T cells transfected with the plasmid constructs shown. (c) Quantification of the data shown in panel b. Data presented as mean ± S.E.M. of n = 3 independent experiments. (d) Western blot of extracts from cells transfected with constructs encoding as a reporter the SV5-tagged membrane protein MHC-Iα fused to each of the two C-terminal 2A* or 2A-P* sequences. Data are representative of n = 3 independent experiments.
Here we characterized how imposing conventional termination immediately after peptide 2A heavily impairs expression of the upstream protein in mammalian cells as a consequence of ribosome stalling at the Gly-STOP-codon boundary, and we report that two different human C-terminal peptides ending in Asn-Pro-Gly-STOP also induce stalling at the STOP-codon, resulting in proteasomal degradation of the terminated polypeptides. We found that the main endoplasmic reticulum (ER) chaperone binding immunoglobulin protein/glucose-regulated protein 78 kDa (BiP/GRP78) have an uncharacterized pivotal involvement in binding ribosome stalled secretory proteins and targeting them to the ER-associated degradation (ERAD) pathway.
Results
C-terminal 2A compromises the expression of upstream protein in human cells
We designed a reporter secretory protein formed by an SV5-tagged single-chain variable fragment (scFv, pr1) fused to the foot and mouth disease virus (FMDV) peptide 2A at the C-terminus, followed by the UAG STOP-codon (*) (Fig. 1b, left panel, construct 2A*). The FMDV-derived 23-aa-long peptide contains the 5 C-terminal residues of VP1 followed by the 18 aa of 2A, as it was shown to be more efficient in driving the independent expression of the upstream and downstream proteins [33]. A second construct with a P-codon positioned between the 2A terminal G-codon and the STOP-codon was also obtained (Fig. 1b, left panel, construct 2A-P*). When transfected in HEK293T cells, high expression levels were obtained from construct 2A-P*, while extremely low levels were obtained from construct 2A* (Fig. 1b, right panel). The product from construct 2A* was produced around 20 times less intracellularly, with this difference increasing to 100 times in the secreted material (Fig. 1c).
As expected, we detected by mass spectrometry analysis the pr1 produced from construct 2A-P* terminated in NPG (approximately 50%) due to the non-conventional termination as well as in NPGP (Supplementary Table 1). The translation termination of construct 2A* was instead only conventional, STOP-codon mediated, as it did not encode the terminal P-codon.
The inhibitory effect of 2A* was also observed with an engineered version of the type I membrane glycoprotein reporter MHC-Iα (Fig. 1d), indicating that expression impairment was not dependent on the reporter protein used.
Since these two initial reporters were translated from membrane-bound ribosomes, we then tested whether 2A* had the same effect also on cytosolic proteins translated by free ribosomes. Two cytosolic reporters were used, the same scFv lacking the ER-targeting leader peptide and EGFP. Expression of both proteins from 2A* constructs (EGFP-2A* and cyt-scFv-2A*) was highly compromised as compared to the corresponding controls (EGFP-2A-P* and cyt-scFv-2A-P*; Supplementary Fig. 1a). Furthermore, mRNA quantification by qPCR did not show a significant reduction in the transcript levels for the 2A* construct compared to 2A-P* one (Supplementary Fig. 1b), indicating that the observed effect was not the consequence of reduced mRNAs levels. Moreover, in vitro translation from T7 polymerase-driven transcripts of construct cyt-scFv-2A* confirmed a reduction of almost 3.5-fold in protein expression with respect to the control, indicating a defect at the translational level (Supplementary Fig. 1c).
Taken together, these results confirm that imposing conventional termination after 2A strongly impairs translation in mammalian cells, regardless of the reporter protein used or the cellular localization, a context consistent with stalling of ribosomes at the STOP-codon.
Ribosomes stalling at the termination codon of 2A in human cells
We then decided to investigate the role of the STOP-codon at the 2A C-terminus. When the 11-amino-acid-long peptide roTag was added immediately after the C-terminal Gly (scheme in Fig. 2a, left panel), the expression of the fusion pr1-2A-roTag was totally rescued. Instead, when Pro was included after 2A (2A-P-roTag*) pr1-2A was produced, as expected, while the fusion product was absent (Fig. 2a, right panel). We can conclude that the expression impairment depends on the presence of a STOP-codon after the 2A-terminal Gly, consistent with a translational defect due to the presence of the STOP-codon in the 2A* construct [32].
Fig. 2.

2A* causes ribosomal stalling at the STOP-codon. (a) Schematic representation of constructs containing the 11-aa-long roTag (left panel) and the corresponding Western blots (right panel, representative of three independent experiments) of supernatants and extracts of transfected cells. Black and red arrowheads indicate conventional and non-conventional translation termination products, respectively. (b) Scheme of constructs (left panel) and the corresponding Western blots (right panel, representative of three independent experiments) of extracts of transfected cells. Red arrowheads indicate non-conventional termination of pr1 in constructs A and B. (c) Autoradiography of anti-SV5 immunoprecipitates obtained from total extracts of cells transfected with the indicated constructs and labeled with [35S]-methionine for 15 min. Quantification is indicated at the bottom of each lane. Representative of n = 2 independent experiments.
It has been described that during translation of a bicistronic mRNA, 2A causes a quick and transient stalling of the ribosome, necessary to allow release of the upstream protein and to resume translation from the downstream P-codon [26]. The presence of the STOP-codon would result in an excessively prolonged stalling of ribosomes at the 2A/STOP-codon boundary. To confirm this point, we obtained two bicistronic constructs (schematically shown in Fig. 2b, left panel) in which the upstream protein (pr1) was produced following 2A non-conventional termination (i.e., 2A followed by Pro), while the downstream protein (pr2, same as pr1 with an extra polypeptide tag) terminated in either 2A* (pr2-2A*, construct B) or, as a control, in 2A-P* (pr2-2A-P*, construct A). We reasoned that, if ribosomes stall at the STOP-codon downstream of pr2-2A in construct B, expression of the upstream pr1 would also be affected, whereas in the absence of ribosome stalling, pr1 would be equally expressed from constructs A and B. The results clearly show that expression of pr1 (as well as pr2) was highly compromised from construct B, but not from the control construct A, which expressed pr1 at even higher levels than pr2 (Fig. 2b, right panel). Thus, compromised expression of pr1 on construct B was consistent with ribosome stalling at the 2A-STOP-codon downstream of pr2.
This was further confirmed in living cells by determining the rate of translation by [35S]-methionine (15 min) labeling experiments, followed by anti-SV5 immunoprecipitation. As shown in Fig. 2c, the protein expressed from construct 2A* was synthesized at a lower rate (around 15-fold lower) than the one from construct 2A-P*, a result consistent with ribosomal stalling.
Ribosome stalling of 2A* peptide is independent of mRNA codon usage but dependent of 2A amino acidic sequence
The translation impairment was not due to the nucleotide sequence of the STOP-codon used, as the same result was obtained when the UAG codon was changed to UAA or UGA (Fig. 3a). Similar result was obtained with all four different G-codons (Fig. 3b). The independence from the nucleotide sequence was further confirmed by mutating all the codons of the 2A peptide (M), which showed impaired expression comparable to the wild type and rescued expression upon inclusion of the P-codon (Fig. 3c). Similar results were obtained with the Teschovirus-derived peptide 2A (three residues different from FMDV; Fig. 3d). Furthermore, the addition of a STOP-codon just upstream of the 2A sequences (constructs *2A*, *2A-P*) completely rescued pr1 expression (Fig. 3e), strongly supporting the hypothesis that the lack of pr1-2A* production was dependent on the translation of the 2A peptide and independent of the nucleotide sequence.
Fig. 3.

Stalling is independent of mRNA codon usage. (a–c) Western blots of cell extracts and/or supernatants, as indicated, derived from cells transfected with the control construct 2A-P* or variants of constructs 2A*, with different STOP-codons (a) and G-codons (b) and with degenerated codons for all amino acids of 2A sequence, shown also for 2A-P* (c). (d) Western blots of extracts and supernatants of cells transfected with constructs containing the Teschovirus-derived 2A sequence. (e) Schematic representation of the two new constructs (*2A-P* and *2A*, left panel) and the corresponding Western blots (right panel) of supernatants of transfected cells. Black arrowheads indicate translation termination. Data in all panels are representative of at least n = 3 independent experiments.
We then investigated which was the minimal 2A length required for the expression inhibitory activity by producing several deletion mutants (Fig. 4a). Deletion of 3 to 5 N-terminal amino acids (constructs ΔN3, ΔN4 and ΔN5) retained strong inhibitory activity, while deletion mutants lacking 6 to 15 aa from the 2A N-terminus (constructs ∆ N6, ∆ N7 and ∆ N15) completely abolished the effect (Fig. 4b). Thus, the minimal length that caused stalling of translation was mapped to the last 13 residues, represented by the sequence LLKLAGDVESNPG* (construct ∆ N5). Interestingly, the minimal length required for 2A peptide non-conventional termination in a bicistronic mRNA was mapped to a very similar length (12 residues) [32].
Fig. 4.

Mapping the minimal 2A amino acid sequence causing ribosomal stalling. (a) Scheme of constructs with 2A variants. (b–e) Western blots of supernatants of cells transfected with the indicated constructs. (f) Scheme of constructs (left panel) and Western blot (right panel) of extracts of cells transfected with the indicated variants. Black and red arrowheads indicate conventional and non-conventional termination, respectively. Data in all panels are representative of at least n = 2 independent experiments.
We next mapped 2A C-terminal residues. In contrast to the inhibitory effect observed with full-length 2A (ending in NPG*), C-terminal deletion mutants lacking the last 1 or 2 aa (∆ G and ∆ PG, respectively) strongly rescued pr1-2A expression (Fig. 4c). In addition, deletion of only NP or P while maintaining the terminal G (∆ NP and ∆ P) also restored expression, although only partially for ∆ P (Fig. 4d). Thus, the C-terminal Pro and Gly residues are essential determinants of the 2A translation inhibition. Indeed, 2A peptides ending with amino acids different from G (A, E, R or W) were efficiently expressed (Fig. 4e). Interestingly, the C-terminal Gly is also essential for 2A non-conventional termination activity [31], [32], [34]. Deletion of 2A C-terminal Gly in construct 2A(ΔG)-P-roTag* produced only pr1-2A-roTag fusion, completely abrogating non-conventional termination represented by product pr1-2A (Fig. 4f).
These data collectively indicated that the 2A last 12 aa and in particular the C-terminal Gly residue are absolutely required not only for the 2A non-conventional termination, but also for the translational stalling at the STOP-codon.
Proteasome degradation of products of stalled ribosomes
In addition to ribosome stalling, which directly affects translation, other mechanisms can concomitantly operate in vivo to further reduce the level of protein expression.
Kinetic studies performed in cells treated with cycloheximide revealed that the secretory reporter protein produced from construct 2A* was fast degraded and hence not secreted, whereas the product from 2A-P* was actively secreted (Fig. 5a). To investigate the degradation pathway of these secretory proteins, cells were treated with the proteasome inhibitor MG132 or with the autophagy inhibitor chloroquine (CQ). Significant accumulation of material was observed after MG132, but not after CQ treatment (Fig. 5b), indicating that polypeptides released from the ER-bound stalled ribosomes were degraded by the proteasome. Cells transfected with HA-tagged ubiquitin and treated with the MG132 also showed significantly higher accumulation of poly-ubiquitinated pr1 expressed from 2A* compared to 2A-P* (Fig. 5c). Furthermore, co-expression with the viral cytosolic deubiquitinylase-like protein OTU (derived from the Congo hemorrhagic fever virus) also caused strong pr1 accumulation from 2A* (Fig. 5d). OTU removes ubiquitin from cytosolic poly-ubiquitinylated proteins targeted to degradation, impairing their engagement by the proteasome and therefore causing accumulation [35], [36]. Similarly, co-expression with the dominant negative mammalian deubiquitinylase YOD1 (YOD1-C160S) [37] or the dominant negative mutant of the AAA p97 ATPase, which lacks ATPase activity (p97QQ, E305 and E578 mutated into Q) [38], blocked degradation of pr1 from construct 2A*, which accumulated intracellularly further confirming degradation through the ubiquitin–proteasome pathway (Fig. 5e). Interestingly, YOD1 deubiquitinylase was previously associated with ERAD and proteasomal degradation of proteins, but not of disposal of ribosome stalled polypeptides. Blocking the ubiquitin–proteasome system with MG132 treatment or co-expression of OTU also showed partial rescue of the MHC-Iα trans-membrane 2A* ribosome stalled protein reporter (Fig. 5f).
Fig. 5.

ER-targeted products of stalled ribosomes are degraded by the proteasome. (a) Western blots of extracts and supernatants of transfected cells treated with cycloheximide (CHX) for the indicated time periods. Representative of n = 2 independent experiments. (b) Western blots of extracts of cells treated with CQ or MG132 (MG) for 4 h. Representative of n = 3 independent experiments. (c) Anti-HA Western blot of anti-SV5 immunoprecipitates of cells co-transfected with the indicated constructs and a HA-tagged ubiquitin encoding plasmid. When indicated, (+) cells were treated with MG132 for 4 h. Representative of n = 2 independent experiments. (d–e) Western blots of extracts of cells co-transfected where indicated (+) with OTU, p97QQ or YOD1-C160S (C160S) encoding plasmids. Representative of n = 3 independent experiments. (f) Western blot of cells transfected with plasmid encoding the 2A-P* or 2A* MHC-Iα membrane protein and treated with MG132 for 4 h or co-transfected with OTU, as indicated. Representative of n = 3 independent experiments. (g) Upper panels: Western blot of extracts of cells transfected with siRNA for Listerin (Ltn) or an irrelevant siRNA (irr) and 48 h later co-transfected with the indicated constructs and EGFP as control. Lower panel: ethidium bromide stained agarose gel showing RT-PCR amplification of a 290-bp fragment of Ltn mRNA from cells transfected with the irrelevant or the Listerin-specific siRNA. Representative of n = 2 independent experiments.
Finally, we explored the involvement of the E3 ubiquitin ligases Listerin (Ltn), found to be associated with stalled ribosomes [11]. Silencing of Ltn partially rescued expression from 2A* constructs (Fig. 5g).
Activation of the ubiquitin–proteasome pathway was also observed when translational stalling occurred on free ribosomes: the cytosolic leader-less scFv protein from construct 2A* accumulated upon MG132 treatment, following co-expression with OTU or p97QQ and upon Ltn silencing (Supplementary Fig. 2).
In conclusion, the reduced protein levels produced from constructs 2A* were the consequence of two concomitant effects: (i) ribosome stalling, which strongly impairs the rate of synthesis, and (ii) engagement of the terminated protein into the proteasomal degradation pathway. These results indicate that molecules translated from membrane-bound ribosomes stalled at the STOP-codon are targeted to the proteasome requiring components of both the ribosomal quality control system and the ERAD pathway.
Stalling of membrane-bound ribosomes triggers ERAD
We further investigated involvement of the ERAD pathway by looking at retro-translocation of polypeptides from the ER lumen to the cytosol using our previously described in vivo retro-translocation assay [39] based on the specific in vivo biotinylation of molecules localized on the cytosolic side. Detection and quantification of the biotinylated (retro-translocated) fraction was carried out in a Western blot-retardation assay in which denatured samples, incubated with streptavidin (StrAv) before electrophoresis, produce retardation of the biotinylated proteins. As shown in Fig. 6a, most of the intracellular secretory reporter produced from 2A* was biotinylated. As expected, the material accumulated from construct 2A* upon proteasome inhibition with MG132, or co-expression with p97QQ or OTU, was mostly biotinylated (Fig. 6b), consistent with active retro-translocation from the ER lumen. Upon co-expression with the dominant negative chaperone BiP/GRP78 (T37G), which binds to newly synthesized protein substrates but is unable to undergo the conformational change upon ATP binding required for protein release, a large accumulation of pr1-2A* was observed, similarly to other ERAD targeted BiP substrates [35], [40]. This material was most likely trapped inside the ER, due to its strong interaction with the BiP mutant, as it was not biotinylated by the cytosolic biotin ligase (Fig. 6c, lanes 3 and 4).
Fig. 6.

Stalling of membrane-bound ribosomes triggers ERAD. (a) Western blot-retardation assay (WB-ra) of extracts of cells co-transfected with BAP-tagged constructs terminating in 2A-P* or 2A* and a cytosolic biotin ligase BirA encoding plasmid. Migration of in vivo biotinylated proteins is retarded in the presence (+) of StrAv. (b) WB-ra as in panel a, in extracts of cells treated with MG132 or co-transfected with OTU or p97QQ, as indicated. (c) WB-ra of extracts of cells co-transfected, where indicated, with the BiP/GRP78 mutant T37G (BiP T37G) encoding plasmid. (d, Scheme of constructs with the N-glycosylation tag (N-2A-P* and N-2A*, left panel) and Western blot upon co-transfection with the BiP T37G encoding plasmid. Arrowheads indicate the glycosylated and de-glycosylated products. All panels are representative of at least three independent experiments.
Consistently, polypeptides with an N-glycosylation tag added to construct 2A* (N-2A*) were found mostly de-glycosylated as a consequence of the cytosolic N-glycanase activity [41], but fully glycosylated when co-expressed with the BiP mutant (Fig. 6d). In contrast, the product of reference construct N-2A-P* was fully glycosylated also in the absence of the mutated chaperone (Fig. 6d).
ERAD targeting of peptides from stalled ribosome is specific
We then assessed that targeting to ERAD was restricted to the product of the stalled ribosome. We used a 2A* construct (pr2-2A*) that was co-expressed with a 2A-P* construct (pr1-2A-P*) encoding the same scFv reporter with a shorter tag (Fig. 7a, left panel). As shown in Fig. 7a (right panel), only expression of pr2 from 2A* was affected, while pr1 from 2A-P* was not. A similar result was obtained with an irrelevant control secretory protein (scFv-ctrl; roTag-tagged), which was also not affected when co-expressed with construct pr2-2A* (Fig. 7b).
Fig. 7.

ERAD targeting is restricted to products of stalled ribosomes. (a) Scheme of constructs (right panel) and Western blots of extracts of cells co-transfected with the indicated plasmids. (b) Western blots of extracts of cells co-transfected with the pr2 constructs shown in panel a and the same scFv lacking 2A sequence and tagged with roTag. Both panels are representative of n = 2 independent experiments.
Thus, proteins translated from membrane-bound stalled ribosomes are exclusively targeted to ERAD, without affecting those same polypeptides originating from non-stalled ribosomes.
The product of membrane-bound stalled ribosomes binds to BiP/GRP78
We then investigated the interactome of polypeptides produced from stalled ribosomes using the BioID2 technique, based on the expression of the Aquifex aeolicus bacterial biotin-ligase with the R40G mutation [42]. This enzyme lacks substrate specificity and is therefore able to biotinylate in vivo the interacting proteins. Constructs with 2A* or 2A-P* at the C-terminus of BioID2 (Fig. 8a) were then independently expressed and the biotinylated proteins were analyzed by Western blot. As shown in Fig. 8a and b, the BioID2 activity was detected by its self-biotinylation. One band of around 78 kDa was found strongly biotinylated in cells expressing BioID2-2A* but not BioID2-2A-P* (Fig. 8a). We suspected that it represented BiP/GRP78, which was confirmed by pull-down with StrAv followed by detection with anti-BiP as well as by BiP immunoprecipitation followed by detection with StrAv-HRP from cells expressing BioID2-2A* (Fig. 8c). Mass spectrometry analysis was then performed on pulled-down biotinylated proteins, confirming detection of BiP only in extracts of cells expressing BioID2-2A*, but not BioID2-2A-P*. Mass spectrometry of BioID2-2A* biotinylated proteins also detected the 23-kDa nascent polypeptide-associated complex α-subunit (NACA) and the 71-kDa cytosolic chaperone HSPA8. A complete list of polypeptides found with both proteins is shown in Supplementary Table 2.
Fig. 8.

The products of membrane-bound stalled ribosomes are bound by BiP/GRP78. (a) Scheme of the secretory version of BioID2 constructs (left panel) and StrAv Western blot of extracts of cells transfected with the indicated plasmids and labeled with biotin for 24 h. The rightmost panel shows a zoom of the 75-kDa region with lower exposure. Black arrow indicates the self-biotinylation of the BioID2 protein, while red arrows indicate bands that appear only in extracts of cells transfected with BioID2-2A*. Representative of n = 3 experiments. (b) Western blots with anti-roTag antibody and StrAv-HRP of extracts shown in panel a. (c) Western blots of extracts of cells mock-transfected or transfected with the indicated BioID2 constructs: upper panel, anti-BiP immunoblotting of proteins pulled-down with StrAv beads; middle and lower panels, StrAv-HRP or anti-BiP blots of proteins immunoprecipited (IP) with anti-BiP. Data are representative of n = 2 independent experiments.
Stalling by non-viral peptides encoded in the human genome
Identification of other sequences capable of inducing stalling of the ribosome at the STOP-codon is challenging. As 2A-like peptides are highly conserved at the last three C-terminal amino acids (NPG), we then searched for human proteins that terminate with the same NPG* sequence in the ENSEMBL data bank. Only five proteins were found [CD99 molecule-like protein 2 (CD99L2), prostate ovary testis-expressed protein family members I and E (POTEI and POTEE), TBCA and SPIRE2, where POTEE and POTEI share the same C-terminal amino acidic sequence]. In most cases, they represent alternative spliced transcripts. We tested the effect exerted on expression of the cytosolic reporter (cyt-scFv) by the four different C-terminal sequences in constructs with and without the addition of the P-codon before the STOP-codon (Fig. 9a). While the C-termini from TBCA and SPIRE2 did not display any effect on the reporter translation, those from CD99L2 and POTE caused a strong expression decrease compared to the reporter without the terminal sequences (Fig. 9b). Of note, expression of proteins terminating with these two sequences was partially rescued by the addition of the C-terminal Pro, similar to what observed with peptide 2A. However, and despite of this, non-conventional translation termination was not observed as shown with a construct elongated with the 11-aa roTag downstream of Pro (shown for POTE, Fig. 9c, left panel).
Fig. 9.

Ribosomal stalling by non-viral sequences. (a) Scheme of the cytosolic scFv reporter constructs showing the four different C-terminal sequences of CD99L2, POTEI/POTEE, TBCA and SPIRE2 and the one from the 3′UTR of AMD1 (C-TAIL) used in place of 2A. The NPG terminal amino acids are shown in red, as well as the entire 18 aa of 2A. (b) Western blots of extracts form cells transfected with the indicated cytosolic constructs including or not a C-terminal proline. (c) Western blots of proline including constructs with POTE C-terminus or C-TAIL sequence including the addition of a C-terminal roTag as indicated. (d) Western blot of extracts of cells transfected with the secretory versions of the CD99L2 and POTE scFv constructs. Lane 1 shows the same reporter without any addition after the SV5 tag. (e) Western blots of extracts of cells transfected with the secretory reporters with CD99L2 or POTE sequences and treated, where indicated (+), with MG132 for 4 h or co-transfected, where indicated (+), with OTU or p97QQ. All panels are representative of at least n = 3 independent experiments.
Recently, a short sequence capable of inducing ribosome stalling was described in an ORF located at the 3′UTR of adenosylmethionine decarboxylase 1 (AMD1) mRNA [43]. This sequence does not terminate in NPG (Fig. 9a), but its 22-aa-long C terminus (C-TAIL) strongly affected expression of the reporter protein to a level comparable to that of 2A (Fig. 9b, right panel). In this case, however, addition of the terminal Pro just upstream of the STOP-codon did not rescue expression and did not promote non-conventional termination in a similar elongated construct, indicating a different stalling mechanism to the one operating with 2A* (Fig. 9b and c, right panels).
We then observed that, as 2A*, also the CD99L2 and POTE sequences caused the same translational effect when tested on the secretory reporter (Fig. 9d).
Interestingly, the secretory reporters produced with CD99L2 or POTE C-termini were also degraded through the same pathway as with 2A*. Both products accumulated upon treatment with MG132 as well as upon co-expression with OTU and p97QQ (Fig. 9e). Collectively, these data suggest that ribosome stalling at the STOP-codon, produced by viral and non-viral C-terminal sequences, represents a way to control gene expression at the translational level.
Discussion
Here we describe the occurrence of ribosome stalling at the STOP-codon in human cells caused by the picornavirus peptide 2A when the P-codon immediately downstream from the C-terminal Gly, required for its non-conventional termination activity, is replaced by a STOP-codon. This 2A-STOP-mediated impairment of translation represents a strong model of ribosome stalling at the STOP-codon, caused by the 2A last 13 aa and, in particular, by the C-terminal NPG sequence. This indicates that stalling is the consequence of the peptide conformation inside the RET. 2A has been described to form an amphipathic α-helix over most of its length, with a reverse turn at its C-terminus [44], [45]. In its normal viral context, this structure has been suggested to cause a transient stalling that favors the non-conventional translation termination of the upstream-encoded protein, determined by the position of the 2A terminal G-codon in the ribosome P site and the P-codon in the ribosome A site [32], [33]. Transient ribosome stalling is thought to occur because of re-orientation of the peptidyl-tRNAGly within the RET that disfavors peptide bond formation with the prolyl-tRNAPro and promotes instead hydrolysis and release of the nascent chain [44], [45].
Instead, when a STOP-codon is found in the A site in place of an aminoacyl-tRNA, stalling becomes more relevant with a profound effect on the expression of the upstream protein, likely because the 2A structure within RET impaired the polypeptide release or the recruitment of release factors.
Previous report showed that an in vitro translated polypeptide from a construct terminating in 2A* can be associated with the tRNA (peptidyl-tRNAGly) [32]. However, in living yeast cells, the majority of ubiquitinated products from stalled ribosomes, recognized by the co-translational quality control pathway, was found already hydrolyzed from the tRNA, owing to the involvement of the yeast release factors Sup45–Sup35 (mammalian eRF1–3 homologues) [46]. In line with this observation, we also detected the residual amount of non-degraded stalled protein as a fully terminated polypeptide mostly hydrolyzed from the tRNA.
As a result of the stalling at the STOP-codon, the ubiquitin–proteasome degradation pathway becomes activated to discard the newly synthesized products. We found that the total levels of the mRNA encoding peptide that cause stalling (constructs 2A*) were not remarkably different from those mRNAs that do not, thus indicating no activation of the RNA decay pathways and suggesting that mechanisms alternative to NGD or NMD are operating in this case. We also found that the reduction in the rate of synthesis of the secretory reporter was about 15-fold, while the total amount of the secreted protein was 100-fold less. Similarly, the cytosolic reporter was expressed 33-fold less. Degradation of polypeptides originated from stalled ribosomes was the main component of the overall reduced expression. A strong, but not complete, rescue of protein expression was obtained upon proteasome inhibition or deubiquitinylation (Fig. 5b–f), consistently with a concomitant decrease of protein synthesis rate and extensive proteasomal degradation of the products.
Although stalling of free ribosomes has been investigated, much less is known on the mechanisms operating on membrane-bound ribosomes. Terminated secretory and membrane polypeptides translocated to the ER lumen are targeted to degradation through the ERAD pathway if recognized as unfolded. ERAD-targeted polypeptides are first retro-translocated to the cytosol and then degraded by the proteasome.
We showed that the luminal proteins derived from stalled ribosome were also targeted to the ERAD pathway and actively retro-translocated to the cytosol, as demonstrated by cytosolic BirA biotinylation, and then polyubiquitinated before proteasomal degradation.
Indeed, experiments with the proteasome inhibitor MG132 indicated stabilization of the proteins resulting from stalled ribosomes. The same conclusions were obtained using the deubiquitinylase-like protein OTU or the YOD1-C160S, a dominant negative mutant of the ERAD-associated deubiquitinylase YOD1 [35], [37], [47] and the dominant negative mutant of the p97 ATPase (p97QQ). We also showed that cytosolic proteins produced from 2A* stalled free ribosomes were degraded by the proteasome. In yeast, the p97 homologous Cdc48 is also part of the ribosome-associated quality control, which recognizes stalled ribosomes in NSD and NGD [13], [14], [15]. The dominant negative mutant of chaperone BiP/GRP78 was able to retain the protein within the ER lumen. This was the first indication that BiP/GRP78 was involved in targeting to degradation polypeptides produced by stalled ER-bound ribosomes. Indeed, we identified BiP/GRP78 as one of the main proteins interacting with the product of the stalled BioID-2A*. Considering that this chaperone is involved in signaling to convey luminal substrates to ERAD [48], it makes it a strong candidate to initiate and re-direct the polypeptide from the stalled ribosome to degradation. It is possible that stalled polypeptides may not be capable to complete folding in the ER lumen or remain too long within the translocon, thus recruiting BiP/GRP78 to initiate engagement into the ERAD pathway. Previous works have found that stalled peptides can obstruct the yeast translocon [49] and translocon components were found in association with components of the ribosome-associated quality control in purified pancreatic rough ER microsomes [12]. 2A-tagged stalled proteins may not activate ERAD targeting because of canonical misfolding, as they are identical to the proteins produced by non-conventional termination. This hypothesis can be suggested by BioID2 activity in the ER lumen when bearing the C-terminal 2A, indicating that ERAD could be activated as a direct consequence of translational stalling rather than protein misfolding per se. However, it is also possible that the domain of the stalled protein close to the translocon exit tunnel is not completely folded and could be the portion of the polypeptide efficiently recognized by BiP. Detection of BioID2 activity during ERAD is consistent with previous works demonstrating that some ERAD substrates can be at least partially folded during retro-translocation to the cytosol before degradation [50], [51], [52].
Of note, the mass spectrometry data of interactors labeled by BioID2-stalled protein also revealed the presence of the α subunit of nascent polypeptide-associated complex (NACA), which is part of the co-translational quality control machinery associated with ribosomes, suggesting that this complex may be also involved in interactions with the polypeptide during or after retro-translocation.
Interestingly, we found the requirement also of the E3 ubiquitin-ligase Listerin, involved in the degradation of polypeptides from ribosomes stalled at the STOP-codon. Consistently, Listerin has been previously described to specifically ubiquitinylate polypeptides originating from both free and membrane-bound stalled ribosomes [11], [12], [13], [53], [54]. However, so far, Listerin has not been associated with canonical ERAD, which instead involves other ubiquitin ligases such as Hrd1 and gp78 [55], [56], [57]. Despite this, we do not exclude involvement of other E3 ligases, as several ubiquitin ligases were found to participate in ERAD in mammalian cells, often with interconnected or overlapping functions [58].
It is important to note that degradation was restricted to products of stalled ribosomes. When co-expressed in the same cells, we observed that only those polypeptides originating from stalled ribosomes were specifically targeted to degradation, without any effect on the same polypeptide produced from non-stalled ribosomes. This is an indication that either the stalled ribosome signals the ER lumen to re-direct the polypeptide to ERAD or, alternatively, that the extended permanence of the terminated polypeptide in the translocon is a signal for degradation, in both cases mediated by BiP/GRP78. The involvement in 2A* degradation of proteins, such BiP/GRP78 and YOD1, of the ER-associated quality control pathway and enzymes, such as Listerin, belonging to the ribosomal quality control system, indicates that translational stalling at the ER membrane activates a poorly described cellular mechanism at the interface of diverse protein quality control surveillance systems.
The general idea that ribosome stalling may represent a mechanism to control protein expression is being increasingly appreciated. There are examples of small coding sequences, some of them having a STOP-codon, like the so-called upstream ORFs (uORF), that can regulate translation initiation of a downstream ORF [2], [20], [59]. In contrast, in our case, the stalling strongly affects expression of the upstream encoded protein. Recently, an independent ORF was found immediately downstream (in the 3′UTR) of the ORF for protein AMD1 and shown to regulate translation of the upstream ORF by causing ribosomal stalling [43]. We found that this sequence, however, produces stalling independently of the STOP-codon.
C-terminal sequences may represent efficient translational regulatory motifs with strong effects on the level of expression of the upstream protein by enacting stalling at the STOP-codon. Our first attempt to find sequences only partially related to the viral 2A (terminating in NPG*) allowed us to identify two of them derived from human proteins that showed stalling activity only at the STOP-codon and that were partially rescued when the P-codon substituted the STOP-codon. Also, more importantly, both proteins induced degradation of the reporter in a manner similar to the 2A sequence, thus confirming that stalling of ribosomes at the STOP-codon activates degradation of their newly made products through the ERAD pathway. In conclusion, we report here the involvement of BiP/GRP78 and the ERAD pathway in the degradation of polypeptides targeted to the ER originating from ribosomes stalled at the STOP-codon, and we speculate that 2A*-like stalling events could regulate protein expression in mammalian cells.
Materials and Methods
Constructs
All new plasmids used are cloned into pcDNA3 vector (Life Technologies). The 2A-P*, 2A*, 2A(M) with mutated codons, T2A, CD99L2, POTE, TBCA, SPIRE2 and C-TAIL coding sequences were inserted by digestion with enzymes KpnI and EcoRI (NEB) in frame at the C-terminus of previously described scFv anti-coronavirus 6A.C3 mAb [60], using oligoes described in Supplementary Table 3. When indicated, SV5-tag (GKPIPNPLLGLD), roTag (SISSSIFKNEG) [61], HA-tag (YPYDVPDYA) and N-glycosylation site (NGT) were engineered at the indicated positions by site-directed mutagenesis (QuikChange Site Directed Mutagenesis Kit; Stratagene). Plasmids encoding secretory proteins bear an immunoglobulin secretion leader peptide [62] at the N-terminus as previously described [61]. MHC-Iα having C-terminal 2A* and 2A-P* sequence was derived from a previously described MHC-Iα plasmid [39]. The coding sequence of BioID2 was previously described [42]. Plasmids expressing cyt-BirA and BiP mutant T37G were previously described [35]. CCHFV-L OTU plasmid was kindly provided by Adolfo García-Sastre, the N-terminal FLAG-tagged human YOD1-C160S plasmid by Christian Schlieker, and the His6-tagged rat p97QQ plasmids by Linda Hendershot.
In vitro protein expression
Protein expression in vitro was performed using the TnT kit (TNT® Quick Coupled Transcription/Translation System) following the manufacture's instruction.
Cell culture and transfection
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (Life Technologies), supplemented with 10% fetal calf serum (Life Technologies). Transfection of 293T was performed in 6-well or 12-well plates (about 5 × 105 or 2.5 × 105 cells) with the calcium phosphate technique. Eighteen hours after transfection, medium was changed, and cells were further incubated for at least 7–8 h, before medium and cell harvesting. Where indicated, the proteasome inhibitor MG132 (Sigma) or CQ (Sigma) was added at a concentration of 20 and 50 μM, respectively, for 4 h. The translation inhibitor cycloheximide (Sigma) was used for the indicated time at the concentration of 100 μg/ml. For biotinylation analysis, 0.15 mM of biotin (Sigma) was added to the media for at least 4 h.
For silencing experiments, HEK293T cells were grown in 12-well plates. Irrelevant (UCGUCUUCUACAACGUCAA) and Ltn-specific (GCAGUGGUGUGAAGAAUUA) siRNA (Sigma) were transfected with Lipofectamine RNAiMAX reagent (Life Technologies) according to the manufacturer's instruction. Forty-eight hours after siRNA transfection, medium was discarded, and cells were transfected with DNA plasmids by the standard calcium phosphate procedure and lysed 24 h post-transfection.
Cell extract preparation, gel retardation assay and Western blotting
Transfected HEK293T cells were washed with phosphate-buffered saline and lysed with SDS-lysis buffer [100 mM Tris–HCl (pH 6.8), 6% SDS, 30 mM NEM (Fluka), 1% protease inhibitors cocktail (Sigma)] and sonicated. For SDS-PAGE, samples were boiled 10 min in SDS-gel loading buffer [25 mM Tris–HCl (pH 6.8), 1% SDS, 10% glycerol, 175 mM β-mercaptoethanol] before gel loading. For Western blot-retardation assays, samples were incubated after boiling without or with 1 μg StrAv (Sigma) for 20 min at room temperature before loading as previously described [39].
Gels were blotted onto PVDF membranes (Millipore), reacted with mouse anti-SV5 (Life Technologies) or mouse anti-roTag mAbs [61] followed by incubation with a HRP-labeled anti-mouse whole IgG antibody (KPL) and developed by ECL reaction. Where indicated, blots were developed using mAb anti-BiP (BD Bioscience), rabbit anti-EGFP serum, StrAv-HRP (Sigma) and anti-HA-HRP (Sigma) and when required by the appropriate HRP-conjugated anti-mouse or anti-rabbit secondary antibody (Jackson). Where indicated, blots were developed with mouse anti-β-tubulin (Calbiochem) or HRP-labeled anti-actin (clone AC-15; Sigma-Aldrich) antibodies, which were used as loading control.
Quantification of bands was performed with the image processing software ImageJ v1.43 (National Institutes of Health) or using UVItec Alliance detection system.
Mass spectrometry analysis
HEK293T cells were transfected with scFv-2A-P* encoding plasmid and lysed 24 h post-transfection in TNN buffer [Tris–HCl 100 mM (pH 8), NaCl 250 mM, NP-40 0.5%], and scFv reporter was immunoprecipitated using mAb anti-SV5. For BioID encoding plasmids, transfection was performed in the presence of biotin and lysed 24 h post-transfection in TNN buffer. Pull-down of biotinylated proteins was performed using StrAv Mag Sepharose™ (GE Healthcare). For mass spectrometry analysis, samples were digested with 200 ng trypsin diluted in 20 mM triethyl ammonium bicarbonate (pH 8.5) for 12 h at room temperature. The supernatants were harvested, and the beads were washed once with digestion buffer. The washes and the supernatants were pooled and purified using STAGE tips. The purified digested samples were subjected to LC–MS/MS analysis using a picofrit nano-bore columns. The sample was loaded in 0.1% formic acid, and the column was developed with a discontinuous gradient and sprayed directly into an Amazon ETD ion trap (Bruker Daltonics). A cycle of one MS scan followed by five MS/MS scans was performed throughout the run. Data were extracted from the runs using Data Analysis (Bruker Daltonics) and analyzed using the X!tandem search engine.
[35S]-Methionine labeling
HEK293T cells were starved for 30 min in methionine/cysteine-free medium supplemented with 10% dialyzed fetal calf serum and then labeled for 15 min with 200 μCi/ml of [35S]-methionine/cysteine (PerkinElmer). Cells were lysed in 100 μl of SDS-lysis buffer, diluted with 400 μl of TNN and digested with DNaseI (Promega) for 1 h at 37 °C. SV5-tagged proteins were immunoprecipitated with anti-SV5 mAb and Protein A agarose (Repligen) and eluted by boiling in SDS-lysis buffer, and samples were resolved on a reducing SDS-PAGE. Gels were fixed in 10% acetic acid and 10% methanol, incubated for 20 min in Amplify fluorographic enhancer (GE Healthcare), dried and exposed for autoradiography on Kodak BioMax XAR films.
RNA isolation, RT-PCR and qRT-PCR
Total RNA from HEK293T cells was isolated with RNeasy mini Kit (Qiagen) following the manufacturer's instructions. DNA was removed by treatment with RNAse-free DNAse I (Fermentas Inc., Waltham, MA, USA). Total RNA was then retro-transcribed to cDNA by Moloney murine leukaemia RT (M-MLV-RT, Invitrogen) in the presence of random hexamers (IDT). qRT-PCR was based on SYBR Green Master Mix technology (Applied Biosystems), and the levels of target gene expression were normalized to those of GAPDH and α-globin. A list of primers used for qRT-PCR and RT-PCR are given in Supplementary Table 3.
Acknowledgments
F.C. and L.S. were supported by ICGEB pre-doctoral fellowships. Plasmid encoding p97QQ was a kind gift of Linda Hendershot (St. Jude Children's Research Hospital). CCHFV-L OTU plasmid and YOD1-C160S plasmids were kindly provided by Adolfo García-Sastre (The Mount Sinai Hospital) and by Christian Schlieker (Yale University), respectively.
Author Contributions: F.C., L.S., G.P. and O.R.B. designed the research; F.C. and L.S. performed research; A.R. identified 2A-like peptides; M.P.M. performed mass spectrometry analysis; F.C., L.S., G.P. and O.R.B. analyzed data; and F.C., L.S., G.P. and O.R.B. wrote the paper.
Conflict of Interest : All authors declare that they have no conflict of interest.
Edited by Sheena Radford
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2018.10.009.
Contributor Information
Gianluca Petris, Email: gianluca.petris@unitn.it.
Oscar R. Burrone, Email: burrone@icgeb.org.
Appendix A. Supplementary data
Supplementary material
References
- 1.Wolff S., Weissman J.S., Dillin A. Differential scales of protein quality control. Cell. 2014;157:52–64. doi: 10.1016/J.CELL.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Yan F., Doronina V.A., Sharma P., Brown J.D. Orchestrating ribosomal activity from inside: effects of the nascent chain on the peptidyltransferase centre. Biochem. Soc. Trans. 2010;38:1576–1580. doi: 10.1042/BST0381576. [DOI] [PubMed] [Google Scholar]
- 3.Wilson D.N., Beckmann R. The ribosomal tunnel as a functional environment for nascent polypeptide folding and translational stalling. Curr. Opin. Struct. Biol. 2011;21:274–282. doi: 10.1016/j.sbi.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Doma M.K., Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graille M., Séraphin B. Surveillance pathways rescuing eukaryotic ribosomes lost in translation. Nat. Rev. Mol. Cell Biol. 2012;13:727–735. doi: 10.1038/nrm3457. [DOI] [PubMed] [Google Scholar]
- 6.Shoemaker C.J., Green R. Translation drives mRNA quality control. Nat. Struct. Mol. Biol. 2012;19:594–601. doi: 10.1038/nsmb.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brandman O., Hegde R.S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016;23:7–15. doi: 10.1038/nsmb.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang J., Hao X., Cao X., Liu B., Nyström T. Spatial sequestration and detoxification of huntingtin by the ribosome quality control complex. elife. 2016;5:1–14. doi: 10.7554/eLife.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chu J., Hong N.A., Masuda C.A., Jenkins B.V., Nelms K.A., Goodnow C.C., Glynne R.J., Wu H., Masliah E., Joazeiro C.A.P., Kay S.A. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 2009;106:2097–2103. doi: 10.1073/pnas.0812819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishimura R., Nagy G., Dotu I., Zhou H., Yang X.-L., Schimmel P., Senju S., Nishimura Y., Chuang J.H., Ackerman S.L. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bengtson M.H., Joazeiro C.A.P. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–473. doi: 10.1038/nature09371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von der Malsburg K., Shao S., Hegde R.S. The ribosome quality control pathway can access nascent polypeptides stalled at the Sec61 translocon. Mol. Biol. Cell. 2015;26:2168–2180. doi: 10.1091/mbc.E15-01-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandman O., Stewart-Ornstein J., Wong D., Larson A., Williams C.C., Li G.-W., Zhou S., King D., Shen P.S., Weibezahn J., Dunn J.G., Rouskin S., Inada T., Frost A., Weissman J.S. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Defenouillère Q., Yao Y., Mouaikel J., Namane A., Galopier A., Decourty L., Doyen A., Malabat C., Saveanu C., Jacquier A., Fromont-Racine M. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc. Natl. Acad. Sci. U. S. A. 2013;110 VN:5046–5051. doi: 10.1073/pnas.1221724110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verma R., Oania R.S., Kolawa N.J., Deshaies R.J. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. elife. 2013;2013:1–17. doi: 10.7554/eLife.00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gandin V., Gutierrez G.J., Brill L.M., Varsano T., Feng Y., Aza-Blanc P., Au Q., McLaughlan S., Ferreira T.A., Alain T., Sonenberg N., Topisirovic I., Ronai Z.A. Degradation of newly synthesized polypeptides by ribosome-associated RACK1/c-Jun N-terminal kinase/eukaryotic elongation factor 1A2 complex. Mol. Cell. Biol. 2013;33:2510–2526. doi: 10.1128/MCB.01362-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pisareva V.P., Skabkin M.A., Hellen C.U.T., Pestova T.V., Pisarev A.V. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J. 2011;30:1804–1817. doi: 10.1038/emboj.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyumkis D., Oliveira Dos Passos D., Tahara E.B., Webb K., Bennett E.J., Vinterbo S., Potter C.S., Carragher B., Joazeiro C.A.P. Structural basis for translational surveillance by the large ribosomal subunit-associated protein quality control complex. Proc. Natl. Acad. Sci. 2014;111:15981–15986. doi: 10.1073/pnas.1413882111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao S., Brown A., Santhanam B., Hegde R.S. Structure and assembly pathway of the ribosome quality control complex. Mol. Cell. 2015;57:433–445. doi: 10.1016/j.molcel.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson D.N., Arenz S., Beckmann R. Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr. Opin. Struct. Biol. 2016;37:123–133. doi: 10.1016/j.sbi.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Seidelt B., Innis C.A., Wilson D.N., Gartmann M., Armache J.-P., Villa E., Trabuco L.G., Becker T., Mielke T., Schulten K., Steitz T.A., Beckmann R. Structural insight into nascent polypeptide chain-mediated translational stalling. Science (80-.) 2009;326:1412–1415. doi: 10.1126/science.1177662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spevak C.C., Ivanov I.P., Sachs M.S. Sequence requirements for ribosome stalling by the arginine attenuator peptide. J. Biol. Chem. 2010;285:40933–40942. doi: 10.1074/jbc.M110.164152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamashita Y., Takamatsu S., Glasbrenner M., Becker T., Naito S., Beckmann R. Sucrose sensing through nascent peptide-meditated ribosome stalling at the stop codon of Arabidopsis bZIP11 uORF2. FEBS Lett. 2017;591:1266–1277. doi: 10.1002/1873-3468.12634. [DOI] [PubMed] [Google Scholar]
- 24.Bhushan S., Meyer H., Starosta A.L., Becker T., Mielke T., Berninghausen O., Sattler M., Wilson D.N., Beckmann R. Structural basis for translational stalling by human cytomegalovirus and fungal arginine attenuator peptide. Mol. Cell. 2010;40:138–146. doi: 10.1016/j.molcel.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Brown A., Shao S., Murray J., Hegde R.S., Ramakrishnan V. Structural basis for stop codon recognition in eukaryotes. Nature. 2015;524:493–496. doi: 10.1038/nature14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doronina V.A., de Felipe P., Wu C., Sharma P., Sachs M.S., Ryan M.D., Brown J.D. Dissection of a co-translational nascent chain separation event. Biochem. Soc. Trans. 2008;36:712–716. doi: 10.1042/BST0360712. [DOI] [PubMed] [Google Scholar]
- 27.Cesaratto F., Burrone O.R., Petris G. Tobacco etch virus protease: a shortcut across biotechnologies. J. Biotechnol. 2016 doi: 10.1016/j.jbiotec.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 28.Piperno G.M., López-Requena A., Predonzani A., Dorvignit D., Labrada M., Zentilin L., Burrone O.R., Cesco-Gaspere M. Recombinant AAV-mediated in vivo long-term expression and antitumour activity of an anti-ganglioside GM3(Neu5Gc) antibody. Gene Ther. 2015;22:960–967. doi: 10.1038/gt.2015.71. [DOI] [PubMed] [Google Scholar]
- 29.Principe M., Ceruti P., Shih N.-Y., Chattaragada M.S., Rolla S., Conti L., Bestagno M., Zentilin L., Yang S.-H., Migliorini P., Cappello P., Burrone O., Novelli F. Targeting of surface alpha-enolase inhibits the invasiveness of pancreatic cancer cells. Oncotarget. 2015;6:11098–11113. doi: 10.18632/oncotarget.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science (80-.) 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doronina V.A., Wu C., de Felipe P., Sachs M.S., Ryan M.D., Brown J.D. Site-specific release of nascent chains from ribosomes at a sense codon. Mol. Cell. Biol. 2008;28 VN-r:4227–4239. doi: 10.1128/MCB.00421-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma P., Yan F., Doronina V.A., Escuin-Ordinas H., Ryan M.D., Brown J.D. 2A peptides provide distinct solutions to driving stop-carry on translational recoding. Nucleic Acids Res. 2012;40:3143–3151. doi: 10.1093/nar/gkr1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donnelly M.L., Hughes L.E., Luke G., Mendoza H., ten Dam E., Gani D., Ryan M.D. The “cleavage” activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring “2A-like” sequences. J. Gen. Virol. 2001;82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- 34.Atkins J.F., Wills N.M., Loughran G., Wu C.-Y., Parsawar K., Ryan M.D., Wang C.-H., Nelson C.C. A case for “StopGo”: reprogramming translation to augment codon meaning of GGN by promoting unconventional termination (Stop) after addition of glycine and then allowing continued translation (Go) RNA. 2007;13:803–810. doi: 10.1261/rna.487907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasset L., Petris G., Cesaratto F., Burrone O.R. The VCP/p97 and YOD1 proteins have different substrate-dependent activities in endoplasmic reticulum-associated degradation (ERAD) J. Biol. Chem. 2015;290:28175–28188. doi: 10.1074/jbc.M115.656660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rumpf S., Jentsch S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol. Cell. 2006;21:261–269. doi: 10.1016/j.molcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 37.Ernst R., Mueller B., Ploegh H.L., Schlieker C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol. Cell. 2009;36:28–38. doi: 10.1016/j.molcel.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okuda-Shimizu Y., Hendershot L.M. Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol. Cell. 2007;28:544–554. doi: 10.1016/j.molcel.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petris G., Vecchi L., Bestagno M., Burrone O.R. Efficient detection of proteins retro-translocated from the ER to the cytosol by in vivo biotinylation. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei J., Gaut J.R., Hendershot L.M. In vitro dissociation of BiP–peptide complexes requires a conformational change in BiP after ATP binding but does not require ATP hydrolysis. J. Biol. Chem. 1995;270:26677–26682. doi: 10.1074/jbc.270.44.26677. ( http://www.ncbi.nlm.nih.gov/pubmed/7592894 (accessed July 12, 2018)) [DOI] [PubMed] [Google Scholar]
- 41.Suzuki T., Huang C., Fujihira H. The cytoplasmic peptide:N-glycanase (NGLY1)—structure, expression and cellular functions. Gene. 2016;577:1–7. doi: 10.1016/j.gene.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim D.I., Jensen S.C., Noble K.A., B. KC, Roux K.H., Motamedchaboki K., Roux K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell. 2016;27:1188–1196. doi: 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yordanova M.M., Loughran G., Alexander V., Mariotti M., Kiniry S.J., Connor P.B.F.O., Andreev D.E., Tzani I., Saffert P., Michel A.M., Gladyshev V.N., Papkovsky D.B., Atkins J.F., Pavel V. AMD1 mRNA employs ribosome stalling as a mechanism for molecular memory formation. Nat. Publ. Gr. 2018;19 doi: 10.1038/nature25174. [DOI] [PubMed] [Google Scholar]
- 44.Donnelly M.L., Luke G., Mehrotra A., Li X., Hughes L.E., Gani D., Ryan M.D. Analysis of the aphthovirus 2A/2B polyprotein “cleavage” mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal “skip”. J. Gen. Virol. 2001;82:1013–1025. doi: 10.1099/0022-1317-82-5-1013. [DOI] [PubMed] [Google Scholar]
- 45.Ryan M.D., Donnelly M., Lewis A., Mehrotra A.P., Wilkie J., Gani D. A model for nonstoichiometric, cotranslational protein scission in eukaryotic ribosomes. Bioorg. Chem. 1999;27:55–79. doi: 10.1006/bioo.1998.1119. [DOI] [Google Scholar]
- 46.Shcherbik N., Chernova T.A., Chernoff Y.O., Pestov D.G. Distinct types of translation termination generate substrates for ribosome-associated quality control. Nucleic Acids Res. 2016;44:6840–6852. doi: 10.1093/nar/gkw566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye Y., Meyer H.H., Rapoport T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–656. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 48.Otero J.H., Lizák B., Hendershot L.M. Life and death of a BiP substrate. Semin. Cell Dev. Biol. 2010;21:472–478. doi: 10.1016/j.semcdb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Izawa T., Tsuboi T., Kuroha K., Inada T., Ichi Nishikawa S., Endo T. Roles of Dom34:Hbs1 in nonstop protein clearance from translocators for normal organelle protein influx. Cell Rep. 2012;2:447–453. doi: 10.1016/j.celrep.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 50.Petris G., Casini A., Sasset L., Cesaratto F., Bestagno M., Cereseto A., Burrone O.R. CD4 and BST-2/tetherin proteins retro-translocate from ER to cytosol as partially folded and multimeric molecules. J. Biol. Chem. 2013:0–21. doi: 10.1074/jbc.M113.512368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiebiger E., Story C., Ploegh H.L., Tortorella D. Visualization of the ER-to-cytosol dislocation reaction of a type I membrane protein. EMBO J. 2002;21:1041–1053. doi: 10.1093/emboj/21.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tirosh B., Furman M.H., Tortorella D., Ploegh H.L. Protein unfolding is not a prerequisite for endoplasmic reticulum-to-cytosol dislocation. J. Biol. Chem. 2003 doi: 10.1074/jbc.M210158200. [DOI] [PubMed] [Google Scholar]
- 53.Shao S., Von der Malsburg K., Hegde R.S. Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol. Cell. 2013;50:637–648. doi: 10.1016/j.molcel.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crowder J.J., Geigges M., Gibson R.T., Fults E.S., Buchanan B.W., Sachs N., Schink A., Kreft S.G., Rubenstein E.M. Rkr1/Ltn1 ubiquitin ligase-mediated degradation of translationally stalled endoplasmic reticulum proteins. J. Biol. Chem. 2015;290:18454–18466. doi: 10.1074/jbc.M115.663559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang T., Xu Y., Liu Y., Ye Y. gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol. Biol. Cell. 2015:5–6. doi: 10.1091/mbc.E15-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ballar P., Shen Y., Yang H., Fang S. The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2006;281:35359–35368. doi: 10.1074/jbc.M603355200. [DOI] [PubMed] [Google Scholar]
- 57.Taxis C., Hitt R., Park S.-H., Deak P.M., Kostova Z., Wolf D.H. Use of modular substrates demonstrates mechanistic diversity and reveals differences in chaperone requirement of ERAD. J. Biol. Chem. 2003;278:35903–35913. doi: 10.1074/jbc.M301080200. [DOI] [PubMed] [Google Scholar]
- 58.Yoshida Y., Tanaka K. Lectin-like ERAD players in ER and cytosol. Biochim. Biophys. Acta Gen. Subj. 2010;1800:172–180. doi: 10.1016/j.bbagen.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 59.Re A., Waldron L., Quattrone A. Control of gene expression by RNA binding protein action on alternative translation initiation sites. PLoS Comput. Biol. 2016;12 doi: 10.1371/journal.pcbi.1005198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bestagno M., Sola I., Dallegno E., Sabella P., Poggianella M., Plana-Duran J., Enjuanes L., Burrone O.R. Recombinant dimeric small immunoproteins neutralize transmissible gastroenteritis virus infectivity efficiently in vitro and confer passive immunity in vivo. J. Gen. Virol. 2007;88:187–195. doi: 10.1099/vir.0.82192-0. [DOI] [PubMed] [Google Scholar]
- 61.Petris G., Bestagno M., Arnoldi F., Burrone O.R. New tags for recombinant protein detection and O-glycosylation reporters. PLoS One. 2014;9 doi: 10.1371/journal.pone.0096700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li E., Pedraza A., Bestagno M., Mancardi S., Sanchez R., Burrone O. Mammalian cell expression of dimeric small immune proteins (SIP) Protein Eng. 1997;10:731–736. doi: 10.1093/protein/10.6.731. ( http://www.ncbi.nlm.nih.gov/pubmed/9278288) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material