

Abstract
Amyloid fibril formation has long been studied because of the variety of proteins that are capable of adopting this structure despite sharing little sequence homology. This makes amyloid fibrils a challenging focus for inhibition studies because the peptides and proteins that form amyloid fibrils cannot be targeted based on a sequence motif. Most peptide inhibitors that target specific amyloidogenic proteins rely heavily on sequence recognition to ensure that the inhibitory peptide is able to bind its target. This approach is limited to targeting one amyloidogenic protein at a time. However, there is increasing evidence of cross-reactivity between amyloid-forming polypeptides. It has therefore become more useful to study the similarities between these proteins that goes beyond their sequence homology. Indeed, the observation that amyloidogenic proteins adopt similar secondary structures along the pathway to fibril formation opens the way to an interesting investigation: the development of inhibitors that could be universal amyloid traps. The review below will analyze two specific amyloidogenic proteins, α-synuclein and human amylin, and introduce a small number of peptides that have been shown to be capable of inhibiting the amyloidogenesis of both of these very dissimilar polypeptides. Some of the inhibitory peptide motifs may indeed, be applicable to Aβ and other amyloidogenic systems.
Keywords: Amyloid, Synuclein, Amylin, Parkinson's disease, Type II diabetes, Inhibitors
abbreviations: Alpha-synuclein, (αS); Human amylin, (hAM); amyloid-beta, (Aβ); (−)-epigallocatechin gallate, (EGCG); tryptophan, (Trp); tyrosine, (Tyr); transthyretin, (TTR)
Graphical abstract

1. Introduction
Amyloidogenic proteins are typically proteins that tend to misfold from their native state to form a β-sheet conformation. These β-sheets undergo self-self-association that results in soluble oligomers. Further oligomerisation creates protofibrils followed by mature amyloid fibrils that are insoluble and can be viewed as plaques [[1], [2], [3]]. (Fig. 1 ) Amyloid fibrils are characterized as long, unbranched fibrils that are made up of strands with well ordered, cross-β-sheet geometry that run perpendicular to the long axis of the fibril [4,5]. Amyloid-forming proteins have been linked to over 40 different diseases including Alzheimer's disease, Parkinson's disease, Type II Diabetes, and Huntington's disease [[6], [7], [8], [9], [10], [11], [12]]. Each of these diseases are said to be caused by different proteins, all of which form amyloid deposits with similar morphology regardless of sequence.
Fig. 1.

A schematic drawing depicting the stages of amyloidogenesis as well as the ways different inhibitors can act on this process. The dashed lines indicate hydrogen bonding between monomers. A soluble oligomer phase (*) has been put forth as the most toxic form of misfolded amyloid proteins in a number of cases.
In the past, amyloid plaques have been accepted to be the cause of disease [[6], [7], [8],13,14]. However, there is mounting evidence that the most toxic form of amyloidogenic peptides occur much earlier on; when they are soluble oligomers [15,16]. This discovery is supported by the observation that the amount of amyloid plaques found in patients with amyloid diseases does not correlate with the severity of the disease [17]. This review focuses on two well-known amyloidogenic proteins; 1. Alpha-synuclein (αS), the protein implicated in the development of Parkinson's disease and 2. Human amylin (hAM), the protein implicated in the development of Type II Diabetes. We will also examine the peptide inhibitors that have been developed to reduce the toxicity of these and other polypeptides (Table 1 ) and how these inhibitors may point to a common thread that connects the misfolding pathway of these proteins.
Table 1.
List of inhibitors discussed in this review. The system that each inhibitor has been shown to act against is listed. For non-sequence homologous peptide inhibitors, peptide sequence is given where available. p = D-Proline.
| Class | Inhibitor | Target system |
|---|---|---|
| Small molecule | 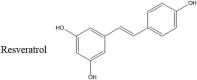 |
αS, hAM |
 |
αS | |
 |
αS, hAM | |
 |
αS | |
 |
αS, hAM | |
| hAM | ||
| Sequence homologous peptides | N-methyl hAM 22–27 (22NFGAIL27) | hAM, Aβ |
| I26P-hAM | hAM | |
| N-methyl αS 68–78 (68GAVVTGVTAVA78) | αS | |
| Non-sequence homologous peptides | AFFITOPE | αS |
| SARS corona virus fragment (TVYVYSRVK-NH2) | hAM | |
| WW2 (KKLTVW-IpGK-WITVSA) | αS, hAM | |
| Cyclo-WW2 cyclo-(K-KLTVW-IpGK-WITVS-IpP) | αS, hAM | |
| RWHCHWE (RWTTHCHRKWE)2 | αS | |
| YY-μPro1 (CH3CO-Y-IpGK-YTG-NH2) | αS | |
| Trp-zip (SWTWEpNKWTWK-NH2) | Aβ, TTR | |
| Aβ-hAM linked hot segment | Aβ, hAM |
1.1. A basic guide to alpha-synuclein
First linked to Parkinson's in 1997, alpha-synuclein (αS) is a 140 amino acid peptide that is found predominantly in neural tissue. Initially assumed to be involved in Alzheimer's disease [18], this protein was later linked to Parkinson's due to a missense mutation in an Italian family that had hereditary Parkinson's disease [10]. Unfortunately the exact function of this protein is not well understood. Several possible roles have been posited including one in dopamine homeostasis, another in the assembly of a soluble NSF attachment protein receptor and an association with synaptic vesicles that has been shown to stabilize the vesicles and inhibit neurotransmitter release [[19], [20], [21]]. When misfolded, this protein has been implicated in the death of dopamine producing cells resulting in the development of Parkinson's disease.
The alpha-synuclein sequence can be divided into three regions: the N-terminal region consisting of residues 1–60, the NAC region consisting of residues 61–99 and the C-terminal tail consisting of residues 100–140 (Fig. 2 A). In its native form, αS exists as a random coil protein that adopts an α-helix configuration when in contact with lipids. This conformational change has been replicated in vitro in the presence of membranes and membrane-like environments [[22], [23], [24]]. However, αS is also capable of misfolding and forming amyloid plaques. In 2008, Vilar et al. put forth a structure of αS in the amyloid state. This structure, derived from quenched deuterium/hydrogen exchange NMR proposes that αS adopts five distinct β-strands from residues 35 to 96 with each strand being connected by a short unstructured loop [5]. (Fig. 2B) Since then, new cryo-EM studies have suggested that αS is capable of adopting more than 5 β-strands. An investigation of the protofilament structure adopted by αS (1–121) has proposed that this truncated form of αS forms 8 distinct β-strands that are broken up by glycine residues [25]. Li et al. on the other hand have used cryo-EM to study the protofilament state of full length αS and their results indicate that αS protofilaments adopt 7 distinct β-strands [26]. A third study done by Jiang and co-workers argues that αS protofilaments are polymorphic and therefore capable of forming different structures [27]. In their study, they were able to identify two such polymorphs in large quantities: the rod and the twister polymorph formed by full-length αS.
Fig. 2.

A) The sequence of αS. The N-terminal region is shown in green, the NAC region is shown in black and the C-terminal region is shown in blue. B) Final form adopted by αS monomers in amyloid fibrils as proposed by Vialr et al. [5] More recent cryo-EM studies of the protofilament stage of αS amymloidogenesis suggest that αS forms polymorphic structures that may have more than 5 β-strands. The strands β1 and β2 of αS have been underlined in A) and highlighted in B) because of the growing interest in this region as a possible pre-amyloid secondary structure formed by αS on the path to amyloid fibril formation. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The misfolding of αS was traditionally assumed to originate from the NAC region of the protein since this region contains the minimum sequence of amino acids that are capable of undergoing amyloidogenesis [28,29]. However, new studies are showing that there may be an earlier event that occurs before the amyloidogenesis of the NAC region. In 2014, Mirecka et al. proposed that a series of residues within the N-terminal region of the peptide may be playing a nucleating role in αS amyloidogenesis [30]. This region, between residues 35 to 56 of αS (Fig. 2A), has been identified as the first two β-strands in the amyloid structure proposed by Vilar et al. It has also been demonstrated that the H50 residue that is located in this region is the first residue to display peak attenuation by solution NMR under amyloid inducing conditions, pointing to a possible initial amyloid event occurring in this region [31].
Salveson et al. showed that this hairpin region of αS was capable of forming higher order structures that display high levels of toxicity to neuronal cells, further supporting the hypothesis that the soluble oligomer conformation of amyloidogenic peptides is the more toxic [32]. The latest cryo-EM studies of the protofilament state of αS have also pinpointed this region as the interface between αS protofilament dimers [25,26]. And finally, the importance of this region within αS can be seen in the point mutations within the SNCA gene that have been linked to autosomal dominant forms of Parkinson's disease. All the mutations with the exception of one occur in this region of αS: A53T, E46K, H50Q and G51D [[33], [34], [35], [36], [37], [38], [39], [40]]. Mutations A53T, E46K and H50Q have all been shown to increase the rate of amyloidogenesis of αS in vitro, providing support that this region may be nucleating amyloidogenesis [[33], [34], [35], [36], [37], [38], [39],41].
The C-terminal region of αS is also of interest. αS has been known to be more compact than other random coil proteins of similar size and this has been attributed to some long range interactions between the C-terminal tail and residues within the NAC region and the N-terminal region [[42], [43], [44]]. These interactions may be exerting protective effects that prevent αS from aggregating since truncation of this region results in higher rates of amyloid formation [45,46]. It has also been shown that this region of αS remains unstructured in the soluble oligomer phase of amyloidogenesis [31].
1.2. Inhibitors of αS amyloidogenesis: what others have discovered
While most Parkinson's disease therapies rely on treating the symptoms of the disease rather than the cause, studies of inhibitors capable of preventing the amyloidogenesis of αS or abrogating the toxicity caused by misfolding have also been reported. Inhibitors of αS typically act in one of four ways; 1. Prevent the misfolding of the protein by stabilizing the native state. 2. Increasing the rate of amyloid formation so that the protein spends little time in the toxic soluble oligomer phase 3. Redirecting amyloidogenesis at an early stage to form non-toxic aggregates and finally 4. Promoting the clearance of toxic forms of the protein.
Most of the small molecules that have been successful inhibitors of αS and/or PD share a common motif, the presence of the phenol moiety [[47], [48], [49]]. Resveratrol, a small molecule found in the skin of grapes and other berries is a polyphenol (Table 1). This small molecule has been shown to have antioxidant properties [50] as well as an ability to confer neuroprotective effects on rats that have been induced with Parkinson's disease [51,52]. The studies done by Jin et al. show that administering resveratrol results in neuroprotection in test subjects in as little as 48 h [51]. However, the mechanism of resveratrol neuroprotection may not involve a direct interaction with αS. In vitro studies show that resveratrol has a very modest effect on αS amyloidogenesis [47]. Instead the action of resveratrol has been attributed to an activation of human sirtuin 1 (SIRT 1) that has been shown to promote the autophagy of proteins in the nucleus and cytoplasm [53,54].
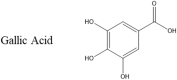
Another well studied small molecule that inhibits αS is gallic acid. In vitro studies have shown that this small molecule is capable of reducing the amount of toxic oligomers formed by αS [49]. At high doses, gallic acid reduces the formation of both toxic and non-toxic oligomers by αS, but at lower doses, gallic acid is less effective at preventing oligomerisation. However, these oligomers were determined to be non-toxic to neuronal cells. From this fact it was concluded that gallic acid redirects the amyloidogenesis of αS and forms off-pathway non-toxic aggregates. Further studies show that gallic acid may be doing this by binding to an intermediate conformation of αS and stabilizing it [49,55]. Size exclusion chromatography shows that the off-pathway oligomers formed by αS when incubated with gallic acid contain gallic acid, indicating a strong interaction between the two molecules.
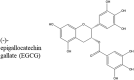
Gallic acid is also part of a derivative known as (−)-epigallocatechin gallate (EGCG) (Table 1) that is a well-known inhibitor of amyloidogenesis. Found in green tea, EGCG has been shown to inhibit other amyloidogenic proteins such as amyloid-beta (Aβ) and tau (both implicated in Alzheimer's disease) [[56], [57], [58]]. When incubated with αS, EGCG has been shown to redirect amyloid formation to off-pathway non-toxic aggregates [59]. While some have proposed that the mechanism of this protection is through the binding of EGCG to the C-terminal tail of monomeric αS [59,60], a separate study shows that this binding may not be specific to the C-terminal tail, but could in fact be occurring throughout the αS sequence [31]. There have also been studies that propose that the binding occurs with the oligomeric state of the protein and destabilize it thus offering protection from αS cytotoxicity [58].
The efficacy of polyphenols as inhibitors has been proposed to be linked to the ability of these molecules to form quinones [61,62]. The quinones have been proposed to interact with aromatic residues within αS and prevent stacking of these residues during self-self-interaction. This hypothesis is supported by the fact that most of these molecules have been shown to interact with the C-terminal tail of αS, where three of the protein's four tyrosine residues are located [63].
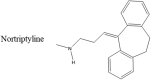
While these small molecules have shown interesting results in vitro, the main problem with these therapies remains the inability of these molecules to cross the blood-brain barrier. However, a recent study has emerged showing a known antidepressant, nortriptyline (NOR) (Table 1) as a possible inhibitor of αS [64]. In vitro studies done by Collier et al. show a dose-dependent decrease of aggregation by αS in the presence of NOR. Further studies done in A30P αS transgenic Drosophila show that the progressive retinal degeneration expressed in the absence of NOR was significantly attenuated when the flies were fed with food supplemented with NOR. The same study also included experiments done with transgenic mice. The mice were given intraperitoneal injections of NOR and αS accumulation in the cortex and hippocampus was analyzed. Analysis showed that NOR inhibited the progression of αS amyloid formation in a dose related fashion. The authors proposed that the mechanism of neuroprotection conferred by NOR arises from an interaction between NOR and the monomer αS. Their results show that in the presence of NOR, monomeric αS is more solvent accessible and therefore less prone to forming oligomers.
Apart from small molecules, peptides have also been shown to reduce the effects of αS cytotoxicity. An example of one category of peptides, known as AFFITOPES (Table 1), has been designed to mimic the structure of the soluble oligomer formed by the C-terminal sequence of αS and generate an immune response [65]. The lack of sequence similarity between the AFFITOPE and native αS prevents the development of a deadly immune response while specifically targeting toxic forms of the protein [66]. Successful mouse model studies have led to a vaccine being tested in clinical trials. The preliminary results show that the vaccine is effective in reducing the worsening of Parkinson's disease symptoms, however there has been a drop in antibody levels over time prompting the need for a booster dose [67,68].
While vaccination is an attractive approach to Parkinson's disease therapy, the most common approach in peptide inhibitor design is to inhibit the transition of monomeric αS to its oligomeric form. The most prevalent approach to design has been to identify and isolate regions of αS that are prone to aggregate and modify these regions to be more soluble and less able to self-associate [69]. One method to prevent continued self-self-association is by N-methylation of residues within the isolated region or by substitution with proline residues. This prevents self-association on both faces of the peptide by eliminating the ability to form stable hydrogen bonds thus preventing the formation of intermolecular β-sheets [[70], [71], [72]]. The sequence homology between the modified peptides and αS allows for recognition but the lack of primary nitrogen on the opposite face of the peptide prevents amyloidogenesis. An example of one such peptide is 68GAVVTGmVTAVA78 [73,74]. The addition of N-methylation at G73 prevents the aggregation of this peptide in vitro. More importantly this peptide is capable of reducing the toxicity of the full length αS [75].
1.3. A basic guide to human amylin
Human amylin was the first amyloidogenic peptide discovered in 1901 by Eugene L. Opie. [76] He attributed the degeneration of islet cells in diabetes patients to amyloid plaques. In 1986, Westermark was finally able to identify the 37 amino acid peptide that made up the fibrils in the amyloid plaques first seen by Opie. [77] It wasn't until a year later that an accurate characterization of hAM was done by Cooper et al. This study showed that hAM has a disulfide bond between the two cysteine residues at the N-terminus and has an amidated C-terminus [11]. (Fig. 3 A) Further studies showed that the peptide is natively random coil but has a helix favoring N-terminus (residues 1–19), and a middle section that is capable of independently forming aggregates and a C-terminal tail that adopts a random coil structure in solution.
Fig. 3.

A) The sequence of human amylin. B) Final form adopted by human amylin monomers in amyloid fibrils. The residues 18–27 of hAM are highlighted in blue in both A) and B) because of the conflicting observations that on the one hand studies show that this is the minimum sequence of hAM capable of forming amyloid fibrils and yet on the other there are indications that in the final amyloid configuration this region has no β-sheet character, rather, it seems to exist as an unstructured loop. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Unlike αS, the function of hAM in the body is better understood. hAM is released by the pancreas in response to the signals that trigger the release of insulin, causing both proteins to be co-secreted at a 20:1 ratio [78]. It is believed that hAM functions in tandem with insulin preventing the release of glucagon, decreasing gastric emptying and stimulating satiety centers of the brain [[78], [79], [80], [81], [82], [83]]. When hAM misfolds, it becomes toxic to the β-cells from which it originates possibly by causing channel or pore formation in the lipid bilayer. This channel then promotes the influx of calcium ions and efflux of potassium and sodium ions [84,85]. Other possible mechanisms of hAM toxicity is through the generation of reactive oxygen species [[86], [87], [88]] and the triggering of apoptosis through the promotion of the expression of genes linked to cell apoptosis such as c-Jun and the Fas-associated death domain [[89], [90], [91], [92]].
The misfolding of hAM proceeds by a similar sequence of events as other amyloidogenic proteins. However, recent studies have posited a possible helical intermediate adopted by hAM before transitioning to β-sheet conformations typically seen in the amyloidogenic pathway [93]. Various spectroscopic studies have detected an increase in the α-helical character of hAM prior to the formation of β-strands [[94], [95], [96], [97], [98], [99], [100]]. A crystal structure analysis of hAM fused to maltose-binding protein (MBP) indicated that the N-terminal helix of hAM was capable of forming strong aromatic stacking between the F15 residues of two hAM monomers [101]. Mutations that improved the helical propensity at this residue resulted in increased rates of amyloidogenesis while residues that favored β-strand formation decreased the rate of aggregation [101,102]. Further evidence of a helical intermediate can be found in the increased propensity of hAM to aggregate in the presence of lipid membranes that have been shown to promote the formation of helices by the N-terminal half of hAM [103,104].
However, the transition from helix to β-strand plays an even more important role in amyloidogenesis of hAM. Upon strongly helix-favoring solvent conditions, hAM does not show signs of amyloidogenesis [105]. This type of “overstabilization” of a hAM secondary state has been proposed to be the mechanism by which insulin prevents the aggregation of hAM in healthy individuals [101]. It has also been linked to the way that rat amylin is able to prevent the cytotoxicity of hAM, with the observation that mutants of rat amylin that form a less stable α-helix are unable to inhibit hAM at the same level as wild type rat amylin [106]. Shifting the equilibrium between the α-helix and β-sheet conformation in favor of the α-helix state has also been shown to decrease the rate of fibril formation by another amyloid forming protein: Aβ [107]. However, the presence of three prolines (A25P/S28P/S29P versus hAM) in rAM may be a more significant feature.
The study of how to maintain hAM solubility was a major area of investigation in the late 1980s and 1990s. It was shown that the use of proline at key positions of hAM was able to maintain the solubility of the protein while having minimal effect on the activity of hAM [108,109]. This discovery was very important because it allowed for the synthesis of a soluble protein known as pramlintide that could serve as an accompaniment to insulin therapies for patients with Type I and Type II Diabetes [[110], [111], [112]]. More recent studies into the therapeutic effect of pramlintide have discovered a possible use for the peptide as a potential therapy to combat obesity [113,114]. Among others, Ravussin et al. showed that patients that were administered pramlintide accompanied by a leptin-analogue, metreleptin, demonstrated significant weight loss [115].
1.4. Some well-known hAM inhibitors
hAM inhibitors have typically fallen into two categories, small molecules and peptides (Table 1). Interestingly, some of the small molecules that are known inhibitors of αS aggregation have also been shown to be effective against hAM. For example, resveratrol has been shown to inhibit hAM amyloid formation in a dose dependent manner [116]. In a study done by Jiang et al. it has been suggested that resveratrol inhibits hAM aggregation by the disruption of the aromatic stacking between hAM monomers [117]. Residues R11, H18, F15 and Y37 (Fig. 3A) in particular were found to be important since mutations of these residues to leucine decreases hAM amyloidogenicity and results in a decrease in resveratrol activity [118].
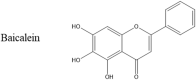
Another small molecule that inhibits both αS and hAM is baicalein (Table 1). This small molecule is a flavonoid that can be found in the roots of certain flowering plants. Studies showed that baicalein was capable of inhibiting hAM aggregation in a dose-dependent manner but that action of the molecule was dependent on the presence of at least two hydroxyl groups, one of which had to be at position 6 on the benzene ring [119]. Once again, a quinone intermediate has been proposed as part of the mechanism of action of baicalein with hAM, similar to that proposed between other phenolic molecules and αS.
Recently however, a new small molecule has been shown to inhibit hAM amyloidogenesis as well as decrease the toxicity related to hAM aggregation. Montane et al. demonstrated that 4-phenylbutyrate (PBA) (Table 1), a drug that is currently used to treat urea-cycle disorders and cystic fibrosis, is capable of restoring the glucose metabolism of transgenic mice that overexpress hAM [120]. The tests show that PBA is able to restore the function of β-cells and prevent the death of β-cells in transgenic mice. In vitro experiments further confirmed that PBA was interacting with hAM, showing a decrease in hAM amyloidogenesis in a dose-dependent manner.
hAM has also been shown to be inhibited by peptides. A common motif employed in the design of peptide inhibitors of hAM is the mutation of key residues to proline [106]. One such mutation, I26P resulted in a peptide that did not undergo aggregation and was able to increase the lag time of hAM aggregation by 20-fold when co-incubated with hAM at equimolar concentrations [121].
Just like with αS, another frequently employed technique is the isolation of amyloid-prone sequences within hAM [122,123] and the development of analogous peptides that contain N-methylated residues. In hAM, the region between residues 23 and 27 has been well studied (Fig. 3A). N-methylations of residues G24 and I26 have been shown to result in peptides that are non-aggregatory and non-toxic while being able to inhibit full length native hAM when incubated in 10 fold excess with hAM [124]. Other studies have targeted a similar region within hAM. Soluble hexapeptides derived from the 20–29 region of hAM have been found to be strong inhibitors of hAM aggregation [125,126].
It is of note that the region being focused on by these studies has been proposed by several studies to not adopt a β-sheet conformation in the final fibril state of hAM. Luca et al. proposed that in the final fibril form that hAM adopts, two β-strands that are connected by a loop consisting of residues 18 to 27 (Fig. 3B) [127]. Wiltzius et al. supported this proposal by studying crystal structures of two fragments of hAM [128]. In this study, residues 28SSTNVG33 were found to adopt a steric zipper while residues 21NNFGAIL27 were found to adopt a loop structure. Shim et al. then proposed that the formation of the two β-strands were independent of one another but that the formation of the loop between residues 16 to 27 was a nucleating event [129]. This theory is further strengthened by a study done by Sivanesam et al. that showed aged samples of 22NFGAILSS29 to be more effective inhibitors of aggregation of full length hAM [130]. One possible explanation for this observation is that this presumably β-oligomeric inhibitor “locks” the 22–29 region of hAM into a β-strand conformation, preventing it from transitioning to the required loop conformation. Preventing this transition could then interfere with the rest of the amyloidogenesis of hAM, further strengthening the hypothesis that the 22–29 region of hAM does not adopt a β-sheet conformation in the final fibril form of hAM.
1.5. Non-sequence homologous peptide inhibitors: universal inhibitors?
The use of peptide inhibitors has typically relied on sequence homologues to facilitate recognition of the inhibitor by the target peptide. There have been a few examples of peptides with no sequence similarity that are capable of interacting with amyloidogenic peptides. For example, Ghosh et al. demonstrated that a nine residue peptide taken from the SARS corona virus (Table 1) was able to inhibit hAM amyloidogenesis [131]. However, it is worth noting that the inhibitor itself was shown to be capable of aggregation albeit at a much slower rate than hAM.
The main drawback of this approach is the lack of universality of the inhibitors that are developed. Inhibitors that are derived from, or optimized for, one amyloidogenic peptide are only effective against that protein. This represents an overly specific type of interaction. However, there is a different approach that has been shown to be effective in the designing of inhibitors that bear no sequence similarity to the target peptide. This approach relies on designing very stable or pre-structured β-sheet peptides that have available edge strands capable of binding to amyloidogenic peptides. This binding is facilitated by the hydrophobic units on the edge strand of the inhibitor β-sheet that favors hydrophobic collapse.
The idea can be traced to efforts by Smith et al., in 2006 [132]. They were interested in building upon studies that had found antibody proteins that were capable of recognizing the soluble oligomers of a variety of amyloidogenic proteins [133]. Their initial experiments involved creating a phage-displayed library that would allow them to determine which randomized mutants of their template protein was capable of interacting strongly with oligomers of Aβ, the protein implicated in Alzheimer's Disease. They found that the mutant containing two tyrosine (Tyr) residue substitutions and two tryptophan (Trp) residue substitutions was the most suitable for detecting their target. Their kinetic assays showed that co-incubation of their protein with Aβ resulted in complete suppression of amyloidogenesis.
Huggins et al. expanded on this work using designed hairpin peptides containing Trp and/or Tyr residues on the exposed face of the peptide [99]. Their initial study showed that these peptides were effective at inhibiting the amyloidogenesis of hAM. They then expanded on this study by testing their most effective inhibitor against a second amyloidogenic system, αS. Their results showed that the peptide, WW2 (Table 1 & Fig. 4 ) was able to increase the lag time of hAM amyloidogenesis by 3 fold when co-incubated at molar equivalent concentrations (Fig. 4). Increments in the concentration of the inhibitor further increased the lag time of hAM aggregation. On the other hand, use of the inhibitor WW2 against αS resulted in an immediate precipitate formation upon addition of the aggregatory stimulus, hexafluoroisopropanol (HFIP). The precipitate was shown to be a non-amyloid aggregate by Congo Red staining. TEM studies showed that the precipitate had shorter thicker fibrils than those seen in amyloid precipitates. This would indicate that the peptide WW2 was slowing down the aggregation of hAM while redirecting the aggregation of αS to off-pathway aggregates. The peptide was also tested against hAM in RIN5fm cells. The results showed that at 1:2 ratios of hAM to inhibitor, there is an increase in cell viability. This increase is dose dependent and can be seen improving as the concentration of inhibitor is increased.
Fig. 4.

A schematic drawing showing the secondary structure of some peptides that show inhibition against α-synuclein. WW2 and cyclo WW2 also show potential as universal amyloid traps since they also show activity against hAM.
The study by Huggins et al. brought forth an interesting target for optimization, the peptide WW2 (Table 1 & Fig. 4). Following this, a new set of aryl rich peptides were tested, including a cyclized version of WW2 (Table 1 & Fig. 4) [31]. This was done to test the hypothesis that the stability of the inhibitor β-sheet would play a role in increasing the potency of the peptide as an inhibitor. Results showed that cyclization definitely improved the potency of WW2 from approximately 50% inhibition at one molar equivalence to approximately 75% inhibition when co-incubated with αS. In the presence of hAM, cyclo-WW2 (Table 1 & Fig. 4) showed even stronger inhibition [100]. Substoichiometric concentrations of cyclo-WW2 were able to completely inhibit the amyloidogenesis of hAM when analyzed by Thioflavin T fluorescence. The effectiveness of cyclo-WW2 as an inhibitor was also tested in cell cytotoxicity assays where 1:0.5 ratio of hAM to cyclo-WW2 was enough to eliminate the cytotoxicity of hAM against RIN5fm cells.
NMR analysis was also performed to determine where these inhibitors were binding to αS [99]. Initial experiments in the absence of any inhibitors showed that the earliest peak attenuation was occurring in the N-terminal section. Of particular interest was the fact that a lot of these peaks belonged to the hairpin-region identified by Mirecka et al. to be part of an early pre-amyloid event that is important to the amyloidogenesis of αS [30]. One of the last portions of αS to show peak attenuation was the C-terminal tail corresponding to this region of αS remaining flexible in the soluble oligomer phase of αS amyloidogenesis. It was therefore quite interesting that the main binding locus of WW2 was located in the C-terminal tail of αS. Some of the more prominent binding shifts observed included Y133 and S129 (Fig. 2A). Y133 is of note because a study done by Ulrih et al. showed that the mutation of this tyrosine to alanine resulted in the complete inhibition of αS amyloidogenesis [134]. It is therefore possible that this residue is playing an important role in amyloid formation, possibly through long range contacts with other parts of the peptide. The other notable shift is S129. S129 is the only residue within αS that has been found to undergo phosphorylation [135]. The effect of phosphorylation on amyloidogenesis is unclear. The initial discovery of S129 phosphorylation seems to indicate that this event favors amyloidogneesis [135]. However, a second study showed that phosphorylation resulted in increased flexibility of αS in this region [136]. While there is a lack of consensus on what exactly phosphorylation does, it does seem to indicate that S129 plays a significant role in the structure and function of αS.
Cyclo-WW2 (Table 1 & Fig. 4) was also analyzed by NMR when co-incubated with αS [31]. The results showed that it had a similar binding locus in the C-terminal of αS. However, a secondary binding locus was identified. Residues G41, V48, H50, V52, A53 and T54 were all observed to have shifted upon cyclo-WW2 titration. Again we note that this region corresponds to the hairpin identified by Mirecka et al. Taken together this data would indicate that this region plays an important early role in the amyloid formation of αS and should be studied in more detail.
Sivanesam et al. also showed that αS inhibition was not limited to the type of β-sheet adopted by a potential peptide inhibitor [31]. A “turnless” β-sheet made up of a dimer connected by a disulfide bond in the middle was tested as an inhibitor of αS amyloidogenesis (Fig. 4). The results showed that at equimolar concentrations the dimeric peptide RWHCHWE was capable of inhibiting αS aggregation up to about 65%. Further increase in the concentration of inhibitor resulted in a corresponding increase in amyloid inhibition.
The study also attempted to determine if the presence of phenols would impact the inhibition of these peptides [31]. A small peptide containing a pair of tryptophans, μPro1 (Table 1) was compared to its tyrosine counterpart, YY- μPro1. Kinetic assays showed that the replacement of Trp with Tyr resulted in an almost 2 fold increase in the percent inhibition. These results support previous studies that have shown αS being inhibited strongly by phenolic compounds.
2. Conclusions
The need for more universal inhibitors of amyloidogenesis is increasing as links between amyloid diseases are found [137]. Cross-amyloid interactions have been identified between amyloid-beta and a number of other proteins including αS and hAM [[138], [139], [140], [141], [142], [143], [144]]. Clinical studies show that patients suffering from Type II Diabetes are at a higher risk of developing Alzheimer's disease and vice versa [[145], [146], [147]]. It is therefore important that inhibitor design be more universal, so that future therapies are not limited to only one target protein. Some small molecules have shown the ability to interact with multiple amyloidogenic peptides and prevent their transition to the toxic oligomer phase. For example, resveratrol has been shown to inhibit both hAM and αS (vide supra) while Congo Red and curcumin have been shown to be effective against αS and the protein responsible for Alzheimer's disease Aβ [148]. EGCG has been reported to be an inhibitor of these proteins and additional systems [[58], [59], [60],149]. However, we focused on peptide inhibitors where the question of sequence similarity and binding motifs can be examined. So far, our investigations have identified a number of peptides capable of inhibiting the amyloidogenesis of both αS and hAM [31,99,100]. Of these, cyclo-WW2 (which bears a resemblance to Trp-zip peptides, vide infra) is effective when co-incubated at sub-stoichiometric concentrations with either αS or hAM. In vivo studies of this peptide with hAM show that the inhibitor is capable of ameliorating the cytotoxicity of hAM in a dose-dependent manner while displaying no sequence similarity to the amyloidogenic target.
Kapurniotu and co-workers have also demonstrated that hAM and Aβ can interact with one another and decrease the rate of amyloidogenesis of both proteins. Their study examined whether a previously tested hAM peptide inhibitor was capable of inhibiting the amyloidogenesis of Aβ as well [150]. These studies showed that when co-incubated at equimolar concentrations, the hAM inhibitor was capable of preventing PC-12 cytotoxicity in the presence of Aβ. Since the hAM inhibitor being tested was in fact an N-methyl modified mimic of hAM between residues 22 to 27, Kapurniotu and co-workers studied if full length hAM was able to interact with Aβ. Their results showed that when co-incubated, both peptides exhibited a decreased rate of amyloidogenesis. An extensive follow up investigation showed that there were 3 regions within Aβ and 2 regions within hAM that bind strongly one to another [151]. They termed these regions hot segments within both peptides. Building upon this, they investigated if an inhibitor derived by linking hot segments from hAM and Aβ was able to inhibit the amyloidogenesis of either protein [152]. They determined that the linked hot segments would be able to inhibit the amyloidogenesis of hAM and Aβ at nanomolar concentrations, making them one of the most potent peptide inihibitors of either peptide. Whether these can be extended to aS and other systems with no sequence homology has yet to be examined.
There is further evidence that certain inhibitors are capable of inhibiting multiple amyloidogenic peptides. Daggett and co-workers investigated if tryptophan-zipper peptides (trp-zip) were capable of inhibiting the amyloidogenesis of Aβ and another amyloidogenic peptide, transthyretin (TTR) [153]. Their studies showed that the stability of the β-sheet structure of the trp-zip peptide played a large role in its ability to inhibit Aβ amyloidogenesis. However, it was the presence of the exposed tryptophan residues that was important in the interaction between trp-zip and TTR since a mutant of trp-zip with all tryptophans mutated to leucine resulted in a decrease in inhibition strength. Given that cyclo-WW2 and a number of other inhibitors (Fig. 4) of αS and hAM also have a Trp-rich β-strand motif, we expect that further cross-reactivities will be found in the future. Pre-structured β-sheet peptides that have available edge strands and aryl residues capable of binding another β-strand may well represent a class of universal amyloid traps.
Unfortunately, none of the recent advances have approached the point of becoming effective therapies. The method of delivery for these peptidic inhibitors has yet to be discovered and the need for high concentrations of inhibitor is still a hurdle. But inhibitor studies have so far yielded interesting information about the amyloidogenesis process itself. By studying where inhibitors bind and how that binding affects the amyloid pathway, a lot of new information has been uncovered about amyloidogenic polypeptides. The region between residues 37–54 of αS is increasingly interesting as a possible pre-amyloid secondary structure. As for hAM, a possible loop feature formed by residues 22–29 in the final structure of the fibrils could be an important target area for further study. It is therefore important that inhibitors, in particular universal inhibitors, continue to be studied, since they might point out heretofore unknown similarities between amyloid proteins that could lead to the development of a therapy that is applicable to all amyloid diseases.
Acknowledgements
The initial studies at UW were supported by a grant from the NIH (GM059658-08S1).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.abb.2019.01.032.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Rochet J.C., Lansbury P.T. Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 2.Padrick S.B., Miranker A.D. Islet amyloid: phase partitioning and secondary nucleation are central to the mechanism of fibrillogenesis. Biochemistry. 2002;41:4694–4703. doi: 10.1021/bi0160462. [DOI] [PubMed] [Google Scholar]
- 3.Ruschak A.M., Miranker A.D. Fiber-dependent amyloid formation as catalysis of an existing reaction pathway. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12341–12346. doi: 10.1073/pnas.0703306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petkova A.T., Ishii Y., Balbach J.J., Antzutkin O.N., Leapman R.D., Delaglio F., Tycko R. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. U.S.A. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vilar M., Chou H.-T., Luhrs T., Maji S.K., Riek-Loher D., Verel R., Manning G., Stahlberg H., Riek R. The fold of alpha-synuclein fibrils. Proc. Natl. Acad. Sci. Unit. States Am. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glenner G.G., Wong C.W. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 7.Masters C.L., Simms G., Weinman N.A., Multhaup G., McDonald B.L., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. U.S.A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masters C.L., Multhaup G., Simms G., Pottgiesser J., Martins R.N., Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4:2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy M.P., LeVine H. Alzheimer's disease and the β-amyloid peptide. J Alzheimers Dis. 2010;19:311. doi: 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A., Pike B., Root H., Rubenstein J., Boyer R., Stenroos E.S., Chandrasekharappa S., Athanassiadou A., Papapetropoulos T., Johnson W.G., Lazzarini A.M., Duvoisin R.C., Di Iorio G., Golbe L.I., Nussbaum R.L. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 11.Cooper G.J., Willis A.C., Clark A., Turner R.C., Sim R.B., Reid K.B. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. U.S.A. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng Z., Diamond M.I. Huntington disease and the huntingtin protein. Prog Mol Biol Transl Sci. 2012;107:189–214. doi: 10.1016/B978-0-12-385883-2.00010-2. [DOI] [PubMed] [Google Scholar]
- 13.Luk K.C., Kehm V., Carroll J., Zhang B., O'Brien P., Trojanowski J.Q., Lee V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pieri L., Madiona K., Bousset L., Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012;102:2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karpinar D.P., Balija M.B.G., Kügler S., Opazo F., Rezaei-Ghaleh N., Wender N., Kim H.-Y., Taschenberger G., Falkenburger B.H., Heise H., Kumar A., Riedel D., Fichtner L., Voigt A., Braus G.H., Giller K., Becker S., Herzig A., Baldus M., Jäckle H., Eimer S., Schulz J.B., Griesinger C., Zweckstetter M. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 2009;28:3256–3268. doi: 10.1038/emboj.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winner B., Jappelli R., Maji S.K., Desplats P.A., Boyer L., Aigner S., Hetzer C., Loher T., Vilar M., Campioni S., Tzitzilonis C., Soragni A., Jessberger S., Mira H., Consiglio A., Pham E., Masliah E., Gage F.H., Riek R. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 2011;108:4194–4199. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DaRocha-Souto B., Scotton T.C., Coma M., Serrano-Pozo A., Hashimoto T., Serenó L., Rodríguez M., Sánchez B., Hyman B.T., Gómez-Isla T. Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J. Neuropathol. Exp. Neurol. 2011;70:360–376. doi: 10.1097/NEN.0b013e318217a118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.George J.M., Clayton D.F. In: Neurodegenerative Diseases. Fiskum G., editor. Springer US; 1996. The non-amyloid-β component of Alzheimer's disease plaque amyloid: comparative analysis suggests a normal function as a synaptic plasticizer; pp. 109–112. [Google Scholar]
- 19.Perez R.G., Waymire J.C., Lin E., Liu J.J., Guo F., Zigmond M.J. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002;22:3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemani V.M., Lu W., Berge V., Nakamura K., Onoa B., Lee M.K., Chaudhry F.A., Nicoll R.A., Edwards R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M.R., Südhof T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eliezer D., Kutluay E., Bussell R., Jr., Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 23.Georgieva E.R., Ramlall T.F., Borbat P.P., Freed J.H., Eliezer D. Membrane-bound alpha-synuclein forms an extended helix: long-distance pulsed ESR measurements using vesicles, bicelles, and rodlike micelles. J. Am. Chem. Soc. 2008;130:12856–12857. doi: 10.1021/ja804517m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jao C.C., Hegde B.G., Chen J., Haworth I.S., Langen R. Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guerrero-Ferreira R., Taylor N.M., Mona D., Ringler P., Lauer M.E., Riek R., Britschgi M., Stahlberg H. In: (Kuriyan J., Scheres S.H., Scheres S.H., Fändrich M., editors. vol. 7. 2018. (Cryo-Em Structure of Alpha-Synuclein Fibrils). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y., Zhao C., Luo F., Liu Z., Gui X., Luo Z., Zhang X., Li D., Liu C., Li X. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 2018;28:897. doi: 10.1038/s41422-018-0075-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B., Ge P., Murray K.A., Sheth P., Zhang M., Nair G., Sawaya M.R., Shin W.S., Boyer D.R., Ye S., Eisenberg D.S., Zhou Z.H., Jiang L. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018;9:3609. doi: 10.1038/s41467-018-05971-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodriguez J.A., Ivanova M.I., Sawaya M.R., Cascio D., Reyes F., Shi D., Sangwan S., Guenther E.L., Johnson L.M., Zhang M., Jiang L., Arbing M.A., Nannega B., Hattne J., Whitelegge J., Brewster A.S., Messerschmidt M., Boutet S., Sauter N.K., Gonen T., Eisenberg D. Structure of the toxic core of α-synuclein from invisible crystals. Nature. 2015;525:486–490. doi: 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giasson B.I., Murray I.V., Trojanowski J.Q., Lee V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 30.Mirecka E.A., Shaykhalishahi H., Gauhar A., Akgül Ş., Lecher J., Willbold D., Stoldt M., Hoyer W. Sequestration of a β-hairpin for control of α-synuclein aggregation. Angew. Chem. Int. Ed. 2014;53:4227–4230. doi: 10.1002/anie.201309001. [DOI] [PubMed] [Google Scholar]
- 31.Sivanesam K., Byrne A., Bisaglia M., Bubacco L., Andersen N. Binding interactions of agents that alter α-synuclein aggregation. RSC Adv. 2015;5:11577–11590. doi: 10.1039/C5RA00325C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salveson P.J., Spencer R.K., Nowick J.S. X-ray crystallographic structure of oligomers formed by a toxic β-hairpin derived from α-synuclein: trimers and higher-order oligomers. J. Am. Chem. Soc. 2016;138:4458–4467. doi: 10.1021/jacs.5b13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giasson B.I., Uryu K., Trojanowski J.Q., Lee V.M.-Y. Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- 34.Conway K.A., Harper J.D., Lansbury P.T., Jr. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 35.Conway K.A., Lee S.J., Rochet J.C., Ding T.T., Williamson R.E., Lansbury P.T. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U.S.A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J., Uversky V.N., Fink A.L. Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry. 2001;40:11604–11613. doi: 10.1021/bi010616g. [DOI] [PubMed] [Google Scholar]
- 37.Choi W., Zibaee S., Jakes R., Serpell L.C., Davletov B., Crowther R.A., Goedert M. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 2004;576:363–368. doi: 10.1016/j.febslet.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 38.Greenbaum E.A., Graves C.L., Mishizen-Eberz A.J., Lupoli M.A., Lynch D.R., Englander S.W., Axelsen P.H., Giasson B.I. The E46K mutation in alpha-synuclein increases amyloid fibril formation. J. Biol. Chem. 2005;280:7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- 39.Ghosh D., Sahay S., Ranjan P., Salot S., Mohite G.M., Singh P.K., Dwivedi S., Carvalho E., Banerjee R., Kumar A., Maji S.K. The newly discovered Parkinson's disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry. 2014;53:6419–6421. doi: 10.1021/bi5010365. [DOI] [PubMed] [Google Scholar]
- 40.Kiely A.P., Asi Y.T., Kara E., Limousin P., Ling H., Lewis P., Proukakis C., Quinn N., Lees A.J., Hardy J., Revesz T., Houlden H., Holton J.L. α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathol. 2013;125:753. doi: 10.1007/s00401-013-1096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Appel-Cresswell S., Vilarino-Guell C., Encarnacion M., Sherman H., Yu I., Shah B., Weir D., Thompson C., Szu-Tu C., Trinh J., Aasly J.O., Rajput A., Rajput A.H., Jon Stoessl A., Farrer M.J. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- 42.Dedmon M.M., Lindorff-Larsen K., Christodoulou J., Vendruscolo M., Dobson C.M. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 43.Bertoncini C.W., Jung Y.-S., Fernandez C.O., Hoyer W., Griesinger C., Jovin T.M., Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. Unit. States Am. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernadó P., Bertoncini C.W., Griesinger C., Zweckstetter M., Blackledge M. Defining long-range order and local disorder in native alpha-synuclein using residual dipolar couplings. J. Am. Chem. Soc. 2005;127:17968–17969. doi: 10.1021/ja055538p. [DOI] [PubMed] [Google Scholar]
- 45.Hoyer W., Cherny D., Subramaniam V., Jovin T.M. Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry. 2004;43:16233–16242. doi: 10.1021/bi048453u. [DOI] [PubMed] [Google Scholar]
- 46.Murray I.V.J., Giasson B.I., Quinn S.M., Koppaka V., Axelsen P.H., Ischiropoulos H., Trojanowski J.Q., Lee V.M.-Y. Role of α-synuclein carboxy-terminus on fibril formation in vitro†. Biochemistry. 2003;42:8530–8540. doi: 10.1021/bi027363r. [DOI] [PubMed] [Google Scholar]
- 47.Caruana M., Högen T., Levin J., Hillmer A., Giese A., Vassallo N. Inhibition and disaggregation of α-synuclein oligomers by natural polyphenolic compounds. FEBS (Fed. Eur. Biochem. Soc.) Lett. 2011;585:1113–1120. doi: 10.1016/j.febslet.2011.03.046. [DOI] [PubMed] [Google Scholar]
- 48.Caruana M., Neuner J., Högen T., Schmidt F., Kamp F., Scerri C., Giese A., Vassallo N. Polyphenolic compounds are novel protective agents against lipid membrane damage by α-synuclein aggregates in vitro. BBA- Biomembranes. 2012;1818:2502–2510. doi: 10.1016/j.bbamem.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 49.Ardah M.T., Paleologou K.E., Lv G., Abul Khair S.B., Kazim A.S., Minhas S.T., Al-Tel T.H., Al-Hayani A.A., Haque M.E., Eliezer D., El-Agnaf O.M.A. Structure activity relationship of phenolic acid inhibitors of α-synuclein fibril formation and toxicity. Front. Aging Neurosci. 2014;6 doi: 10.3389/fnagi.2014.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia N., Daiber A., Förstermann U., Li H. Antioxidant effects of resveratrol in the cardiovascular system. Br. J. Pharmacol. 2017;174:1633–1646. doi: 10.1111/bph.13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin F., Wu Q., Lu Y.-F., Gong Q.-H., Shi J.-S. Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson's disease in rats. Eur. J. Pharmacol. 2008;600:78–82. doi: 10.1016/j.ejphar.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 52.Blanchet J., Longpré F., Bureau G., Morissette M., DiPaolo T., Bronchti G., Martinoli M.-G. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog. Neuro Psychopharmacol. Biol. Psychiatr. 2008;32:1243–1250. doi: 10.1016/j.pnpbp.2008.03.024. [DOI] [PubMed] [Google Scholar]
- 53.Albani D., Polito L., Batelli S., De Mauro S., Fracasso C., Martelli G., Colombo L., Manzoni C., Salmona M., Caccia S., Negro A., Forloni G. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by α-synuclein or amyloid-β (1-42) peptide. J. Neurochem. 2009;110:1445–1456. doi: 10.1111/j.1471-4159.2009.06228.x. [DOI] [PubMed] [Google Scholar]
- 54.Cheung Z.H., Ip N.Y. The emerging role of autophagy in Parkinson's disease. Mol. Brain. 2009;2:29. doi: 10.1186/1756-6606-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y., Carver J.A., Calabrese A.N., Pukala T.L. Gallic acid interacts with α-synuclein to prevent the structural collapse necessary for its aggregation. Biochim. Biophys. Acta. 2014;1844:1481–1485. doi: 10.1016/j.bbapap.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 56.Wobst H.J., Sharma A., Diamond M.I., Wanker E.E., Bieschke J. The green tea polyphenol (−)-epigallocatechin gallate prevents the aggregation of tau protein into toxic oligomers at substoichiometric ratios. FEBS Lett. 2015;589:77–83. doi: 10.1016/j.febslet.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cascella M., Bimonte S., Muzio M.R., Schiavone V., Cuomo A. The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer's disease: an overview of pre-clinical studies and translational perspectives in clinical practice. Infect. Agents Canc. 2017;12 doi: 10.1186/s13027-017-0145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bieschke J., Russ J., Friedrich R.P., Ehrnhoefer D.E., Wobst H., Neugebauer K., Wanker E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ehrnhoefer D.E., Bieschke J., Boeddrich A., Herbst M., Masino L., Lurz R., Engemann S., Pastore A., Wanker E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 60.Lorenzen N., Nielsen S.B., Yoshimura Y., Vad B.S., Andersen C.B., Betzer C., Kaspersen J.D., Christiansen G., Pedersen J.S., Jensen P.H., Mulder F.A.A., Otzen D.E. How epigallocatechin gallate can inhibit α-synuclein oligomer toxicity in vitro. J. Biol. Chem. 2014 doi: 10.1074/jbc.M114.554667. jbc.M114.554667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu M., Rajamani S., Kaylor J., Han S., Zhou F., Fink A.L. The flavonoid baicalein inhibits fibrillation of alpha-synuclein and disaggregates existing fibrils. J. Biol. Chem. 2004;279:26846–26857. doi: 10.1074/jbc.M403129200. [DOI] [PubMed] [Google Scholar]
- 62.Taniguchi S., Suzuki N., Masuda M., Hisanaga S., Iwatsubo T., Goedert M., Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 63.Meng X., Munishkina L.A., Fink A.L., Uversky V.N. Molecular mechanisms underlying the flavonoid-induced inhibition of α-synuclein fibrillation. Biochemistry. 2009;48:8206–8224. doi: 10.1021/bi900506b. [DOI] [PubMed] [Google Scholar]
- 64.Collier T.J., Srivastava K.R., Justman C., Grammatopoulous T., Hutter-Paier B., Prokesch M., Havas D., Rochet J.-C., Liu F., Jock K., de Oliveira P., Stirtz G.L., Dettmer U., Sortwell C.E., Feany M.B., Lansbury P., Lapidus L., Paumier K.L. Nortriptyline inhibits aggregation and neurotoxicity of alpha-synuclein by enhancing reconfiguration of the monomeric form. Neurobiol. Dis. 2017;106:191–204. doi: 10.1016/j.nbd.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneeberger A., Mandler M., Mattner F., Schmidt W. AFFITOME® technology in neurodegenerative diseases: the doubling advantage. Hum. Vaccine. 2010;6:948–952. doi: 10.4161/hv.6.11.13217. [DOI] [PubMed] [Google Scholar]
- 66.Mandler M., Valera E., Rockenstein E., Weninger H., Patrick C., Adame A., Santic R., Meindl S., Vigl B., Smrzka O., Schneeberger A., Mattner F., Masliah E. Next-generation active immunization approach for synucleinopathies: implications for Parkinson's disease clinical trials. Acta Neuropathol. 2014;127:861–879. doi: 10.1007/s00401-014-1256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuhl M.M. 2014, July 31. Foxfeed Blog. Parkinson's Vaccine Safe In Phase I Trial. [Google Scholar]
- 68.Kuhl M.M. 2016, September 7. ) Foxfeed Blog. Vaccine For Parkinson's Reports Positive Results From Boost Study. [Google Scholar]
- 69.El-Agnaf O.M.A., Paleologou K.E., Greer B., Abogrein A.M., King J.E., Salem S.A., Fullwood N.J., Benson F.E., Hewitt R., Ford K.J., Martin F.L., Harriott P., Cookson M.R., Allsop D. A strategy for designing inhibitors of alpha-synuclein aggregation and toxicity as a novel treatment for Parkinson's disease and related disorders. FASEB J. 2004;18:1315–1317. doi: 10.1096/fj.03-1346fje. [DOI] [PubMed] [Google Scholar]
- 70.Gordon D.J., Tappe R., Meredith S.C. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits Abeta1-40 fibrillogenesis. J. Pept. Res. 2002;60:37–55. doi: 10.1034/j.1399-3011.2002.11002.x. [DOI] [PubMed] [Google Scholar]
- 71.Adessi C., Frossard M.-J., Boissard C., Fraga S., Bieler S., Ruckle T., Vilbois F., Robinson S.M., Mutter M., Banks W.A., Soto C. Pharmacological profiles of peptide drug candidates for the treatment of Alzheimer's disease. J. Biol. Chem. 2003;278:13905–13911. doi: 10.1074/jbc.M211976200. [DOI] [PubMed] [Google Scholar]
- 72.Kokkoni N., Stott K., Amijee H., Mason J.M., Doig A.J. N-Methylated peptide inhibitors of beta-amyloid aggregation and toxicity. Optimization of the inhibitor structure. Biochemistry. 2006;45:9906–9918. doi: 10.1021/bi060837s. [DOI] [PubMed] [Google Scholar]
- 73.Bodles A.M., Guthrie D.J.S., Harriott P., Campbell P., Irvine G.B. Toxicity of non-Aβ component of Alzheimer's disease amyloid, and N-terminal fragments thereof, correlates to formation of β-sheet structure and fibrils. Eur. J. Biochem. 2000;267:2186–2194. doi: 10.1046/j.1432-1327.2000.01219.x. [DOI] [PubMed] [Google Scholar]
- 74.Bodles A.M., Guthrie D.J.S., Greer B., Irvine G.B. Identification of the region of non-Aβ component (NAC) of Alzheimer's disease amyloid responsible for its aggregation and toxicity. J. Neurochem. 2001;78:384–395. doi: 10.1046/j.1471-4159.2001.00408.x. [DOI] [PubMed] [Google Scholar]
- 75.Bodles A.M., El-Agnaf O.M.A., Greer B., Guthrie D.J.S., Irvine G.B. Inhibition of fibril formation and toxicity of a fragment of alpha-synuclein by an N-methylated peptide analogue. Neurosci. Lett. 2004;359:89–93. doi: 10.1016/j.neulet.2003.12.077. [DOI] [PubMed] [Google Scholar]
- 76.Opie E.L. The relation of diabetes mellitus to lesions of the pancreas. Hyaline degeneration of the islands of langerhahns. J. Exp. Med. 1901;5:527–540. doi: 10.1084/jem.5.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Westermark P., Wernstedt C., Wilander E., Sletten K. A novel peptide in the calcitonin gene related peptide family as an amyloid fibril protein in the endocrine pancreas. Biochem. Biophys. Res. Commun. 1986;140:827–831. doi: 10.1016/0006-291x(86)90708-4. [DOI] [PubMed] [Google Scholar]
- 78.Martin C. The physiology of amylin and insulin: maintaining the balance between glucose secretion and glucose uptake. Diabetes Educat. 2006;32(Suppl 3):101S–104S. doi: 10.1177/0145721706288S237. [DOI] [PubMed] [Google Scholar]
- 79.Cooper G.J. Amylin compared with calcitonin gene-related peptide: structure, biology, and relevance to metabolic disease. Endocr. Rev. 1994;15:163–201. doi: 10.1210/edrv-15-2-163. [DOI] [PubMed] [Google Scholar]
- 80.Scherbaum W.A. The role of amylin in the physiology of glycemic control. Exp. Clin. Endocrinol. Diabetes. 1998;106:97–102. doi: 10.1055/s-0029-1211958. [DOI] [PubMed] [Google Scholar]
- 81.Kruger D.F., Gatcomb P.M., Owen S.K. Clinical implications of amylin and amylin deficiency. Diabetes Educat. 1999;25:389–397. doi: 10.1177/014572179902500310. quiz 398. [DOI] [PubMed] [Google Scholar]
- 82.Silvestre R.A., Rodríguez-Gallardo J., Jodka C., Parkes D.G., Pittner R.A., Young A.A., Marco J. Selective amylin inhibition of the glucagon response to arginine is extrinsic to the pancreas. Am. J. Physiol. Endocrinol. Metab. 2001;280:E443–E449. doi: 10.1152/ajpendo.2001.280.3.E443. [DOI] [PubMed] [Google Scholar]
- 83.Akesson B., Panagiotidis G., Westermark P., Lundquist I. Islet amyloid polypeptide inhibits glucagon release and exerts a dual action on insulin release from isolated islets. Regul. Pept. 2003;111:55–60. doi: 10.1016/s0167-0115(02)00252-5. [DOI] [PubMed] [Google Scholar]
- 84.Mirzabekov T.A., Lin M., Kagan B.L. pore formation by the cytotoxic islet amyloid peptide amylin. J. Biol. Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 85.Kayed R., Sokolov Y., Edmonds B., McIntire T.M., Milton S.C., Hall J.E., Glabe C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 86.Mattson M.P., Goodman Y. Different amyloidogenic peptides share a similar mechanism of neurotoxicity involving reactive oxygen species and calcium. Brain Res. 1995;676:219–224. doi: 10.1016/0006-8993(95)00148-j. [DOI] [PubMed] [Google Scholar]
- 87.Schubert D., Behl C., Lesley R., Brack A., Dargusch R., Sagara Y., Kimura H. Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. U. S. A. 1995;92:1989–1993. doi: 10.1073/pnas.92.6.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tomiyama T., Kaneko H., Kataoka K.i, Asano S., Endo N. Rifampicin inhibits the toxicity of pre-aggregated amyloid peptides by binding to peptide fibrils and preventing amyloid-cell interaction. Biochem. J. 1997;322(Pt 3):859–865. doi: 10.1042/bj3220859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lorenzo A., Razzaboni B., Weir G.C., Yankner B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 90.Zhang S., Liu J., Saafi E.L., Cooper G.J. Induction of apoptosis by human amylin in RINm5F islet beta-cells is associated with enhanced expression of p53 and p21WAF1/CIP1. FEBS Lett. 1999;455:315–320. doi: 10.1016/s0014-5793(99)00894-7. [DOI] [PubMed] [Google Scholar]
- 91.Zhang S., Liu J., MacGibbon G., Dragunow M., Cooper G.J.S. Increased expression and activation of c-Jun contributes to human amylin-induced apoptosis in pancreatic islet beta-cells. J. Mol. Biol. 2002;324:271–285. doi: 10.1016/s0022-2836(02)01044-6. [DOI] [PubMed] [Google Scholar]
- 92.Zhang S., Liu H., Yu H., Cooper G.J.S. Fas-associated death receptor signaling evoked by human amylin in islet beta-cells. Diabetes. 2008;57:348–356. doi: 10.2337/db07-0849. [DOI] [PubMed] [Google Scholar]
- 93.Williamson J.A., Miranker A.D. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kayed R., Bernhagen J., Greenfield N., Sweimeh K., Brunner H., Voelter W., Kapurniotu A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J. Mol. Biol. 1999;287:781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 95.Goldsbury C., Goldie K., Pellaud J., Seelig J., Frey P., Müller S.A., Kistler J., Cooper G.J., Aebi U. Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol. 2000;130:352–362. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 96.Lopes D.H.J., Meister A., Gohlke A., Hauser A., Blume A., Winter R. Mechanism of islet amyloid polypeptide fibrillation at lipid interfaces studied by infrared reflection absorption spectroscopy. Biophys. J. 2007;93:3132–3141. doi: 10.1529/biophysj.107.110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yonemoto I.T., Kroon G.J.A., Dyson H.J., Balch W.E., Kelly J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47:9900–9910. doi: 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ling Y.L., Strasfeld D.B., Shim S.-H., Raleigh D.P., Zanni M.T. Two-dimensional infrared spectroscopy provides evidence of an intermediate in the membrane-catalyzed assembly of diabetic amyloid. J. Phys. Chem. B. 2009;113:2498–2505. doi: 10.1021/jp810261x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huggins K.N.L., Bisaglia M., Bubacco L., Tatarek-Nossol M., Kapurniotu A., Andersen N.H. Designed hairpin peptides interfere with amyloidogenesis pathways: fibril formation and cytotoxicity inhibition, interception of the preamyloid state. Biochemistry. 2011;50:8202–8212. doi: 10.1021/bi200760h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sivanesam K., Shu I., Huggins K.N.L., Tatarek-Nossol M., Kapurniotu A., Andersen N.H. Peptide Inhibitors of the amyloidogenesis of IAPP: verification of the hairpin-binding geometry hypothesis. FEBS Lett. 2016;590:2575–2583. doi: 10.1002/1873-3468.12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wiltzius J.J.W., Sievers S.A., Sawaya M.R., Eisenberg D. Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 2009;18:1521–1530. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tu L.-H., Raleigh D.P. Role of aromatic interactions in amyloid formation by islet amyloid polypeptide. Biochemistry. 2013;52:333–342. doi: 10.1021/bi3014278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Evers F., Jeworrek C., Tiemeyer S., Weise K., Sellin D., Paulus M., Struth B., Tolan M., Winter R. Elucidating the mechanism of lipid membrane-induced IAPP fibrillogenesis and its inhibition by the red wine compound resveratrol: a synchrotron X-ray reflectivity study. J. Am. Chem. Soc. 2009;131:9516–9521. doi: 10.1021/ja8097417. [DOI] [PubMed] [Google Scholar]
- 104.Zhang X., Clair St, R J., London E., Raleigh D.P. Islet amyloid polypeptide membrane interactions: effects of membrane composition. Biochemistry. 2017;56:376–390. doi: 10.1021/acs.biochem.6b01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Higham C.E., Jaikaran E.T., Fraser P.E., Gross M., Clark A. Preparation of synthetic human islet amyloid polypeptide (IAPP) in a stable conformation to enable study of conversion to amyloid-like fibrils. FEBS Lett. 2000;470:55–60. doi: 10.1016/s0014-5793(00)01287-4. [DOI] [PubMed] [Google Scholar]
- 106.Cao P., Meng F., Abedini A., Raleigh D.P. The ability of rodent islet amyloid polypeptide to inhibit amyloid formation by human islet amyloid polypeptide has important implications for the mechanism of amyloid formation and the design of inhibitors. Biochemistry. 2010;49:872–881. doi: 10.1021/bi901751b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Soto C., Castaño E.M., Frangione B., Inestrosa N.C. The alpha-helical to beta-strand transition in the amino-terminal fragment of the amyloid beta-peptide modulates amyloid formation. J. Biol. Chem. 1995;270:3063–3067. doi: 10.1074/jbc.270.7.3063. [DOI] [PubMed] [Google Scholar]
- 108.Cooper G.J., Leighton B., Dimitriadis G.D., Parry-Billings M., Kowalchuk J.M., Howland K., Rothbard J.B., Willis A.C., Reid K.B. Amylin found in amyloid deposits in human type 2 diabetes mellitus may be a hormone that regulates glycogen metabolism in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 1988;85:7763–7766. doi: 10.1073/pnas.85.20.7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chantry A., Leighton B., Day A.J. Cross-reactivity of amylin with calcitonin-gene-related peptide binding sites in rat liver and skeletal muscle membranes. Biochem. J. 1991;277(Pt 1):139–143. doi: 10.1042/bj2770139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kolterman O.G. Amylin and glycaemic regulation: a possible role for the human amylin analogue pramlintide. Diabet. Med. 1997;14(Suppl 2):S35–S38. doi: 10.1002/(sici)1096-9136(199706)14:2+<s35::aid-dia402>3.3.co;2-#. [DOI] [PubMed] [Google Scholar]
- 111.Kolterman O.G., Gottlieb A., Moyses C., Colburn W. Reduction of postprandial hyperglycemia in subjects with IDDM by intravenous infusion of AC137, a human amylin analogue. Diabetes Care. 1995;18:1179–1182. doi: 10.2337/diacare.18.8.1179. [DOI] [PubMed] [Google Scholar]
- 112.Thompson R.G., Gottlieb A., Organ K., Koda J., Kisicki J., Kolterman O.G. Pramlintide: a human amylin analogue reduced postprandial plasma glucose, insulin, and C-peptide concentrations in patients with type 2 diabetes. Diabet. Med. 1997;14:547–555. doi: 10.1002/(SICI)1096-9136(199707)14:7<547::AID-DIA390>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 113.Rössger K., Charpin-El-Hamri G., Fussenegger M. A closed-loop synthetic gene circuit for the treatment of diet-induced obesity in mice. Nat. Commun. 2013;4:2825. doi: 10.1038/ncomms3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hoogwerf B.J., Doshi K.B., Diab D. Pramlintide, the synthetic analogue of amylin: physiology, pathophysiology, and effects on glycemic control, body weight, and selected biomarkers of vascular risk. Vasc. Health Risk Manag. 2008;4:355–362. doi: 10.2147/vhrm.s1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ravussin E., Smith S.R., Mitchell J.A., Shringarpure R., Shan K., Maier H., Koda J.E., Weyer C. Enhanced weight loss with pramlintide/metreleptin: an integrated neurohormonal approach to obesity pharmacotherapy. Obesity. 2009;17:1736–1743. doi: 10.1038/oby.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mishra R., Sellin D., Radovan D., Gohlke A., Winter R. Inhibiting islet amyloid polypeptide fibril formation by the red wine compound resveratrol. Chembiochem. 2009;10:445–449. doi: 10.1002/cbic.200800762. [DOI] [PubMed] [Google Scholar]
- 117.Jiang P., Li W., Shea J.-E., Mu Y. Resveratrol inhibits the formation of multiple-layered β-sheet oligomers of the human islet amyloid polypeptide segment 22-27. Biophys. J. 2011;100:1550–1558. doi: 10.1016/j.bpj.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tu L.-H., Young L.M., Wong A.G., Ashcroft A.E., Radford S.E., Raleigh D.P. Mutational analysis of the ability of resveratrol to inhibit amyloid formation by islet amyloid polypeptide: critical evaluation of the importance of aromatic-inhibitor and histidine-inhibitor interactions. Biochemistry. 2015;54:666–676. doi: 10.1021/bi501016r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Velander P., Wu L., Ray W.K., Helm R.F., Xu B. Amylin amyloid inhibition by flavonoid baicalein: key roles of its vicinal dihydroxyl groups of the catechol moiety. Biochemistry. 2016;55:4255–4258. doi: 10.1021/acs.biochem.6b00578. [DOI] [PubMed] [Google Scholar]
- 120.Montane J., de Pablo S., Castaño C., Rodríguez-Comas J., Cadavez L., Obach M., Visa M., Alcarraz-Vizán G., Sanchez-Martinez M., Nonell-Canals A., Parrizas M., Servitja J.-M., Novials A. Amyloid-induced β-cell dysfunction and islet inflammation are ameliorated by 4-phenylbutyrate (PBA) treatment. FASEB J. 2017;31:5296–5306. doi: 10.1096/fj.201700236R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Abedini A., Meng F., Raleigh D.P. A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrillization inhibitor. J. Am. Chem. Soc. 2007;129:11300–11301. doi: 10.1021/ja072157y. [DOI] [PubMed] [Google Scholar]
- 122.Tenidis K., Waldner M., Bernhagen J., Fischle W., Bergmann M., Weber M., Merkle M.-L., Voelter W., Brunner H., Kapurniotu A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties 1. J. Mol. Biol. 2000;295:1055–1071. doi: 10.1006/jmbi.1999.3422. [DOI] [PubMed] [Google Scholar]
- 123.Mazor Y., Gilead S., Benhar I., Gazit E. Identification and characterization of a novel molecular-recognition and self-assembly domain within the islet amyloid polypeptide. J. Mol. Biol. 2002;322:1013–1024. doi: 10.1016/s0022-2836(02)00887-2. [DOI] [PubMed] [Google Scholar]
- 124.Kapurniotu A., Schmauder A., Tenidis K. Structure-based design and study of non-amyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxicity 1. J. Mol. Biol. 2002;315:339–350. doi: 10.1006/jmbi.2001.5244. [DOI] [PubMed] [Google Scholar]
- 125.Tatarek-Nossol M., Yan L.-M., Schmauder A., Tenidis K., Westermark G., Kapurniotu A. Inhibition of hIAPP amyloid-fibril formation and apoptotic cell death by a designed hIAPP amyloid- core-containing hexapeptide. Chem. Biol. 2005;12:797–809. doi: 10.1016/j.chembiol.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 126.Yan L.-M., Tatarek-Nossol M., Velkova A., Kazantzis A., Kapurniotu A. Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2046–2051. doi: 10.1073/pnas.0507471103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Luca S., Yau W.-M., Leapman R., Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wiltzius J.J.W., Sievers S.A., Sawaya M.R., Cascio D., Popov D., Riekel C., Eisenberg D. Atomic structure of the cross-beta spine of islet amyloid polypeptide (amylin) Protein Sci. 2008;17:1467–1474. doi: 10.1110/ps.036509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shim S.-H., Gupta R., Ling Y.L., Strasfeld D.B., Raleigh D.P., Zanni M.T. Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6614–6619. doi: 10.1073/pnas.0805957106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sivanesam K., Andersen N.H. Inhibition of human amylin amyloidogenesis by human amylin-fragment peptides: exploring the effects of serine residues and oligomerization upon inhibitory potency. Biochemistry. 2017;56:5373–5379. doi: 10.1021/acs.biochem.7b00739. [DOI] [PubMed] [Google Scholar]
- 131.Ghosh A., Pithadia A.S., Bhat J., Bera S., Midya A., Fierke C.A., Ramamoorthy A., Bhunia A. Self-assembly of a nine-residue amyloid-forming peptide fragment of SARS corona virus E-protein: mechanism of self aggregation and amyloid-inhibition of hIAPP. Biochemistry. 2015;54:2249–2261. doi: 10.1021/acs.biochem.5b00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Smith T.J., Stains C.I., Meyer S.C., Ghosh I. Inhibition of β-amyloid fibrillization by directed evolution of a β-sheet presenting miniature protein. J. Am. Chem. Soc. 2006;128:14456–14457. doi: 10.1021/ja065557e. [DOI] [PubMed] [Google Scholar]
- 133.Kayed R., Head E., Thompson J.L., McIntire T.M., Milton S.C., Cotman C.W., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 134.Ulrih N.P., Barry C.H., Fink A.L. Impact of Tyr to Ala mutations on alpha-synuclein fibrillation and structural properties. Biochim. Biophys. Acta. 2008;1782:581–585. doi: 10.1016/j.bbadis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 135.Fujiwara H., Hasegawa M., Dohmae N., Kawashima A., Masliah E., Goldberg M.S., Shen J., Takio K., Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 136.Paleologou K.E., Schmid A.W., Rospigliosi C.C., Kim H.-Y., Lamberto G.R., Fredenburg R.A., Lansbury P.T., Fernandez C.O., Eliezer D., Zweckstetter M., Lashuel H.A. Phosphorylation at ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of α-synuclein. J. Biol. Chem. 2008;283:16895–16905. doi: 10.1074/jbc.M800747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Giasson B.I., Lee V.M.-Y., Trojanowski J.Q. Interactions of amyloidogenic proteins. NeuroMolecular Med. 2003;4:49–58. doi: 10.1385/NMM:4:1-2:49. [DOI] [PubMed] [Google Scholar]
- 138.Luo J., Wärmländer S.K.T.S., Gräslund A., Abrahams J.P. Cross-interactions between the alzheimer disease amyloid-β peptide and other amyloid proteins: a further aspect of the amyloid cascade hypothesis. J. Biol. Chem. 2016;291:16485–16493. doi: 10.1074/jbc.R116.714576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Seeliger J., Evers F., Jeworrek C., Kapoor S., Weise K., Andreetto E., Tolan M., Kapurniotu A., Winter R. Cross-amyloid interaction of Aβ and IAPP at lipid membranes. Angew. Chem., Int. Ed. Engl. 2012;51:679–683. doi: 10.1002/anie.201105877. [DOI] [PubMed] [Google Scholar]
- 140.Moreno-Gonzalez I., Edwards G., Iii, Salvadores N., Shahnawaz M., Diaz-Espinoza R., Soto C. Molecular interaction between type 2 diabetes and Alzheimer's disease through cross-seeding of protein misfolding. Mol. Psychiatr. 2017;22:1327–1334. doi: 10.1038/mp.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Clinton L.K., Blurton-Jones M., Myczek K., Trojanowski J.Q., LaFerla F.M. Synergistic interactions between Aβ, tau, and α-synuclein: acceleration of neuropathology and cognitive decline. J. Neurosci. 2010;30:7281–7289. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rezaei-Ghaleh N., Andreetto E., Yan L.-M., Kapurniotu A., Zweckstetter M. Interaction between Amyloid Beta Peptide and an Aggregation Blocker Peptide Mimicking Islet Amyloid Polypeptide. PLoS One. 2011;6 doi: 10.1371/journal.pone.0020289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ono K., Takahashi R., Ikeda T., Yamada M. Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 2012;122:883–890. doi: 10.1111/j.1471-4159.2012.07847.x. [DOI] [PubMed] [Google Scholar]
- 144.Jose J.C., Chatterjee P., Sengupta N. Cross dimerization of amyloid-β and αSynuclein proteins in aqueous environment: a molecular dynamics simulations study. PLoS One. 2014;9 doi: 10.1371/journal.pone.0106883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li X., Song D., Leng S.X. Link between type 2 diabetes and Alzheimer's disease: from epidemiology to mechanism and treatment. Clin. Interv. Aging. 2015;10:549–560. doi: 10.2147/CIA.S74042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sridhar G.R., Lakshmi G., Nagamani G. Emerging links between type 2 diabetes and Alzheimer's disease. World J. Diabetes. 2015;6:744–751. doi: 10.4239/wjd.v6.i5.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Janson J., Laedtke T., Parisi J.E., O'Brien P., Petersen R.C., Butler P.C. Increased risk of type 2 diabetes in alzheimer disease. Diabetes. 2004;53:474–481. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 148.Nie Q., Du X., Geng M. Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer's disease. Acta Pharmacol. Sin. 2011;32:545–551. doi: 10.1038/aps.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Meng F., Abedini A., Plesner A., Verchere C.B., Raleigh D.P. The flavanol (−)-Epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry. 2010;49:8127–8133. doi: 10.1021/bi100939a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yan L.-M., Velkova A., Tatarek-Nossol M., Andreetto E., Kapurniotu A. IAPP mimic blocks Abeta cytotoxic self-assembly: cross-suppression of amyloid toxicity of Abeta and IAPP suggests a molecular link between Alzheimer's disease and type II diabetes. Angew. Chem., Int. Ed. Engl. 2007;46:1246–1252. doi: 10.1002/anie.200604056. [DOI] [PubMed] [Google Scholar]
- 151.Andreetto E., Yan L.-M., Tatarek-Nossol M., Velkova A., Frank R., Kapurniotu A. Identification of hot regions of the Abeta-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angew. Chem., Int. Ed. Engl. 2010;49:3081–3085. doi: 10.1002/anie.200904902. [DOI] [PubMed] [Google Scholar]
- 152.Andreetto E., Malideli E., Yan L.-M., Kracklauer M., Farbiarz K., Tatarek-Nossol M., Rammes G., Prade E., Neumüller T., Caporale A., Spanopoulou A., Bakou M., Reif B., Kapurniotu A. A hot-segment-based approach for the design of cross-amyloid interaction Surface mimics as inhibitors of amyloid self-assembly. Angew. Chem. Int. Ed. 2015;54:13095–13100. doi: 10.1002/anie.201504973. [DOI] [PubMed] [Google Scholar]
- 153.Hopping G., Kellock J., Caughey B., Daggett V. Designed trpzip-3 β-hairpin inhibits amyloid formation in two different amyloid systems. ACS Med. Chem. Lett. 2013;4:824–828. doi: 10.1021/ml300478w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.