

Abstract
The cryptophycins are a family of macrocyclic depsipeptide natural products that display exceptionally potent anti-proliferative activity against drug-resistant cancers. Unique challenges facing the synthesis and derivatization of this complex group of molecules motivated us to investigate a chemoenzymatic synthesis designed to access new analogs for biological evaluation. The cryptophycin thioesterase (CrpTE) and the cryptophycin epoxidase (CrpE) are a versatile set of enzymes that catalyze macrocyclization and epoxidation of over twenty natural cryptophycin metabolites. Thus, we envisioned a drug development strategy involving their use as standalone biocatalysts for production of unnatural derivatives. Herein, we developed a scalable synthesis of 12 new unit A-B-C-D linear chain elongation intermediates containing heterocyclic aromatic groups as alternatives to the native unit A benzyl group. N-acetyl cysteamine activated forms of each intermediate were assessed for conversion to macrocyclic products using wild type CrpTE, which demonstrated the exceptional flexibility of this enzyme. Semi-preparative scale reactions were conducted for isolation and structural characterization of new cryptophycins. Each was then evaluated as a substrate for CrpE P450 and its ability to generate the epoxidized products from these substrates that possess altered electronics at the unit A styrenyl double bond position. Finally, biological evaluation of the new cryptophycins revealed a des-β-epoxy analog with low picomolar potency, previously limited to cryptophycins bearing epoxide functionality.
Polyketide synthases (PKSs), non-ribosomal peptide synthetases (NRPSs), and their hybrids (PKS/NRPS) are modular proteins that generate a vast array of complex natural products. These proteins select a group of relatively simple chemical building blocks (malonate, malonate derivatives, amino acids, and/or amino acid derivatives) and construct diverse scaffolds with a wide range of biological activities.1, 2 A large class of these natural products, macrocycles,3 contain a constrained ring structure which locks them into their biologically active conformation and protects the constituent peptide bonds from degradation.4 The biosynthesis of these important molecules is typically terminated by a thioesterase (TE) domain, an α,β-hydro-lase that utilizes a serine, histidine, aspartic acid catalytic triad to affect regio- and stereospecific cyclization.4, 5 Finally, these macrocyclic structures are often further tailored by cytochromes P450 to produce the mature, biologically active natural product.
An interesting family of macrocycles, the cryptophycins are a class of 16-membered ring depsipeptides natural products generated by a mixed PKS/NRPS biosynthetic system.6, 7 These molecules were first identified in Nostoc sp. ATCC 53789 as antifungals,8 and were subsequently rediscovered in Nostoc sp. GSV 224 as one of the most potent anti-proliferative, microtubule binding agents.9 An initial medicinal chemistry screening effort indicated that the β-epoxide functionality was necessary for maximal activity and that most modifications were detrimental to potency. A synthetic analog, cryptophycin-52 (Figure 1) that contains a geminal dimethyl functionality in unit C entered clinical trials for both the treatment of platinum resistant ovarian cancer and non-small cell lung cancer.10–12 Despite showing significant disease stabilization and an overall positive result, the trials were discontinued due to dose limiting peripheral neuropathy and a lack of broad in vivo efficacy.10, 13 Although the cryptophycin-52 clinical trial was terminated, this class of metabolites continues to represent a compelling scaffold for further lead optimization as it is particularly effective in difficult-to-treat, drug-resistant cancers.14 Recently, the cryptophycins have been investigated extensively as potential payloads for antibody drug conjugates (as well as other direct targeting agents including conjugation with RGD peptides15 and folic acid16), as this could circumvent the systemic toxicity observed with these compounds.17–21 Although there is still significant interest in cryptophycins as anticancer agents, challenges in lead optimization as well as costly synthetic efforts for their production have stymied exploration.
Figure 1.

Select cryptophycin molecules.
The penultimate step in cryptophycin biosynthesis is the stereospecific macrocyclization of the chain elongation intermediate via the cryptophycin thioesterase (CrpTE).6 Finally, the styrene moiety of the cryptophycin macrocycle is further tailored by an epoxidizing cytochrome P450 (CrpE) to produce the main isolate of this class cryptophycin 1 (Figure 1). In a previous study, we demonstrated that excised CrpTE is able to catalyze facile cyclization to varying degrees of native and modified substrates to generate cryptophycin 3, 51 (Figure 1) and an unnatural cryptophycin containing a terminal olefin in unit A, originally synthesized as LY404291 at Eli Lilly.22 Moreover, the CrpTE was employed in conjunction with the late stage, P450 epoxidase (CrpE) to provide both native and non-native structures.7, 23 These initial studies demonstrated the unique, inherent flexibility of both CrpTE and CrpE, and their potential as a versatile biocatalysts for the production of novel cryptophycin analogs of medicinal importance. Synthetically, the two most difficult steps in the formation of complex natural products are regio- and stereo- specific macrocyclization and epoxidation. Despite excellent methodology for macrocyclization via cross metathesis, biocatalytic head to tail cyclization would be an excellent addition to the synthetic toolbox as chemically, this method is often complicated by low yields. In the cryptophycins, the most difficult synthetic step is epoxidation as current methodologies display inefficient stereoselectivity towards the β isomer for this late stage transformation, demonstrating the need for alternative methodologies, including biocatalytic ones that can efficiently complete this regio- and stereo-specific process. Thus, we embarked on an effort to generate a series of novel cryptophycin chain elongation intermediates designed to probe substrate tolerance for both CrpTE and CrpE, and to produce high value analogs that may address some of the limitations identified previously (dose limiting peripheral neuropathy and broad in vivo efficacy).10, 13 Herein, we report the synthesis of a series of heterocyclic cryptophycin A subunits, elaboration of synthetic unit A-B-C-D N-acetyl cysteamine (NAc) chain elongation intermediates, analysis of CrpTE’s ability to catalyze macrolactonization, semi-preparative scale-up, and biological evaluation of the corresponding macrocyclic products. The cryptophycin analogs were then assessed as substrates for the CrpE P450, giving us valuable insight into the chemical and biochemical features that likely govern its catalytic competency. In this process, we identified a new styrenyl cryptophycin derivative that possesses low picomolar activity against human colorectal carcinoma (HCT-116), previously observed only in β-epoxy cryptophycins.
RESULTS AND DISCUSSION
Based on the progress made from our previous work,22, 23 we initially sought to continue interrogating the selectivity of the CrpTE towards unnatural substrates with the goal of expanding to late stage oxidation via the CrpE upon successfully generated macrolactones. Pioneering work on PKS and NRPS TEs comprised of both structural studies and in vitro biochemical analysis have yielded important insights into the complex catalytic mechanisms that mediate cyclization.24–31 PKS TEs have recently been implicated as one of the major bottlenecks in formation of unnatural polyketide macrocycles.32 However, these results contrast with earlier studies on NRPS TEs that identified inherent substrate pre-organization as a major driving force in cyclization of certain peptides.28, 33, 34 Despite the identification of different catalytic mechanisms employed for cyclization between different TE subtypes, little is known about the factors employed by mixed PKS/NRPS systems, underscoring the importance of continued investigations into this enzyme family. The unusual aryl starter unit of cryptophycins served as an intriguing site of exploration for the CrpTE and CrpE enzymes as it is exocyclic to the macrocycle yet conjugated to the double bond that is epoxidized by CrpE. The unit A terminus also represents an underexplored site for cryptophycin structure-activity relationship studies (SAR). Previously, alkylated, halogenated, and phenolic moieties of unit A have been explored with little emphasis placed on heterocyclic aryl groups.35–38 Moreover, it represents an attractive position for use of these as tactical bioisosteres that could increase water solubility, leading to better in vivo efficacy as well as potentially decreased toxicity.11, 13
Synthesis of Seco Cryptophycin Chain Elongation Intermediates.
In order to test the substrate scope of the CrpTE as well as produce novel cryptophycin macrolactones, a scalable synthesis of the N-Acetylcysteamine-activated (NAc) linear chain elongation intermediate was developed that was amenable to late-stage diversification of the unit A aryl group. Analogs were synthesized using two key intermediates, including unit AB (Scheme 1A, 8) and unit CD-NAc (Scheme 1B, 14a or b). Formulation of units AB took advantage of chiral auxiliary chemistry as well as a Suzuki coupling strategy reported previously.39 A final Horner Wadsworth Emmons olefination (HWE) was employed to form the key junction between units A and B. Units CD with the NAc recognition element was generated via peptide coupling of commercially and readily manipulated amino acid derivatives.
Scheme 1.
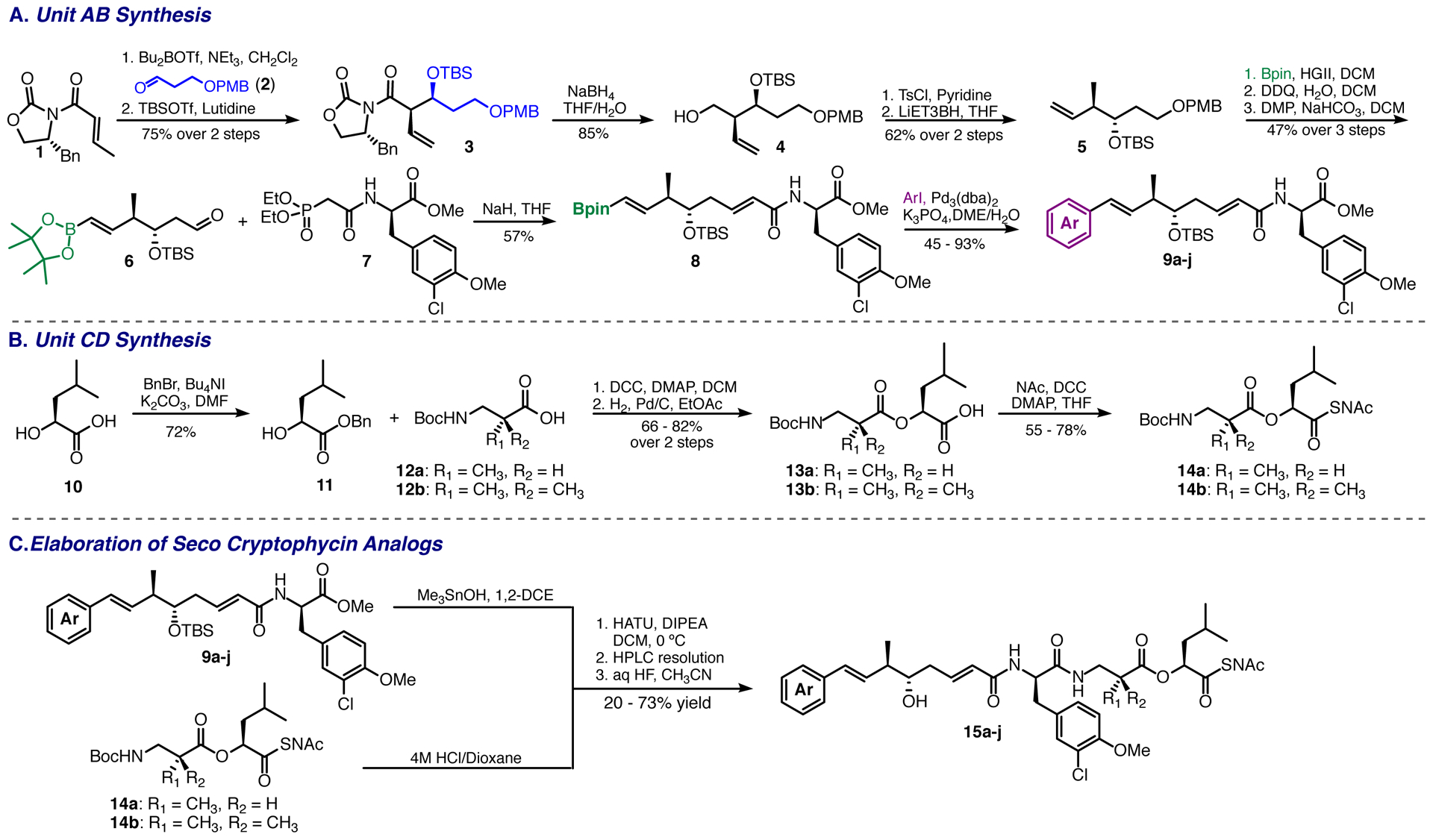
A. Diversifiable synthesis of unit AB intermediates with altering aryl groups. B. Synthesis of units CD incorporating either native β-alanine or the gem dimethyl analog. C. Elaboration of seco-cryptophycins from unit AB and unit CD intermediates.
As a starting point, unit A was synthesized (Scheme 1A) using an Evans asymmetric aldol with N-crotonyl oxazolidinone 1 and aldehyde 2, which proceeded in excellent yields with a high dr (>20:1) to afford the desired (2R, 3S) adduct. Subsequent silation with TBS trifluoromethanesulfonate produced 3, which was reductively cleaved form the chiral auxiliary furnishing 4. The primary alcohol was then tosylated and a consecutive reductive deoxygenation with lithium triethyl borohydride furnished the desired intermediate 5.40 At this stage, a vinyl pinacol boronic ester was introduced via Hoveyda-Grubbs cross metathesis to give the desired Suzuki handle for future diversification.39 Removal of the p-methoxy benzyl (PMB) protecting group with DDQ and subsequent Des Martin Periodinane (DMP) oxidation to 6 furnished the unit A aldehyde fragment necessary for the HWE olefination.41 Utilizing phosphonate 7, HWE olefination conditions were explored, yielding optimal conversion with sodium hydride in THF, which furnished the diversifiable unit AB fragment 8 in a 57% yield of the correct isomer (70% overall yield, with ~ 5:1 E:Z product ratio).
Unit CD-NAc was readily synthesized beginning from commercially available leucic acid 10 (Scheme 1B). Initial benzyl protection of the acid functionality produced 11, which was subsequently coupled with β-amino acids 12 (a or b) to produce the desired esters. These were readily deprotected via H2/Pd hydrogenolysis to furnish acids 13 (a or b).42 Finally, these molecules were coupled with N-acetyl cysteamine, to yield the desired unit CD fragments 14 (a or b).
With diversifiable substrates 8 in hand, coupling with 14 (a or b) and successive Suzuki diversification were investigated. Labile functional groups and racemization precluded diversification prior to the final peptide coupling. Screening Suzuki conditions for substrate 8, yielded a procedure that utilized Pd2(dba)3 and K3PO4 to produce a suite of novel unit-AB cryptophycin analogs 9a-j (Scheme 1A), in good to excellent yields with no detectable racemization. Elaboration of the final seco cryptophycin chain elongation intermediates was accomplished via the coupling of units AB with units CD (Scheme 1C). Saponification of the methyl esters 9a-j also proved to be susceptible to racemization and a screen of different hydrolysis procedures yielded trimethyltin hydroxide, which provided the corresponding acids in good yield with no detectable racemization.43 Simultaneous Boc deprotection of 14 (a or b) with 4M HCl/Dioxane and subsequent peptide coupling at 0 °C with HATU produced the TBS protected intermediates in good yields with minimal racemization (7:1 to 12:1 dr). Diastereomers were resolved using reverse phase HPLC prior to removal of the TBS group. Finally, deprotection of the silyl group using aqueous HF in acetontrile furnished the desired secocryptophycin NAc analogs (Scheme 1C, 15a-j).
Analytical Substrate Conversion Assay of CrpTE with Unit A Heterocycles.
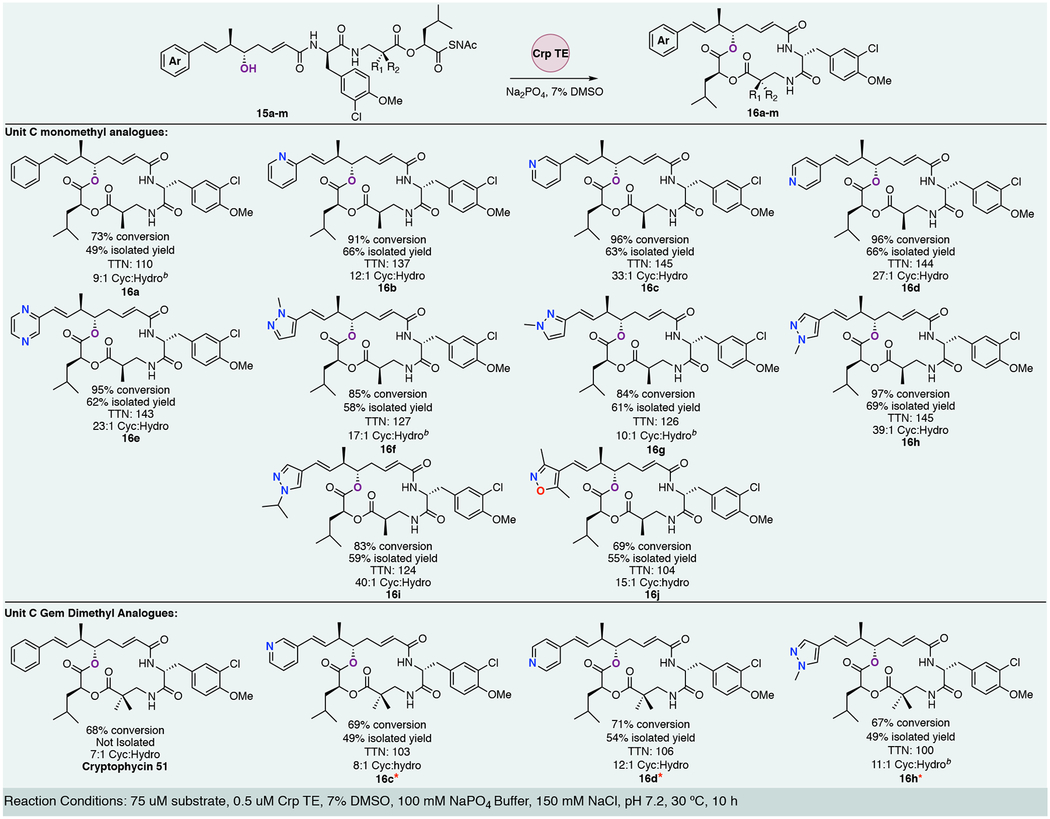
With a diversifiable synthesis in hand we began to explore the flexibility of CrpTE against the newly generated unit A chain elongation intermediates (Figure 1 15a-j). The initial set of CrpTE analogs contained six membered ring heterocycles in place of the native benzene ring (Figure 2, 15be). Analytical scale reactions revealed an increase in conversion to product as well as higher total turnover numbers (TTN) compared to the native benzyl substrate for the four alternate six membered aromatic rings tested. The native benzyl substrate (Table 1, 16a) shows an overall conversion of 73% and TTN of 110. Unreacted starting material as well as the competing hydrolysis reaction observed in a 9:1 ratio in comparison to cyclization account for all peaks observed. In contrast, the 2-, 3-, 4-pyridyl and pyrazine substrates showed nearly complete substrate consumption as well as nearly undetectable levels of hydrolytic byproducts (Figure 2, 15b-e). This is reflected in higher percent conversion, TTNs, and cyclization:hydrolysis (Table 1, 16b-e), all of which are significantly greater (91 −96% conversion, >10:1 cyclization:hydrolysis ratio) when compared to the native substrate 16a.
Figure 2.
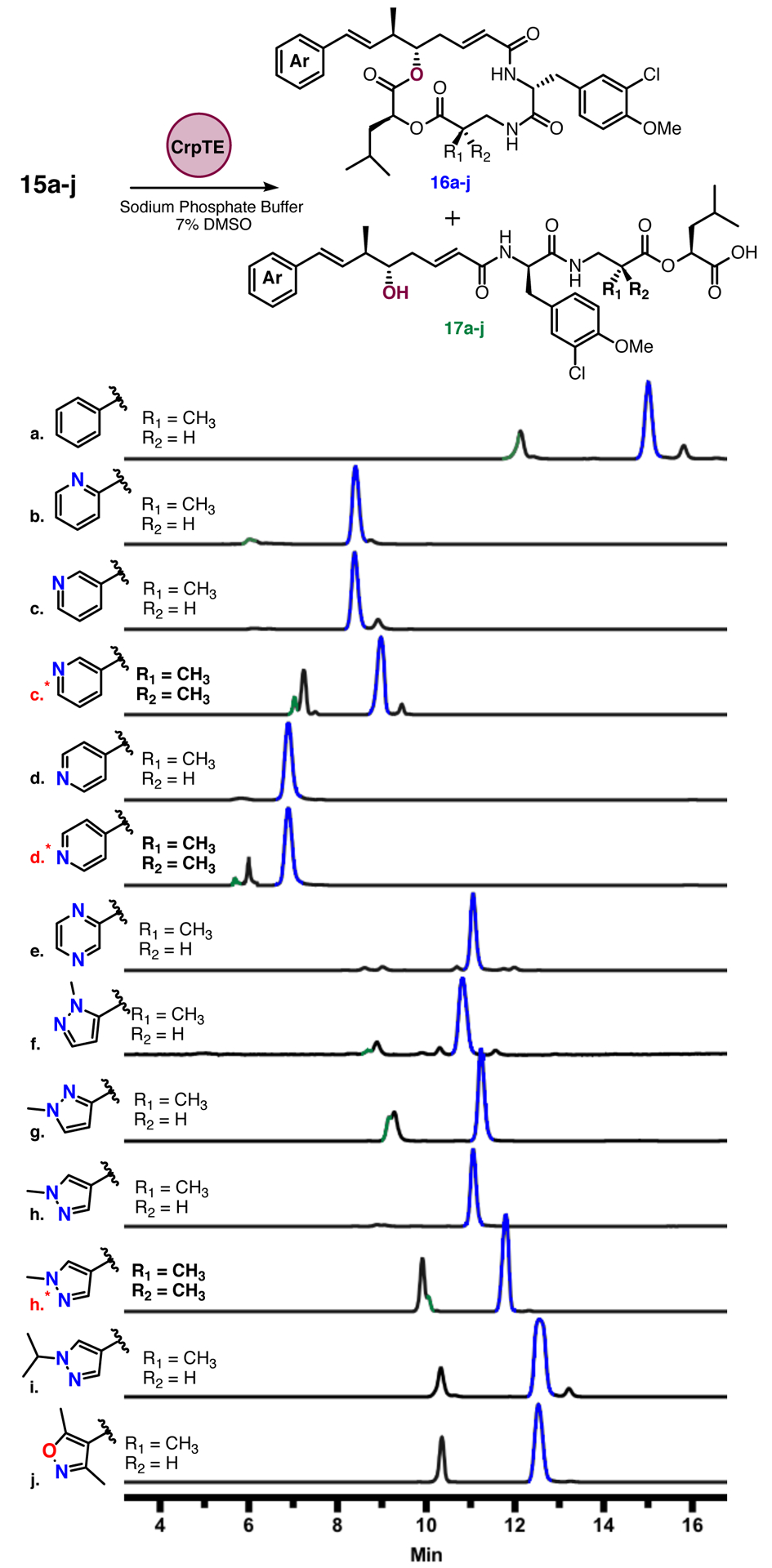
Analysis of analytical scale CrpTE cyclization reactions for analogs 14a-m with starting material peaks in black, product peaks in blue, and hydrolytic byproducts in green as assessed by TOF-MS. Minor peaks in traces a, c, c*, and i represent likely diastereomers but were not characterized. * Indicate gem dimethyl analogs.
Table 1.
Percent conversion, isolation yield, total turnover numbers (TTN)a, and cyclization to hydrolysis analysis of CrpTE reactions.
|
TTN = moles of product / moles of TE, bnumbers estimated from extrapolated peaks due to co-elution of starting material and hydrolysis peaks
The set of unnatural unit A analog was expanded to include five membered ring aromatic heterocycles with varying alkyl chains. The 2-, 3-, and 4-methyl pyrazole derivatives (15f-j) were synthesized and tested in the same analytical assay, utilizing the benzyl substrate 15a as a control. The 2-methyl pyrazole (16f) and 3-methyl pyrazole (16g) groups showed slightly lower percent conversion than the previous six membered ring heterocycles at 85% and 84%, with unreacted starting material accounting for the lower conversion (Table 1, 16f and g). Interestingly, incorporation of a 4-methyl pyrazole ring (15h), showed nearly complete conversion to product with no measurable starting substrate or hydrolytic byproducts.
Incorporating a larger alkyl chain, the 4-isopropyl pyrazole group (Figure 2, 16i) provided important insight into potential size restrictions of the CrpTE binding pocket. Although hydrolytic products were not observed, incomplete consumption of substrate after conclusion of the reaction produced a lower percent conversion (83%) compared to its methyl counterpart, which showed almost quantitative conversion to product. Although the enzyme showed a slight preference for the smaller alkyl substituent overall conversion observed with this analog rivals the native indicating the preference is not substantial. Finally, a dimethyl isoxazole substrate 15j was investigated. This compound showed similar percent conversion and TTN compared with native substrate (Table 1, 16j) with a significant amount of starting substrate remaining.
Isolation and Characterization of Unit A Cryptophycin Analogs.
All reactions were conducted on semi-preparative scale (using the same conditions as the analytical reactions) of 10 mg in order to obtain isolated yields, full structural characterization, and for subsequent CrpE analysis as well as biological evaluation. These results corresponded closely with percent conversions observed in the analytical reactions. The six membered heterocycles 16b-e were isolated in good yields from 62 – 66% (Table 1). The five membered rings 16f-j were also conducted on 10 mg scale and isolated in yields varying from 55% – 69% (Table 1). All novel cryptophycin analogs generated from these chemoenzymatic reactions were confirmed by HRMS, 1H NMR, and 13C NMR.
Evaluation of CrpE with Newly Generated, Unit A Heterocyclic Analogs.
Following the generation of nine new cryptophycin unit A analogs 16b-j with the CrpTE, we next sought to investigate their binding and conversion to β-epoxy derivatives using the wild type CrpE P450. Initial characterization of this enzyme was performed using a CrpE-MBP fusion protein23 however, for this study, an optimized, histidine tagged form of CrpE was generated (Supplemental Figure X). This new variant was recharacterized utilizing the native substrate cryptophycin 3 (16a) which demonstrated a Soret band maximum at 390–394 nm resulting in a high-spin (HS) state (the so-called type I shift).44 A titration of this substrate gave a Kd value of 0.53 ± 0.15 μM, lower than that of the MBP-fusion previously characterized (Figure 3A). Next, Kd values were calculated for analogs 16b-j to determine each compounds viability as substrates. All but one analog, 16d, displayed characteristic type I binding, with varied Kd values. The binding of the 4-pyridyl containing cryptophycin 16d displayed the opposite spectrum with a maximum at 430 nm and a negative minimum at 415 nm (the so-called type II shift) suggesting the formation of a six-coordinated complex with the ligand, generally considered as a P450 inhibitor (Figure 3B).45
Figure 3.
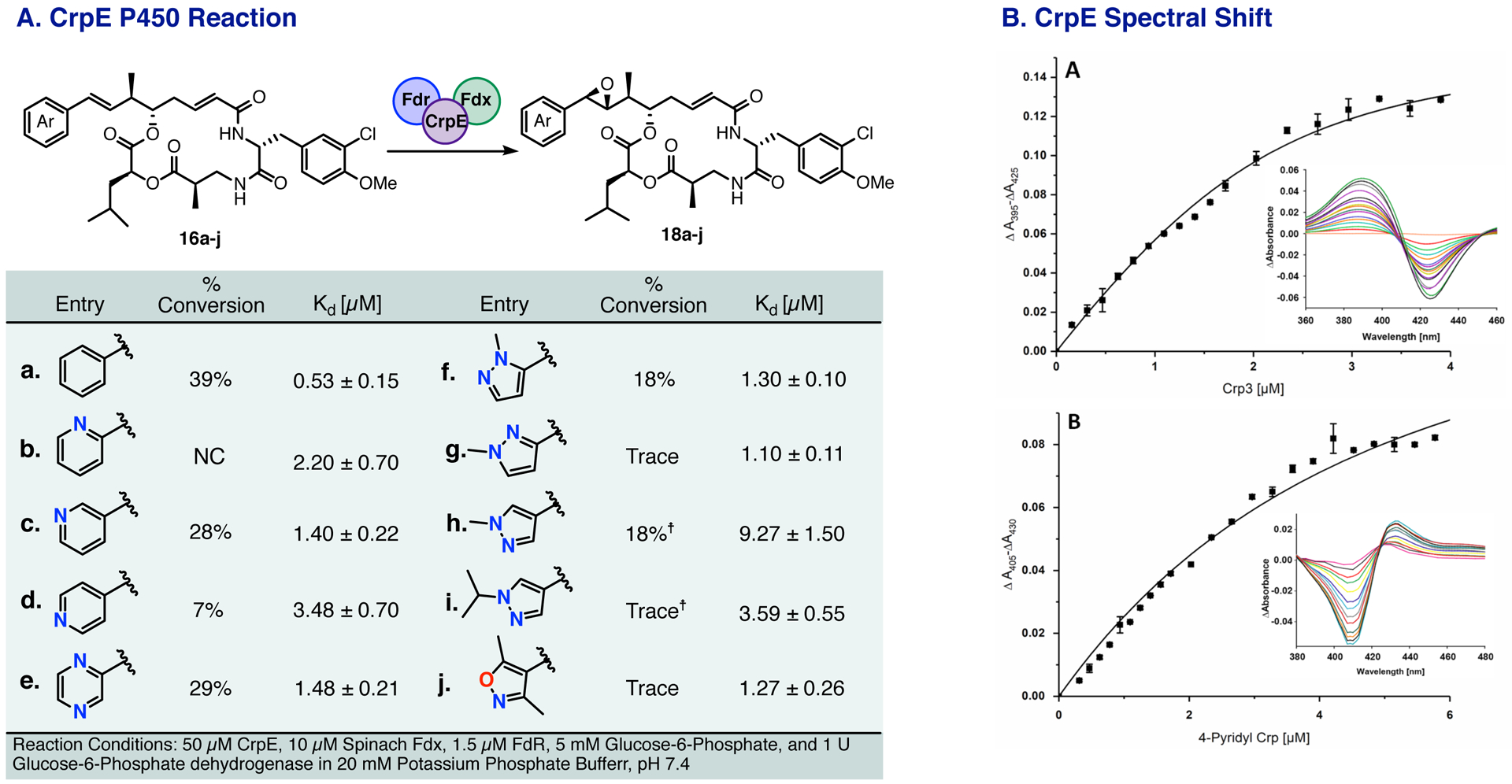
A. CrpE epoxidation reaction with percent conversion and Kd values for CrpE epoxidation depicted, ☨ indicates [Mepoxidation+H2O] B. Spectral shifts induced by binding of A) Crp3 and B) 4-pyridyl Crp to the P450 CrpE. CrpE displays a type I shift with a peak maximum at 395 nm and a minimum at 425 nm (A), whereas for 4-pyridyl Crp the peak maximum and minimum were 430 and 405, respectively, representing the type II shift.
With the knowledge that eight of the new analogs bind as potential substrates, we sought to conduct an in vitro conversion assay. In the absence of autologous redox partners of CrpE from Nostoc. sp. ATCC 53789, we investigated two separate surrogate systems: the RhFRED system previously described46 as well as spinach ferredoxin and ferredoxin reductase for in vitro reconstitution. These initial tests demonstrated no conversion with the RhFRED system however, significant conversion (38%) was observed with the spinach redox system on cryptophycin 3 (Figure 3A, 16a).
Analysis of CrpE P450 with the heterocyclic cryptophycin analogs resulted in distinct epoxidized products with varying percent conversions. Substantial turnover to peaks corresponding to the epoxidized product were observed with the 3-pyridyl, pyrazine, and 2-methyl pyrazole cryptophycins with varying percent conversions (Figure 3A, 16c, e, and f) and trace conversion seen with the 3-pyridyl and isoxazole containing substrates (Figure 3C, 16g and 16j). Interestingly, despite the 4-pyridyl analog (16d) displaying a type II shift (Kd = 3.50 μM), a small amount of epoxidized product was observed indicating that some of the type II ligand can also act as a substrate.47 The 4-methyl pyrazole substrate (h) also displayed significant turnover, although to the presumed diol product (epoxidized mass plus water) that appears to be produced upon the mechanical stress of the HPLC. In order to confirm this result chemical epoxidation was attempted using mCPBA. The crude reaction product appeared to contain a mixture of the desired α and β epoxide product that upon HPLC purification decomposed to the diol products. This same decomposition product is observed in trace quantities with the isopropyl-pyrazole (16i) substrate as well.
Closer analysis of the correlation between binding affinity and percent conversion give insight into the biochemical basis for the differences in reactivities displayed. Predominantly, higher binding affinity appears to correlate with higher percent conversion, however this appears to break down in some instances. The 3-methyl pyrazole analog 16g displays a similar Kd to that of the 2-methyl pyrazole, pyrazine, and 3-pyridyl analogs. These three compounds showed the highest turnover however, only trace product could be detected for the 3-methyl pyrazole despite a similar Kd value. The opposite is observed with the 4-methyl pyridyl analog, in which its binding affinity is the lowest of the set however its percent conversion is similar to those with the lower binding affinity. Substrate binding alone does not appear to confer activity indicating either electronics or double bond geometry plays a crucial role. Despite the overall low turnover displayed with this enzyme, product peaks are observed for the majority of the analogs indicating a more appropriate redox partner in conjunction with possible engineering efforts could afford epoxidized products in high yield for a variety of substrates that alter the double bond electronics.
Biological Evaluation of Novel Styrene Cryptophycin Analogs.
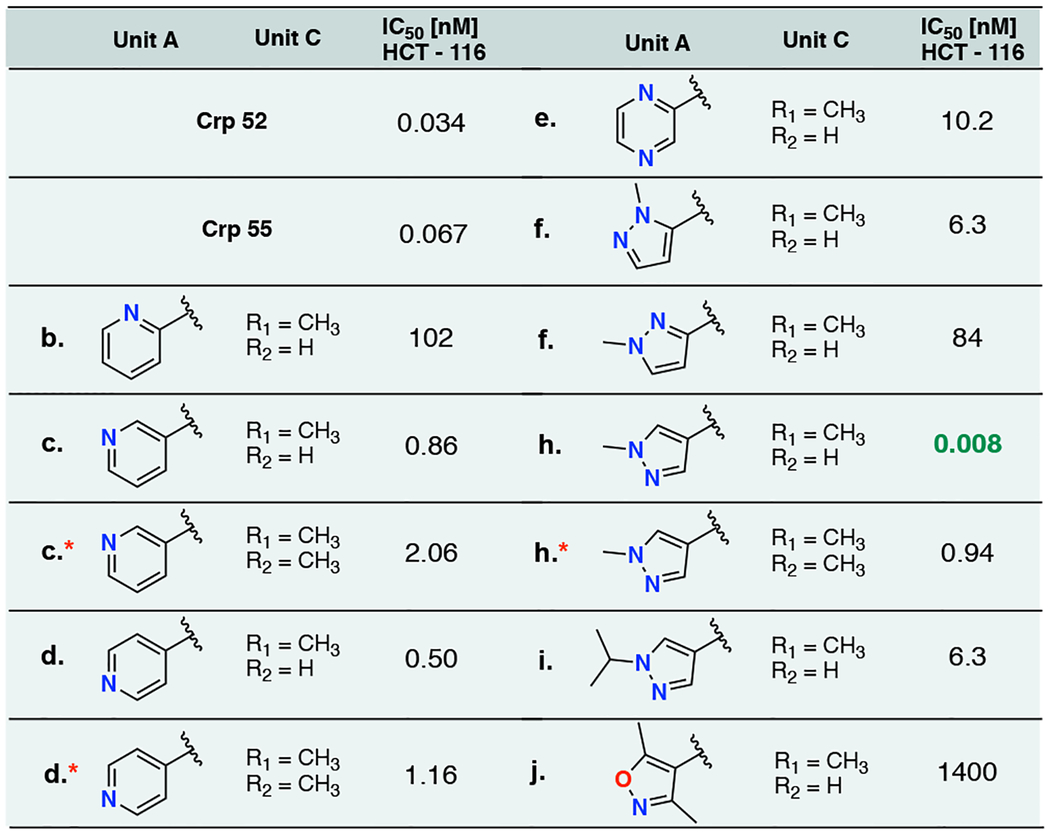
Each of the cryptophycin analogs were initially assessed using an in vitro disk diffusion assay which examines the differential cell killing as zones over a variety of both murine and human solid tumor cell lines and leukemia.48 This data indicates these are all very potent in both solid tumors and leukemia cell lines, with the isoxazole containing heterocycle (16j) being the clear outlier in terms of potency over all cell lines tested (Table S1). Next, each analog was assessed for half minimal inhibitory concentration (IC50) values in HCT-116 human colorectal cancer cell line. The potency of the initial monomethyl unit C analogs displayed significant variability in activities depending on the heterocyclic ring present (Table 2). For the cryptophycin analog bearing six membered ring heterocycles, the IC50 values vary dramatically with the seemingly small change of nitrogen migration around the ring. Within the pyridyl set of analogs, the 2-pyridyl is the least potent with an IC50 of 102 nM, which is over two orders of magnitude higher than the 3-pyridyl (16c) and 4-pyridyl (16d) containing analog (0.860 nM and 0.51 nM, Table 2). Cryptophycin analog bearing five membered rings show even larger differences in IC50 values spanning over 5 orders of magnitude. The inclusion of an isoxazole ring (16j) greatly diminished activity with an IC50 value of 1.4 μM. Most notably, the introduction of a 4-methyl pyrazole ring (16h) provided an analog with single digit picomolar activity, making it one of the most potent cryptophycins known to date. In a direct comparison, our des-epoxy analog displays lower IC50 values than that of the clinically evaluated cryptophycin 52 (as well as the glycinate pro drug ester cryptophycin 55, Table 2) in this cell line. Interestingly, the previous SAR of these compounds demonstrated the importance of the β-epoxide moiety to achieve low picomolar activity, however incorporation of a 4-methyl pyrazole ring maintains this activity in the absence of an epoxide. All substrates tested against CrpE P450 (Figure 3) were conducted on analytical scale and biological evaluation of products was not conducted.
Table 2.
IC50 valuesa for cryptophycin analogs in HCT-116 human colorectal carcinoma
|
IC50 values represented as an average of two replicates
Chemoenzymatic Synthesis and Biological Evaluation of Gem-dimethyl Analogs.
Utilizing the above data to guide our design of analogs to test with the CrpTE biocatalyst, the gem-dimethyl unit C analogs were synthesized utilizing the chemistry described above for our top three analogs: unit A bearing a terminal 3-pyridyl, 4-pyridyl or 4-methyl pyrazole. The ester linkage between units C and D is known to be metabolically unstable and addition of a second methyl group adjacent to this labile position was utilized in the case of the clinically utilized cryptophycin 52 to improve drug half-life.49, 50 In our previous work, we showed there was an increase in hydrolytic byproducts (6:1 cyclization to hydrolysis versus 10:1) with the gem-dimethyl (unit C) present, and the native benzyl containing unit A.22 Our new analogs (15c*, d*, and h*) were first tested on an analytical scale for direct comparison of hydrolysis:cyclization ratios as well as percent conversion to the monomethyl counterparts. All three showed an increase in hydrolytic activity when incubated with CrpTE (Figure 2 and Table 1) as well as a higher percentage of unreacted substrate, consistent with our previous findings. Despite the lower overall conversion to macrocycle, the corresponding chain elongation intermediates showed similar TTNs to that of the native substrate, further demonstrating the remarkable flexibility of CrpTE against substrates containing non-native functional groups in both the PKS and NRPS derived portions of the molecule.
These dimethyl cryptophycin analogs were evaluated for biological activity using both the in vitro disk diffusion assay described above and IC50 determination in the HCT-116 cell line. Incorporation of the gem-dimethyl moiety resulted in attenuated potency for all three analogs in this cell line, indicating alternative methods for blocking metabolism at this site is not universally tolerated and alternative methods could be explored.
Conclusion.
In this work we have described the synthesis, biocatalytic cyclization and epoxidation of a library of cryptophycin analogs. This work has demonstrated the utility of these enzymes as standalone biocatalysts for production of a new set of unit A variants of these potent tubulin-binding compounds. Furthermore, we have demonstrated the viability of this biocatalytic strategy as a useful addition to the medicinal chemistry toolbox. Biological evaluation of the newly generated analogs has enabled identification of one of the most potent cryptophycin analogs to date, which contains a styrene functionality and obviates the need for the β-epoxide group to achieve low picomolar potency. The valuable insights regarding selectivity and specificity of CrpTE and CrpE P450 toward unnatural substrates will facilitate future efforts to engineer more robust and broader scope biocatalysts. These enzymes are especially attractive for mediating late-stage assembly of complex chain elongation intermediates derived through synthetic chemistry, or as a part of larger, enzymatic cascade for the production of complex bioactive molecules.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank Dr. K. L. Bolduc and Dr. A. N. Lowell for helpful discussions and technical assistance with synthetic procedures. We would also like to thank Dr. W. Feng for maintaining and technical assistance with LCMS and NMR experiments.
Funding Sources
The authors thank the National Institutes of Health (R35 GM118101) and the Hans W. Vahlteich Professorship for financial support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://doi.org/10.1021/acschembio.9b00998
Full synthetic and experimental details, NMR spectra, tables, and figures.
The authors declare no competing financial interest.
REFERENCES
- 1.Weissman KJ, Genetic engineering of modular PKSs: from combinatorial biosynthesis to synthetic biology. Nat Prod Rep 2016, 33, 203–230. [DOI] [PubMed] [Google Scholar]
- 2.Walsh CT, Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat Prod Rep 2016, 33, 127–135. [DOI] [PubMed] [Google Scholar]
- 3.Driggers EM; Hale SP; Lee J; Terrett NK, The exploration of macrocycles for drug discovery - an underexploited structural class. Nat. Rev. Drug Discov 2008, 7, 608–624. [DOI] [PubMed] [Google Scholar]
- 4.Horsman ME; Hari TP; Boddy CN, Polyketide synthase and non-ribosomal peptide synthetase thioesterase selectivity: logic gate or a victim of fate? Nat Prod Rep 2016, 33, 183–202. [DOI] [PubMed] [Google Scholar]
- 5.Kohli RM; Walsh CT, Enzymology of acyl chain macrocyclization in natural product biosynthesis. ChemComm 2003, 7, 297–307. [DOI] [PubMed] [Google Scholar]
- 6.Magarvey NA; Beck ZQ; Golakoti T; Ding Y; Huber U; Hemscheidt TK; Abelson D; Moore RE; Sherman DH, Biosynthetic characterization and chemoenzymatic assembly of the cryptophycins. Potent anticancer agents from cyanobionts. ACS Chem Biol 2006, 1, 766–779. [DOI] [PubMed] [Google Scholar]
- 7.Ding Y; Rath CM; Bolduc KL; Hakansson K; Sherman DH, Chemoenzymatic synthesis of cryptophycin anticancer agents by an ester bond-forming non-ribosomal peptide synthetase module. J Am Chem Soc 2011, 133, 14492–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz RE; Hirsch CF; Sesin DF; Flor JE; Chartrain M; Fromtling RE; Harris GH; Salvatore MJ; Liesch JM; Yudin K, Pharmaceuticals from Cultured Algae. J Ind Microbiol 1990, 5, 113–123. [Google Scholar]
- 9.Smith CD; Zhang XQ; Mooberry SL; Patterson GML; Moore RE, Cryptophycin - a New Antimicrotubule Agent Active against Drug-Resistant Cells. Cancer Res 1994, 54, 3779–3784. [PubMed] [Google Scholar]
- 10.D’Agostino G; del Campo J; Mellado B; Izquierdo MA; Minarik T; Cirri L; Marini L; Perez-Gracia JL; Scambia G, A multicenter phase II study of the cryptophycin analog LY355703 in patients with platinum-resistant ovarian cancer. Int J Gynecol Cancer 2006, 16, 71–76. [DOI] [PubMed] [Google Scholar]
- 11.Stevenson JP; Sun W; Gallagher M; Johnson R; Vaughn D; Schuchter L; Algazy K; Hahn S; Enas N; Ellis D; Thornton D; O’Dwyer PJ, Phase I trial of the cryptophycin analogue LY355703 administered as an intravenous infusion on a day 1 and 8 schedule every 21 days. Clin Cancer Res 2002, 8, 2524–2529. [PubMed] [Google Scholar]
- 12.Sessa C; Weigang-Kohler K; Pagani O; Greim G; Mora O; De Pas T; Burgess M; Weimer I; Johnson R, Phase I and pharmacological studies of the cryptophycin analogue LY355703 administered on a single intermittent or weekly schedule. Eur J Cancer 2002, 38, 2388–2396. [DOI] [PubMed] [Google Scholar]
- 13.Edelman MJ; Gandara DR; Hausner P; Israel V; Thornton D; DeSanto J; Doyle LA, Phase 2 study of cryptophycin 52 (LY355703) in patients previously treated with platinum based chemotherapy for advanced non-small cell lung cancer. Lung Cancer 2003, 39, 197–199. [DOI] [PubMed] [Google Scholar]
- 14.Eggen M; Georg GI, The cryptophycins: their synthesis and anticancer activity. Med Res Rev 2002, 22, 85–101. [DOI] [PubMed] [Google Scholar]
- 15.Nahrwold M; Weiss C; Bogner T; Mertink F; Conradi J; Sammet B; Palmisano R; Royo Gracia S; Preusse T; Sewald N, Conjugates of modified cryptophycins and RGD-peptides enter target cells by endocytosis. J. Med. Chem 2013, 56, 1853–1864. [DOI] [PubMed] [Google Scholar]
- 16.Leamon CP; Wang Y; Vlahov IR; You F; Kleindl PJ; Santhapuram HKR Conjugates containing hydrophilic spacer linkers. WO2009002993A1, 2008/12/31/, 2008.
- 17.Su D; Kozak KR; Sadowsky J; Yu SF; Fourie-O’Donohue A; Nelson C; Vandlen R; Ohri R; Liu LN; Ng C; He JT; Davis H; Lau J; Del Rosario G; Cosino E; dela Cruz-Chuh J; Ma Y; Zhang DL; Darwish M; Cai WW; Chen CJ; Zhou HX; Lu JW; Liu YC; Kaur S; Xu KY; Pillow TH, Modulating Antibody-Drug Conjugate Payload Metabolism by Conjugation Site and Linker Modification. Bioconjug Chem 2018, 29, 1155–1167. [DOI] [PubMed] [Google Scholar]
- 18.Verma VA; Pillow TH; DePalatis L; Li G; Phillips GL; Polson AG; Raab HE; Spencer S; Zheng B, The cryptophycins as potent payloads for antibody drug conjugates. Bioorg. Med. Chem. Lett 2015, 25, 864–868. [DOI] [PubMed] [Google Scholar]
- 19.Bigot A; Bouchard H; Brun M-P; Clerc F; Zhang J Novel Cryptophycin Compounds and Conjugates, Their Preparation and Their Therapeutic Use. WO2017076998 (A1), 2017/05/11/, 2017.
- 20.Steinkuhler MC; Gallinari MP; Osswald B; Sewald N; Ritzefeld M; Frese M Cryptophycin-Based Antibody-Drug Conjugates with Novel Self-Immolative Linkers. WO2016146638 (A1), 2016/09/22/, 2016.
- 21.Bouchard H; Brun M-P; Commercon A; Zhang J Novel Conjugates, Preparation Thereof, and Therapeutic Use Thereof. WO2011001052 (A1), 2011/01/06/, 2011.
- 22.Beck ZQ; Aldrich CC; Magarvey NA; Georg GI; Sherman DH, Chemoenzymatic synthesis of cryptophycin/arenastatin natural products. Biochemistry 2005, 44, 13457–13466. [DOI] [PubMed] [Google Scholar]
- 23.Ding Y; Seufert WH; Beck ZQ; Sherman DH, Analysis of the cryptophycin P450 epoxidase reveals substrate tolerance and cooperativity. J Am Chem Soc 2008, 130, 5492–5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruner SD; Weber T; Kohli RM; Schwarzer D; Marahiel MA; Walsh CT; Stubbs MT, Structural basis for the cyclization of the lipopeptide antibiotic surfactin by the thioesterase domain SrfTE. Structure 2002, 10, 301–310. [DOI] [PubMed] [Google Scholar]
- 25.Samel SA; Wagner B; Marahiel MA; Essen LO, The thioesterase domain of the fengycin biosynthesis cluster: A structural base for the macrocyclization of a non-ribosomal lipopeptide. J. Mol. Biol 2006, 359, 876–889. [DOI] [PubMed] [Google Scholar]
- 26.Kopp F; Grunewald J; Mahlert C; Marahiel MA, Chemoenzymatic design of acidic lipopeptide hybrids: New insights into the structure-activity relationship of daptomycin and A54145. Biochemistry 2006, 45, 10474–10481. [DOI] [PubMed] [Google Scholar]
- 27.Yeh E; Lin HN; Clugston SL; Kohli RM; Walsh CT, Enhanced macrocyclizing activity of the thioesterase from tyrocidine synthetase in presence of nonionic detergent. Chem Biol 2004, 11, 1573–1582. [DOI] [PubMed] [Google Scholar]
- 28.Trauger JW; Kohli RM; Mootz HD; Marahiel MA; Walsh CT, Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature 2000, 407, 215–218. [DOI] [PubMed] [Google Scholar]
- 29.Pinto A; Wang M; Horsman M; Boddy CN, 6-Deoxyerythronolide B Synthase Thioesterase-Catalyzed Macrocyclization Is Highly Stereoselective. Org Lett 2012, 14, 2278–2281. [DOI] [PubMed] [Google Scholar]
- 30.Tsai SC; Miercke LJW; Krucinski J; Gokhale R; Chen JCH; Foster PG; Cane DE; Khosla C; Stroud RM, Crystal structure of the macrocycle-forming thioesterase domain of the erythromycin polyketide synthase: Versatility from a unique substrate channel. Proc Natl Acad Sci USA 2001, 98, 14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen DA; Koch AA; Sherman DH, Identification of a Thioesterase Bottleneck in the Pikromycin Pathway through Full-Module Processing of Unnatural Pentaketides. J Am Chem Soc 2017, 139, 13450–13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch AA; Hansen DA; Shende VV; Furan LR; Houk KN; Jimenez-Oses G; Sherman DH, A Single Active Site Mutation in the Pikromycin Thioesterase Generates a More Effective Macrocyclization Catalyst. J Am Chem Soc 2017, 139, 13456–13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohli RM; Trauger JW; Schwarzer D; Marahiel MA; Walsh CT, Generality of peptide cyclization catalyzed by isolated thioesterase domains of nonribosomal peptide synthetases. Biochemistry 2001, 40, 7099–7108. [DOI] [PubMed] [Google Scholar]
- 34.Trauger JW; Kohli RM; Walsh CT, Cyclization of backbone-substituted peptides catalyzed by the thioesterase domain from the tyrocidine nonribosomal peptide synthetase. Biochemistry 2001, 40, 7092–7098. [DOI] [PubMed] [Google Scholar]
- 35.Eissler S; Bogner T; Nahrwold M; Sewald N, Efficient synthesis of cryptophycin-52 and novel paraalkoxymethyl unit A analogues. Chemistry 2009, 15, 11273–87. [DOI] [PubMed] [Google Scholar]
- 36.Al-Awar RS; Corbett TH; Ray JE; Polin L; Kennedy JH; Wagner MM; Williams DC, Biological evaluation of cryptophycin 52 fragment A analogues: effect of the multidrug resistance ATP binding cassette transporters on antitumor activity. Molecular cancer therapeutics 2004, 3, 1061–7. [PubMed] [Google Scholar]
- 37.Al-Awar RS; Ray JE; Schultz RM; Andis SL; Kennedy JH; Moore RE; Liang J; Golakoti T; Subbaraju GV; Corbett TH, A convergent approach to cryptophycin 52 analogues: synthesis and biological evaluation of a novel series of fragment a epoxides and chlorohydrins. J Med Chem 2003, 46, 2985–3007. [DOI] [PubMed] [Google Scholar]
- 38.deMuys JM; Rej R; Nguyen D; Go B; Fortin S; Lavallee JF, Synthesis and in vitro cytotoxicity of cryptophycins and related analogs. Bioorg. Med. Chem. Lett 1996, 6, 1111–1116. [Google Scholar]
- 39.Bolduc KL; Larsen SD; Sherman DH, Efficient, divergent synthesis of cryptophycin unit A analogues. ChemComm 2012, 48, 6414–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnamurthy S, Rapid Reduction of Alkyl Tosylates with Lithium Triethylborohydride - Convenient and Advantageous Procedure for Deoxygenation of Simple and Hindered Alcohols - Comparison of Various Hydride Reagents. J Organomet Chem 1978, 156, 171–181. [Google Scholar]
- 41.Ghosh AK; Swanson L, Enantioselective synthesis of (+)-cryptophycin 52 (LY355703), a potent antimitotic antitumor agent. J Org Chem 2003, 68, 9823–9826. [DOI] [PubMed] [Google Scholar]
- 42.Mast CA; Eissler S; Stoncius A; Stammler HG; Neumann B; Sewald N, Efficient and versatile stereoselective synthesis of cryptophycins. Chemistry 2005, 11, 4667–4677. [DOI] [PubMed] [Google Scholar]
- 43.Nicolaou KC; Estrada AA; Zak M; Lee SH; Safina BS, A mild and selective method for the hydrolysis of esters with trimethyltin hydroxide. Angew Chem Int Edit 2005, 44, 1378–1382. [DOI] [PubMed] [Google Scholar]
- 44.Denisov IG; Makris TM; Sligar SG; Schlichting I, Structure and chemistry of cytochrome P450. Chem Rev 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- 45.Gigon PL; Gram TE; Gillette JR, Studies on the rate of reduction of hepatic microsomal cytochrome P-450 by reduced nicotinamide adenine dinucleotide phosphate: effect of drug substrates. Mol Pharmacol 1969, 5, 109–122. [PubMed] [Google Scholar]
- 46.Li SY; Podust LM; Sherman DH, Engineering and analysis of a self-sufficient biosynthetic cytochrome P450 PikC fused to the RhFRED reductase domain. J Am Chem Soc 2007, 129, 12940–12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearson JT; Hill JJ; Swank J; Isoherranen N; Kunze KL; Atkins WM, Surface plasmon resonance analysis of antifungal azoles binding to CYP3A4 with kinetic resolution of multiple binding orientations. Biochemistry 2006, 45, 6341–6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Valeriote FA; Tenney K; Media J; Pietraszkiewicz H; Edelstein M; Johnson TA; Amagata T; Crews P, Discovery and development of anticancer agents from marine sponges: perspectives based on a chemistry-experimental therapeutics collaborative program. J Exp Ther Oncol 2012, 10, 119–134. [PubMed] [Google Scholar]
- 49.Wagner MM; Paul DC; Shih C; Jordan MA; Wilson L; Williams DC, In vitro pharmacology of cryptophycin 52 (LY355703) in human tumor cell lines. Cancer Chemother Pharmacol 1999, 43, 115–125. [DOI] [PubMed] [Google Scholar]
- 50.Golakoti T; Ogino J; Heltzel CE; LeHusebo T; Jensen CM; Larsen LK; Patterson GML; Moore RE; Mooberry SL; Corbett TH; Valeriote FA, Structure determination, conformational analysis, chemical stability studies, and antitumor evaluation of the cryptophycins. Isolation of 18 new analogs from Nostoc sp strain GSV 224. J Am Chem Soc 1996, 117, 12030–12049. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.