

Drug hope for SARS
The SARS (severe acute respiratory syndrome) epidemic of 2003 caused almost 800 deaths, many of them due to acute respiratory distress syndrome (ARDS) as a complication. There are no effective drugs available for treating ARDS, but new work in mice suggests that ACE2 (angiotensin-converting enzyme 2) might be an option. ACE2 can protect mice from lung injury in an ARDS-like syndrome, whereas other components of the renin–angiotensin system for controlling blood pressure and salt balance actually make the condition worse. ACE2 is expressed in the healthy lung but downregulated by lung injury and it was shown recently (Nature 426, 450–454; 2003) to be a receptor for the SARS coronavirus.
Supplementary information
The online version of this article (doi:10.1038/nature03712) contains supplementary material, which is available to authorized users.
Abstract
Acute respiratory distress syndrome (ARDS), the most severe form of acute lung injury, is a devastating clinical syndrome with a high mortality rate (30–60%) (refs 1–3). Predisposing factors for ARDS are diverse1,3 and include sepsis, aspiration, pneumonias and infections with the severe acute respiratory syndrome (SARS) coronavirus4,5. At present, there are no effective drugs for improving the clinical outcome of ARDS1,2,3. Angiotensin-converting enzyme (ACE) and ACE2 are homologues with different key functions in the renin–angiotensin system6,7,8. ACE cleaves angiotensin I to generate angiotensin II, whereas ACE2 inactivates angiotensin II and is a negative regulator of the system. ACE2 has also recently been identified as a potential SARS virus receptor and is expressed in lungs9,10. Here we report that ACE2 and the angiotensin II type 2 receptor (AT2) protect mice from severe acute lung injury induced by acid aspiration or sepsis. However, other components of the renin–angiotensin system, including ACE, angiotensin II and the angiotensin II type 1a receptor (AT1a), promote disease pathogenesis, induce lung oedemas and impair lung function. We show that mice deficient for Ace show markedly improved disease, and also that recombinant ACE2 can protect mice from severe acute lung injury. Our data identify a critical function for ACE2 in acute lung injury, pointing to a possible therapy for a syndrome affecting millions of people worldwide every year.
Supplementary information
The online version of this article (doi:10.1038/nature03712) contains supplementary material, which is available to authorized users.
Main
The renin–angiotensin system has an important role in maintaining blood pressure homeostasis, as well as fluid and salt balance11,12,13. ACE2 is a homologue of ACE, and functions a negative regulator of the renin–angiotensin system6,7,8. Although ACE2 is expressed in the lungs of humans10 and mice (see Supplementary Fig. 1a, b), nothing is known about its function in the lungs. However, mortality following SARS coronavirus infections approaches almost 10% owing to the development of ARDS14,15,16. To elucidate the role of ACE2 in acute lung injury, we examined the effect of Ace2 gene deficiency in mouse experimental models that mimic the common lung failure pathology observed in several human diseases, including sepsis, acid aspiration and pneumonias such as SARS and avian influenza A17.
Aspiration of gastric contents with a low pH is a frequent cause of acute lung injury/ARDS1,2,3. Acid aspiration in wild-type mice, which mimics human acute lung injury18,19, resulted in rapid impairment of lung functions assessed by increased lung elastance (a measure of the change in pressure achieved per unit change in volume, representing the stiffness of the lungs) (Fig. 1a), decreased blood oxygenation (Fig. 1b) and the development of pulmonary oedema (Fig. 1c). Acid aspiration resulted in increased alveolar wall thickness, oedema, bleeding, inflammatory cell infiltrates and formation of hyaline membranes (Fig. 1d). Notably, acid-treated Ace2 knockout mice8 showed significantly greater lung elastance compared with control wild-type mice, but there were no differences in lung elastance between saline-treated Ace2 knockout and wild-type mice (Fig. 1a). Moreover, loss of Ace2 resulted in worsened oxygenation (Fig. 1b), massive lung oedema (Fig. 1c), increased inflammatory cell infiltration and hyaline membrane formations (Fig. 1d) in response to acid aspiration. It should be noted that ACE2 protein expression is typically downregulated in wild-type mice following acid challenge (Fig. 1e).
Figure 1. Loss of ACE2 worsens acid aspiration-induced acute lung injury.

a, Lung elastance after acid or saline treatment in wild type (WT) and Ace2 knockout (Ace2 KO) mice (n = 10 for acid-treated groups, n = 6 for saline-treated groups). P < 0.05 for the whole time course comparing acid-treated WT and Ace2 knockout mice. b, Partial pressure of oxygen in arterial blood () in acid-induced acute lung injury. c, Wet-to-dry weight ratios of lungs 3 h after acid injury. Single asterisk, P < 0.05; double asterisk, P < 0.01. d, Lung histopathology. Note the enhanced hyaline membrane formation, inflammatory cell infiltration and lung oedema in acid-treated Ace2 knockout mice (H&E staining, ×200). e, ACE and ACE2 protein expression in total lysates from control lungs and lungs 3 h after acid injury. Error bars indicate s.e.m.
Sepsis is the most common cause of acute lung injury/ARDS1,2,3. We therefore examined the effect of Ace2 gene deficiency on sepsis-induced acute lung injury using caecal ligation and perforation (CLP)20. CLP causes lethal peritonitis and sepsis due to a polymicrobial infection that is accompanied by acute lung failure20. Whereas all CLP-treated wild-type mice survived, only two out of ten CLP-treated Ace2 knockout mice survived the 6 h experimental observation period (Fig. 2a). CLP resulted in lung failure defined by increased elastance (Fig. 2a), pulmonary oedema (Fig. 2b) and leukocyte accumulation (Fig. 2c) in wild-type mice. CLP-treated Ace2 knockout mice had a marked worsening of lung functions (Fig. 2a), increased oedema (Fig. 2b) and leukocyte accumulation (Fig. 2c) compared with wild-type mice. In addition, Ace2 knockout mice also developed markedly enhanced acute lung injury after endotoxin challenge18 (see Supplementary Fig. 2a–c). Ace2 maps to the X chromosome, and it should be noted that loss of ACE2 expression resulted in equally severe acute lung injury phenotypes in male (Ace2-/y) and female (Ace2-/-) mice. Our data from three different acute lung injury models show that loss of Ace2 expression precipitates severe acute lung failure.
Figure 2. ACE2 controls acute lung failure.

a, Lung elastance after acute lung injury in WT and Ace2 knockout (KO) mice induced by caecal ligation perforation (CLP). Eighteen hours after sham or CLP surgery, animals received mechanical ventilation for 6 h (n = 10 in CLP-treated groups, n = 6 in sham-treated groups). As 8/10 CLP-treated Ace2 knockout mice died at 4–4.5 h, only data up to 4 h are shown. CLP-treated Ace2 knockout mice had significantly higher elastance than CLP-treated WT mice (P < 0.01). b, c, Wet-to-dry weight ratios of lungs (b) and lung histopathology (c) in sham or CLP-treated WT and Ace2 knockout mice determined after 4 h of ventilation. Asterisk denotes a significant difference (P < 0.05) between CLP-treated WT and Ace2 knockout mice. Note the enhanced lung oedema and inflammatory infiltrates in Ace2 knockout mice (H&E staining, ×200). d, e, Lung elastance (d) and wet-to-dry weight ratios (e) after acid or saline instillation of Ace2 knockout mice injected intraperitoneally with recombinant human ACE2 protein (rhuACE2; 0.1 mg kg-1), mutant rhuACE2 (mut-rhuACE2; 0.1 mg kg-1) or vehicle (n = 6 per group). Asterisk denotes a significant difference (P < 0.05) comparing rhuACE2-treated Ace2 knockout mice with mut-rhuACE2-treated and vehicle-treated Ace2 knockout mice at 3 h. f, Lung elastance after acid instillation in WT mice treated with rhuACE2 protein (0.1 mg kg-1), mut-rhuACE2 protein (0.1 mg kg-1) or vehicle (n = 6–8 per group). Asterisk denotes a significant difference (P < 0.05) between WT mice treated with rhuACE2 and mut-rhuACE2 or with vehicle at 3 h. Errors bars indicate s.e.m.
To test whether loss of ACE2 is essential for disease pathogenesis, we performed a rescue experiment using recombinant human ACE2 protein (rhuACE2) (see Supplementary Fig. 3a, b). Injection of rhuACE2 into acid-treated Ace2 knockout mice decreased the degree of acute lung injury, as assessed by lung elastance (Fig. 2d) and pulmonary oedema formation (Fig. 2e). When we injected rhuACE2 protein into acid-treated wild-type mice, lung function (Fig. 2f) and oedema formation (see Supplementary Fig. 3c) were also rescued. In saline-treated wild-type or Ace2 knockout mice, injections of rhuACE2 did not affect pulmonary functions (Fig. 2d–f). Catalytically inactive ACE2 protein (mut-rhuACE2) (see Supplementary Fig. 3a, b) did not rescue the severe lung phenotype in Ace2 knockout mice (Fig. 2d, e) and had no effect on the severity of acute lung injury in wild-type animals (Fig. 2f and Supplementary Fig. 3c). These results show that the catalytic activity of ACE2 can directly protect lungs from acute lung injury.
ACE2 is a homologue of ACE, both of which are central enzymes in the renin–angiotensin system6,7,8. ACE cleaves the decapeptide angiotensin (Ang)I into the octapeptide AngII (refs 11, 12). ACE2 cleaves a single residue from AngI to generate Ang1–9 (refs 6, 7), and a residue from AngII to generate Ang1–7 (ref. 6). In this way, ACE2 negatively regulates the renin–angiotensin system by inactivating AngII (Fig. 3a). Acid aspiration in wild-type mice resulted in marked downregulation of ACE2 protein, but ACE levels remained constant (Fig. 1e). Moreover, only catalytically active ACE2 improved the acute lung injury phenotype in mutant and wild-type mice (Fig. 2d–f). To clarify whether acute lung injury shifts the functional equilibrium between ACE and ACE2, we measured AngII levels in acid-treated and control mice. Acid aspiration markedly increased AngII levels in lungs (Fig. 3b) and plasma (see Supplementary Fig. 4a) of wild-type mice. We observed a further, significant increase in AngII levels in the lungs (Fig. 3b) and plasma (see Supplementary Fig. 4a) of acid-treated Ace2 knockout mice. Thus, acute lung injury results in decreased ACE2 expression and increased production of AngII.
Figure 3. ACE deficiency reduces the severity of acute lung injury.
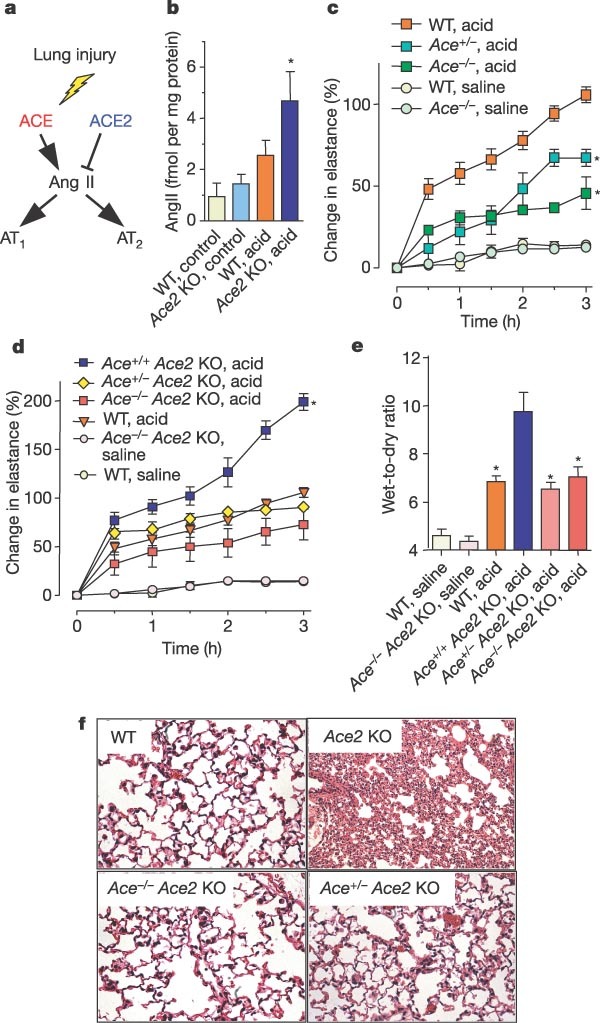
a, Schematic diagram of the renin–angiotensin system. b, Lung levels of AngII in control and acid-treated WT and Ace2 knockout (KO) mice determined at 3 h by enzyme immunoassay (n = 3–5 per group). Asterisk denotes a significant difference (P < 0.05) between acid-treated WT and Ace2 knockout mice. c, Lung elastance after acid instillation in Ace+/+ (WT), Ace+/- and Ace-/- mice (n = 4–6 mice per group). Asterisk denotes a significant difference (P < 0.05) comparing Ace+/+ with Ace+/- and Ace-/- mice at 3 h. d, e, Lung elastance (d) and wet-to-dry lung weight ratios (e) in acid- or saline-treated Ace+/+Ace2 KO, Ace+/-Ace2 KO, Ace-/-Ace2 KO and WT mice (n = 5 per group). Asterisk denotes a significant difference (P < 0.05) comparing Ace2 KO with WT, Ace+/-Ace2 KO or Ace-/-Ace2 KO mice 3 h after acid-treatment. f, Lung histopathology. Severe lung interstitial oedema and leukocyte infiltration in Ace2 KO mice are attenuated by homozygous (Ace-/-) or heterozygous (Ace+/-) mutations of Ace (H&E staining, ×200). Error bars indicate s.e.m.
On the basis of these results we speculated that, in contrast to ACE2, ACE promotes disease pathogenesis through increased AngII production (Fig. 3a). Indeed, genetic inactivation of Ace on an Ace2 wild-type or Ace2 knockout background markedly decreased AngII levels in lung and plasma in our acid injury model (see Supplementary Fig. 4b, c). Moreover, treatment with rhuACE2 protein attenuated lung injury (Fig. 2d–f) and further reduced AngII levels in the lungs of acid-treated mice (Supplementary Fig. 4d). In contrast to Ace2 knockout mice, Ace-/- mice21 were partly protected against acute lung injury induced by acid-aspiration (Fig. 3c and Supplementary Fig. 5). These effects were dependent on gene dosage and were observed to a lesser extent in Ace+/- mice. In addition, inactivation of Ace on an Ace2 knockout background rescued the severe lung failure (Fig. 3d), oedema formation (Fig. 3e) and histological changes (Fig. 3f) compared with Ace2 knockout mice. Similarly, in endotoxin-induced acute lung injury, the severe lung impairments in Ace2 knockout mice were reversed by additional Ace gene deficiency (see Supplementary Fig. 6). Thus, ACE promotes acute lung injury pathology and ACE2 alleviates it.
Both ACE and ACE2 are non-specific proteases that cleave additional substrates11,12. Thus, although increased levels of AngII have been correlated with Ace2 deficiency, it has not been shown that upregulation of the AngII pathway accounts for the observed phenotypes of Ace2 knockout mice in vivo. The receptors for AngII in mice are angiotensin II type 1a (AT1a) receptor22, the type 1b (AT1b) receptor and the type 2 (AT2) receptor23. AT1a and AT2, but not AT1b receptor expression is found in the lungs25. We therefore explored which AngII receptor subtypes are responsible for ACE/ACE2 regulated acute lung injury, and whether AngII signalling through its receptors is responsible for ACE2-regulated lung pathology (Fig. 3a). Compared with wild-type mice, genetic loss of AT1a receptor expression in Agtr1a-/- mice24 markedly improved lung function (Fig. 4a) and reduced oedema formation (see Supplementary Fig. 7a). In contrast, inactivation of the AT2 receptor (Agtr2-/y)25 aggravated acute lung injury (Fig. 4a and Supplementary Fig. 7a). AngII levels induced by acid aspiration in both Agtr1a-/- and Agtr2-/y mice were comparable to those in wild-type controls (not shown).
Figure 4. The AngII receptor AT1a controls acute lung injury severity and pulmonary vascular permeability.
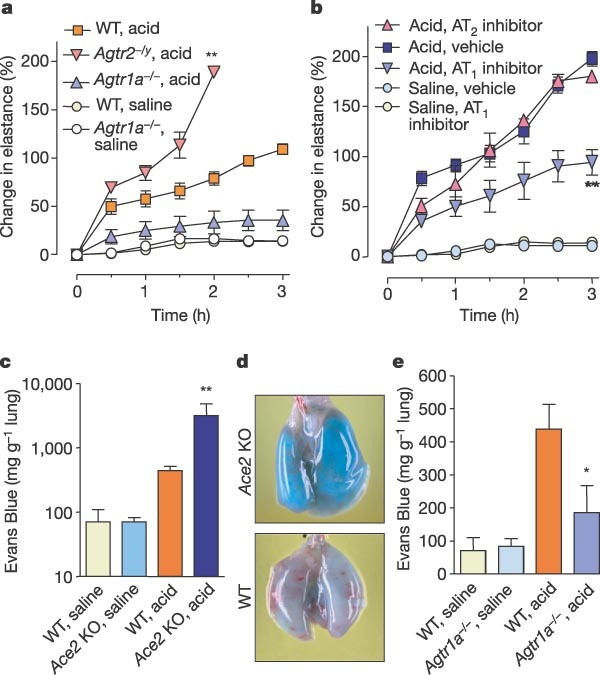
a, Lung elastance measurements in Agtr1a-/- mice, Agtr2-/y mice and WT mice after acid aspiration (n = 4–6 per group). All acid-treated Agtr2-/y mice died after 2 h. There is a significant difference (P < 0.01) between acid-treated WT and acid-treated Agtr1a-/- mice over the whole time course. Double asterisk denotes a significant difference (P < 0.01) between WT and Agtr2-/y mice at 2 h. b, Lung elastance measurements in Ace2 knockout mice treated with vehicle or inhibitors to AT1 (Losartan, 15 mg kg-1) or AT2 (PD123.319, 15 mg kg-1) after acid or saline instillation (see Methods, n = 4–6 per group). Double asterisk denotes a significant difference (P < 0.01) comparing Ace2 knockout mice treated with AT1 inhibitor with vehicle or AT2 inhibitor treatment at 3 h. c, Pulmonary vascular permeability as determined by intravenous injection of Evans Blue. Extravascular Evans Blue in lungs was measured in WT and Ace2 knockout mice 3 h after acid injury (n = 5 per group). Double asterisk denotes a significant difference (P < 0.01) between acid-treated WT and Ace2 knockout mice. d, Representative images of Evans Blue-injected lungs of WT and Ace2 knockout mice 3 h after acid aspiration. e, Extravascular Evans Blue in lungs of WT and Agtr1a-/- mice 3 h after acid injury (n = 5 per group). Asterisk denotes a significant difference (P < 0.05) between acid-treated WT and Agtr1a-/- mice at 3 h. Error bars indicate s.e.m.
We next attempted to rescue acute lung injury in Ace2 knockout mice using specific AT1 and AT2 receptor blockers. Pharmacological inhibition of AT1 attenuated the severity of acid-induced lung injury in Ace2 knockout mice (Fig. 4b and Supplementary Fig. 7b). Inhibition of AT2 had no apparent effect on the acute lung injury phenotypes of Ace2 knockout mice (Fig. 4b). These data show that the AT1a and AT2 receptors have opposite functions in controlling the severity of acute lung injury, and that actions of AngII through the AT1a receptor have a causative role in acute lung failure.
Pulmonary oedema could arise from increased hydrostatic pressure (due to pulmonary vascular constriction) and/or enhanced microvascular permeability26. We first tested whether AngII can increase hydrostatic pressure using isolated, perfused murine lungs ex vivo27. In this system, pulmonary perfusion pressures were comparable between wild-type and Ace2 knockout mice under baseline control conditions (wild-type 3.0 ± 1.9 cm H2O, n = 6 versus Ace2 knockout 1.8 ± 1.6 cm H2O, n = 9; mean ± s.e.m,), and these values were not changed by either acid-treatment or continuous perfusion of the bacterial endotoxin lipopolysaccharide (LPS). Pulmonary perfusion pressures generated by AngI or AngII injection into lungs of acid-instilled animals or into lungs perfused with LPS were also similar between wild-type and Ace2 knockout mice (see Supplementary Fig. 8a, b). Moreover, fractional shortening using echocardiography (an indicator of left ventricular systolic function) and mean arterial pressures were comparable between Ace2 knockout and wild-type mice during the experimental period (see Supplementary Fig. 9a, b). Thus, the severe lung oedemas in Ace2 knockout mice do not seem to be secondary to systemic haemodynamic alterations.
As enhanced pulmonary vascular permeability is a hallmark of acute lung injury/ARDS in humans2, we examined whether loss of Ace2 results in increased vascular permeability using Evans Blue dye injections as an in vivo indicator of albumin leakage28. Acid aspiration increased vascular permeability in wild-type mice. In Ace2 knockout mice, pulmonary Evans Blue accumulation was greatly increased after acid aspiration (Fig. 4c, d). These results were confirmed using fluorescein isothiocyanate (FITC)-conjugated dextran (40 kDa) as another marker to assess vascular leakage of macromolecules (data not shown). Vascular permeability was significantly attenuated in the lungs of Agtr1a-/- mice (Fig. 4e). We suggest that loss of ACE2 expression in acute lung injury leads to leaky pulmonary blood vessels through AT1a receptor stimulation. However, hydrostatic oedemas cannot be excluded, and the effects of local AngII production on lung blood vessels require further investigation27,29.
ARDS is the most severe form of a wide spectrum of pathological processes designated as acute lung injury2. ARDS is characterized by pulmonary oedema due to increased vascular permeability, the accumulation of inflammatory cells and severe hypoxia2. Predisposing factors for ARDS include sepsis, aspiration and pneumonias (including infections with SARS coronavirus1,2,3,4,5 or avian and human influenza viruses17). Our data show that acute lung injury results in a marked downregulation of ACE2, a key enzyme involved in the regulation of the renin–angiotensin system.
It has been previously shown that an insertion/deletion ACE polymorphism that affects ACE activity is associated with ARDS susceptibility and outcome30. Our data provide a mechanistic explanation for these clinical findings and indicate that, in the pathogenesis of acute lung injury, AngII is upregulated by ACE and drives severe lung failure through the AT1a receptor. On the other hand, ACE2 and the AT2 receptor protect against lung injury. Exogenous recombinant human ACE2 attenuates acute lung failure in Ace2 knockout as well as in wild-type mice. This combination of genetic, pharmacological and protein rescue experiments defines a new and critical role for the renin–angiotensin system in the pathogenesis of acute lung injury, and show that ACE2 is a key molecule involved in the development and progression of acute lung failure.
Methods
For detailed methods please refer to the Supplementary Information.
Animals
Ace2, Ace, Agtr1a and Agtr2 mutant mice have previously been described2,4,5,6. Sex-, age-, and background-matched mice were used as controls. Basal lung functions and lung structure were comparable among all the mice tested. Mice were handled in accordance with institutional guidelines.
Experimental murine models of acute lung injury
For acid aspiration-induced acute lung injury, anaesthesized mice were intratracheally instilled with HCl (pH 1.5; 2 ml kg-1) and ventilated for 3 h (refs 18, 19). To study sepsis-induced acute lung injury, we performed caecal ligation perforation (CLP)20. Sham-operated mice underwent the same procedure without ligation and puncture of the caecum. Eighteen hours after sham/CLP surgery, animals were subjected to mechanical ventilation for up to 6 h. For endotoxin-induced acute lung injury, anaesthetized mice received LPS and zymosan intratracheally immediately after starting mechanical ventilation and 1 h later, respectively18. In all acute lung injury models, total positive end expiratory pressure (PEEPt) and plateau pressure (Pplat) were measured at the end of expiratory and inspiratory occlusion, respectively. Elastance was calculated as (Pplat - PEEPt) divided by tidal volume (VT) every 30 min during the ventilation periods.
Blood oxygenation, pulmonary oedema, pulmonary vascular permeability and histology
Blood samples were obtained from the left heart ventricle and partial pressure of oxygen in arterial blood () was measured. To assess pulmonary oedemas, the lung wet-to-dry weight ratios were calculated. Pulmonary vascular permeability was assessed by measuring the pulmonary extravasation of Evans Blue. For histological analysis, 5-µm thick sections were cut and stained with haematoxylin and eosin (H&E).
Recombinant ACE2 and AT1/AT2 receptor inhibitors
Thirty minutes before acid instillation, mice received intraperitoneal injections of recombinant human ACE2 (rhuACE2) protein (0.1 mg kg-1) (R&D Systems or our own rhuACE2 preparation), catalytically inactive (H374N, H378N)10 mutant recombinant human ACE2 (mut-rhuACE2) or vehicle (0.1% BSA/PBS). All animals were then ventilated for 3 h. RhuACE2 protein and mut-rhuACE2 protein were purified from transfected CHO cells by affinity chromatography. The catalytic activities of purified recombinant ACE2 proteins were measured using the fluorogenic peptide Substrate VI (R&D Systems). Mut-rhuACE2-Fc showed >95% loss of catalytic activity (see Supplementary Fig. 3a). For inhibitor studies, mice received intraperitoneal injections of the AT1 inhibitor Losartan (15 mg kg-1), the AT2 inhibitor PD123.319 (15 mg kg-1) or control vehicle 30 min before surgical procedures.
Angiotensin II peptide levels and western blotting
AngII peptide levels were measured as described8. For western blotting, rabbit polyclonal anti-ACE2 antibody8 and rabbit polyclonal anti-mouse ACE antibody (R&D Systems) were used.
Statistical analyses
All data are shown as mean ± s.e.m.. Measurements at single time points were analysed by analysis of variance (ANOVA). Time courses were analysed by repeated measurements ANOVA with Bonferroni post-tests.
Supplementary information
Supplementary Figures S1-S9 (PPT 3809 kb)
Legends for Supplementary Figures S1-S9 and Supplementary Methods. (RTF 163 kb)
Acknowledgements
We thank M. Chappell, C. Richardson, B. Seed, U. Eriksson, J. Ishida and all members of our laboratory for discussions and reagents. This work is supported by the Institute for Molecular Biotechnology of the Austrian Academy of Sciences (IMBA) and the Jubilaeumsfonds of the Austrian National Bank. This work is in part supported by the Canadian Institutes of Health Research (CIHR) and the Canada Foundation for Innovation (CFI). K.K. is supported by a Marie Curie Fellowship from the EU. C.J. is supported a Beijing Committee of Science and Technology grant and the Natural Science Fundation of China. L.H. is supported by the Deutsche Forschungsgemeinschaft.
Competing interests
J.M.P. declares personal financial interests.
Footnotes
Yumiko Imai and Keiji Kuba: *These authors contributed equally to this work
References
- 1.Hudson LD, Milberg JA, Anardi D, Maunder RJ. Clinical risks for development of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995;151:293–301. doi: 10.1164/ajrccm.151.2.7842182. [DOI] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. The acute respiratory distress syndrome. N. Engl. J. Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 3.Vincent JL, Sakr Y, Ranieri VM. Epidemiology and outcome of acute respiratory failure in intensive care unit patients. Crit. Care Med. 2003;31(suppl.):S296–S299. doi: 10.1097/01.CCM.0000057906.89552.8F. [DOI] [PubMed] [Google Scholar]
- 4.Ksiazek TG, et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 5.Drosten C, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 6.Donoghue M, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87:E1–E9. doi: 10.1161/01.RES.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 7.Tipnis SR, et al. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 8.Crackower MA, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 9.Li W, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamming I, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004;203:631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skeggs LT, Dorer FE, Levine M, Lentz KE, Kahn JR. The biochemistry of the renin-angiotensin system. Adv. Exp. Med. Biol. 1980;130:1–27. doi: 10.1007/978-1-4615-9173-3_1. [DOI] [PubMed] [Google Scholar]
- 12.Corvol P, Williams TA, Soubrier F. Peptidyl dipeptidase A: angiotensin I-converting enzyme. Methods Enzymol. 1995;248:283–305. doi: 10.1016/0076-6879(95)48020-X. [DOI] [PubMed] [Google Scholar]
- 13.Boehm M, Nabel EG. Angiotensin-converting enzyme 2—a new cardiac regulator. N. Engl. J. Med. 2002;347:1795–1797. doi: 10.1056/NEJMcibr022472. [DOI] [PubMed] [Google Scholar]
- 14.Tsang KW, et al. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1977–1985. doi: 10.1056/NEJMoa030666. [DOI] [PubMed] [Google Scholar]
- 15.Lee N, et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 16.Poutanen SM, et al. Identification of severe acute respiratory syndrome in Canada. N. Engl. J. Med. 2003;348:1995–2005. doi: 10.1056/NEJMoa030634. [DOI] [PubMed] [Google Scholar]
- 17.Tran TH, et al. Avian influenza A (H5N1) in 10 patients in Vietnam. N. Engl. J. Med. 2004;350:1179–1188. doi: 10.1056/NEJMoa040419. [DOI] [PubMed] [Google Scholar]
- 18.Nagase T, et al. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nature Immunol. 2000;1:42–46. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 19.Imai Y, et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. J. Am. Med. Assoc. 2003;289:2104–2112. doi: 10.1001/jama.289.16.2104. [DOI] [PubMed] [Google Scholar]
- 20.Martin EL, et al. Negative impact of tissue inhibitor of metalloproteinase-3 null mutation on lung structure and function in response to sepsis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;285:L1222–L1232. doi: 10.1152/ajplung.00141.2003. [DOI] [PubMed] [Google Scholar]
- 21.Krege JH, et al. Male–female differences in fertility and blood pressure in ACE-deficient mice. Nature. 1995;375:146–148. doi: 10.1038/375146a0. [DOI] [PubMed] [Google Scholar]
- 22.Inagami T, et al. Cloning, expression and regulation of angiotensin II receptors. Adv. Exp. Med. Biol. 1995;377:311–317. doi: 10.1007/978-1-4899-0952-7_21. [DOI] [PubMed] [Google Scholar]
- 23.Gasc JM, Shanmugam S, Sibony M, Corvol P. Tissue-specific expression of type 1 angiotensin II receptor subtypes. An in situ hybridization study. Hypertension. 1994;24:531–537. doi: 10.1161/01.HYP.24.5.531. [DOI] [PubMed] [Google Scholar]
- 24.Sugaya T, et al. Angiotensin II type 1a receptor-deficient mice with hypotension and hyperreninemia. J. Biol. Chem. 1995;270:18719–18722. doi: 10.1074/jbc.270.32.18719. [DOI] [PubMed] [Google Scholar]
- 25.Hein L, Barsh GS, Pratt RE, Dzau VJ, Kobilka BK. Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor in mice. Nature. 1995;377:744–747. doi: 10.1038/377744a0. [DOI] [PubMed] [Google Scholar]
- 26.Plante GE, Chakir M, Ettaouil K, Lehoux S, Sirois P. Consequences of alteration in capillary permeability. Can. J. Physiol. Pharmacol. 1996;74:824–833. doi: 10.1139/y96-090. [DOI] [PubMed] [Google Scholar]
- 27.Roy BJ, Pitts VH, Townsley MI. Pulmonary vascular response to angiotensin II in canine pacing-induced heart failure. Am. J. Physiol. 1996;271:H222–H227. doi: 10.1152/ajpheart.1996.271.1.H222. [DOI] [PubMed] [Google Scholar]
- 28.Goggel R, et al. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nature Med. 2004;10:155–160. doi: 10.1038/nm977. [DOI] [PubMed] [Google Scholar]
- 29.Hansen TN, Le Blanc AL, Gest AL. Hypoxia and angiotensin II infusion redistribute lung blood flow in lambs. J. Appl. Physiol. 1985;58:812–818. doi: 10.1152/jappl.1985.58.3.812. [DOI] [PubMed] [Google Scholar]
- 30.Marshall RP, et al. Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2002;166:646–650. doi: 10.1164/rccm.2108086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1-S9 (PPT 3809 kb)
Legends for Supplementary Figures S1-S9 and Supplementary Methods. (RTF 163 kb)