

Abstract
Multiple sclerosis, an inflammatory disease of the central nervous system, is characterized by primary destruction of myelin. This review covers recent advances in neuropathology, immunogenetics, neuroimmunology, and neurovirology that have provided insights regarding its pathogenesis. Three hypotheses are discussed: (1) autoimmunity, (2) “bystander” demyelination, and (3) immune destruction of persistently infected oligodendrocytes. A paradigm for induction of primary demyelination is proposed in which immune cells recognize “foreign” antigens on the surface of oligodendrocytes in the context of major histocompatibility complex gene products. The final result of this scheme may be “dying-back gliopathy,” the alteration being noted first in the most distal extension of the oligodendrocyte—that is, the myelin sheaths.
PATHOLOGY
Multiple sclerosis (MS) affects scattered areas of the central nervous system with a predilection for periventricular white matter, brainstem, spinal cord, and optic nerves.1 The plaques are characterized by primary demyelination (destruction of myelin sheaths with preservation of axons) and death of oligodendrocytes (myelin-producing cells) within the center of the lesion. During the early evolution of the plaque, perivascular inflammatory cells (lymphocytes, plasma cells, macrophages) invade the substance of the white matter and are thought to play a critical role in myelin destruction.2 This process is followed by extensive gliosis by astrocytes and aberrant attempts at remyelination with oligodendrocytes proliferating at the edges of the plaque.1 In addition, immunoglobulins are deposited within each plaque.3
Whether the principal effector cells that mediate demyelination are T cells or macrophages is unknown. Attempts to type the cells that infiltrate the brain in MS have yielded conflicting results.4, 5, 6, 7 Both CD8+ (cytotoxic/suppressor) T lymphocytes and CD4+ (helper/inducer) T lymphocytes surround the MS plaque. The relative proportions of T-cell subsets are controversial. Some investigators have found an excess of CD8+ cells in the perivascular cuffs at the edge of the lesion.7 Others have found an excess of CD4+ cells.4 These discrepancies probably result from examination of the plaques at different stages of their evolution.
Recent experiments have analyzed antigens of the major histocompatibility complex (MHC) in the brains of patients with MS.8 The MHC is a set of closely linked genes that play a central role in the control of immune responses to self and foreign antigens. This function is of particular importance because CD4+ and CD8+ T cells recognize foreign antigen in the context of class II (HLA-DR, la) or class I (HLA-ABC) MHC gene products, respectively. The class II gene products, found primarily on macrophages and B cells, are important in presentation of antigen to T cells. Class I gene products are on the majority of cells in the body and are important in the generation of the cytotoxic response against viruses.
The central nervous system is unique because MHC antigens normally are not present on neurons and glial cells.8 In MS, however, class I and class II MHC-positive astrocytes are found with high frequency in active lesions. The class I-reactive glia are primarily associated with T-cell infiltrates, whereas class II-reactive astrocytes are found in many lesions, independently of the composition of inflammatory cells. Thus, class I and II MHC-positive astrocytes might play a role in local antigen presentation to T cells. In addition, the simultaneous presence of high numbers of class I MHC-positive astrocytes and CD8+ cells in acute lesions suggests the possibility that CD8+ cells play a role as cytotoxic T cells during early development of the lesion.
The final consequence—that is, demyelination—in chronic active plaques may be attributable to several immunologic mechanisms, including receptor-mediated endocytosis by macrophages, cytotoxicity by T cells, or lymphokine release.
IMMUNOGENETICS AND IMMUNOLOGY
One piece of evidence that provides an important clue to the pathogenesis of MS is the association between susceptibility and specific MHC haplotypes.9 Northern Caucasians with MS have an overrepresentation of the A3, B7, DR2, and Dw2 histocompatibility alleles with relative risks of 2 to 3 for the class I MHC alleles (A3, B7) and 4 to 5 for class II MHC alleles (DR2, Dw2). Because linkage disequilibria exist between these class I alleles and DR2, the possible increased representation of one group of alleles may merely reflect this phenomenon.
The indefinite association between susceptibility and MHC haplotypes suggests that either more than one gene is involved or a strong environmental agent “breaks through” to disease in the absence of the MS susceptibility allele. The frequency of occurrence of A2, B12, DR7, and Dw7 is decreased in patients with MS.9 The data that pertain to this occurrence are in much better agreement, suggesting that some MHC alleles may protect against MS. Investigators have also reported that some MHC alleles (DR2) are associated with more progressive disease whereas others (DR3) are associated with more benign disease.10 Studies in various populations (American blacks, Japanese, Arabs, Israeli Jews) have also shown an association with MHC but with different alleles in each case.
The risk of MS developing among first-degree relatives of patients with MS is increased 15- to 20-fold over the risk in the general population.9 This finding may reflect either genetic factors or shared environmental agents. Attempts to analyze linkage between disease and HLA haplotypes in siblings affected by MS have substantiated a loose link between MS and MHC but do not permit conclusions about the mode of inheritance.11 For more distant relatives, concordance for MS is lower but exceeds chance expectation. In contrast, for non-blood-relatives living together (for example, husband and wife), the concordance for MS is not increased above chance. Studies in twins have shown greater concordance between monozygotic twins than between dizygotic twins.12, 13, 14, 15 Many monozygotic pairs, however, are discordant for MS, a strong argument that genetic influence is insufficient for the development of MS. Also, the severity and expression of the disease vary greatly between members of concordant pairs.
Diseases that have been linked to the HLA complex have numerous similar features, including chronic course, inflammatory component, and weak genetic predisposition that does not obey mendelian genetics. One theory is that these disorders may have an autoimmune basis. The development of experimental autoimmune encephalomyelitis (EAE) as an animal model of MS has prompted investigation of the autoimmune basis of human demyelinating diseases.16 EAE can be induced by injection of central nervous system myelin or its components along with adjuvants. From the EAE model has evolved the concept that the primary target in MS may be myelin and not the oligodendrocyte.17 In addition, the disease can be transferred to naive animals with cells enriched for T lymphocytes reactive to the basic protein component of myelin.17, 18, 19 Therefore, attempts have been made to determine whether an immune response to normal myelin develops in patients with MS. To date, efforts to measure such a response in patients with MS have failed even though these responses can be detected in patients with postinfectious encephalomyelitis.20
Although certain immunologic abnormalities have been reported in MS, many of the findings are still controversial.21 CD8+ lymphocytes apparently are decreased in the blood of patients with active MS and in those with a progressive course,22, 23, 24, 25 a situation that leads to a relative increase in the CD4/CD8 ratio. Disease activity is not always associated with these T-cell changes, however. The data on T-cell subsets in the cerebrospinal fluid are more discordant.26, 27, 28 Some investigators have reported a corresponding decrease in CD8+ cells in the cerebrospinal fluid,28 whereas others have found no change.26, 27 Some investigators have argued that the loss of CD8+ cells from the blood represents a disorder of immunoregulation.22 Others have suggested that the imbalance is the result of preferential sequestration of CD8+ cells in the central nervous system or lymphoid organs.25
Recent experiments suggest a decrease in the suppressor/inducer subset (CD4+, 2H4+) in serum of patients with progressive MS.29 These suppressor/inducer cells are not found in the cerebrospinal fluid, an indication that the loss of such cells is not due to migration into the central nervous system. Suppressor T-cell function is also defective during active disease, as measured by concanavalin A-induced T-cell suppression30, 31 or by polyclonal IgG stimulation of peripheral blood mononuclear cells by pokeweed mitogen.32 Natural killer cell activity against tumor or virally infected targets may be decreased in MS,33 but these results have not been confirmed.34, 35
VIRUSES
Attempts to identify a single infectious agent as the cause of MS have been unsuccessful thus far.36 Many infectious agents have been isolated in cultures of specimens from patients with MS, but the majority represent contaminants or noncausal agents. In addition, direct inoculation of brain material into primates, so successful in identifying a transferable agent in Creutzfeldt-Jakob disease, has been negative or inconclusive. The more sensitive technique of looking for “footprints” of virus infection in brain tissue by nucleic acid hybridization with radiolabeled DNA probes or RNA probes (riboprobes) complementary to viral genomes has suggested that some brains of subjects with MS harbor measles virus.37 The finding of measles virus genome in several control subjects without MS, however, suggested that viruses may reside in the nervous system without causing disease.37
Analysis of the cerebrospinal fluid of patients with MS shows increased IgG levels along with oligoclonal Ig bands.38, 39, 40 Attempts to identify the antigen to which the IgG is directed have been unsuccessful.40 In serum samples from patients with MS, titers of antibody to measles virus are increased.40, 41 In many patients with MS, however, antibody titers to two or more viruses are increased in the cerebrospinal fluid. In diseases that are known to be caused by a virus (subacute sclerosing panencephalitis [measles] and mumps meningitis), IgG and oligoclonal bands in the cerebrospinal fluid are directed almost exclusively against the infectious agent.42, 43 Because this is not the case with MS, some investigators have suggested that, in MS, the IgG in the central nervous system is “nonsense antibody” that represents a dysfunction in immune regulation. The alternative hypothesis is that the humoral immune response is to an unrecognized infectious agent that is the cause of MS.
Two approaches to distinguish whether antibody is “nonsense” or “sense” have been used: analysis of banding patterns of IgG eluted from different MS plaques in the same patient44 and study of idiotypes (combining sites of antibody molecules) of the oligoclonal IgG in cerebrospinal fluid from multiple patients.45, 46 The first approach showed that each MS plaque may have unique IgG banding patterns. The second approach showed that anti-idiotypic antisera raised against cerebrospinal fluid IgG fail to cross-react with cerebrospinal fluid from other patients with MS. These results are consistent with the possibility that the antibody is, indeed, a “nonsense antibody.”
An important impetus to continuation of the search for a viral cause for MS has come from study of several naturally occurring model diseases in animals in which viruses cause demyelination.47 These diseases include canine distemper virus (paramyxovirus) in dogs,48 visna virus (nononcogenic retrovirus) in sheep,49 JHM virus (coronavirus) in mice50 and rats,51 Semliki Forest virus (togavirus) in mice,52 and Theiler's virus (picornavirus) in mice.53 The mechanisms of the demyelination in these viral models are beginning to be understood. For example, in JHM virus infection in mice the demyelination seems to be the result of direct cytopathic injury of oligodendrocytes, the myelin-producing cells.50 In contrast, in rats the demyelination by JHM may be a consequence of autoimmune mechanisms by which sensitized T cells recognize myelin antigens.51 In visna virus, macrophages that are persistently infected are actively involved in the demyelinating process.53 With infection by Theiler's virus, viral antigens have been detected on the inner and outer glial loops of oligodendrocytes such that an immune response seems to be directed against viral or “novel” antigens on the surface of these glial cells.54, 55, 56 These experiments emphasize that viruses from different “families” may induce primary demyelination, probably by unique mechanisms.44
The recent discovery, in Japan57 and in the Caribbean,58 that chronic progressive myelopathy may be the result of persistent infection with human T-lymphotropic virus type I (HTLV-I) has stimulated speculation that viruses may be implicated in MS. Koprowski and associates59 reported that serum and spinal fluid samples from patients with MS in Sweden and in Key West, Florida, contained antibodies reactive with viral protein (p24) of HTLV-I. Others have failed to detect any antibody to HTLV-I, II, or III in patients with MS.60, 61 In Japan, however, investigators62 found that 11 of 46 patients with MS reacted to purified HTLV-I proteins by Western blot analysis. The discrepancies in the results may be due to differences in sensitivity of various methods. Recently, Reddy and associates63 amplified and molecularly cloned HTLV-I sequences from DNA of lymphocytes from patients with MS. Confirmation of these results by other investigators will be necessary before a firm statement can be made about its role in pathogenesis.
IMMUNE HYPOTHESES FOR THE CAUSE OF MS
Several hypotheses have been proposed to explain the clinical and experimental features of this demyelinating disorder, three of which will be discussed. They are relevant to the possibility of an immune-mediated demyelination triggered by viral infection.
Autoimmunity.
The most widely considered hypothesis is that MS is the result of an immune reaction directed against self myelin antigens.64 T cells are thought to enter the central nervous system through endothelial cells and to react with normal white matter. The myelin antigens may be presented to helper T cells (CD4+) by endothelial cells65 or astrocytes,66 which are known to carry class II MHC antigens in MS lesions. As a result of T-cell activation, lymphokines and macrophages could mediate myelin destruction.3
This hypothesis is supported by elegant experiments, using EAE, that demonstrated that T-cell clones (CD4+) sensitized to myelin proteins can mediate demyelination in the central nervous system.18, 19 In addition, the linkage of the disease with class II MHC gene products (DR2) is further evidence that this mechanism may be important. A series of crucial experiments by Hafler and associates20 on T-cell clones isolated from the cerebrospinal fluid of patients with MS, however, failed to identify a single clone that was reactive for myelin antigens despite the ongoing demyelination. In contrast, myelin-reactive clones were demonstrated in patients with postinfectious encephalomyelitis.
“Bystander” Hypothesis.
One important hypothesis being considered is that in MS the myelin is an “innocent bystander” that is destroyed as a consequence of an immune response occurring within the nervous system.47 This hypothesis may help explain why different viruses can induce demyelination in the nervous system of rodents and why different “etiologic” agents have been isolated from MS-involved brains. The scenario would be that viruses or other infectious agents frequently invade the central nervous system. During the defense against this infection, T lymphocytes and macrophages are recruited to the lesion. Subsequently, in the process of clearance of virus by T cells, myelin is destroyed nonspecifically by lymphokines or neutral proteases released by activated macrophages.
Experiments by Wisniewski and Bloom67 support this hypothesis by demonstrating that animals previously sensitized to purified protein derivative (PPD) of tuberculin will undergo demyelination if challenged by this antigen in the nervous system. In addition, myelin basic protein can be degraded by neutral proteases in vitro.68 The concept of bystander demyelination, however, is not supported by the observation that demyelination is a rare consequence of an immune response to viral or fungal encephalitides. If bystander demyelination were more prominent, then demyelination would occur each time the immune system interacts with viruses that infect the central nervous system (that is, measles, mumps, and herpes). It may be argued, however, that genetic factors control whether specific lymphokines that could induce demyelination are released.
Immune Destruction of Persistently Infected Oligodendrocytes.
On the basis of the paradigm of demyelination in mice induced by Theiler's virus, Rodriguez and colleagues56 proposed that the demyelination in MS may be the result of immune-mediated destruction of virus-infected oligodendrocytes. This hypothesis would incorporate epidemiologic data (exogenous agent) and a contribution of immunogenetics to pathogenesis. Immunogenetic data in MS implicate that specific genes within the MHC protect against the development of MS. Genes may play a role in host resistance against the exogenous agent that is acquired early in life.
In resistant hosts, viral antigens would be recognized in the context of class I or class II MHC gene products, and viral replication would be limited as a consequence of a protective immune response. Virus would be cleared from the central nervous system without long-term sequelae.
In susceptible hosts, absence of specific MHC gene products may result in failure of clearance of the virus; thus, the virus persists in glial cells. During the course of infection, the virus could infect oligodendrocytes in a manner such that novel antigens are present on the surface of these cells (Fig. 1 ). These novel antigens may be virus antigens or “up-regulated” normal host antigens that are seen as foreign in the context of specific MHC alleles. The virus itself may be capable of inducing demyelination by interfering with the function of the myelin-producing cell. The current evidence suggests, however, that immune cells must also play a role in demyelination because immunosuppression seems to have a favorable effect on the course of MS. The presence of a novel antigen on an oligodendrocyte or on myelin could result in immunoglobulin-directed killing (injury by complement, antibody-dependent cell-mediated cytotoxicity, or activation of macrophages). Processed viral antigens may be recognized by CD8+ cells (class I-restricted cytotoxic T cells) or CD4+ cells (class II-restricted cytotoxic T cells), which could be effectors in demyelination.
Fig. 1.
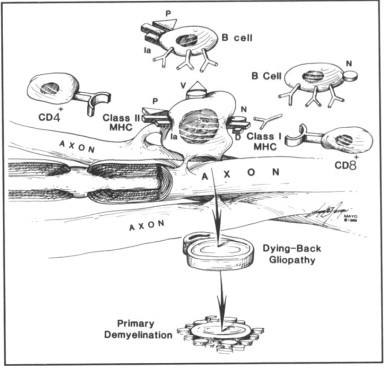
Hypothesis by which immune cells injure persistently infected oligodendrocytes and produce primary demyelination. During the course of viral infections of the central nervous system, specific viruses in “susceptible” patients may not be cleared by the immune system. Viruses may then persist in oligodendrocytes (myelin-producing cells) and injure these cells either by direct effects or by becoming targets of immune-mediated destruction. Oligodendrocytes infected by virus may express viral antigen (V), processed viral antigen (P), or novel antigens (N) on their surfaces. Immunoglobulins secreted by B cells may be directed at viral or novel antigens and thus result in injury to oligodendrocytes by complement, antibody-dependent cell-mediated cytotoxicity, or receptor-mediated phagocytosis by macrophages. Processed viral antigens (peptides) may be expressed on surface of oligodendrocytes and be presented to CD4+ cells in context of class II major histocompatibility complex (MHC) antigens (la antigens). These CD4+ cells (class II-restricted cytotoxic T cells or “helper” T cells) could directly injure oligodendrocytes or help B cells. Novel virus-induced antigens may be expressed on surface of oligodendrocytes and be seen in context of class I MHC antigens by CD8+ cells (cytotoxic T cells). These CD8+ cells may injure oligodendrocytes in an effort to clear viral infection or by interacting with normal host proteins. The final consequence of these mechanisms would be injury to the oligodendrocyte. The initial pathologic process could be in the inner and outer glial loops of myelin-producing cells such that destruction of myelin would occur before visible structural damage to the oligodendrocyte (“dying-back gliopathy”). This process would result in primary demyelination. Therapeutic strategies to deplete B cells, CD4+ cells, or CD8+ cells and to “down-regulate” expression of class I or class II MHC gene products may prove beneficial.
The final result would be injury to the myelin-producing cell or to the myelin sheath. This result may occur as a dying-back gliopathy,69, 70 the alteration being noted first in the most distal extension of the oligodendrocytes (that is, the glial loops and myelin sheaths) and interfering with the differential function of the oligodendrocyte (that is, the maintenance of myelin).
CONCLUSION
The evidence suggests that an exogenous agent (that is, a virus) may be important in triggering demyelination in MS. If multiple exogenous agents are able to induce this pathologic process, however, then identifying the offending agent may prove to be difficult. If common pathogens can produce demyelination, then distinguishing these etiologic agents from the viruses that “normally” reside in the central nervous system may be impossible. Alternatively, attempts to interfere specifically with various arms of the immune response by use of monoclonal antibody therapy may be beneficial without the requirement of knowing the causative agent. The scheme proposed in Figure 1 may provide the basis for designing specific immunotherapy to interfere with demyelination.
ACKNOWLEDGMENT
The artwork for Figure 1 was done by Floyd E. Hosmer, Section of Medical Graphics.
Footnotes
This investigation was supported in part by Grants NS 849 and NS 24180 from the National Institutes of Health, Public Health Service, and Grant RG 1878 A-1 from the Multiple Sclerosis Society.
Individual reprints of this article are not available. The entire Symposium on Multiple Sclerosis will be available for purchase as a bound booklet from the Proceedings Circulation Office in July.
REFERENCES
- 1.Prineas JW. The neuropathology of multiple sclerosis. In: Vinken PJ, Bruyn GW, Klawans HL, Koetsier JC, editors. Handbook of Clinical Neurology. Vol 47: Demyelinating Diseases. Elsevier Science Publishing Company; New York: 1985. pp. 213–257. [Google Scholar]
- 2.Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab Invest. 1978;38:409–421. [PubMed] [Google Scholar]
- 3.Prineas JW, Graham JS. Multiple sclerosis: capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann Neurol. 1981;10:149–158. doi: 10.1002/ana.410100205. [DOI] [PubMed] [Google Scholar]
- 4.Traugott U, Reinherz EL, Raine CS. Multiple sclerosis: distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J Neuroimmunol. 1983;4:201–221. doi: 10.1016/0165-5728(83)90036-x. [DOI] [PubMed] [Google Scholar]
- 5.Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci. 1983;62:219–232. doi: 10.1016/0022-510x(83)90201-0. [DOI] [PubMed] [Google Scholar]
- 6.Traugott U. Characterization and distribution of lymphocyte subpopulations in multiple sclerosis plaques versus autoimmune demyelinating lesions. Springer Semin Immunopathol. 1985;8:71–95. doi: 10.1007/BF00197248. [DOI] [PubMed] [Google Scholar]
- 7.Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- 8.Traugott U. Multiple sclerosis: relevance of class I and class II MHC-expressing cells to lesion development. J Neuroimmunol. 1987;16:283–302. doi: 10.1016/0165-5728(87)90082-8. [DOI] [PubMed] [Google Scholar]
- 9.Oger JJF, Arnason BGW. Immunogenetics of multiple sclerosis. In: Panayi GS, David CS, editors. Immunogenetics. Butterworths; Boston: 1984. pp. 177–206. [Google Scholar]
- 10.Duquette P, Décary F, Pleines J, Boivin D, Lamoureux G, Cosgrove JBR, Lapierre Y. Clinical sub-groups of multiple sclerosis in relation to HLA: DR alleles as possible markers of disease progression. Can J Neurol Sci. 1985;12:106–110. doi: 10.1017/s0317167100046795. [DOI] [PubMed] [Google Scholar]
- 11.Stewart GJ, McLeod JG, Basten A, Bashir HV. HLA family studies and multiple sclerosis: a common gene, dominantly expressed. Hum Immunol. 1981;3:13–29. doi: 10.1016/0198-8859(81)90040-9. [DOI] [PubMed] [Google Scholar]
- 12.Bobowick AR, Kurtzke JF, Brody JA, Hrubec Z, Gillespie M. Twin study of multiple sclerosis: an epidemiologic inquiry. Neurology. 1978;28:978–987. doi: 10.1212/wnl.28.10.978. [DOI] [PubMed] [Google Scholar]
- 13.Williams A, Eldridge R, McFarland H, Houff S, Krebs H, McFarlin D. Multiple sclerosis in twins. Neurology. 1980;30:1139–1147. doi: 10.1212/wnl.30.11.1139. [DOI] [PubMed] [Google Scholar]
- 14.Ebers GC, Bulman DE, Sadovnick AD, Paty DW, Warren S, Hader W, Murray TJ, Seland TP, Duquette P, Grey T, Nelson R, Nicolle M, Brunet D. A population-based study of multiple sclerosis in twins. N Engl J Med. 1986;315:1638–1642. doi: 10.1056/NEJM198612253152603. [DOI] [PubMed] [Google Scholar]
- 15.Kinnunen E, Koskenvuo M, Kaprio J, Aho K. Multiple sclerosis in a nationwide series of twins. Neurology. 1987;37:1627–1629. doi: 10.1212/wnl.37.10.1627. [DOI] [PubMed] [Google Scholar]
- 16.Raine CS, Traugott U. Experimental autoimmune demyelination: chronic relapsing models and their therapeutic implications for multiple sclerosis. Ann NY Acad Sci. 1984;436:33–51. doi: 10.1111/j.1749-6632.1984.tb14774.x. [DOI] [PubMed] [Google Scholar]
- 17.Raine CS. Experimental allergic encephalomyelitis and experimental allergic neuritis. In: Vinken PJ, Bruyn GW, Klawans HL, Koetsier JC, editors. Handbook of Clinical Neurology. Vol 47: Demyelinating Diseases. Elsevier Science Publishing Company; New York: 1985. pp. 429–466. [Google Scholar]
- 18.Mokhtarian F, McFarlin DE, Raine CS. Adoptive transfer of myelin basic protein-sensitized T cells produces chronic relapsing demyelinating disease in mice. Nature. 1984;309:356–358. doi: 10.1038/309356a0. [DOI] [PubMed] [Google Scholar]
- 19.Sakai K, Namikawa T, Kunishita T, Yamanouchi K, Tabira T. Studies of experimental allergic encephalomyelitis by using encephalitogenic T cell lines and clones in euthymic and athymic mice. J Immunol. 1986;137:1527–1531. [PubMed] [Google Scholar]
- 20.Hafler DA, Benjamin DS, Burks J, Weiner HL. Myelin basic protein and proteolipid protein reactivity of brain- and cerebrospinal fluid-derived T cell clones in multiple sclerosis and postinfectious encephalomyelitis. J Immunol. 1987;139:68–72. [PubMed] [Google Scholar]
- 21.Bach MA. Immunoregulatory T cells in multiple sclerosis: markers and functions. Springer Semin Immunopathol. 1985;8:45–56. doi: 10.1007/BF00197246. [DOI] [PubMed] [Google Scholar]
- 22.Paty DW, Kastrukoff L, Morgan N, Hiob L. Suppressor T-lymphocytes in multiple sclerosis: analysis of patients with acute relapsing and chronic progressive disease. Ann Neurol. 1983;14:445–449. doi: 10.1002/ana.410140408. [DOI] [PubMed] [Google Scholar]
- 23.Hauser SL, Reinherz EL, Hoban CJ, Schlossman SF, Weiner HL. Immunoregulatory T-cells and lymphocytotoxic antibodies in active multiple sclerosis: weekly analysis over a six-month period. Ann Neurol. 1983;13:418–425. doi: 10.1002/ana.410130408. [DOI] [PubMed] [Google Scholar]
- 24.Kastrukoff LF, Paty DW. A serial study of peripheral blood T lymphocyte subsets in relapsing-remitting multiple sclerosis. Ann Neurol. 1984;15:250–256. doi: 10.1002/ana.410150308. [DOI] [PubMed] [Google Scholar]
- 25.Hafler DA, Fox DA, Manning ME, Schlossman SF, Reinherz EL, Weiner HL. In vivo activated T lymphocytes in the peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. N Engl J Med. 1985;312:1405–1411. doi: 10.1056/NEJM198505303122201. [DOI] [PubMed] [Google Scholar]
- 26.Hauser SL, Reinherz EL, Hoban CJ, Schlossman SF, Weiner HL. CSF cells in multiple sclerosis: monoclonal antibody analysis and relationship to peripheral blood T-cell subsets. Neurology. 1983;33:575–579. doi: 10.1212/wnl.33.5.575. [DOI] [PubMed] [Google Scholar]
- 27.Hafler DA, Buchsbaum M, Johnson D, Weiner HL. Phenotypic and functional analysis of T cells cloned directly from the blood and cerebrospinal fluid of patients with multiple sclerosis. Ann Neurol. 1985;18:451–458. doi: 10.1002/ana.410180407. [DOI] [PubMed] [Google Scholar]
- 28.Noronha A, Richman DP, Arnason BGW. Multiple sclerosis: activated cells in cerebrospinal fluid in acute exacerbations. Ann Neurol. 1985;18:722–725. doi: 10.1002/ana.410180617. [DOI] [PubMed] [Google Scholar]
- 29.Morimoto C, Hafler DA, Weiner HL, Letvin NL, Hagan M, Daley J, Schlossman SF. Selective loss of the suppressor-inducer T-cell subset in progressive multiple sclerosis: analysis with anti-2H4 monoclonal antibody. N Engl J Med. 1987;316:67–72. doi: 10.1056/NEJM198701083160202. [DOI] [PubMed] [Google Scholar]
- 30.Antel JP, Arnason BGW, Medof ME. Suppressor cell function in multiple sclerosis: correlation with clinical disease activity. Ann Neurol. 1979;5:338–342. doi: 10.1002/ana.410050406. [DOI] [PubMed] [Google Scholar]
- 31.Antel JP, Rosenkoetter M, Reder A, Oger JJ-F, Arnason BGW. Multiple sclerosis: relation of in vitro IgG secretion to T suppressor cell number and function. Neurology. 1984;34:1155–1160. doi: 10.1212/wnl.34.9.1155. [DOI] [PubMed] [Google Scholar]
- 32.Goust J-M, Hogan EL, Arnaud P. Abnormal regulation of IgG production in multiple sclerosis. Neurology. 1982;32:228–234. doi: 10.1212/wnl.32.3.228. [DOI] [PubMed] [Google Scholar]
- 33.Hauser SL, Ault KA, Levin MJ, Garovoy MR, Weiner HL. Natural killer cell activity in multiple sclerosis. J Immunol. 1981;127:1114–1117. [PubMed] [Google Scholar]
- 34.Rice GPA, Casali P, Merigan TC, Oldstone MBA. Natural killer cell activity in patients with multiple sclerosis given α interferon. Ann Neurol. 1983;14:333–338. doi: 10.1002/ana.410140312. [DOI] [PubMed] [Google Scholar]
- 35.Hirsch RL, Johnson KP. The effect of recombinant alpha2-interferon on defective natural killer cell activity in multiple sclerosis. Neurology. 1985;35:597–600. doi: 10.1212/wnl.35.4.597. [DOI] [PubMed] [Google Scholar]
- 36.Johnson RT. Viral Infections of the Nervous System. Raven Press; New York: 1982. [Google Scholar]
- 37.Haase AT, Ventura P, Gibbs CJ, Jr, Tourtellotte WW. Measles virus nucleotide sequences: detection by hybridization in situ. Science. 1981;212:672–675. doi: 10.1126/science.7221554. [DOI] [PubMed] [Google Scholar]
- 38.Hershey LA, Trotter JL. The use and abuse of the cerebrospinal fluid IgG profile in the adult: a practical evaluation. Ann Neurol. 1980;8:426–434. doi: 10.1002/ana.410080415. [DOI] [PubMed] [Google Scholar]
- 39.Miller JR, Burke AM, Bever CT. Occurrence of oligoclonal bands in multiple sclerosis and other CNS diseases. Ann Neurol. 1983;13:53–58. doi: 10.1002/ana.410130112. [DOI] [PubMed] [Google Scholar]
- 40.Vartdal F, Vandvik B, Norrby E. Viral and bacterial antibody responses in multiple sclerosis. Ann Neurol. 1980;8:248–255. doi: 10.1002/ana.410080305. [DOI] [PubMed] [Google Scholar]
- 41.Adams JM, Imagawa DT. Measles antibodies in multiple sclerosis. Proc Soc Exp Biol Med. 1962;111:562–566. doi: 10.3181/00379727-111-27855. [DOI] [PubMed] [Google Scholar]
- 42.Vandvik B, Norrby E. Oligoclonal IgG antibody response in the central nervous system to different measles virus antigens in subacute sclerosing panencephalitis. Proc Natl Acad Sci USA. 1973;70:1060–1063. doi: 10.1073/pnas.70.4.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Link H, Laurenzi MA, Frydén A. Viral antibodies in oligoclonal and polyclonal IgG synthesized within the central nervous system over the course of mumps meningitis. J Neuroimmunol. 1981;1:287–298. doi: 10.1016/0165-5728(81)90032-1. [DOI] [PubMed] [Google Scholar]
- 44.Mattson DH, Roos RP, Arnason BGW. Isoelectric focusing of IgG eluted from multiple sclerosis and subacute sclerosing panencephalitis brains. Nature. 1980;287:335–337. doi: 10.1038/287335a0. [DOI] [PubMed] [Google Scholar]
- 45.Gerhard W, Taylor A, Wroblewska Z, Sandberg-Wollheim M, Koprowski H. Analysis of a predominant immunoglobulin population in the cerebrospinal fluid of a multiple sclerosis patient by means of an anti-idiotypic hybridoma antibody. Proc Natl Acad Sci USA. 1981;78:3225–3229. doi: 10.1073/pnas.78.5.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerhard W, Taylor A, Sandberg-Wollheim M, Koprowski H. Longitudinal analysis of three intrathecally produced immunoglobulin subpopulations in an MS patient. J Immunol. 1985;134:1555–1560. [PubMed] [Google Scholar]
- 47.Dal Canto MC, Rabinowitz SG. Experimental models of virus-induced demyelination of the central nervous system. Ann Neurol. 1982;11:109–127. doi: 10.1002/ana.410110202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCullough B, Krakowka S, Koestner A. Experimental canine distemper virus-induced demyelination. Lab Invest. 1974;31:216–222. [PubMed] [Google Scholar]
- 49.Haase AT. Pathogenesis of lentivirus infections. Nature. 1986;322:130–136. doi: 10.1038/322130a0. [DOI] [PubMed] [Google Scholar]
- 50.Lampert PW, Sims JK, Kniazeff AJ. Mechanism of demyelination in JHM virus encephalomyelitis: electron microscopic studies. Acta Neuropathol (Berl) 1973;24:76–85. doi: 10.1007/BF00691421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watanabe R, Wege H, ter Meulen V. Adoptive transfer of EAE-like lesions from rats with coronavirus-induced demyelinating encephalomyelitis. Nature. 1983;305:150–153. doi: 10.1038/305150a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suckling AJ, Pathak S, Jagelman S, Webb HE. Virus-associated demyelination: a model using avirulent Semliki Forest virus infection of mice. J Neurol Sci. 1978;39:147–154. doi: 10.1016/0022-510x(78)90195-8. [DOI] [PubMed] [Google Scholar]
- 53.Lipton HL. Theiler's virus infection in mice: an unusual biphasic disease process leading to demyelination. Infect Immun. 1975;11:1147–1155. doi: 10.1128/iai.11.5.1147-1155.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez M, Oleszak E, Leibowitz J. Theiler's murine encephalomyelitis: a model of demyelination and persistence of virus. CRC Crit Rev Immunol. 1987;7:325–365. [PubMed] [Google Scholar]
- 55.Rodriguez M, Leibowitz JL, Lampert PW. Persistent infection of oligodendrocytes in Theiler's virus-induced encephalomyelitis. Ann Neurol. 1983;13:426–433. doi: 10.1002/ana.410130409. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez M, Pease LR, David CS. Immune-mediated injury of virus-infected oligodendrocytes: a model of multiple sclerosis. Immunol Today. 1986;7:359–363. doi: 10.1016/0167-5699(86)90025-3. [DOI] [PubMed] [Google Scholar]
- 57.Osame M, Matsumoto M, Usuku K, Izumo S, Ijichi N, Amitani H, Tara M, Igata A. Chronic progressive myelopathy associated with elevated antibodies to human T-lymphotropic virus type I and adult T-cell leukemialike cells. Ann Neurol. 1987;21:117–122. doi: 10.1002/ana.410210203. [DOI] [PubMed] [Google Scholar]
- 58.Vernant JC, Maurs L, Gessain A, Barin F, Gout O, Delaporte JM, Sanhadji K, Buisson G, de-Thé G. Endemic tropical spastic paraparesis associated with human T-lymphotropic virus type I: a clinical and seroepidemiological study of 25 cases. Ann Neurol. 1987;21:123–130. doi: 10.1002/ana.410210204. [DOI] [PubMed] [Google Scholar]
- 59.Koprowski H, DeFreitas EC, Harper ME, Sandberg-Wollheim M, Sheremata WA, Robert-Guroff M, Saxinger CW, Feinberg MB, Wong-Staal F, Gallo RC. Multiple sclerosis and human T-cell lymphotropic retroviruses. Nature. 1985;318:154–160. doi: 10.1038/318154a0. [DOI] [PubMed] [Google Scholar]
- 60.Karpas A, Kämpf U, Sidèn Ã, Koch M, Poser S. Lack of evidence for involvement of known human retroviruses in multiple sclerosis (letter to the editor) Nature. 1986;322:177–178. doi: 10.1038/322177a0. [DOI] [PubMed] [Google Scholar]
- 61.Hauser SL, Aubert C, Burks JS, Kerr C, Lyon-Caen O, de The G, Brahic M. Analysis of human T-lymphotropic virus sequences in multiple sclerosis tissue (letter to the editor) Nature. 1986;322:176–177. doi: 10.1038/322176a0. [DOI] [PubMed] [Google Scholar]
- 62.Ohta M, Ohta K, Mori F, Nishitani H, Saida T. Sera from patients with multiple sclerosis react with human T cell lymphotropic virus-I gag proteins but not env proteins—Western blotting analysis. J Immunol. 1986;137:3440–3443. [PubMed] [Google Scholar]
- 63.Reddy EP, Sandberg-Wollheim M, Mettus RV, Ray PE, DeFreitas E, Koprowski H. Amplification and molecular cloning of HTLV-I sequences from DNA of multiple sclerosis patients. Science. 1989;243:529–533. doi: 10.1126/science.2536193. [DOI] [PubMed] [Google Scholar]
- 64.Arnason BGW. Relevance of experimental allergic encephalomyelitis to multiple sclerosis. Neurol Clin. August 1983;1:765–782. [PubMed] [Google Scholar]
- 65.McCarron RM, Kempski O, Spatz M, McFarlin DE. Presentation of myelin basic protein by murine cerebral vascular endothelial cells. J Immunol. 1985;134:3100–3103. [PubMed] [Google Scholar]
- 66.Hofman FM, von Hanwehr RI, Dinarello CA, Mizel SB, Hinton D, Merrill JE. Immunoregulatory molecules and IL 2 receptors identified in multiple sclerosis brain. J Immunol. 1986;136:3239–3245. [PubMed] [Google Scholar]
- 67.Wisniewski HM, Bloom BR. Primary demyelination as a nonspecific consequence of a cell-mediated immune reaction. J Exp Med. 1975;141:346–359. doi: 10.1084/jem.141.2.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cammer W, Bloom BR, Norton WT, Gordon S. Degradation of basic protein in myelin by neutral proteases secreted by stimulated macrophages: a possible mechanism of inflammatory demyelination. Proc Natl Acad Sci USA. 1978;75:1554–1558. doi: 10.1073/pnas.75.3.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ludwin SK, Johnson ES. Evidence for a “dying-back” gliopathy in demyelinating disease. Ann Neurol. 1981;9:301–305. doi: 10.1002/ana.410090316. [DOI] [PubMed] [Google Scholar]
- 70.Rodriguez M. Virus-induced demyelination in mice: “dying back” of oligodendrocytes. Mayo Clin Proc. 1985;60:433–438. doi: 10.1016/s0025-6196(12)60865-9. [DOI] [PubMed] [Google Scholar]