

Mutations in angiotensin-converting enzyme 2 are shown to predispose mice to colitis as a consequence of neutral amino acid malabsorption and a change in the resident microbiota; these results could explain how protein malnutrition — affecting up to one billion people — leads to intestinal inflammation.
Supplementary information
The online version of this article (doi:10.1038/nature11228) contains supplementary material, which is available to authorized users.
Subject terms: Inflammation, Pathogenesis, Gastrointestinal diseases, Malnutrition
Hunger pangs: how malnutrition leads to intestinal disorders
Malnutrition affects many millions of people in the developing world and remains a problem in wealthy nations, especially for disadvantaged groups. In many cases, it is the associated diarrhoea and intestinal inflammation that cause morbidity and death. A study published in this issue presents a molecular explanation for the increased susceptibility to intestinal inflammation in malnutrition. Angiotensin converting enzyme 2 (ACE2), which has a central role in blood-pressure regulation and has been implicated in diabetes, heart failure and viral infection, is shown to influence dietary amino-acid homeostasis, innate immunity, gut microbial ecology and susceptibility to colitis. Mice deficient in this enzyme show impaired tryptophan metabolism and develop colitis, which is alleviated by dietary tryptophan and its metabolite, nicotinamide. This surprising result explains nutritional effects that have been known for centuries and provides a molecular link between malnutrition and the intestinal microbiome.
Supplementary information
The online version of this article (doi:10.1038/nature11228) contains supplementary material, which is available to authorized users.
Abstract
Malnutrition affects up to one billion people in the world and is a major cause of mortality1,2. In many cases, malnutrition is associated with diarrhoea and intestinal inflammation, further contributing to morbidity and death2. The mechanisms by which unbalanced dietary nutrients affect intestinal homeostasis are largely unknown. Here we report that deficiency in murine angiotensin I converting enzyme (peptidyl-dipeptidase A) 2 (Ace2), which encodes a key regulatory enzyme of the renin-angiotensin system (RAS), results in highly increased susceptibility to intestinal inflammation induced by epithelial damage. The RAS is known to be involved in acute lung failure3, cardiovascular functions4 and SARS infections5. Mechanistically, ACE2 has a RAS-independent function, regulating intestinal amino acid homeostasis, expression of antimicrobial peptides, and the ecology of the gut microbiome. Transplantation of the altered microbiota from Ace2 mutant mice into germ-free wild-type hosts was able to transmit the increased propensity to develop severe colitis. ACE2-dependent changes in epithelial immunity and the gut microbiota can be directly regulated by the dietary amino acid tryptophan. Our results identify ACE2 as a key regulator of dietary amino acid homeostasis, innate immunity, gut microbial ecology, and transmissible susceptibility to colitis. These results provide a molecular explanation for how amino acid malnutrition can cause intestinal inflammation and diarrhoea.
Supplementary information
The online version of this article (doi:10.1038/nature11228) contains supplementary material, which is available to authorized users.
Main
The RAS has now been studied for more than a century. It is regulated by the opposing actions of two key carboxypeptidases, angiotensin converting enzyme (ACE) and ACE2 (refs 4, 6, 7). In addition to its catalytic activity, we and others have recently found that ACE2 associates with the neutral amino acid transporter B0AT1 and that ACE2 is required for expression of this transporter on the luminal surface of intestinal epithelial cells8,9. Variants in the gene that encodes B0AT1 (SLC6A19) have been identified as a cause of Hartnup’s disease10, a rare autosomal recessive disorder associated with pellagra-like symptoms including diarrhoea that manifests under conditions of malnutrition11. However, the in vivo function of ACE2 in the gut epithelium remained to be investigated.
Morphological and ultra-structural analysis of the small and large intestine of Ace2 knockout mice (see below) did not reveal any alterations. When we challenged such mice with dextran sodium sulphate (DSS), an irritant that disrupts the intestinal epithelial barrier and results in colitis12, a profoundly increased inflammatory reaction was observable compared to wild-type littermates. We detected an enhanced infiltration of inflammatory cells, significant shortening of the colon length, increased intestinal bleeding, crypt damage, weight loss and severe diarrhoea, resulting in an overall increased disease activity13 (Fig. 1a–c, Supplementary Fig. 1). Similar results were obtained using trinitrobenzene sulphonic acid (TNBS)-induced colitis (Supplementary Fig. 2). Thus, genetic inactivation of the key RAS enzyme ACE2 results in severe colitis following intestinal injury. We note that Ace2 deficiency was investigated in male mice (Ace2−/y, the Ace2 gene being located on the X chromosome) throughout this study, however, we observed a similar phenotype in female Ace2−/− mice (not shown).
Figure 1. Ace2 deficiency and protein malnutrition worsen DSS-induced colitis.
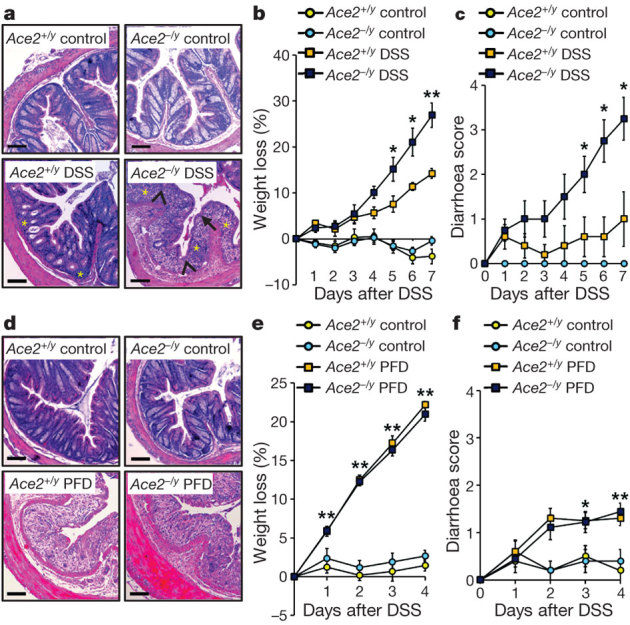
a, Colon histopathology, b, percentage weight loss, and c, diarrhoea scores in control and DSS-treated Ace2+/y and Ace2−/y littermates. In a, note crypt damage (arrowheads), ulcerations (arrow), and infiltration of inflammatory cells (asterisks) in DSS-treated Ace2−/y mice. Haematoxylin and eosin staining on day 7 after DSS challenge. Scale bars, 100 μm. d, Colon histopathology (haematoxylin and eosin staining, day 4 after DSS challenge; scale bars, 100 μm), e, percentage weight loss, and f, diarrhoea scores of DSS-treated Ace2+/y and Ace2−/y littermates fed either normal chow (Control) or a protein free diet (PFD; <0.2% protein). All values are mean ± s.e.m. of 5–9 mice per group. *P < 0.05, **P < 0.01 comparing DSS-treated Ace2+/y with Ace2−/y littermates, or Ace2+/y mice on normal diet with those on PFD (paired-t-test).
To test whether the severe colitis phenotype is due to the catalytic activity of ACE2, we treated mice with recombinant soluble ACE2 (rsACE2). We and others have shown that rsACE2 rescues virtually all previously reported in vivo ACE2 functions within the RAS system3,14. As it lacks the transmembrane domain, soluble ACE2 cannot associate with the neutral amino acid transporter B0AT1 and thus cannot stabilize its cell-surface localization8. Treatment of mice with rsACE2 did not rescue the severe DSS-induced colitis phenotype of Ace2 mutant mice (Supplementary Fig. 3a–h). One in vivo consequence of Ace2 deletion within the RAS is accumulation of angiotensin II (ANGII), which then primarily acts on the G-protein-coupled AT1a receptor (Agtr1a)3. We indeed observed increased AngII levels in the colon of DSS-treated Ace2 mutant mice that were reduced to background levels after treatment with rsACE2 (Supplementary Fig. 3i). To rule out the possibility that locally increased AngII might cause severe colitis, we generated Agtr1a−/−Ace2−/y mice. Deletion of Agtr1 did not rescue the severe DSS-induced colitis (Supplementary Fig. 4), indicating that these effects are independent of the classical RAS system. In addition to cleaving AngII, ACE2 exhibits catalytic activity towards a second peptide system, Apelin15. However, DSS-induced colitis was not altered in mice carrying genetic mutations in Apelin (Supplementary Fig. 5) or its receptor Apj (Supplementary Fig. 6). Thus, the catalytic activity of ACE2, essential for its function in the RAS and Apelin cleavage, has no overt role in DSS-induced intestinal inflammation.
It had been reported that the RAS can control immune functions16. However, in unchallenged Ace2 mutant mice, we did not observe any apparent differences in immune cell populations of the colon and small intestine (not shown). TLR1–9 mRNA levels in jejunum, ileum, and colon were largely similar in Ace2 deficient and wild-type mice, with the exception of TLR5 and TLR9 in the colon (Supplementary Fig. 7). Transplantation of bone marrow from Ace2 mutant mice (knock-out, KO) into wild-type (WT) mice (KO→WT) did not result in any obvious effect on DSS-induced colitis as compared to WT→WT transplant recipients. By contrast, reconstitution of Ace2 mutant mice with WT bone-marrow (WT→KO) led to an increased severity of DSS-induced colitis that was indistinguishable from the KO→KO control group (Supplementary Fig. 8). Thus, ACE2 exerts its role on intestinal inflammation in non-haematopoietic cells.
ACE2 expression was primarily mapped to the luminal surface of differentiated small intestinal epithelial cells, whereas lower ACE2 expression is observed in crypt cells and the colon (Supplementary Figs 9a, b and 10a). Ace2 inactivation had no overt effect on basal proliferation of small intestinal or colon crypt cells (Supplementary Fig. 10a, b) nor did it affect apoptosis rates of intestinal epithelial cells (Supplementary Fig. 10a, c). ACE levels were slightly, albeit not significantly, increased in the jejunum of Ace2−/y mice (Supplementary Fig. 9c, d). As reported previously8, protein expression of the neutral amino acid transporter B0AT1 was absent in the small intestine of Ace2 mutant mice, whereas mRNA expression was not affected (Supplementary Figs 9e, f and 11a). Owing to the lack of intestinal B0AT1 protein expression, serum levels of the neutral amino acids valine (Val), threonine (Thr) and tyrosine (Tyr), and the essential amino acid tryptophan (Trp) were markedly reduced in Ace2−/y mice (Supplementary Fig. 11b). The marked reduction in tryptophan correlates with human data, as patients suffering from Hartnup’s disease caused by Slc6a19 mutations or malnutrition such as in anorexia nervosa exhibit reduced tryptophan serum levels17,18.
ACE2 is a chimaeric protein that emerged from the duplication and fusion of two genes: it is homologous with ACE at the catalytic domain and with collectrin (TMEM27) in the membrane proximal domain19. Inactivation of collectrin in mice (Tmem27−/−) results in a near complete downregulation of apical amino acid transporters such as B0AT1 in the kidney, thereby regulating renal amino acid re-absorption19. By contrast, ACE2 associates with B0AT1 on the luminal surface of intestinal epithelial cells, that is, ACE2 and B0AT1 regulate the uptake of neutral amino acids in the intestine where collectrin is not expressed (Supplementary Figs 11a, c and 12). In both Tmem27−/− and Ace2−/y mutant mice, tryptophan is reduced in the serum (Supplementary Fig 11b, and ref. 19). To test whether impaired re-absorption of amino acids in the kidney might also result in altered susceptibility to colitis, we challenged Tmem27−/− mice with DSS. However, genetic ablation of Tmem27 did not affect DSS-induced colitis (Supplementary Fig. 13). Although genetic inactivation of both Ace2 or Tmem27 result in a similar deregulation of amino acids in the serum, functionally only the loss of ACE2 affects the susceptibility to intestinal inflammation.
We, therefore, speculated that the function of ACE2 in intestinal uptake of dietary amino acids might underlie the observed phenotype. To test this hypothesis, we fed Ace2 mutant and control mice a protein free-diet (PFD) to eliminate dietary amino acids and challenged these mice with DSS at a dose (1%) that triggers only very mild colitis. Basal weight loss was comparable among control and Ace2 mutant mice during the period of the PFD (Supplementary Fig. 14a). Moreover, average food intake did not show a significant difference between Ace2 deficient and control mice fed PFD or normal chow (Supplementary Fig. 14b). Importantly, PFD markedly worsened DSS-triggered colitis in wild-type mice to levels seen in Ace2 mutants (Fig. 1d–f; Supplementary Fig. 14c–g). Thus, protein malnutrition alters the severity of DSS-induced intestinal inflammation.
The general lack of dietary protein might induce broad effects on several organ systems. We, therefore, set out to define whether a specific amino acid might be responsible for the severe inflammatory phenotype. Because serum tryptophan was markedly decreased in Ace2 mutant mice, we focused on this essential amino acid. Tryptophan is required for the in vivo generation of nicotinamide (also known as vitamin B3 or niacin)17, and insufficient niacin or tryptophan in the diet is the cause of pellagra, a disease still endemic in many countries with protein malnutrition20. More than 90% of pellagra patients develop colitis11 and for nearly 80 years nicotinamide has been used as treatment for pellagra. In Ace2 mutant mice, nicotinamide almost completely alleviated the severe colitis and diarrhoea (Fig. 2a-c; Supplementary Fig. 15). To demonstrate that the severe colitis in Ace2 deficient mice is due to impaired tryptophan uptake, we provided tryptophan in form of a Gly-Trp dipeptide to bypass the loss of the single amino acid transporter B0AT1. Dietary dipeptidic tryptophan (a Trp+ diet) restored serum tryptophan levels (Supplementary Fig. 16a) and rescued the enhanced DSS-susceptibility of Ace2−/y mice (Fig. 2d–f; Supplementary Fig. 16b–d). A tryptophan-free (Trp−) diet resulted in a very marked increase in susceptibility to DSS-induced inflammation (Supplementary Fig. 17a–h). In addition, a Trp− diet markedly worsened weight loss in wild-type mice challenged with TNBS (Supplementary Fig. 17i–l). Our results show that deficiency of Ace2 causes a critical impairment of local tryptophan homeostasis which alters the susceptibility to intestinal inflammation.
Figure 2. Rescue of severe colitis with nicotinamide or tryptophan di-peptides.
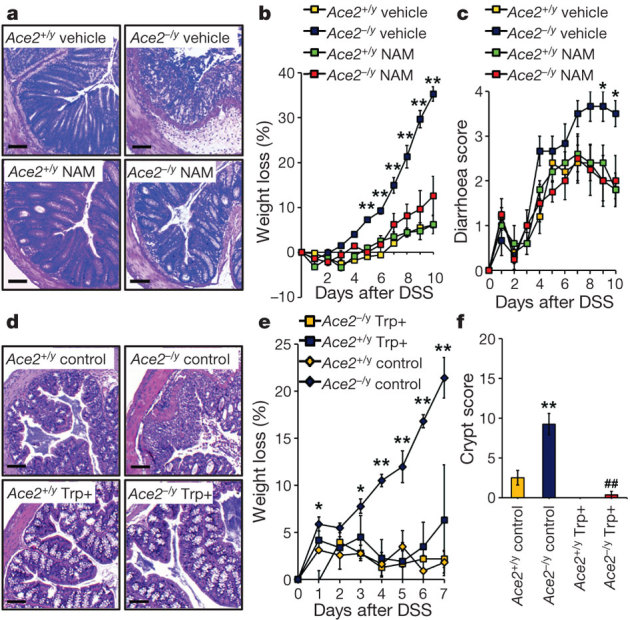
a, Colon histopathology (haematoxylin and eosin, day 10 after DSS challenge; scale bars, 100 μm), b, percentage weight loss, and c, diarrhoea scores of DSS-treated Ace2+/y and Ace2−/y littermates that received vehicle or nicotinamide (NAM) in their drinking water. Nicotinamide treatment was started 3 days before DSS challenge. d, Colon histopathology (haematoxylin and eosin, day 7; scale bars, 100 μm), e, percentage weight loss, and f, crypt injury scores of Ace2+/y and Ace2−/y mice fed a di-peptidic tryptophan diet (Trp+) or normal chow (Control). Values are mean ± s.e.m. of 3–10 mice per group. *P < 0.05, **P < 0.01 comparing Ace2−/y mice on a normal diet with those on Trp+ diet, or vehicle- versus nicotinamide-treated Ace2−/y mice. ##P < 0.01 comparing Ace2+/y versus Ace2−/y mice (paired-t-test).
It is conceivable that an altered amino acid availability in the epithelium could lead to decreased regenerative responses and repair mechanisms after epithelial injury21,22. However, proliferation rates and cell death of small intestinal and colon epithelial cells were comparable between control and Ace2 mutant mice following DSS challenge (Supplementary Fig. 18). However, expression of multiple antimicrobial peptides was markedly reduced in isolated gut ileal epithelial cells from unchallenged Ace2 mutant mice (Fig. 3a; Supplementary Fig. 19). A Trp− diet for wild-type mice also resulted in a significant downregulation of antimicrobial peptides (Fig. 3b). Similar downregulation of antimicrobial peptides was observed on feeding wild-type mice a PFD (Supplementary Fig. 20a). By contrast, dietary nicotinamide (Supplementary Fig. 20b–g) and a Trp+ diet (Fig. 3c and Supplementary Fig. 20h, i) triggered induction of antimicrobial peptides in intestinal epithelial cells from Ace2 mutant and wild-type mice. Thus, dietary tryptophan controls expression of small intestinal antimicrobial peptides.
Figure 3. Tryptophan controls antimicrobial peptides and mTOR activity.
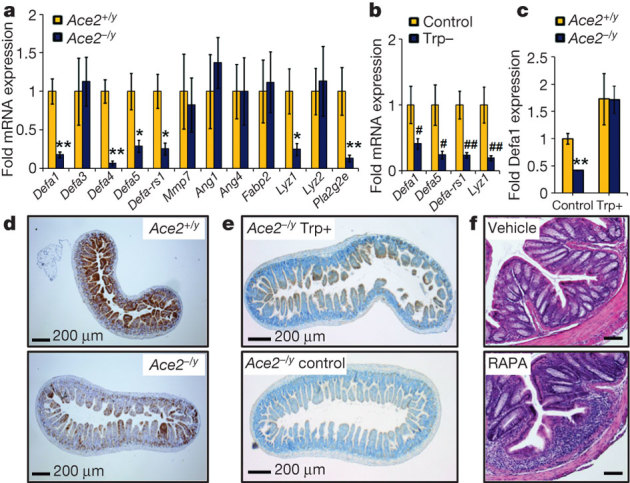
a, b, mRNA expression levels of antimicrobial peptides in epithelial cells isolated from the small intestine of a, unchallenged Ace2+/y and Ace2−/y littermates, and b, Ace2+/y mice fed a tryptophan-free diet (Trp−) or normal chow (Control). c, mRNA expression levels of antimicrobial peptide Defa1 in Ace2+/y and Ace2−/y littermates fed a Trp+ diet or normal chow (Control) for 10 days. d, e, Immunohistochemistry to detect levels of phosphorylated S6 (brown) in the small intestine of d, unchallenged Ace2+/y and Ace2−/y littermates or e, Ace2−/y mice fed a Trp+ or normal chow diet (Control). Scale bars, 200 μm. f, Colon histopathology (haematoxylin and eosin, day 8; scale bars, 100 μm) of DSS treated wild-type mice receiving vehicle or rapamycin (RAPA) i.p., initiated 6 days before DSS challenge. Values are mean ± s.e.m. of 5–6 mice per group. *P < 0.05, **P < 0.01 comparing Ace2+/y with Ace2−/y mice; #P < 0.05, ##P < 0.01 comparing Ace2+/y mice on normal diet with those on Trp− diet (paired-t-test).
Amino acids and nicotinamide can activate mTOR, which is involved in cell proliferation, survival, protein synthesis and transcription23. In epithelial intestinal cells from unchallenged Ace2 mutant mice, we observed markedly impaired p70S6 kinase activity (Supplementary Fig. 21a) as well as reduced S6 phosphorylation (Fig. 3d and Supplementary Fig. 21b), indicative of reduced mTOR activity. A similar reduction of mTOR activity was recently reported in mice lacking B0AT1 (ref. 24). Administration of a Trp+ diet resulted in increased mTOR activity in the small intestine of Ace2 mutant mice (Fig. 3e). In vivo inhibition of mTOR with rapamycin in wild-type mice resulted in a significant downregulation of antimicrobial peptide expression, an effect that could not be rescued by nicotinamide (Supplementary Fig. 22a). Moreover, administration of rapamycin before the first challenge with DSS increased the severity of colitis (Fig. 3f; Supplementary Fig. 22b–h). Notably, although acute mTOR inhibition may have beneficial effects in murine colitis models25, mTOR blockade has not been proven successful in human clinical trials in inflammatory bowel disease26. Thus, at the molecular level, nicotinamide and dietary tryptophan appear to exert their effects on intestinal antimicrobial peptides and colitis via the mTOR pathway.
How does ACE2 regulated uptake of tryptophan in the small intestine affect DSS-induced inflammation in the colon? It has been shown that α-defensins secreted into the ileum lumen persist in a functional state throughout the entire gut27 and that alterations in antimicrobial peptides can affect the ecology of the small and large bowel microbiota28. We therefore performed deep profiling of the intestinal microbiome of Ace2 mutant and wild-type littermates using 16S rDNA fingerprinting. The luminal ileocaecal microbiome of Ace2 mutant mice was markedly altered, as shown by weighted (Bray–Curtis) (Fig. 4a) and unweighted (Unifrac) (Fig. 4b) analyses. Distinct operational taxonomical units (OTUs) are overrepresented in Ace2 mutant mice (Fig. 4c; Supplementary Tables 1 and 2). Rapamycin treatment resulted in a distinct alteration of the ileocaecal gut microbiome in wild-type animals; but the altered microbiome was more closely related to that found in untreated wild-type animals than that found in untreated Ace2−/y animals (Supplementary Fig. 23; Supplementary Tables 3 and 4). Rapamycin acts on many different cell types that in addition to the changes in intestinal epithelial cells could alter microbiota composition29. Importantly, both Trp+ diet and nicotinamide treatment reverted the composition of the intestinal microbiota of Ace2 mutant mice to be more similar to that of untreated wild-type littermates (Fig. 4a–c, Supplementary Fig. 24, Supplementary Tables 1, 2, 5 and 6). Thus, de-regulation of tryptophan amino acid homeostasis in Ace2 mutant mice alters the intestinal microbiome.
Figure 4. Altered gut bacteria from Ace2 mutant mice can confer susceptibility to colitis.
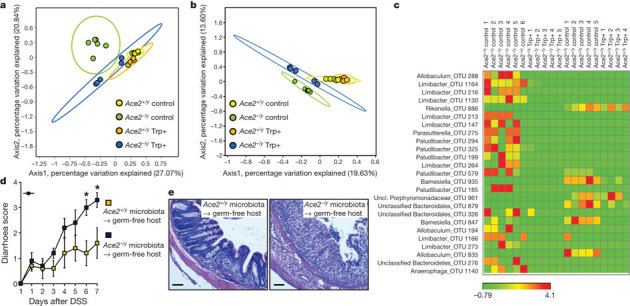
a, b, Principal coordinate analysis plots; a, calculated by Bray–Curtis algorithm and b, based on unweighted UniFrac analysis. Plots show the similarity among ileocaecal bacterial communities in Ace2+/y and Ace2−/y mice fed a Trp+ diet or normal chow (Control) for 10 days. Only the two axes with high R2 values are shown (axis 1, R2 = 0.335; axis 2, R2 = 0.8116). Each dot represents data from an individual animal. c, Comparison of microbial communities in Ace2+/y and Ace2−/y mice fed a Trp+ diet or normal chow (Control). The heat map depicts abundance of the top 25 species level OTUs contributing significantly to the axis shown in the weighted principal coordinate analysis plot (a). d, Diarrhoea scores and e, colon histopathology (haematoxylin and eosin, day 7; scale bars, 100 μm) of DSS challenged germ-free mice that received intestinal microbiota from Ace2+/y or Ace2−/y littermates. Values are mean ± s.e.m. of 4–6 mice per group. *P < 0.05 (paired-t-test).
Consistent with an alteration in the intestinal microbiome, antibiotic treatment of Ace2 mutant mice alleviated the severe colitis (Supplementary Fig. 25). Finally, we performed gut microbiome transplants from control and Ace2 mutant mice into germ-free hosts followed by DSS challenge. Transplantation of the ileocaecal gut microbiota from Ace2 mutant mice transmitted the inflammatory phenotype to wild-type hosts, which was not the case for mice that were colonized with the microbiota of Ace2+/y mice (Fig. 4d, e; Supplementary Fig. 26a–f). Expression of antimicrobial peptides in wild-type mice did not change significantly upon transplantation of Ace2+/y or Ace2−/y microbiota within the timeframe of analysis (Supplementary Fig. 26g).
We propose the following scenario for how malnutrition and tryptophan deficiency can cause diarrhoea and colitis. Dietary tryptophan is primarily absorbed via the B0AT1/ACE2 transport pathway on the luminal surface of small intestinal epithelial cells. This results in the activation of mTOR, either directly through nutrient sensing and/or through the tryptophan-nicotinamide pathway. mTOR then regulates expression of antimicrobial peptides, which affect the intestinal composition of the gut microbiota. Whether pathways other than mTOR and antimicrobial peptides also contribute to the altered microbiome needs to be explored. Our results further indicate that under conditions of intestinal injury, such an altered microbiome contributes to the severity of colitis. Importantly, our data indicate that the essential amino acid tryptophan and its metabolite nicotinamide are key regulators of gut microbiota and of propensity to inflammation.
Malnutrition is a major global health burden, affecting up to one billion people in the world and its consequences such as colitis and diarrhoea are often fatal1,2. In addition, chronic inflammatory conditions of the intestine, cachexia, anorexia nervosa, or amino acid mal-absorption constitute severe clinical problems30. Our results provide novel mechanistic insights into how protein malnutrition can lead to colitis and diarrhoea. Our data also identify a molecular crosstalk between the RAS system and intestinal amino acid homeostasis via ACE2—and provide a direct link between dietary amino acid metabolism and innate immunity, the composition of the intestinal microbiota, and susceptibility to colitis.
Methods Summary
Colitis models
Ace2, Tmem27, Agtr1, Apj and Apelin mutant mice have been described. Mice were given protein-free and tryptophan-free diet, tryptophan di-peptides, nicotinamide, rapamycin, or were treated with catalytically active murine ACE2 protein and challenged with DSS or TNBS. Bone marrow transplantations were performed in lethally irradiated Ace2 mutant and wild-type mice. For microbiota transplantation, caecal and ileum extracts from Ace2+/y or Ace2−/y mice were transplanted to germ-free hosts. Severity of colitis was monitored as described13. All experiments were performed in accordance with institutional guidelines.
Expression analyses
Small and large intestinal epithelial cells and mucosal cells were purified and gene expression assessed using quantitative RT–PCR. Protein expression was determined by western blot and localized using immunohistochemistry. Immune cell populations were determined by FACS.
Microbiota sequencing
Genomic DNA was extracted from the terminal ileum, amplified by PCR, and sequenced using Roche 454 Titanium chemistry. All sequence reads were filtered for quality and length using PANGEA. Sequences were clustered using average neighbour algorithm into species level operational taxonomical units (OTUs). Distance matrices between samples were generated based on weighted (Bray–Curtis similarity) and non-weighted (unweighted UniFrac) algorithms and shown as principal coordinate analysis (PCoA). Spearman correlations and similarity percentage analysis (SIMPER) were performed to determine significant differences between bacterial communities among genotype and diet groups.
Online Methods
Mice
Ace2 (ref. 4), Agtr1a (ref. 31), Apelin (ref. 32), Apj−/− (ref. 33) and Collectrin (ref. 19) mutant mice have been described previously. All mutant strains were backcrossed onto a C57BL/6 background more than 6 generations. Apj−/− mice were on a mixed C57BL/6 × CBA background. In all experiments, only sex-, age- and background-matched mice were used as controls. Mice were handled and all experiments performed in accordance with institutional guidelines.
Colitis models
Acute colitis in C57BL/6 background mice was induced with 1.5% (w/v) DSS (molecular mass 36–50 kDa; MP Biologicals) dissolved in drinking water ad libitum for the experimental days 1–7 followed by normal drinking water until the end of the experiment. For mixed C57BL/6 × CBA background mice, colitis was induced by feeding mice 5% (w/v) DSS. The DSS solutions were made fresh every day. TNBS colitis was induced by intrarectal administration of 0.1 ml TNBS (5 mg per mouse in 40% ethanol), through a feeding needle (Fine Science Tools) 3.5 cm proximal to the anal verge as described previously34. Body weight, diarrhoea, and occult blood in stool (Hemoccult test) were determined daily. Diarrhoea scores were: 0, no diarrhoea; 1, mild diarrhoea; 2, severe watery diarrhoea; 3, mild diarrhoea with blood; 4, severe watery diarrhoea with blood. Colon length was determined at the end of the experiments. Crypt scores, inflammation scores, and disease activity indexes were determined as previously described13. For PFD experiment, normal chow (20.8% protein) and PFD (<0.2% protein) were obtained from Ssniff GmbH. Mice were fed the protein-free, isocaloric diet for 3 days before start of the 1% (w/v) DSS treatment. Owing to the severe diarrhoea in PFD fed mice, the experiment had to be stopped for ethical reasons on day 4. For nicotinamide (NAM) treatment, mice were given NAM (Sigma) in their drinking water (0.4 g l−1) from 3 days before the first DSS treatment until the end of the experiment. For rapamycin (RAPA) treatment, rapamycin (2 mg kg−1 d−1; LC Laboratories) or vehicle were administered intraperitoneally (i.p.) daily for 6 days with or without NAM treatment. The solvent for rapamycin was 0.2% sodium carboxymethylcellulose, 0.25% polysorbate-80 in water. For tryptophan rescue experiments, standard food was supplemented with glycyl-l-tryptophan (Gly-Trp) hydrate (G0144, TCI Europe) (10 mg per g dry food) daily for 10 days followed by 1.5% (w/v) DSS treatment. Tryptophan-free diet was purchased from Oriental Yeast and was fed for 18 days before DSS challenge. For antibiotic treatment, 4-week-old mice were placed on broad spectrum antibiotics ampicillin (1 g l−1) and neomycin (0.5 g l−1) in drinking water for 4 weeks followed by 1.5% (w/v) DSS treatment. For microbiota transplantation, caecal and ileum extracts were pooled from Ace2 mutant mice and ACE2 expressing littermates, suspended in 3 ml PBS, and administered (0.1 ml per mouse) immediately to sterile-packed 4-week-old Swiss-Webster germ-free mice (Taconic). Transplanted mice were maintained in sterile cages for 2 weeks followed by 5% (w/v) DSS treatment. For ACE2 reconstitution, mice received intraperitoneal injections of recombinant mouse ACE2 protein (0.1 mg kg−1) (R&D) daily during 1.5% (w/v) DSS treatment.
Serum amino acid analyses
Heparinized serum was mixed with an equal part of a solution containing 20 µg ml−1 each of norleucine (internal standard for Ala, Gly and Val), norvaline (for Leu, Ile and Pro) and parachloro-phenylalanine (for all other amino acids) and deproteinized by ultrafiltration through a 10 kDa cutoff regenerated cellulose ultrafiltration membrane (Millipore). Amino acids were then purified by cation exchange, converted to their chloroformate derivatives and separated and quantified by GC/MS in the selected ion monitoring (SIM) mode. Tryptophan ELISA kit (LDN, BA E-2700) was used to assess Trp serum levels upon Gly-Trp treatment.
Purification of intestinal epithelial cells and mucosal cells
After washing with PBS, segments of small intestines were placed in 1.5 mM EDTA in Hank’s balanced salt solution (HBSS) without calcium and magnesium and tumbled for 10 min at 37 °C. The mucosa was incubated again with HBSS/EDTA for 10 min at 37 °C. The supernatant containing the crypts was collected. The number and viability of the crypt epithelial cells were determined by 0.1% trypan blue exclusion. Purity of the epithelial cell preparation was checked by haematoxylin and eosin staining. For mucosa preparations jejunum, ileum, or colon were inverted and mucosa was scraped off with a scalpel.
Quantitative RT–PCR
Total RNA was prepared from isolated small intestinal epithelial cells using the RNeasy Mini Kit (Qiagen) in accordance with the manufacturer’s instructions. Total RNA (0.2 μg) was subjected to reverse transcription using random hexamers (Roche) and SuperscriptII (Invitrogen) followed by quantitative PCR analysis. The following primers were used: GAPDH, 5′-TCATCAACGGGAAGCCCATCAC-3′ (forward) and 5′-AGACTCCACGACATACTCAGCACCG-3′ (reverse); β-actin, 5′-GGCTGTATTCCCCTCCATCG-3′ (forward) and 5′-CCAGTTGGTAACAATGCCATGT-3′ (reverse); DEFA1, 5′-TCCTCCTCTCTGCCCTYGTCCTG-3′ (forward) and 5′-CTCTTCTCCTGGCTGCTCCTC-3′ (reverse); DEFA3, 5′-GCTAGGGAGCACTTGTTTGC-3′ (forward) and 5′-TTGTTTGAGGAAAGGAGGCA-3′ (reverse); DEFA4, 5′-CCAGGGGAAGATGACCAGGCTG-3′ (forward) and 5′-TGCAGCGACGATTTCTACAAAGGC-3′ (reverse); DEFA5, 5′-AGGCTGATCCTATCCACAAAACAG-3′ (forward) and 5′-TGAAGAGCAGACCCTTCTTGGC-3′ (reverse); DEFA-RS1, 5′-CACCACCCAAGCTCCAAATACACAG-3′ (forward) and 5′-ATCGTGAGGACCAAAAGCAAATGG-3′ (reverse); MMP7, 5′-ACCCTGTTCTGCTTTGTGTGTC-3′ (forward) and 5′-TCTGAGCCTGTTCCCACTGATG-3′ (reverse); Angiogenin1 (ANG1), 5′-CCATGACGCCAAGCCAAAGG-3′ (forward) and 5′-GCTTCCATTCGCTCCACAGATG-3′ (reverse); Angiogenin4 (ANG4), 5′-GCTGGGTCTGGTTGTGATTCC-3′ (forward) and 5′-AGGCGAGGTTAGCTTTCTTTCC-3′ (reverse); FABP2, 5′-AGCTCGGTGTAAACTTTCCCTAC-3′ (forward) and 5′-AAACCTCTCGGACAGCAATCAG-3′ (reverse); LYZ1, 5′-GCCAAGGTCTACAATCGTTGTGAGTTG-3′ (forward) and 5′-CAGTCAGCCAGCTTGACACCACG-3′ (reverse); LYZ2, 5′-GGCTGGCTACTATGGAGTCAGCCTG-3′ (forward) and 5′-GCATTCACAGCTCTTGGGGTTTTG-3′ (reverse); PLA2G2E, 5′-AGGATTCCCCCAAGGATGCCAC-3′ (forward) and 5′-CAGCCGTTTCTGACAGGAGTTCTGG -3′ (reverse); ACE, 5′-TCATCATCCAGTTCCAGTTCCA-3′ (forward) and 5′-CGGTGACGAGCCATTCTGT-3′ (reverse); ACE2, 5′-TGGTCTTCTGCCATCCGATT-3′ (forward) and 5′-CCATCCACCTCCACTTCTCTAA-3′ (reverse); B0AT1, 5′-AACGCTCATGTATAGCATCTGG-3′ (forward) and 5′-CAGCCACAGTGACCACAAC-3′ (reverse); TLR1, 5′-AAGAACTCAGGCGAGCAGAG-3′ (forward) and 5′-TCTGGTAAGGTTTGCGTTTGG-3′ (reverse); TLR2, 5′-CACTTCCAGGTCTTCAGTCTTC-3′ (forward) and 5′-ACTATGATTGCGGACACATCTC-3′ (reverse); TLR3, 5′-TTGCGTTGCGAAGTGAAGAA-3′ (forward) and 5′-AGACAGATACCTCAGGCTTGG-3′ (reverse); TLR4, 5′-CAGAACTTCAGTGGCTGGATT-3′ (forward) and 5′-TGTACTAGGTTCGTCAGATTGG-3′ (reverse); TLR5, 5′-GAGTGAGGTCAGTCCTGGAG-3′ (forward) and 5′-GAGATGAGGCGTCTGGAGAG-3′ (reverse); TLR6, 5′-TACCGTCAGTGCTGGAAATAGA-3′ (forward) and 5′-TTCTGTCTTGGCTCATGTTGC-3′ (reverse); TLR7, 5′-AGAACCACTCTATGCCTTCAAG-3′ (forward) and 5′-GTCTCTTCCGTGTCCACATC-3′ (reverse); TLR8, 5′-CCTTCCTTTGTCTATAGAACATGGA-3′ (forward) and 5′-CACAAGGATAGCTTCTGGAATAGT-3′ (reverse); TLR9, 5′-AGAATCCTCCATCTCCCAACAT-3′ (forward) and 5′-CAGACTTCAGGAACAGCCAAT-3′ (reverse).
Western blotting and ELISA
Western blotting was performed with standard protocols using isolated primary small intestinal epithelial cells. Primary antibodies reactive to phosphorylated S6 (2215; Cell Signaling), S6 (2217; Cell Signaling), and β-actin (Sigma) were used. Blots were washed three times in TBST for 30 min, incubated with horseradish peroxidase-conjugated secondary antibodies (1:5,000 dilution; Jackson ImmunoResearch) for 20 min, washed three times in TBST, and visualized with enhanced chemiluminescence. Small intestine was collected for ELISA assays to detect phosphorylated p70S6K or total p70S6K (DYC896, DYC8962; R&D) and colon was collected to determine angiotensin II levels (EK-002-12; Phoenix). To analyse membrane proteins, total membranes were prepared using mannitol buffer (200 mM mannitol, 80 mM HEPES, 41 mM KOH, pH 7.5 in the presence of protease inhibitor cocktail, Sigma). After homogenization of the samples using MagNA Lyser green beads (Roche), total membranes were attained by dual consecutive centrifugation steps (100g and 100,000g) at 4 °C. 50 µg of total membrane were used per lane for western blotting. The following primary antibodies were used: anti-Ace2 and anti-Ace (R&D Systems, 1:1,000), anti-B0AT1 (Pineda, 1:1,000), anti-mouse β-actin (Sigma, 1:10,000). Antibody binding was detected with Immobilon Western Chemiluminescent HRP or AP substrates (Millipore) and chemiluminescence visualized with a DIANA III camera (Raytest, Dietikon).
Histology and immunohistochemistry
For histological analysis, 2–4-μm-thick sections were stained with haematoxylin and eosin. Immunohistochemistry was performed using the automated Ventana system. Sections were incubated with antibodies against phosphorylated S6 (Cell Signaling), Ace2 (R&D), Ki67 (Novacastra), and cleaved caspase 3 (Cell Signaling). For phosphorylated S6 and cleaved caspase 3 staining, a multimer based detection system (UltraMap) was used. In addition, immunofluorescence was performed on cryosections of the kidney and small intestine using anti-Ace2 antibodies at 1:100, anti-B0AT1 at 1:200, and anti-collectrin at 1:1,000 followed by Alexa488-conjugated anti-goat IgG or Alexa594-labelled anti-rabbit IgG.
DNA extraction, pyrosequencing, sequence analysis
Genomic DNA from terminal ileum was extracted by MO BIO PowerSoil DNA Isolation kit with modifications: solution C1 and 20 µl of Proteinase K were added to faeces and incubated for 2 h at 50°C to enhance the lysis. Extracted DNA was quantified by using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). Variable region V1-V2 of the 16S rRNA gene was amplified using the forward primer 5′-CTATGCGCCTTGCCAGCCCGCtcagTCAGAGTTTGATCCTGGCTCAG-3′. Sequence in bold is 454 Life Sciences primer B, the underlined sequence represents the broadly conserved bacterial primer 27F. A two-base linker sequence (‘TC’) and a four base key (lower case) were added. The reverse primer (5′- CGTATCGCCTCCCTCGCGCCAtcagXXXXXXXXXXCATGCTGCCTCCCGTAGGAGT-3′) contained the 454 Life Sciences primer A sequence (bold) a unique 10 base multiplex identifier (MIDs designated as XXXXXXXXXX) to tag each PCR product, the broad-range bacterial primer 338R, and a ‘CA’ linker sequence inserted between the MIDs and the rRNA gene primer. In addition, a four base key (lower case) was added before the MIDs as described earlier35. Replicate PCR reactions were performed for each sample and the respective negative controls. Each reaction consisted of 1 μl (10 pmol μl−1) of each primer, 40 ng of DNA, 1 μl of dNTPs mix (10 mM stock), 1.0 units of Phusion Hot Start II High-Fidelity DNA Polymerase, and 10 μl of 5× Phusion HF buffer. The final volume of reaction was adjusted to 50 μl. Amplification was performed using an initial denaturation of 3 min at 98 °C followed by 30 cycles, denaturation at 98 °C for 10 s, annealing at 55 °C for 30 s and elongation at 72 °C for 30 s. Final extension was at 72 °C for 10 min. Amplified products were run on an agarose gel, specific bands excised and amplicons purified using the Qiagen gel purification kit (Qiagen). Concentrations of the eluted and purified amplicons were measured using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). Equal amounts of PCR products were mixed in a single tube and sequenced using Roche 454 Titanium chemistry. To generate negative control templates for the PCR reaction, water was taken as a sample during DNA extraction; in all cases, the PCR reactions from these templates were negative.
Sequence analysis
After pyrosequencing, all sequence reads were screened and filtered for quality and length using PANGEA36. Sequences were trimmed and binned by samples using the specific MIDs as barcode. Sequences not meeting the following quality criteria were excluded from further analysis: no perfect match with MID sequences or specific primers; reads shorter than 200 bases; mean quality score ≥25; the presence of any ambiguous base; and/or more than 8 homopolymers. Remaining sequences were aligned using the curated SILVA seed database (bacterial reference alignment) as a template with the Needleman-Wunsch algorithm37. Sequences not matching with defined core regions and putative chimaeric sequences were detected by using the uchime38 command implemented in Mothur39 and were subsequently removed. Sequences were further confirmed to originate from bacteria using the Ribosomal Data base project (RDP) classifier with 60% bootstrap threshold40. For all the downstream analysis, 1,000 sequences per samples were randomly sampled to normalize the number of reads distribution. In order to reduce the overestimation of distances between sequences, leading and trailing periods from aligned files were removed. Pairwise distances between sequences were calculated, gaps of any length were treated as single event or mismatch. Sequences with ≥97% similarity were clustered using average neighbour algorithm in to species level operational taxonomical units (OTUs)41. Phylogenetic affiliation of each OTU was performed by using RDP taxonomy and a template file. To visualize the microbial composition and structure in related on host genotype or treatment, distance matrices between samples were generated based on weighted (Bray–Curtis similarity) and non-weighted (unweighted UniFrac) algorithms and visualized using principal coordinate analysis (PCoA). OTUs contributing for the shifting of the samples along the two axes were ascertained by calculating the Spearman correlation of each OTU with the two axes in the PCoA39. Similarity percentage analysis (SIMPER)42 was performed to determine the driving OTUs imparting significant differences among bacterial community composition among genotype and diet groups. The analysis was performed on Bray–Curtis similarity coefficient with the program PAST (Paleontological Statistics, ver. 2.14)43.
Bone marrow transplantation
Eight week old Ace2 mutant and Ace2 wild-type mice were lethally irradiated with 980 cGy (Gammacell 40). Bone marrow cells were harvested from 8–9-week-old donor Ace2 mutant or Ace2 wild-type mice by flushing the femurs and tibias with DMEM. Recipient mice received 5 × 106 bone marrow cells in 250 μl DMEM from male donor mice by tail vein injection 6 h after irradiation. Six weeks after transplantation, the genotypes of haematopoietic cells were determined by PCR using peripheral blood. The PCR primer set for the mutant Ace2 allele was as follows: 5′-CCAGCTCATTCCTCCCACTC-3′ and 5′-CCGGCTGCTCTTTGAGAGGACA-3′. The PCR primer set for wild-type Ace2 was: 5′-CTTCATTGGCTCCGTTTCTTAGC-3′ and 5′-CCGGCTGCTCTTTGAGAGGACA-3′.
Calorimetry cages
Average food intake was measured using indirect calorimetry cages from TSE Systems over a period of seven days following 48 h of acclimatization. Data were analysed with proprietary Phenomaster Analysis Software (TSE Systems) and Microsoft Excel.
Statistical analyses
All data are shown as mean ± s.e.m. Measurements at single time points were analysed by ANOVA and if significant, further analysed by a two-tailed t-test. P < 0.05 was considered to indicate statistical significance.
Supplementary information
This file contains Supplementary Figures 1-26. (PDF 12306 kb)
This file contains Supplementary Tables 1-6. Please note Table 1 comprises pages 1-38, Table 2 pages 39-69, Table 3 pages 70-108, Table 4 pages 109-137, Table 5 pages 138-178 and Table 6 pages 179-223. (PDF 3908 kb)
Acknowledgements
We thank all members of our laboratories for discussions. T. Hashimoto was supported by grants from the European Respiratory Society, EuGeneHeart, and SENSHIN. T.P. was supported by a Marie Curie IIF. J.M.P. was supported by grants from IMBA, the Austrian Ministry of Sciences, the Austrian Academy of Sciences, GEN-AU (AustroMouse), an EU ERC Advanced Grant, and the EU network grants EuGeneHeart, ApoSys and INFLA-Care. P.R. was supported by the BMBF Network ‘Systematic genomics of chronic inflammation’, the DFG Cluster of Excellence Inflammation at Interfaces, SPP1399, SFB877, and the DFG project RO1394.
PowerPoint slides
Author Contributions
T. Hashimoto and T.P. performed most experiments, together with J.T., H.I., M.P., V.S., R.H. and T. Hanada; A. Rehman, P.R., S.L. and S.S. performed sequencing and analysis of the gut microbiome. S.M.R.C., D.S, F.V. and H.C. analysed collectrin and ACE2 expression in gut and kidney. A. Richter and B.W. performed amino acid analysis in serum. K.K. and A.F. provided essential mouse strains. J.M.P. and P.R. coordinated the project, and together with T. Hashimoto and T.P. wrote the manuscript and designed the experiments.
Competing interests
The authors declare no competing financial interests.
Footnotes
Tatsuo Hashimoto and Thomas Perlot: These authors contributed equally to this work.
Contributor Information
Philip Rosenstiel, Email: p.rosenstiel@mucosa.de.
Josef M. Penninger, Email: josef.penninger@imba.oeaw.ac.at
References
- 1.Khan Y, Bhutta ZA. Nutritional deficiencies in the developing world: current status and opportunities for intervention. Pediatr. Clin. North Am. 2010;57:1409–1441. doi: 10.1016/j.pcl.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 2.Weisstaub G, Araya M. Acute malnutrition in Latin America: the challenge of ending avoidable deaths. J. Pediatr. Gastroenterol. Nutr. 2008;47:S10–S14. doi: 10.1097/MPG.0b013e3181818e78. [DOI] [PubMed] [Google Scholar]
- 3.Imai Y, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112–116. doi: 10.1038/nature03712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crackower MA, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 5.Kuba K, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nature Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donoghue M, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ. Res. 2000;87:E1–E9. doi: 10.1161/01.RES.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 7.Tipnis SR, et al. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 8.Camargo SM, et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with Hartnup mutations. Gastroenterology. 2009;136:872–882. doi: 10.1053/j.gastro.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kowalczuk S, et al. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008;22:2880–2887. doi: 10.1096/fj.08-107300. [DOI] [PubMed] [Google Scholar]
- 10.Kleta R, et al. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nature Genet. 2004;36:999–1002. doi: 10.1038/ng1405. [DOI] [PubMed] [Google Scholar]
- 11.Segal I, et al. Rectal manifestations of pellagra. Int. J. Colorectal Dis. 1986;1:238–243. doi: 10.1007/BF01648345. [DOI] [PubMed] [Google Scholar]
- 12.Okayasu I, et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-H. [DOI] [PubMed] [Google Scholar]
- 13.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 14.Osterreicher CH, et al. Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice. Hepatology. 2009;50:929–938. doi: 10.1002/hep.23104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vickers C, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 16.Swirski FK, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darby WJ, McNutt KW, Todhunter EN. Niacin. Nutr. Rev. 1975;33:289–297. doi: 10.1111/j.1753-4887.1975.tb05075.x. [DOI] [PubMed] [Google Scholar]
- 18.Attia E, Wolk S, Cooper T, Glasofer D, Walsh BT. Plasma tryptophan during weight restoration in patients with anorexia nervosa. Biol. Psychiatry. 2005;57:674–678. doi: 10.1016/j.biopsych.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 19.Danilczyk U, et al. Essential role for collectrin in renal amino acid transport. Nature. 2006;444:1088–1091. doi: 10.1038/nature05475. [DOI] [PubMed] [Google Scholar]
- 20.Stratigos JD, Katsambas A. Pellagra: a still existing disease. Br. J. Dermatol. 1977;96:99–106. doi: 10.1111/j.1365-2133.1977.tb05197.x. [DOI] [PubMed] [Google Scholar]
- 21.Nenci A, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 22.Chalaris A, et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010;207:1617–1624. doi: 10.1084/jem.20092366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS ONE. 2010;5:e9199. doi: 10.1371/journal.pone.0009199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bröer A, et al. Impaired nutrient signaling and body weight control in a Na+ neutral amino acid cotransporter (Slc6a19)-deficient mouse. J. Biol. Chem. 2011;286:26638–26651. doi: 10.1074/jbc.M111.241323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farkas S, et al. Rapamycin decreases leukocyte migration in vivo and effectively reduces experimentally induced chronic colitis. Int. J. Colorectal Dis. 2006;21:747–753. doi: 10.1007/s00384-005-0793-7. [DOI] [PubMed] [Google Scholar]
- 26.Reinisch W, et al. A multicenter, randomized, double-blind trial of everolimus versus azathioprine and placebo to maintain steroid-induced remission in patients with moderate-to-severe active Crohn's disease. Am. J. Gastroenterol. 2008;103:2284–2292. doi: 10.1111/j.1572-0241.2008.02024.x. [DOI] [PubMed] [Google Scholar]
- 27.Mastroianni JR, Ouellette AJ. Alpha-defensins in enteric innate immunity: functional Paneth cell alpha-defensins in mouse colonic lumen. J. Biol. Chem. 2009;284:27848–27856. doi: 10.1074/jbc.M109.050773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salzman NH, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nature Immunol. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weichhart T. Mammalian target of rapamycin: a signaling kinase for every aspect of cellular life. Methods Mol. Biol. 2012;821:1–14. doi: 10.1007/978-1-61779-430-8_1. [DOI] [PubMed] [Google Scholar]
- 30.Abu-Qurshin R, et al. Crohn's disease associated with pellagra and increased excretion of 5-hydroxyindolacetic acid. Am. J. Med. Sci. 1997;313:111–113. doi: 10.1097/00000441-199702000-00008. [DOI] [PubMed] [Google Scholar]
- 31.Ito M, et al. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc. Natl Acad. Sci. USA. 1995;92:3521–3525. doi: 10.1073/pnas.92.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuba K, et al. Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ. Res. 2007;101:e32–e42. doi: 10.1161/CIRCRESAHA.107.158659. [DOI] [PubMed] [Google Scholar]
- 33.Ishida J, et al. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J. Biol. Chem. 2004;279:26274–26279. doi: 10.1074/jbc.M404149200. [DOI] [PubMed] [Google Scholar]
- 34.McCafferty DM, Miampamba M, Sihota E, Sharkey KA, Kubes P. Role of inducible nitric oxide synthase in trinitrobenzene sulphonic acid induced colitis in mice. Gut. 1999;45:864–873. doi: 10.1136/gut.45.6.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rehman A, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut. 2011;60:1354–1362. doi: 10.1136/gut.2010.216259. [DOI] [PubMed] [Google Scholar]
- 36.Giongo A, et al. PANGEA: pipeline for analysis of next generation amplicons. ISME J. 2010;4:852–861. doi: 10.1038/ismej.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pruesse E, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schloss PD, Handelsman J. Status of the microbial census. Microbiol. Mol. Biol. Rev. 2004;68:686–691. doi: 10.1128/MMBR.68.4.686-691.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clarke KR. Nonparametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993;18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 43.Hammer, Ø., Harper, D. A. T. & Ryan, P. D. Past: paleontological statistics software package for education and data analysis. Palaeontologia Electronica4, art. 4. (2004)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Supplementary Figures 1-26. (PDF 12306 kb)
This file contains Supplementary Tables 1-6. Please note Table 1 comprises pages 1-38, Table 2 pages 39-69, Table 3 pages 70-108, Table 4 pages 109-137, Table 5 pages 138-178 and Table 6 pages 179-223. (PDF 3908 kb)