

Abstract
Background
Acute lung injury and the acute respiratory distress syndrome continue to be significant causes of morbidity and mortality in the intensive care setting. The failure of patients to resolve the alveolar edema associated with these conditions is a major contributing factor to mortality; hence there is continued interest to understand the mechanisms of alveolar edema fluid clearance.
Discussion
The accompanying review by Vadász et al. details our current understanding of the signaling mechanisms and cellular processes that facilitate clearance of edema fluid from the alveolar compartment, and how these signaling processes may be exploited in the development of novel therapeutic strategies. To complement that report this review focuses on how intact organ and animal models and clinical studies have facilitated our understanding of alveolar edema fluid clearance in acute lung injury and acute respiratory distress syndrome. Furthermore, it considers how what we have learned from these animal and organ models and clinical studies has suggested novel therapeutic avenues to pursue.
Keywords: Acute respiratory distress syndrome; Edema; Sodium transport; Isolated lung; Epithelial sodium channel; Na,K-ATPase
Introduction
Acute lung injury (ALI), and its more severe form, acute respiratory distress syndrome (ARDS), are important causes of morbidity and mortality in critically ill patients [1]. One of the hallmarks of ALI/ARDS is the accumulation of protein-rich edema fluid in the alveolar compartment of the lung [1], caused by increased fluid influx into the airspaces, and decreased fluid transport out of these airspaces. In the case of the latter, impaired alveolar fluid clearance (AFC) is a key underlying cause of alveolar edema persistence [2, 3], and the ability of ALI/ARDS patients to clear edema fluid is correlated with a shorter stay in the intensive care unit, and reduced mortality [4]. Much effort has therefore been focused on identifying pathogenic mechanisms underlying perturbed AFC in ALI/ARDS patients, and how we can potentiate AFC, which may form the basis of novel or improved therapeutic strategies. The accompanying review by Vadász et al. (http://dx.doi.org/10.1007/s00134-007-0661-8) details our current understanding of the signaling mechanisms that facilitate AFC. To complement that report this review focuses on how intact organ and animal models and clinical studies have facilitated our understanding of AFC in ALI/ARDS. We also discuss how some of these studies have led to novel therapeutic approaches.
Edema fluid is transported out of the alveolar airspaces into the interstitium, where it is cleared by the lymphatic drainage. Alternatively, fluid can also be transported into the vasculature, where it is cleared by the circulation. Maintenance of an optimum alveolar fluid volume results from a finely balanced influx of fluid into the lung, and fluid clearance out of the lung; therefore, endothelial and epithelial barrier integrity is essential for optimal fluid balance [3]. In patients with ALI/ARDS, the integrity of both the endothelial and epithelial barriers may be compromised, leading to accelerated fluid influx into the alveolar compartment and impaired AFC [3]. In healthy lungs the alveolar epithelium is considerably less permeable than the endothelium [5]. Approximately 90% of the alveolar epithelium surface area is composed of flat alveolar type I cells, with the remaining 10% accounted for by cuboidal type II cells which produce surfactant and are progenitor cells that regenerate the epithelium after injury. Historically, type II cells are accredited with a key role in AFC. It is widely accepted that AFC is driven by sodium transport across the airway epithelium, which is affected by the concerted action of two sodium transport systems: the epithelial sodium channel (ENaC) [6] and the Na+, K+ transporting adenosine-5′-triphosphatase (Na,K-ATPase) [7] as well as the cystic fibrosis transmembrane conductance regulator (CFTR; a chloride channel) [8] and other, as yet uncharacterized channels [9]. Impaired function of any one of these ion transport systems can perturb AFC. Diffuse alveolar damage, including severe alveolar epithelial damage, is one of the hallmarks of ALI/ARDS [1, 10], where extensive type I cell necrosis occurs, leaving an intact but denuded basement membrane which is repopulated by hyperplastic type II cells that regenerate the alveolar epithelium [11]. Given the established importance of the type II cell in AFC [6] and the emerging importance of the type I cell in AFC with the recent discovery that type I cells also contain functional sodium and chloride channels [12], this epithelial damage is likely to massively impact AFC in ALI/ARDS patients. This idea is strengthened by the observation that in patients with hydrostatic pulmonary edema, which may result from congestive heart failure or acute myocardial infarction, both the alveolar epithelial barrier and AFC remain intact [13].
Animal and organ models have proved particularly useful to study AFC, because the three-compartment (alveolar airspace-interstitium-vasculature) structure of the lung is preserved [14–16]. Studies in humans have relied heavily on the assessment of extravascular lung water (EVLW) [17]. More recently, nasal potential difference (NPD) has been employed as a surrogate measurement for transepithelial ion transport [18], although this measurement is clearly limited in its usefulness when studying diseases that exclusively impair alveolar ion transport. The aspects of impaired AFC in ALI/ARDS that have been examined in animal or organ models and clinical studies are summarized in Fig. 1.
Fig. 1.
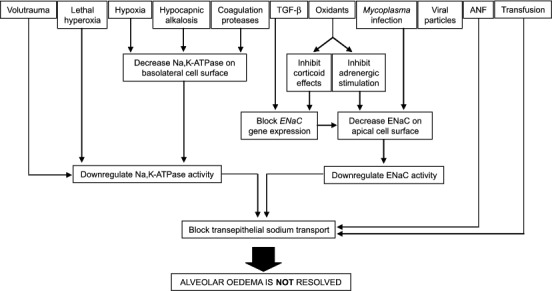
Factors that cause impaired alveolar fluid clearance in ALI/ARDS that have been investigated in animal and organ models and in clinical studies. ANF, Atrial natriuretic factor; ENaC, epithelial sodium channel; Na,K-ATPase, Na+, K+ transporting adenosine-5′-triphosphatase, TGF-β, transforming growth factor β
Studies in live, anesthetized, ventilated sheep provided the first evidence that alveolar fluid relied on active ion transport, where clearance of salt and water occurred against an increase protein concentration in the alveolar lining fluid [19]. Supporting these data, fluid clearance was impeded at low temperature in in situ perfused goat lungs (maintained at 18 °C) [20], and in isolated, perfused liquid-filled rat lungs [21] consistent with the inhibition of an active transport process.
Active sodium transport in particular was implicated in AFC, since amiloride, a potent and specific inhibitor ENaC inhibited 40–70% of basal fluid clearance in sheep, rabbits, rats, mice, and in the human lung [3]. O'Brodovich and colleagues [22] have further demonstrated that application of amiloride to newborn guinea pig lungs caused respiratory distress, hypoxemia and elevated extravascular lung water, implicating active sodium transport in AFC in neonates. Consistent with this idea, targeted deletion of both ENaCα alleles in mice caused death within 40 h of birth as a consequence of impaired AFC [23]. Transgenic overexpression of the ENaCα gene in an ENaCα–/– null mouse background rescued this pulmonary phenotype [24]. Conversely, overexpression of ENaC in the mouse lower airways accelerated sodium absorption, and depleted the volume of fluid coating the airway epithelium [25]. Together, these data implicate both active transepithelial sodium transport and ENaC itself in AFC. Using small interfering RNA directed against ENaCα applied to the airways of live rats, other non-ENaC type ion channels are also likely to play an important role in transepithelial sodium transport and fluid reabsorption, particularly baseline fluid reabsorption [9].
While ENaC located on the apical surface of alveolar epithelial cells acts as a sodium channel, investigations in live animals and isolated organ models have revealed that the driving force behind ENaC-mediated sodium uptake is the Na,K-ATPase [7]. Ouabain, a potent and specific inhibitor of the Na,K-ATPase, inhibited AFC in isolated, perfused fluid-filled mouse lungs [26]. Furthermore, adenoviral-mediated gene transfer of the Na,K-ATPase β1 subunit into the lungs of rats augmented AFC [27]. Furthermore, overexpression of the Na,K-ATPase β1 subunit in the alveolar epithelium restored active transepithelial sodium transport and AFC in a rat model of acute hydrostatic pulmonary edema [28]. Gene knockout strategies have also addressed a role for the two Na,K-ATPase α subunits in AFC, where mice heterozygous for each of the Na,K-ATPase α subunits (α1 +/–/α2 +/–) exhibit reduced cyclic-adenosine monophosphate (cAMP) dependent AFC [29].
In addition to a role for sodium transport, it has recently emerged that chloride transport via the CFTR channel also plays a role in AFC. Using glibenclamide, an inhibitor of potassium and CFTR channels, fluid clearance from in situ perused and unperfused mouse lungs and ex vivo human lungs was impaired [8]. These data were supported using a cystic fibrosis (ΔF508) mouse in a model of acute hydrostatic edema, where chloride transport by the CFTR channel was demonstrated to play a major role in AFC [8]. Further supporting this idea, adenoviral-mediated transfer of the CFTR gene into the lungs of live rats and mice significantly increased AFC, an effect that was blocked in the presence the chloride channel blockers 5-nitro-2-(3-phenylpropylamino) benzoic acid, bumetanide, and glibenclamide [30]. Animal and organ models have thus confirmed invaluable in the identification of transepithelial active sodium transport as the basis of AFC, and in the validation of ENaC and other channels and the Na,K-ATPase, as key components that drive this process.
Mechanisms of impaired fluid clearance in ALI/ARDS: what we have learned from intact organ and animal models, and clinical studies
Hypoxia and hyperoxia
Hypoxia is a feature of both ARDS and high-altitude pulmonary edema (HAPE). In isolated, ventilated rat lungs perfused at constant pressure (therefore independent of pressure changes caused by adaptation to hypoxic pulmonary vasoconstriction), exposure to hypoxia (1.5–14% O2, for 2 h) increased the permeability of the alveolo-capillary barrier, causing alveolar flooding that was exacerbated by a hypoxia-mediated block of AFC [31]. Alveolar fluid clearance in live rats was decreased by 36% after 48 h of exposure to 10% O2 [32] while transepithelial sodium transport assessed by NPD was decreased by 24% in live rats exposed to 10% O2 for 24 h [33]. Both observations were attributed to reduced Na,K-ATPase activity. It remains unclear whether hypoxia influenced expression of sodium transporter genes, since different groups have reported downregulation [34], unchanged expression [33], and upregulation [35] of sodium transport machinery in response to hypoxia; however, all groups reported impaired AFC in response to hypoxia. The rapid reversibility of impaired AFC by β2-agonists suggested that hypoxia alters intracellular trafficking of sodium transport machinery [35]. This idea is supported by the observed hypoxia-induced endocytosis of Na,K-ATPase from the basolateral membrane of alveolar epithelial cells [36], which slows down transepithelial sodium transport, and hence, AFC [37]. Irrespective of the mechanism at play, the ability of hypoxia to block AFC is now regarded as a key mechanism of barrier dysfunction and edema persistence in HAPE [38] and in ALI/ARDS [39].
In contrast to hypoxia, exposure of rats to sublethal hyperoxia (85% O2) increased AFC by upregulating gene expression of both Na,K-ATPase [40] and ENaC [41], which are induced to limit hyperoxic lung damage. However, higher oxygen concentrations (100% O2) impaired AFC in live rats and caused significant mortality [42–44]. This effect was attributed to impaired transepithelial active sodium transport [42] occurring as a consequence of downregulation of the Na,K-ATPase activity [43], and was corrected by adenovirus-mediated transfer of the Na,K-ATPase into the rat lungs [43]. These and other observations introduced the possibility of using gene therapy to augment AFC and treat ALI/ARDS.
Mechanical ventilation
Patients with ALI/ARDS frequently require mechanical ventilation to facilitate breathing. However, mechanical ventilation can directly induce ALI (ventilator-induced lung injury, VILI), by promoting alveolo-capillary barrier permeability [1, 45], inducing proinflammatory cytokines [46], or by directly inhibiting AFC. Supporting the latter idea, Lecuona et al. [47] demonstrated that ventilation of rats with high tidal volumes (VT) of 40 ml/kg and a peak airway pressure of 35 cmH2O rapidly impaired AFC, accompanied by a significant decrease in activity (but not expression) of Na,K-ATPase. Thus high VT ventilation can directly block AFC by inhibiting Na,K-ATPase-driven sodium transport, and hence AFC.
Mechanical ventilation can also worsen preexisting ALI (ventilator-associated lung injury, VALI). In a canine model of acid aspiration-induced ALI Corbridge et al. [45] demonstrated that ventilation with high VT (30 ml/kg) but a fixed, low positive end-expiratory pressure (PEEP; 3 cmH2O) increased EVLW to a greater extent than did ventilation with lower VT (15 ml/kg) but a fixed, higher PEEP (12 cmH2O). Similar trends were observed in an acid aspiration-induced model of ALI in rats, where a progressive reduction in VT from 12 to 6 to 3 ml/kg, keeping PEEP fixed at 10 to 12 cm H2O, was accompanied by a progressive decrease in EVLW accumulation [48]. In both instances a low VT/high PEEP strategy proved beneficial. Thus in addition to reducing both permeability and proinflammatory cytokine release, a low VT/high PEEP ventilation strategy might also promote AFC and has formed the basis of important advances in ventilation strategies for ALI/ARDS patients that are in routine use today.
Hypocapnia and hypocapnic alkalosis
Hyperventilation and hypocapnic alkalosis are often found together in patients with ALI/ARDS [49]. In an isolated, buffer-perfused ventilated rabbit lung, exposure to hypocapnia (> 3 h) increased vascular permeability, and hence promoted alveolar edema [50]. Further to this, using an isolated, perfused, fluid-filled rat lung, Myrianthefs et al. [51] demonstrated that hypocapnia dramatically impaired AFC. This block was reversible upon restoration of normal CO2 levels and was not induced by metabolic alkalosis. The AFC block was attributed to impaired sodium transport resulting from decreased membrane abundance of the Na,K-ATPase in rat lungs [51]. Hypocapnia can therefore promote both the formation of alveolar edema and impair the resolution of this edema by blocking transepithelial sodium transport, and hence AFC.
Coagulation
Procoagulant pathways are upregulated in ALI/ARDS, while fibrinolysis is suppressed [1]. Coagulation proteases such as thrombin have well documented roles in the development of ALI/ARDS where thrombin can directly increase vascular permeability and promote alveolar flooding [1]. Recent work by Vadász et al. [52] demonstrated that thrombin applied to the vascular compartment of isolated, ventilated and perfused rabbit lungs also impaired AFC. This was attributed to a block in transepithelial sodium and hence fluid transport across the alveolocapillary barrier. It was subsequently demonstrated that thrombin promoted the endocytosis of the Na,K-ATPase, thereby reducing Na,K-ATPase activity (see the accompanying review by Vadász et al. (http://dx.doi.org/10.1007/s00134-007-0661-8). Thus coagulation proteases play a dual role in impaired AFC. In addition to causing a permeability edema, thrombin, by virtue of its signaling properties, directly blocks fluid reabsorption. In contrast to the thrombin-signaling effects on Na,K-ATPase trafficking, serine proteases can directly enhance β-adrenergic-stimulated ENaC activity in alveolar epithelial cells and enhance β-adrenergic-stimulated AFC in live mice [53]. Thus serine proteases appear to have opposing effects on different components of the sodium transport machinery.
Infection and inflammatory mediators
Infection and purified endotoxin promote vascular permeability and pulmonary edema by inflammatory mechanisms involving granulocytes [14]. Interestingly, AFC was upregulated after intratracheal administration of endotoxin to live rats and in a rat model of septic shock [54], possibly as a protective mechanism induced during septic shock to overcome alveolar flooding. However, both cell-surface ENaC expression and AFC were downregulated in an experimental Mycoplasma pneumoniae infection in live mice, an effect mediated by reactive oxygen-nitrogen intermediates [55]. This phenomenon is not limited to bacterial pathogens, since in live rats, the influenza virus can directly impair ENaC activity by reducing the open probability (P o) of the channel, and thus downregulate AFC, independently of viral entry into the epithelium [56]. Thus it appears that lung infections can impair AFC, although there appear to be multiple underlying causes.
Acute lung injury may develop rapidly in recipients of transfused whole plasma [1]. This transfusion-associated acute lung injury (TRALI) is caused by biologically active lipids, and antileukocyte antibodies, which causea pronounced vascular leak [1]. In a new mouse model of TRALI Looney et al. [57] have demonstrated that AFC is also impaired in TRALI, although the molecular mechanism has not been described.
Levels of proinflammatory factors are elevated in bronchoalveolar lavage fluids from ALI/ARDS patients, including interleukin (IL) 1β, IL-6, IL-8, tumor necrosis factor (TNF)-α, and transforming growth factor (TGF) β [58]. Based on studies in rats Fukuda et al. [59] demonstrated that different domains of TNF-α have opposing effects on sodium transport and AFC, where interaction with the TNF-α receptor caused inflammation and increased permeability, while the TNF-α lectin-like domain directly activated ENaC and potentiated AFC. In contrast, atrial natriuretic factor applied to the vasculature of isolated, perfused lungs [60], and TGF-β applied intratracheally to live rats [61] impaired transepithelial sodium transport and blocked AFC. It was subsequently demonstrated that TGF-β downregulated ENaC gene expression [61]. Together these data indicate new and emerging roles for polypeptide growth factors and other hormones in the regulation of AFC.
Targeting fluid clearance in ALI/ARDS: what we have learned from intact organ and animal models, and clinical studies
Ventilation strategies
Studies in animal models demonstrated that low VT and high PEEP ventilation strategies potentiated AFC in acid aspiration-induced ALI [45, 48], suggesting a therapeutic benefit of this ventilation strategy in the management of ALI/ARDS. Several small phase III clinical trials addressing the potential benefit of low VT (which is lung protective) vs. traditional higher VT (providing better oxygenation) yielded conflicting results (reviewed in [62]). A subsequent multicenter, randomized, controlled trial of 861 patients conducted under the auspices of the National Heart, Lung and Blood Institute ARDS Network demonstrated that a 6 ml/kg VT ventilation strategy yielded a significant reduction in mortality compared with a 12 ml/kg VT ventilation strategy (with a plateau pressure of < 30 cmH2O) [63]. While EVLW was not assessed in this trial, the 6 ml/kg VT ventilation strategy also increased the number of ventilator-free days, indicative of improved lung function. To date this remains the only intervention with a confirmed benefit on clinical outcome, and clearly demonstrates how studies on ALI/ARDS in animal and organ models have translated into a successful therapeutic strategy.
Catecholamines
Catecholamine-stimulated transepithelial sodium transport is the most intensely explored possibility of manipulating AFC in a therapeutic context. It was demonstrated in 1978 that epinephrine stimulated AFC from the airspaces of newborn mammals [64], implicating β-adrenergic receptors in AFC. Both epinephrine and terbutaline stimulated AFC in anesthetized sheep [65], and terbutaline stimulated transepithelial sodium transport in an isolated, buffer-perfused rat lung [66] and dramatically enhanced alveolar fluid clearance in a resected human lung [67]. β-Adrenergic agonists also reduce high vascular pressure-induced vascular permeability in isolated rat lungs [68], indicating that β-adrenergic agonsist may influence both reduce barrier permeability and potentiate AFC. The ability of β-adrenergic agonists to stimulate AFC is attributed in part to recruitment from intracellular pools (a) of Na,K-ATPase to the basolateral membrane [69] and (b) of ENaC to the apical membrane [70] of alveolar epithelial cells (reviewed in [71]). Furthermore, β-adrenergic stimulation can clear edema in hypoxia-induced ALI [35] and ventilator-associated lung injury [72] in animals, suggesting a potentially exciting therapeutic potential for β-adrenergic agonists in the treatment of ALI/ARDS. This idea was further supported by the observations that adenovirus-mediated transfer of β-adrenergic receptor genes to live rats improved AFC due to increased sensitivity to endogenous catecholamines and consequent upregulation of Na,K-ATPase activity and ENaC protein expression the lung [73]. More recently a role for the CFTR channel in cAMP-stimulated AFC has been proposed since AFC in cystic fibrosis (ΔF508) mice, which lack CFTR activity was not stimulated with β-adrenergic agonists [8]. Furthermore, administration of a CFTR inhibitor to live mice blocked cAMP-stimulated AFC [14]. These studies provide strong evidence of an important role for the CFTR channel, along with the transepithelial sodium transport system, in β-adrenergic-stimulated AFC.
Among the neurotransmitter catecholamines, dopamine stimulated AFC in isolated, perfused rat lungs, by activation of the dopamine D1 receptor, which stimulated exocytosis of the Na,K-ATPase [74], and by activation of the D2 receptor, which induced Na,K-ATPase gene expression [75]. Dopamine also promoted edema clearance in rats when ALI was induced either by hyperoxia or mechanical ventilation [74]. Although not assessed in animal or organ models, dopamine also exerted an enhancing effect on ENaC activity [76], where dopamine increased the P o of ENaC, without altering ENaC density on the apical surface of L2 cells. Thus, separate from the β-adrenergicsystem, the dopamine system provides an alternative opportunity for the pharmacological manipulation of AFC.
Some β-adrenergic agonists, for example, albuterol, can be deposited into the lungs of patients with pulmonary edema at therapeutic concentrations by aerosolization [77]. In a double-blind, randomized, placebo-controlled study Sartori et al. [78] assessed the effect of inhalation of the β-agonist salmeterol (125 μg every 12 h) on the incidence of pulmonary edema in 37 HAPE-prone subjects at high altitude. Prophylactic salmeterol significantly reduced the incidence of HAPE by more than 50%, without any change in pulmonary hemodynamics. Furthermore, this study demonstrated that transepithelial sodium transport in the nasal epithelium was reduced in HAPE-prone subjects. Together, these data suggest a therapeutic benefit of inhaled β-agonists in the treatment of HAPE.
The β-Agonist Lung Injury Trial (BALTI), a single-center, double-blind, randomized controlled trial, assessed the effect of sustained infusion of a β-agonist (in this case, 15 μg/kg salbutamol per hour) on the resolution of pulmonary edema in 40 ALI/ARDS patients [79], using EVLW as a primary endpoint. The BALTI trial demonstrated that sustained treatment with intravenous β-agonists was generally well tolerated, although patients receiving salbutamol exhibited a trend towards higher heart rates, and patients in the salbutamol group exhibited higher incidence (26% vs. 10% in the placebo group) of supraventricular arrhythmias. Patients receiving salbutamol demonstrated a significant reduction in EVLW in comparison to placebo-treated patients. The trial was not, however, powered to detect a mortality benefit. Therefore a second multicenter, randomized, double-blind, placebo-controlled trial (BALTI-2; International Standard Randomized Controlled Trial number 38366450) is currently underway to assess the influence of intravenous salbutamol in ARDS patients on 28-day mortality.
Anti-inflammatory agents
Although glucocorticoids have been employed in the management of ARDS to reduce inflammation (in the early phase), and fibrosis (in the late phase) [1], their use is controversial. The potential beneficial effects of glucocorticoid use are attributed in part to the ability of glucocorticoids to influence AFC, by upregulating protein expression of the sodium transporting machinery. Preterm infants with respiratory distress exhibit reduced expression of ENaC relative to healthy, full-term infants [80]. Administration of dexamethasone to these infants upregulated ENaC expression, restoring ENaC expression to levels observed in healthy infants [80]. Working in adult rats, Noda et al. [81] demonstrated that a single intraperitoneal injection of dexamethasone dramatically increased AFC and reversed hypoxemia induced by an intratracheal fluid challenge, 48–72 h after dexamethasone administration. This was attributed to an increase in ENaC mRNA levels. While Na,K-ATPase mRNA levels were not altered, Na,K-ATPase activity was increased. These data suggest that glucocorticoids may be of use in the treatment of ARDS by potentiating AFC.
A number of clinical trials have addressed the use of glucocorticoids in early and late phase ARDS. Notable among these, in a randomized, double-blind, placebo-controlled trial involving 24 patients with unresolving ARDS, Meduri et al. [82] demonstrated that prolonged methlyprednisolone administration significantly improved lung function and reduced mortality. A larger randomized, multicenter, placebo-controlled trial of 180 patients with unresolving ARDS (the “Late Steroid Rescue Study”, LaSRS) that used 60-day mortality as the primary end-point did not support the use of methylprednisolone for unresolving ARDS [83]. However, methylprednisolone did increase the number of ventilator-free days during the first 28 days, accompanied by improved compliance and oxygenation. The EVLW was not assessed in either study.
Cytokines, growth factors, and hormones
Several polypeptide growth factors can potentiate AFC and protect against ALI induced in experimental animal models, notable among them, keratinocyte growth factor (reviewed in [84]). Keratinocyte growth factor upregulated alveolar fluid clearance in anesthetized, ventilated rats [85], and upregulated transepithelial sodium transport in isolated, ventilated, and perfused lungs from healthy rats as well as in rats in which ALI has been induced with α-naphthylurea [86]. This protective effect has been attributed to the ability of keratinocyte growth factor (KGF) to upregulate expression of components of the sodium transporting machinery, primarily the Na,K-ATPase, as well as its mitogenic properties on alveolar type II cells, since the alveolar epithelium is may be denuded in ALI/ARDS. An upregulation of AFC attributable to elevated Na,K-ATPase levels has also been reported for epidermal growth factor (EGF) instilled into lungs of live rats, where EGF was proposed to upregulate expression levels of Na,K-ATPase [87]. Other nonpeptide hormones can also influence AFC. For example, 3,3′,5-triiodo-L-thyronine upregulated AFC in live adult rats, apparently by upregulation of transepithelial sodium transport [88].
While no clinical trial has yet been initiated, recombinant human KGF and liposome-mediated KGF gene delivery into mouse lungs afforded significant protection against oleic acid induced lung injury by improving arterial oxygenation and lung compliance in comparison to the vehicle-treated group [89]. It was not reported in that study, however, whether AFC was improved in the KGF-treated group. However, given the ability of KGF upregulate AFC in an isolated organ model, and to repopulate denuded areas of the alveolar epithelium by virtue of its mitogenic properties on type II cells, further evaluation of the therapeutic potential of KGF appears warranted.
The lectin-like domain of TNF-α enhanced AFC in ventilated rats [59]. Mouse TNF-α promoted fluid reabsorption in wild-type mice and in mice deficient in both TNF-α receptors, indicating that the effect of TNF-α on AFC was independent of both TNF-α receptors [90]. Intratracheal application of a synthetic peptide that was based on the lectinlike domain sequence to an isolated, ventilated, autologous blood-perfused rat lung model caused a significant reduction in lung water and improvement in lung compliance, in comparison to lungs treated with a scrambled peptide of the same length [90]. Thus at least two growth factors, KGF and the lectin-like domain of TNF-α, present us with novel opportunities for augmentation of AFC.
Antioxidants
Granulocyte-derived reactive oxygen and nitrogen species are believed to play an important role in ALI/ARDS (reviewed in [91]), and increased levels of the stable byproducts of nitric oxide decomposition, nitrite, and nitrate are observed in edema fluid from patients with ALI/ARDS [92]. These and other data have suggested a therapeutic benefit of antioxidants, including N-acetylcysteine (NAC), procysteine, and albumin [62]. Antioxidant therapy could also augment AFC. Oxidants such as hydrogen peroxide suppress glucocorticoid-induced ENaC gene transcription, an effect reversed by reactive oxygen scavengers, including thioredoxin. In a rat model of hemorrhagic shock Modelska et al. [93] demonstrated that catecholamines were ineffective at upregulating AFC. However, after either intravenous administration of NAC, intratracheal administration of reduced glutathione or neutrophil depletion with vinblastine, the effect of catecholamines was restored. These data provided in vivo evidence that neutrophil-mediated oxidative injury impaired AFC in ALI. Building on this study, Lee et al. [94] demonstrated that the induction of hemoxygenase I (HO-1), a potent antioxidant, restores normal AFC after hemorrhagic shock by blocking iNOS-mediated NO release. Therefore with regard to edema resolution antioxidant therapy would most likely augment glucocorticoid or catecholamine-based efforts to upregulate AFC.
Clinical trials involving antioxidants in ARDS have yielded equivocal results. In one randomized, placebo-controlled, double-blind trial of 61 ARDS patients Suter et al. [95] demonstrated that intravenous NAC (40 mg/kg per day) improved systemic oxygenation and reduced the need for ventilatory support. In contrast, Domenighetti et al. [96] in a comparable trial (although employing a higher dose: 190 mg/kg per day) demonstrated no improvement in systemic oxygenation or a reduction in the need for ventilatory support. Neither study reported a survival benefit, nor was EVLW assessed. Clearly additional trials are warranted, although a recent, much larger phase III double-blind, placebo-controlled, clinical trial evaluating procysteine in ALI/ARDS was prematurely discontinued due to mortality concerns in the intervention group [62].
Anticoagulation
To date, a single animal study has explored the effect of anticoagulation on AFC, where the natural anticoagulant, activated protein C (APC) was applied in a Pseudomonas-induced ALI model in rats [97]. In that study AFC was potentiated in a TNFα-dependent manner; however, activated protein C did not enhance AFC. Given the recent report of Vadász et al. [52] which demonstrated that thrombin impaired AFC by blocking transepithelial sodium transport mediated by Na,K-ATPase, it appears likely that anticoagulant therapy would improve edema resolution by upregulating Na,K-ATPase-mediated AFC. To date, no randomized, placebo-controlled trials have been conducted with anticoagulants in ALI/ARDS patients, although a randomized, phase II clinical trial of APC is currently in progress at the University of California in San Fransisco (ClinicalTrials.gov Identifier NCT00112164), employing the number of ventilator-free days measured at day 28 as the primary end-point.
Gene therapy to improve AFC
The lung is an organ that is particularly amenable to local delivery of DNA, making it an attractive target for gene therapy studies. In the context of ALI/ARDS this possibility is particularly attractive since ALI/ARDS does not impair virus-mediated gene delivery to the alveolar epithelium [98]. Several studies have highlighted the potential benefit of augmenting AFC by local delivery of genes into the lungs of live animals. The potential therapeutic benefit of upregulating Na,K-ATPase and CFTR channels in the lung as well as the delivery of KGF and HO-1 to the lung have already been discussed. All four systems have been explored in the context of gene therapy for ALI.
Adenovirus-mediated transfer of Na,K-ATPase genes to lungs of live rats increased AFC [27]. Upregulation of Na,K-ATPase expression in the alveolar epithelium by gene transfer improved edema resolution in rat models of ALI induced by elevated left arterial pressure [28] and hyperoxia [43]. Furthermore, transfer of β2-adrenergic receptor genes to the lungs of adult rats increased their sensitivity to exogenous catecholamines, and upregulated AFC due to increased delivery of both ENaC and Na,K-ATPase to epithelial cell membranes [73]. Transfer of the CTFR gene to mouse and rat lungs also augmented AFC and appeared to affect the expression and function of components of the sodium transport machinery [30]. These data together suggest that the sodium transporting and β-adrenergic signaling systems in the lung are candidate targets for intervention by gene therapy in ALI/ARDS. Clearly, enough epithelial cells would have to be present in the damaged epithelium for gene therapy to be effective, therefore, regeneration of the damaged epithelium with growth factors such as KGF has also been addressed by gene therapy. Liposome-mediated KGF gene delivery into mouse lungs afforded protection in an oleic-acid induced lung injury model [89]. Similarly, in the case of antioxidants adenovirus-mediated gene delivery of HO-1 into the lungs of live mice afforded significant protection against influenza virus- and lipopolysaccharide-induced ALI [99]. While AFC was not specifically addressed in the KGF or HO-1 studies, the documented role of KGF and HO-1 in augmenting AFC suggests that this phenomenon contributed to the beneficial effect observed.
Conclusion
Organ and animal models of ALI together with clinical studies have helped us understand the contribution of impaired AFC to the development and persistence of ALI/ARDS (Fig. 2). Perturbations to the alveolo-capillary barrier can both promote the formation and prevent the resolution of alveolar edema associated with ALI/ARDS. Indeed, edema resolution is critical for the ALI/ARDS patient to survive. Animal and organ models of edema resolution and ALI have proved irreplaceable in the development of novel therapeutic strategies that augment AFC, the most obvious case being the low tidal volume ventilation strategy for the management of ARDS, a strategy that has now been validated in a large multicenter, randomized, controlled clinical trial. Due to our focus on AFC several other important ALI/ARDS candidate therapies currently under evaluation have been omitted from this review, including other ventilation strategies, surfactant replacement, fluid management, inhaled vasodilators, and nonglucocorticoid anti-inflammatory agents. The reader is referred to other excellent reviews on these topics [62, 100]. Animal and organ models and clinical studies will no doubt continue to prove instrumental in the further development of several new and emerging ideas designed to augment AFC in ALI, notable among these are anticoagulation, stimulation of the β-adrenergic system, and local gene delivery to the alveolar epithelium.
Fig. 2.
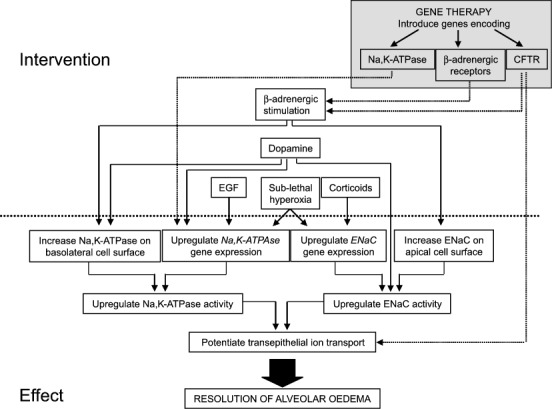
Methods of potentiating alveolar fluid clearance in ALI/ARDS that have been validated in animal and organ models or in clinical studies. Interventions involving gene therapy are indicated in the shaded box. CFTR, Cystic fibrosis transmembrane conductance regulator; D 1, dopamine D1 receptor; D 2, dopamine D2 receptor; EGF, epidermal growth factor; ENaC, epithelial sodium channel; KGF, keratinocyte growth factor; Na,K-ATPase, Na+, K+ transporting adenosine -5′-triphosphatase; T 3, 3,3′,5-triiodo-L-thyronine; TNF-α, tumor necrosis factor α
Acknowledgements
The authors are supported by the Deutsche Forschungsgemeinschaft (SFB547 “Kardiopulmonales Gefäßsystem” and KLIFO “Lungenfibrose”) and the European Commission Sixth European Framework Programme “Pulmonary Hypertension”.
Footnotes
This article refers to the articles available at: http://dx.doi.org/10.1007/s00134-007-0625-z and http://dx.doi.org/10.1007/s00134-007-0661-8
References
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 3.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- 4.Sznajder JI. Alveolar edema must be cleared for the acute respiratory distress syndrome patient to survive. Am J Respir Crit Care Med. 2001;163:1293–1294. doi: 10.1164/ajrccm.163.6.ed1801d. [DOI] [PubMed] [Google Scholar]
- 5.Matthay MA. Function of the alveolar epithelial barrier under pathologic conditions. Chest. 1994;105:67–74. doi: 10.1378/chest.105.3_Supplement.67S. [DOI] [PubMed] [Google Scholar]
- 6.Matalon S, O'Brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol. 1999;61:627–661. doi: 10.1146/annurev.physiol.61.1.627. [DOI] [PubMed] [Google Scholar]
- 7.Sznajder JI, Factor P, Ingbar DH. Invited review: lung edema clearance: role of Na (+)-K (+)-ATPase. J Appl Physiol. 2002;93:1860–1866. doi: 10.1152/japplphysiol.00022.2002. [DOI] [PubMed] [Google Scholar]
- 8.Fang X, Fukuda N, Barbry P, Sartori C, Verkman AS, Matthay MA. Novel role for CFTR in fluid absorption from the distal airspaces of the lung. J Gen Physiol. 2002;119:199–207. doi: 10.1085/jgp.119.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li T, Folkesson HG. RNA interference for alpha-ENaC inhibits rat lung fluid absorption in vivo. Am J Physiol Lung Cell Mol Physiol. 2006;290:L649–L660. doi: 10.1152/ajplung.00205.2005. [DOI] [PubMed] [Google Scholar]
- 10.Bachofen M, Weibel ER. Alterations of the gas exchange apparatus in adult respiratory insufficiency associated with septicemia. Am Rev Respir Dis. 1977;116:589–615. doi: 10.1164/arrd.1977.116.4.589. [DOI] [PubMed] [Google Scholar]
- 11.Geiser T. Mechanisms of alveolar epithelial repair in acute lung injury-a translational approach. Swiss Med Wkly. 2003;133:586–590. doi: 10.4414/smw.2003.10267. [DOI] [PubMed] [Google Scholar]
- 12.Johnson MD, Bao HF, Helms MN, Chen XJ, Tigue Z, Jain L, Dobbs LG, Eaton DC. Functional ion channels in pulmonary alveolar type I cells support a role for type I cells in lung ion transport. Proc Natl Acad Sci U S A. 2006;103:4964–4969. doi: 10.1073/pnas.0600855103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verghese GM, Ware LB, Matthay BA, Matthay MA. Alveolar epithelial fluid transport and the resolution of clinically severe hydrostatic pulmonary edema. J Appl Physiol. 1999;87:1301–1312. doi: 10.1152/jappl.1999.87.4.1301. [DOI] [PubMed] [Google Scholar]
- 14.Matthay MA, Robriquet L, Fang X. Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc Am Thorac Soc. 2005;2:206–213. doi: 10.1513/pats.200501-009AC. [DOI] [PubMed] [Google Scholar]
- 15.Ghofrani HA, Kohstall MG, Weissmann N, Schmehl T, Schermuly RT, Seeger W, Grimminger F. Alveolar epithelial barrier functions in ventilated perfused rabbit lungs. Am J Physiol Lung Cell Mol Physiol. 2001;280:L896–904. doi: 10.1152/ajplung.2001.280.5.L896. [DOI] [PubMed] [Google Scholar]
- 16.Vadász I, Morty RE, Kohstall MG, Olschewski A, Grimminger F, Seeger W, Ghofrani HA. Oleic acid inhibits alveolar fluid reabsorption: a role in acute respiratory distress syndrome? Am J Respir Crit Care Med. 2005;171:469–479. doi: 10.1164/rccm.200407-954OC. [DOI] [PubMed] [Google Scholar]
- 17.Lange NR, Schuster DP. The measurement of lung water. Crit Care (Lond) 1999;3:R19–R24. doi: 10.1186/cc342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knowles M, Gatzy J, Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med. 1981;305:1489–1495. doi: 10.1056/NEJM198112173052502. [DOI] [PubMed] [Google Scholar]
- 19.Matthay MA, Landolt CC, Staub NC. Differential liquid and protein clearance from the alveoli of anesthetized sheep. J Appl Physiol. 1982;53:96–104. doi: 10.1152/jappl.1982.53.1.96. [DOI] [PubMed] [Google Scholar]
- 20.Serikov VB, Grady M, Matthay MA. Effect of temperature on alveolar liquid and protein clearance in an in situ perfused goat lung. J Appl Physiol. 1993;75:940–947. doi: 10.1152/jappl.1993.75.2.940. [DOI] [PubMed] [Google Scholar]
- 21.Rutschman DH, Olivera W, Sznajder JI. Active transport and passive liquid movement in isolated perfused rat lungs. J Appl Physiol. 1993;75:1574–1580. doi: 10.1152/jappl.1993.75.4.1574. [DOI] [PubMed] [Google Scholar]
- 22.O'Brodovich H, Hannam V, Seear M, Mullen JB. Amiloride impairs lung water clearance in newborn guinea pigs. J Appl Physiol. 1990;68:1758–1762. doi: 10.1152/jappl.1990.68.4.1758. [DOI] [PubMed] [Google Scholar]
- 23.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet. 1996;12:325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 24.Egli M, Duplain H, Lepori M, Cook S, Nicod P, Hummler E, Sartori C, Scherrer U. Defective respiratory amiloride-sensitive sodium transport predisposes to pulmonary oedema and delays its resolution in mice. J Physiol (Lond) 2004;560:857–865. doi: 10.1113/jphysiol.2004.066704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 26.Icard P, Saumon G. Alveolar sodium and liquid transport in mice. Am J Physiol. 1999;277:L1232–1238. doi: 10.1152/ajplung.1999.277.6.L1232. [DOI] [PubMed] [Google Scholar]
- 27.Factor P, Saldías F, Ridge K, Dumasius V, Zabner J, Jaffe HA, Blanco G, Barnard M, Mercer R, Perrin R, Sznajder JI. Augmentation of lung liquid clearance via adenovirus-mediated transfer of a Na,K-ATPase beta1 subunit gene. J Clin Invest. 1998;102:1421–1430. doi: 10.1172/JCI3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azzam ZS, Dumasius V, Saldías FJ, Adir Y, Sznajder JI, Factor P. Na,K-ATPase overexpression improves alveolar fluid clearance in a rat model of elevated left atrial pressure. Circulation. 2002;105:497–501. doi: 10.1161/hc0402.102848. [DOI] [PubMed] [Google Scholar]
- 29.Looney MR, Sartori C, Chakraborty S, James PF, Lingrel JB, Matthay MA. Decreased expression of both the alpha1- and alpha2-subunits of the Na-K-ATPase reduces maximal alveolar epithelial fluid clearance. Am J Physiol Lung Cell Mol Physiol. 2005;289:L104–110. doi: 10.1152/ajplung.00464.2004. [DOI] [PubMed] [Google Scholar]
- 30.Mutlu GM, Adir Y, Jameel M, Akhmedov AT, Welch L, Dumasius V, Meng FJ, Zabner J, Koenig C, Lewis ER, Balagani R, Traver G, Sznajder JI, Factor P. Interdependency of beta-adrenergic receptors and CFTR in regulation of alveolar active Na+ transport. Circ Res. 2005;96:999–1005. doi: 10.1161/01.RES.0000164554.21993.AC. [DOI] [PubMed] [Google Scholar]
- 31.Dehler M, Zessin E, Bärtsch P, Mairbäurl H. Hypoxia causes permeability oedema in the constant-pressure perfused rat lung. Eur Respir J. 2006;27:600–606. doi: 10.1183/09031936.06.00061505. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki S, Noda M, Sugita M, Ono S, Koike K, Fujimura S. Impairment of transalveolar fluid transport and lung Na (+)-K (+)-ATPase function by hypoxia in rats. J Appl Physiol. 1999;87:962–968. doi: 10.1152/jappl.1999.87.3.962. [DOI] [PubMed] [Google Scholar]
- 33.Carpenter TC, Schomberg S, Nichols C, Stenmark KR, Weil JV. Hypoxia reversibly inhibits epithelial sodium transport but does not inhibit lung ENaC or Na-K-ATPase expression. Am J Physiol Lung Cell Mol Physiol. 2003;284:L77–83. doi: 10.1152/ajplung.00181.2002. [DOI] [PubMed] [Google Scholar]
- 34.Wodopia R, Ko HS, Billian J, Wiesner R, Bärtsch P, Mairbäurl H. Hypoxia decreases proteins involved in epithelial electrolyte transport in A549 cells and rat lung. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1110–1119. doi: 10.1152/ajplung.2000.279.6.L1110. [DOI] [PubMed] [Google Scholar]
- 35.Vivona ML, Matthay M, Chabaud MB, Friedlander G, Clerici C. Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: reversal by beta-adrenergic agonist treatment. Am J Respir Cell Mol Biol. 2001;25:554–561. doi: 10.1165/ajrcmb.25.5.4420. [DOI] [PubMed] [Google Scholar]
- 36.Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest. 2003;111:1057–1064. doi: 10.1172/JCI16826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dada LA, Sznajder JI. Mechanisms of pulmonary edema clearance during acute hypoxemic respiratory failure: role of the Na,K-ATPase. Crit Care Med. 2003;31:S248–252. doi: 10.1097/01.CCM.0000057895.22008.EC. [DOI] [PubMed] [Google Scholar]
- 38.Höschele S, Mairbäurl H. Alveolar flooding at high altitude: failure of reabsorption? News Physiol Sci. 2003;18:55–59. doi: 10.1152/nips.01421.2002. [DOI] [PubMed] [Google Scholar]
- 39.Vadász I, Sznajder J. Hypoxia-induced alveolar epithelial dysfunction. Journal of Organ Dysfunction. 2006;2:244–249. doi: 10.1080/17471060600763377. [DOI] [Google Scholar]
- 40.Johnson CR, Guo Y, Helton ES, Matalon S, Jackson RM. Modulation of rat lung Na+, K (+)-ATPase gene expression by hyperoxia. Exp Lung Res. 1998;24:173–188. doi: 10.3109/01902149809099581. [DOI] [PubMed] [Google Scholar]
- 41.Yue G, Russell WJ, Benos DJ, Jackson RM, Olman MA, Matalon S. Increased expression and activity of sodium channels in alveolar type II cells of hyperoxic rats. Proc Natl Acad Sci U S A. 1995;92:8418–8422. doi: 10.1073/pnas.92.18.8418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardiman KM, Lindsey JR, Matalon S. Lack of amiloride-sensitive transport across alveolar and respiratory epithelium of iNOS (–/–) mice in vivo. Am J Physiol Lung Cell Mol Physiol. 2001;281:L722–731. doi: 10.1152/ajplung.2001.281.3.L722. [DOI] [PubMed] [Google Scholar]
- 43.Factor P, Dumasius V, Saldías F, Brown LA, Sznajder JI. Adenovirus-mediated transfer of an Na+/K+-ATPase beta1 subunit gene improves alveolar fluid clearance and survival in hyperoxic rats. Hum Gene Ther. 2000;11:2231–2242. doi: 10.1089/104303400750035753. [DOI] [PubMed] [Google Scholar]
- 44.Mutlu GM, Dumasius V, Burhop J, McShane PJ, Meng FJ, Welch L, Dumasius A, Mohebahmadi N, Thakuria G, Hardiman K, Matalon S, Hollenberg S, Factor P. Upregulation of alveolar epithelial active Na+ transport is dependent on beta2-adrenergic receptor signaling. Circ Res. 2004;94:1091–1100. doi: 10.1161/01.RES.0000125623.56442.20. [DOI] [PubMed] [Google Scholar]
- 45.Corbridge TC, Wood LD, Crawford GP, Chudoba MJ, Yanos J, Sznajder JI. Adverse effects of large tidal volume and low PEEP in canine acid aspiration. Am Rev Respir Dis. 1990;142:311–315. doi: 10.1164/ajrccm/142.2.311. [DOI] [PubMed] [Google Scholar]
- 46.Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1999;282:54–61. doi: 10.1001/jama.282.1.54. [DOI] [PubMed] [Google Scholar]
- 47.Lecuona E, Saldías F, Comellas A, Ridge K, Guerrero C, Sznajder JI. Ventilator-associated lung injury decreases lung ability to clear edema in rats. Am J Respir Crit Care Med. 1999;159:603–609. doi: 10.1164/ajrccm.159.2.9805050. [DOI] [PubMed] [Google Scholar]
- 48.Frank JA, Gutierrez JA, Jones KD, Allen L, Dobbs L, Matthay MA. Low tidal volume reduces epithelial and endothelial injury in acid-injured rat lungs. Am J Respir Crit Care Med. 2002;165:242–249. doi: 10.1164/ajrccm.165.2.2108087. [DOI] [PubMed] [Google Scholar]
- 49.Laffey JG, Kavanagh BP. Hypocapnia. N Engl J Med. 2002;347:43–53. doi: 10.1056/NEJMra012457. [DOI] [PubMed] [Google Scholar]
- 50.Laffey JG, Engelberts D, Kavanagh BP. Injurious effects of hypocapnic alkalosis in the isolated lung. Am J Respir Crit Care Med. 2000;162:399–405. doi: 10.1164/ajrccm.162.2.9911026. [DOI] [PubMed] [Google Scholar]
- 51.Myrianthefs PM, Briva A, Lecuona E, Dumasius V, Rutschman DH, Ridge KM, Baltopoulos GJ, Sznajder JI. Hypocapnic but not metabolic alkalosis impairs alveolar fluid reabsorption. Am J Respir Crit Care Med. 2005;171:1267–1271. doi: 10.1164/rccm.200408-998OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vadász I, Morty RE, Olschewski A, Konigshoff M, Kohstall MG, Ghofrani HA, Grimminger F, Seeger W. Thrombin impairs alveolar fluid clearance by promoting endocytosis of Na+, K+-ATPase. Am J Respir Cell Mol Biol. 2005;33:343–354. doi: 10.1165/rcmb.2004-0407OC. [DOI] [PubMed] [Google Scholar]
- 53.Planès C, Leyvraz C, Uchida T, Angelova MA, Vuagniaux G, Hummler E, Matthay M, Clerici C, Rossier B. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1099–1109. doi: 10.1152/ajplung.00332.2004. [DOI] [PubMed] [Google Scholar]
- 54.Pittet JF, Wiener-Kronish JP, McElroy MC, Folkesson HG, Matthay MA. Stimulation of lung epithelial liquid clearance by endogenous release of catecholamines in septic shock in anesthetized rats. J Clin Invest. 1994;94:663–671. doi: 10.1172/JCI117383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hickman-Davis JM, McNicholas-Bevensee C, Davis IC, Ma HP, Davis GC, Bosworth CA, Matalon S. Reactive species mediate inhibition of alveolar type II sodium transport during Mycoplasma infection. Am J Respir Crit Care Med. 2006;173:334–344. doi: 10.1164/rccm.200501-155OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen XJ, Seth S, Yue G, Kamat P, Compans RW, Guidot D, Brown LA, Eaton DC, Jain L. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol. 2004;287:L366–373. doi: 10.1152/ajplung.00011.2004. [DOI] [PubMed] [Google Scholar]
- 57.Looney MR, Su X, Van Ziffel JA, Lowell CA, Matthay MA. Neutrophils and their Fc receptors are essential in a mouse model of transfusion-related acute lung injury. J Clin Invest. 2006;116:1615–1623. doi: 10.1172/JCI27238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med. 1999;27:304–312. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- 59.Fukuda N, Jayr C, Lazrak A, Wang Y, Lucas R, Matalon S, Matthay MA. Mechanisms of TNF-alpha stimulation of amiloride-sensitive sodium transport across alveolar epithelium. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1258–1265. doi: 10.1152/ajplung.2001.280.6.L1258. [DOI] [PubMed] [Google Scholar]
- 60.Olivera W, Ridge K, Wood LD, Sznajder JI. ANF decreases active sodium transport and increases alveolar epithelial permeability in rats. J Appl Physiol. 1993;75:1581–1586. doi: 10.1152/jappl.1993.75.4.1581. [DOI] [PubMed] [Google Scholar]
- 61.Frank J, Roux J, Kawakatsu H, Su G, Dagenais A, Berthiaume Y, Howard M, Canessa CM, Fang X, Sheppard D, Matthay MA, Pittet JF. Transforming growth factor-beta1 decreases expression of the epithelial sodium channel alphaENaC and alveolar epithelial vectorial sodium and fluid transport via an ERK1/2-dependent mechanism. J Biol Chem. 2003;278:43939–43950. doi: 10.1074/jbc.M304882200. [DOI] [PubMed] [Google Scholar]
- 62.Cepkova M, Matthay MA. Pharmacotherapy of acute lung injury and the acute respiratory distress syndrome. J Intensive Care Med. 2006;21:119–143. doi: 10.1177/0885066606287045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.ARDSNet Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 64.Walters DV, Olver RE. The role of catecholamines in lung liquid absorption at birth. Pediatr Res. 1978;12:239–242. doi: 10.1203/00006450-197803000-00017. [DOI] [PubMed] [Google Scholar]
- 65.Berthiaume Y, Staub NC, Matthay MA. Beta-adrenergic agonists increase lung liquid clearance in anesthetized sheep. J Clin Invest. 1987;79:335–343. doi: 10.1172/JCI112817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crandall ED, Heming TA, Palombo RL, Goodman BE. Effects of terbutaline on sodium transport in isolated perfused rat lung. J Appl Physiol. 1986;60:289–294. doi: 10.1152/jappl.1986.60.1.289. [DOI] [PubMed] [Google Scholar]
- 67.Sakuma T, Okaniwa G, Nakada T, Nishimura T, Fujimura S, Matthay MA. Alveolar fluid clearance in the resected human lung. Am J Respir Crit Care Med. 1994;150:305–310. doi: 10.1164/ajrccm.150.2.8049807. [DOI] [PubMed] [Google Scholar]
- 68.Parker JC, Ivey CL. Isoproterenol attenuates high vascular pressure-induced permeability increases in isolated rat lungs. J Appl Physiol. 1997;83:1962–1967. doi: 10.1152/jappl.1997.83.6.1962. [DOI] [PubMed] [Google Scholar]
- 69.Bertorello AM, Ridge KM, Chibalin AV, Katz AI, Sznajder JI. Isoproterenol increases Na+-K+-ATPase activity by membrane insertion of alpha-subunits in lung alveolar cells. Am J Physiol. 1999;276:L20–27. doi: 10.1152/ajplung.1999.276.1.L20. [DOI] [PubMed] [Google Scholar]
- 70.Planès C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and beta 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem. 2002;277:47318–47324. doi: 10.1074/jbc.M209158200. [DOI] [PubMed] [Google Scholar]
- 71.Mutlu GM, Sznajder JI. Mechanisms of pulmonary edema clearance. Am J Physiol Lung Cell Mol Physiol. 2005;289:L685–695. doi: 10.1152/ajplung.00247.2005. [DOI] [PubMed] [Google Scholar]
- 72.Saldías FJ, Lecuona E, Comellas AP, Ridge KM, Rutschman DH, Sznajder JI. beta-adrenergic stimulation restores rat lung ability to clear edema in ventilator-associated lung injury. Am J Respir Crit Care Med. 2000;162:282–287. doi: 10.1164/ajrccm.162.1.9809058. [DOI] [PubMed] [Google Scholar]
- 73.Dumasius V, Sznajder JI, Azzam ZS, Boja J, Mutlu GM, Maron MB, Factor P. beta (2)-adrenergic receptor overexpression increases alveolar fluid clearance and responsiveness to endogenous catecholamines in rats. Circ Res. 2001;89:907–914. doi: 10.1161/hh2201.100204. [DOI] [PubMed] [Google Scholar]
- 74.Bertorello AM, Sznajder JI. The dopamine paradox in lung and kidney epithelia: sharing the same target but operating different signaling networks. Am J Respir Cell Mol Biol. 2005;33:432–437. doi: 10.1165/rcmb.2005-0297TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guerrero C, Lecuona E, Pesce L, Ridge KM, Sznajder JI. Dopamine regulates Na-K-ATPase in alveolar epithelial cells via MAPK-ERK-dependent mechanisms. Am J Physiol Lung Cell Mol Physiol. 2001;281:L79–85. doi: 10.1152/ajplung.2001.281.1.L79. [DOI] [PubMed] [Google Scholar]
- 76.Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L710–L722. doi: 10.1152/ajplung.00486.2004. [DOI] [PubMed] [Google Scholar]
- 77.Atabai K, Ware LB, Snider ME, Koch P, Daniel B, Nuckton TJ, Matthay MA. Aerosolized beta (2)-adrenergic agonists achieve therapeutic levels in the pulmonary edema fluid of ventilated patients with acute respiratory failure. Intensive Care Med. 2002;28:705–711. doi: 10.1007/s00134-002-1282-x. [DOI] [PubMed] [Google Scholar]
- 78.Sartori C, Allemann Y, Duplain H, Lepori M, Egli M, Lipp E, Hutter D, Turini P, Hugli O, Cook S, Nicod P, Scherrer U. Salmeterol for the prevention of high-altitude pulmonary edema. N Engl J Med. 2002;346:1631–1636. doi: 10.1056/NEJMoa013183. [DOI] [PubMed] [Google Scholar]
- 79.Perkins GD, McAuley DF, Thickett DR, Gao F. The beta-agonist lung injury trial (BALTI): a randomized placebo-controlled clinical trial. Am J Respir Crit Care Med. 2006;173:281–287. doi: 10.1164/rccm.200508-1302OC. [DOI] [PubMed] [Google Scholar]
- 80.Helve O, Pitkanen OM, Andersson S, O'Brodovich H, Kirjavainen T, Otulakowski G. Low expression of human epithelial sodium channel in airway epithelium of preterm infants with respiratory distress. Pediatrics. 2004;113:1267–1272. doi: 10.1542/peds.113.5.1267. [DOI] [PubMed] [Google Scholar]
- 81.Noda M, Suzuki S, Tsubochi H, Sugita M, Maeda S, Kobayashi S, Kubo H, Kondo T. Single dexamethasone injection increases alveolar fluid clearance in adult rats. Crit Care Med. 2003;31:1183–1189. doi: 10.1097/01.CCM.0000059640.77535.29. [DOI] [PubMed] [Google Scholar]
- 82.Meduri GU, Headley AS, Golden E, Carson SJ, Umberger RA, Kelso T, Tolley EA. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: a randomized controlled trial. JAMA. 1998;280:159–165. doi: 10.1001/jama.280.2.159. [DOI] [PubMed] [Google Scholar]
- 83.Steinberg KP, Hudson LD, Goodman RB, Hough CL, Lanken PN, Hyzy R, Thompson BT, Ancukiewicz M. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354:1671–1684. doi: 10.1056/NEJMoa051693. [DOI] [PubMed] [Google Scholar]
- 84.Ware LB, Matthay MA. Keratinocyte and hepatocyte growth factors in the lung: roles in lung development, inflammation, and repair. Am J Physiol Lung Cell Mol Physiol. 2002;282:L924–940. doi: 10.1152/ajplung.00439.2001. [DOI] [PubMed] [Google Scholar]
- 85.Wang Y, Folkesson HG, Jayr C, Ware LB, Matthay MA. Alveolar epithelial fluid transport can be simultaneously upregulated by both KGF and beta-agonist therapy. J Appl Physiol. 1999;87:1852–1860. doi: 10.1152/jappl.1999.87.5.1852. [DOI] [PubMed] [Google Scholar]
- 86.Guery BP, Mason CM, Dobard EP, Beaucaire G, Summer WR, Nelson S. Keratinocyte growth factor increases transalveolar sodium reabsorption in normal and injured rat lungs. Am J Respir Crit Care Med. 1997;155:1777–1784. doi: 10.1164/ajrccm.155.5.9154891. [DOI] [PubMed] [Google Scholar]
- 87.Sznajder JI, Ridge KM, Yeates DB, Ilekis J, Olivera W. Epidermal growth factor increases lung liquid clearance in rat lungs. J Appl Physiol. 1998;85:1004–1010. doi: 10.1152/jappl.1998.85.3.1004. [DOI] [PubMed] [Google Scholar]
- 88.Folkesson HG, Norlin A, Wang Y, Abedinpour P, Matthay MA. Dexamethasone and thyroid hormone pretreatment upregulate alveolar epithelial fluid clearance in adult rats. J Appl Physiol. 2000;88:416–424. doi: 10.1152/jappl.2000.88.2.416. [DOI] [PubMed] [Google Scholar]
- 89.Ulrich K, Stern M, Goddard ME, Williams J, Zhu J, Dewar A, Painter HA, Jeffery PK, Gill DR, Hyde SC, Geddes DM, Takata M, Alton EW. Keratinocyte growth factor therapy in murine oleic acid-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1179–1192. doi: 10.1152/ajplung.00450.2004. [DOI] [PubMed] [Google Scholar]
- 90.Elia N, Tapponnier M, Matthay MA, Hamacher J, Pache JC, Brundler MA, Totsch M, De Baetselier P, Fransen L, Fukuda N, Morel DR, Lucas R. Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-alpha. Am J Respir Crit Care Med. 2003;168:1043–1050. doi: 10.1164/rccm.200206-618OC. [DOI] [PubMed] [Google Scholar]
- 91.Lang JD, McArdle PJ, O'Reilly PJ, Matalon S. Oxidant-antioxidant balance in acute lung injury. Chest. 2002;122:314–320. doi: 10.1378/chest.122.6_suppl.314S. [DOI] [PubMed] [Google Scholar]
- 92.Zhu S, Ware LB, Geiser T, Matthay MA, Matalon S. Increased levels of nitrate and surfactant protein a nitration in the pulmonary edema fluid of patients with acute lung injury. Am J Respir Crit Care Med. 2001;163:166–172. doi: 10.1164/ajrccm.163.1.2005068. [DOI] [PubMed] [Google Scholar]
- 93.Modelska K, Matthay MA, Brown LA, Deutch E, Lu LN, Pittet JF. Inhibition of beta-adrenergic-dependent alveolar epithelial clearance by oxidant mechanisms after hemorrhagic shock. Am J Physiol. 1999;276:L844–857. doi: 10.1152/ajplung.1999.276.5.L844. [DOI] [PubMed] [Google Scholar]
- 94.Lee H, Pespeni M, Roux J, Dennery PA, Matthay MA, Pittet JF. HO-1 induction restores c-AMP-dependent lung epithelial fluid transport following severe hemorrhage in rats. FASEB J. 2005;19:287–289. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- 95.Suter PM, Domenighetti G, Schaller MD, Laverriere MC, Ritz R, Perret C. N-acetylcysteine enhances recovery from acute lung injury in man. A randomized, double-blind, placebo-controlled clinical study. Chest. 1994;105:190–194. doi: 10.1378/chest.105.1.190. [DOI] [PubMed] [Google Scholar]
- 96.Domenighetti G, Suter PM, Schaller MD, Ritz R, Perret C. Treatment with N-acetylcysteine during acute respiratory distress syndrome: a randomized, double-blind, placebo-controlled clinical study. J Crit Care. 1997;12:177–182. doi: 10.1016/S0883-9441(97)90029-0. [DOI] [PubMed] [Google Scholar]
- 97.Robriquet L, Collet F, Tournoys A, Prangère T, Nevière R, Fourrier F, Guery BP. Intravenous administration of activated protein C in Pseudomonas-induced lung injury: impact on lung fluid balance and the inflammatory response. Respir Res. 2006;7:41. doi: 10.1186/1465-9921-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dumasius V, Mendez M, Mutlu GM, Factor P. Acute lung injury does not impair adenoviral-mediated gene transfer to the alveolar epithelium. Chest. 2002;121:33–34. doi: 10.1378/chest.121.3_suppl.33S. [DOI] [PubMed] [Google Scholar]
- 99.Shinohara T, Kaneko T, Nagashima Y, Ueda A, Tagawa A, Ishigatsubo Y. Adenovirus-mediated transfer and overexpression of heme oxygenase 1 cDNA in lungs attenuates elastase-induced pulmonary emphysema in mice. Hum Gene Ther. 2005;16:318–327. doi: 10.1089/hum.2005.16.318. [DOI] [PubMed] [Google Scholar]
- 100.Guidot DM, Folkesson HG, Jain L, Sznajder JI, Pittet JF, Matthay MA. Integrating Acute Lung Injury and Regulation of Alveolar Fluid Clearance. Am J Physiol Lung Cell Mol Physiol. 2006;291:L301–306. doi: 10.1152/ajplung.00153.2006. [DOI] [PubMed] [Google Scholar]