

Abstract
Here we focus on how neutrophils have a key regulatory role in vascular inflammation. Recent studies using advanced imaging techniques have yielded new insights into the mechanisms by which neutrophils contribute to defense against bacterial infections and also against sterile injury. In these settings, neutrophils are recruited by various mechanisms depending on the situation. We also describe how these processes may be disrupted in systemic infections, with a particular emphasis on mouse models of sepsis. Neutrophils are often immobilized in the lungs and liver during systemic infections, and this immobilization may be a mechanism through which bacteria can evade the innate immune response or allow neutrophils to form neutrophil extracellular traps that trap and kill bacteria in blood. The platelet is also an important player in sepsis, and we describe how it collaborates with neutrophils in the formation of neutrophil extracellular traps.
Supplementary information
The online version of this article (doi:10.1038/nm.2514) contains supplementary material, which is available to authorized users.
Subject terms: Neutrophils, Sepsis, Bacterial infection
Main
The neutrophil patrols the blood in continuous search of prey, which presents itself primarily in the form of bacteria or dead and dying host cells. Because more than 4 trillion bacteria have colonized each human, particularly at the mucosal and skin surfaces, the task of preventing pathogen invasion is not trivial. In more than half a million people in North America each year, this system fails and severe sepsis ensues, killing approximately one-third of these people. Therefore, understanding the behavior of the immune system in response to pathogens, particularly the role of neutrophils, is essential. There is much urgency in this regard, as antibiotic-resistant bacteria such as Staphylococcus aureus and multidrug-resistant Gram-negative bacilli are on the rise, and viruses (such as the SARS corona virus and H5N1 and H1N1 influenza viruses) that make the host more susceptible to secondary infections are also becoming more prominent. Tweaking host immunity may be a way to fight these emerging pathogens.
In addition to the inflammation that occurs in blood vessels as a result of infection, inflammation can also occur intravascularly as a consequence of cell death or sterile injury. Whether the neutrophil can discriminate between a sterile injury and an infection is not clear, but the molecular mechanisms mediating neutrophil recruitment can be quite variable. Ultimately, to perform their functions, neutrophils need to know where to go. Therefore, efficient neutrophil guidance through the vasculature and into the affected tissue site is crucial and is normally governed by chemotactic gradients. During systemic infection, this system is altered, as the highest concentrations of chemokines and other chemotactic agents are found in plasma. This reverse chemotactic gradient results in activated neutrophils within the vasculature. For many years, the prevailing view was that these neutrophils get stuck in the capillary beds of lungs and liver where they release their cytotoxic contents, which induce inappropriate tissue injury leading to multiorgan failure. However, recently, other explanations have been put forth that suggest that sequestration of neutrophils in lungs and liver is beneficial to host survival. New and somewhat unorthodox ways by which neutrophils may fight bacterial infections in the blood have been described. As one example, neutrophils have been shown to expel their DNA to form neutrophil extracellular traps (NETs) to ensnare bacteria before dying. Interactions between platelets and neutrophils are essential for this process to occur in the vasculature.
This review discusses recent work pertaining to neutrophil recruitment and functions during both sterile injury and infectious stimuli. We use sepsis as an example of the war between neutrophils and bacteria and the intriguing approaches that are implemented as the host and pathogen each attempt to get the upper hand. We also discuss the emerging roles of neutrophils during tissue repair and angiogenesis. Many of these discoveries rely on experimental mouse models of inflammation and newly evolving imaging technology, and we pay particular attention to these studies. However, it is noteworthy that the relevance of many of these observations in mice remains to be confirmed in humans.
Neutrophil recruitment to sites of local infection
Over the last few years, the multistep process of neutrophil recruitment from blood to tissue has been extensively studied using various in vivo microscopy approaches (summarized in Fig. 1). In vivo spinning-disk confocal, laser-scanning confocal and two-photon microscopy have enabled the refined, high-resolution examination of neutrophil behavior in the context of other cells and allowed researchers to follow the interactions of neutrophils with the surrounding endothelium and interendothelial junctions, as well as their behavior as they emigrate from the vasculature.
Figure 1. The neutrophil recruitment cascade.
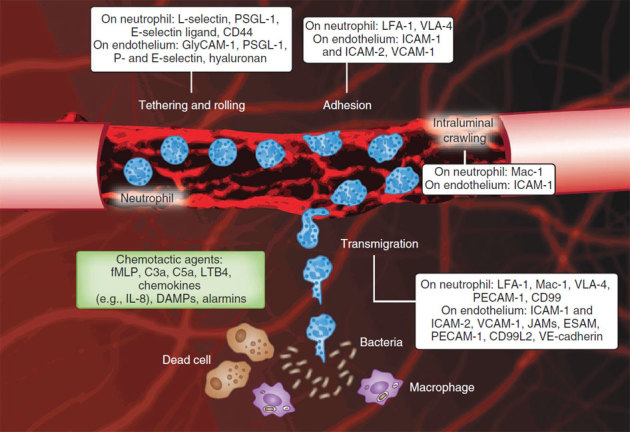
Intravital confocal microscopy image of a cremasteric postcapillary venule with added cartoon cells illustrating the consecutive steps of the recruitment of circulating neutrophils to localized inflammation (infection or necrosis). The white boxes show the adhesion molecules involved in each step. Endothelial cell junctions are stained with monoclonal antibodies to CD31 (red). Endothelial upregulation of adhesion molecules results in interactions between selectins and their ligands on neutrophils, leading to neutrophil tethering and rolling. Chemokines sequestered on the luminal endothelium induce conformation changes of neutrophil β2 integrins, which results in neutrophil adhesion and crawling. Mechanotactic and chemotactic guidance signals direct crawling neutrophils to junctional transmigration sites closer to the source of chemotactic agent, examples of which are given in the green box.
Localized tissue infections are thought to activate sentinel cells, which include dendritic cells, macrophages, mast cells, endothelial cells and perhaps all parenchymal cells, inducing the recruitment of circulating neutrophils to the infected tissue. In addition, monocytes crawling along the lumen of blood vessels may infiltrate tissues early in infection to then recruit neutrophils1. The upregulation of adhesion molecules, such as selectins, on endothelium is essential to initiate neutrophil recruitment. P-selectin, stored in Weibel-Palade bodies within resting endothelial cells, and E-selectin, which is synthesized de novo, are translocated to the apical cell membrane where they transiently bind ligands on neutrophils, including P-selectin glycoprotein ligand-1 (PSGL-1), E-selectin ligand-1 (ESL-1), CD44 and other glycosylated ligands (Fig. 1). Transient interactions between selectins and their ligands on neutrophils result in neutrophil tethering and rolling, which are the first steps of the leukocyte recruitment cascade2,3.
Molecular guidance signals (chemoattractants or chemokines) originating from the infection or released by tissue leukocytes form gradients that guide leukocytes to the site of infection. G protein–coupled receptors on rolling neutrophils bind chemokines sequestered on the apical endothelium, leading to 'inside-out' signals that induce conformational changes in β2 integrins expressed by neutrophils that increase their avidity and affinity, thus resulting in neutrophil arrest4 and subsequent intravascular crawling to transmigration sites5. Neutrophils express high levels of the β2 integrins CD11a (ITGAL)-CD18 (also known as LFA-1 or αLβ2) and CD11b (ITGAM)-CD18 (also known as Mac-1 or αMβ2), and the distinct roles of these molecules have been recently described1,5. CD11a-CD18 is involved in firm adhesion, and CD11b-CD18 is crucial for the intravascular crawling of neutrophils5 but not for crawling mononuclear cells1.
Shear contributes to the directional crawling of neutrophils, as adherent neutrophils in venules immediately start to crawl perpendicular to blood flow, a behavior that was confirmed in vitro when shear was applied to adherent neutrophils6. Teleologically, because endothelial cells are elongated in the direction of flow, perpendicular crawling allows a neutrophil to find an endothelial junction in the shortest period of time. This inherent response to shear might be important for rapid transmigration, as neutrophils have been shown to preferentially transmigrate through junctions5,6,7,8,9,10,11 unless crawling was specifically inhibited5. In CD11b-deficient mice, neutrophils did not crawl within the vasculature, and, consequently, transcellular transmigration through endothelial cells predominated5. Endothelial cells are very active and responsive during transmigration and form docking structures (endothelial projections)12,13,14 that, in vivo, resulted in domes surrounding the entire neutrophil15,16,17. Endothelial membrane proteins, including PECAM-1, ESAM-1 and CD99, aid neutrophil emigration, and additional adhesive molecules help neutrophils move across the basement membrane through areas with low expression of laminin, collagen and nidogen-2 (ref. 18). Reverse migration back into the vessel is prevented by endothelial JAM-C11,19.
During tissue infection, numerous neutrophils are recruited locally from blood and are also mobilized from bone marrow, which results in blood neutrophilia to ensure an adequate neutrophil supply. Key molecules during neutrophil mobilization from bone marrow include leukotriene B4, C5a and the CXC chemokine interleukin-8 (IL-8) (in rodents, the key molecules are CXCL1 (also known as KC) and CXCL2 (also known as MIP-2))20, whereas CXCL12 (stromal cell–derived factor-1 or SDF-1) and its receptor CXCR4 are involved in neutrophil retention in bone marrow21. However, CXCL12 is also crucial for neutrophil mobilization during infection. During sepsis, production of CXCL12 by bone marrow is reduced and its systemic production is increased, resulting in an inverted gradient that directs neutrophils out of the bone marrow and into the circulation22.
How do chemokine signals get established in flowing blood? Early work proposed that proinflammatory molecules such as platelet-activating factor were embedded in the membrane of activated endothelium and functioned in a juxtacrine manner to activate and polarize neutrophils23. Evidence is now emerging that chemokines also form gradients within the vascular compartment. Experiments in which a chemokine source was placed on one side of a blood vessel showed that the crawling neutrophils were directed to emigrate from that side of the vessel and that heparan sulfate was necessary for this process24. The heparan sulfate residues on the surface of endothelium prevent dissolution of the chemokine gradients into the blood and provide localized directional cues for crawling neutrophils24. These observations were made using CXCL1 and CXCL2 chemokines, but it is notable that different chemokines bind with different affinities to heparan sulfate25,26, so whether all chemokines can establish these heptotactic gradients remains unclear.
Numerous chemoattractants can be released to form gradients during a bacterial infection, partly from the bacteria themselves or from complement fragments emanating from the bacteria but also from nearby activated macrophages and endothelial cells. Neutrophils have been shown to respond to such guidance signals in a hierarchical manner, preferring 'end-target' chemoattractant factors such as bacterial products (for example, N-formyl-methionine-leucine-phenylalanine (fMLP)) and C5a over 'intermediate' chemokines such as IL-8 (ref. 27). This preference raised the possibility that separate signaling pathways were activated in neutrophils in response to fMLP and IL-8. Notably, both fMLP and IL-8 activated the same pathway by activating the class I isoform of phosphatidylinositol 3-kinase (PI3K), which phosphorylates the membrane lipid phosphatidylinositol-3,4-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-triphosphate (PIP3) at the leading edge of the neutrophil (Fig. 2). At the same time, the phosphatase and tensin homolog (PTEN) is localized to the sides and back of the crawling cell, thereby converting PIP3 back to PIP2 in areas of the cell away from the leading edge28. It is worth mentioning that there is a body of work that suggests that SHIP rather than PTEN is the important phosphatase in this process29. This mechanism is thought to allow the neutrophil to recognize and amplify shallow gradients of only a few molecules along its length to know that it must move in the direction of PIP3. In opposing gradients, PTEN and PIP2 surround the entire cell circumference (Fig. 2c), and no gradient is recognized. However, if the same pathway is activated by all chemoattractants and PIP2 surrounds the entire cell, then how does a neutrophil ever detect a gradient? A number of groups have shown that fMLP but not IL-8 also activates p38 mitogen-activated protein kinase (MAPK) (Fig. 2b)30,31, and that the fMLP-induced p38 MAPK allowed the neutrophil to respond to the bacterial product (Fig. 2c)32. Additional molecules that contribute to chemoattractant signaling in migrating cells ranging from Dictyostelium to neutrophils have been described recently, for example, PLA2 (ref. 33), cGMP34 and DOCK2/plus phosphatidic acid35, but whether they have redundant or distinct functions and how they fit into the hierarchical nature of neutrophil chemotaxis remains unclear.
Figure 2. Proposed model of PTEN, PI3K and p38 MAPK function during neutrophil chemotaxis.
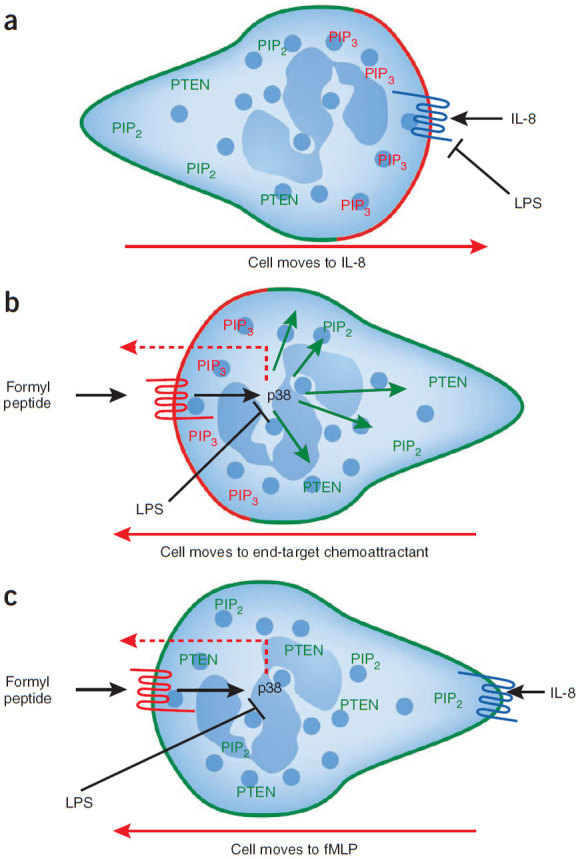
(a) The localizations of PIP3 (red), PTEN and PIP2 (green) are shown in a neutrophil migrating toward an intermediary chemoattractant (CXCR2 ligand (CXCL2–MIP-2 or CXCL8-IL-8)). LPS inhibits IL-8–induced chemotaxis. (b) The localizations of PIP3 (red), PTEN and PIP2 (green) are shown in a cell migrating toward an end-target chemoattractant (formylpeptide). p38 MAPK mediates both PTEN localization (green arrows) as well as chemotaxis (red dotted arrow) in response to end-target chemoattractants. LPS inhibits p38 MAPK and thereby inhibits chemotaxis. (c) The localizations of PTEN and PIP2 (green) are shown in a neutrophil migrating in opposing gradients of end-target (formylpeptide) and intermediary (CXCR2 ligand) chemoattractants. Because PTEN antagonizes any PIP3 accumulation, all chemotaxis occurs through p38 MAPK (red dotted arrow). LPS stops chemotaxis by overriding the effects of the chemoattractants through p38 MAPK inhibition.
Although no formal evidence of multiple in vivo gradients of chemoattractants exists, it is hard to imagine how neutrophils could move through numerous compartments over a substantial distance using only a single gradient. When one considers that a neutrophil adheres to the endothelium after being activated by endothelially produced and apically sequestered IL-8, unless a second chemoattractant is released from the affected tissue, it is difficult to understand why neutrophils would move out of the vasculature and away from the vessel. Once they have emigrated, neutrophils need to migrate toward the bacterial focus or the dying cells without being distracted by what would then be an opposing IL-8 gradient coming from the vasculature. Receptor internalization of IL-8, allowing a second chemoattractant to dominate in a sequential manner, could occur36. Alternatively, a hierarchical response may take place in which fMLP and other end-target chemoattractants inhibit the IL-8 signal30. Recently, an in vivo experiment showed that neutrophils in sinusoidal vessels initially crawled along an MIP-2 gradient but then ignored this signal and followed a new gradient of fMLP receptor ligands laid over the MIP-2 gradient, thus gaining access to a precise tissue site37 (Fig. 3). Therefore, growing evidence indicates that neutrophil recruitment in vivo results from the release of multiple chemoattractants and that synergistic and opposing effects of the involved signaling molecules are likely to govern neutrophil recruitment into the afflicted tissues.
Figure 3. Neutrophils move to sites of sterile injury by intravascular crawling.
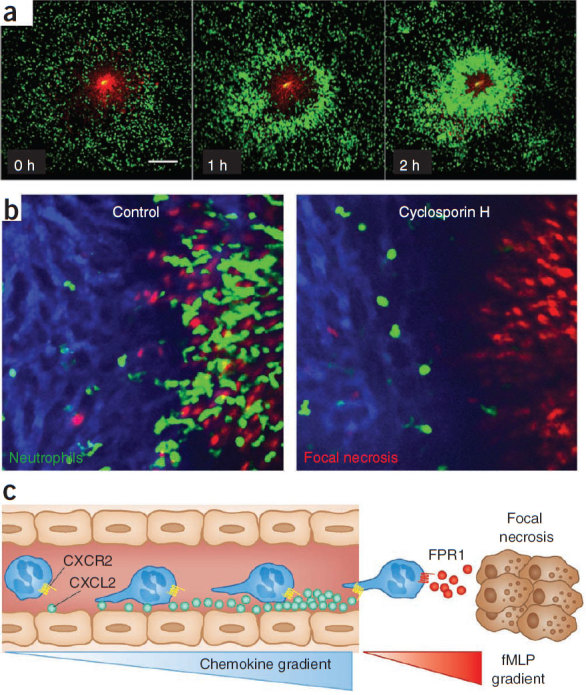
(a) Time-lapse images using spinning-disk confocal intravital microscopy show the response of neutrophils (green) to focal hepatic necrosis (red, propiodium iodide). Scale bar, 200 μm (ref. 37). (b) Images of neutrophils homing (green) to sterile injury (red, propiodium iodide) in the liver of untreated mice (left) or after treatment with the fMLP receptor inhibitor cyclosporine H (right). (c) Neutrophils within the liver vasculature can prioritize an fMLP gradient when they are subjected to competing gradients of CXCL2 and fMLP, thus allowing them to chemotax toward the site of focal necrosis.
Neutrophil recruitment is different in systemic infections
The highly regulated mechanisms that control neutrophils during chemotaxis are altered in systemic infection and are perhaps disrupted in mouse models of endotoxemia and sepsis in which systemic infection is presumed to occur. As a corollary, neutrophil migration defects have consistently been reported in septic individuals38. Sepsis is clinically defined by the presence of infection plus some of the following: fever, tachypnea, edema and increased or decreased white blood cell counts, high serum concentrations of chemokines and C-reactive protein, and hemodynamic alterations. When multiple organs begin to fail, this leads to severe sepsis39. Severe sepsis has one of the highest mortality rates of any disease state. Most importantly, anyone of any age is susceptible to sepsis (although the very young and very old are at the highest risk), and any delay in the immune response is likely to increase mortality, making rapid neutrophil recruitment a key endogenous host defense mechanism. Indeed, neutropenic individuals are highly susceptible to sepsis40. Despite the clear need for effective neutrophil function, overexuberant neutrophils can potentially injure 'bystander' tissues that are not part of the infected site. In fact, for many years, neutrophils have been a therapeutic target in sepsis. Total elimination of neutrophils would lead to death, but altering or modifying their actions may have potential clinical benefits.
During endotoxemia and sepsis in humans and in experimental models, the whole chemotactic process may be disrupted, as the highest concentrations of chemokines and other chemotactic agents are found circulating in the plasma, and they activate the endothelium as well as neutrophils within blood vessels. In addition, the uncontrolled exposure of neutrophils to plasma chemokines results in downregulation of chemokine receptors in individuals with severe sepsis41 as well as downregulation of CD11b in lipopolysaccharide (LPS)-treated mice42, which further impairs chemotaxis and results in insufficient leukocyte recruitment to the source of infection. Furthermore, molecules such as LPS activate p38 MAPK, the key signaling pathway in fMLP-induced chemotaxis, through Toll-like receptor 4 (TLR4), thus disrupting this chemotactic process.
Although LPS is sometimes mistaken for a chemotactic molecule, on its own it does not induce chemotaxis but instead functions as a stop signal that overrides chemoattractants (Fig. 2)30, a function that has also been observed for tumor necrosis factor-α (TNF-α)43. In fact, LPS downregulates CXCR2 on human and mouse neutrophils, thereby inhibiting chemotaxis toward IL-8 in humans and toward KC and MIP-2 in mice44. This downregulation is prevented by interleukin-33 (IL-33) through inhibition of receptor internalization44. Lipoteichoic acid, a component of Gram-negative cell walls and a TLR2 ligand, has been shown to cause downregulation of CXCR2, which would reduce neutrophil chemotaxis45. It is clear that bacterial products that activate TLRs may disrupt neutrophil chemotaxis. These data are consistent with the improved bacterial localization and clearance observed in mice that express TLRs only on endothelium and not on neutrophils or other immune cells46. In wild-type mice, TLRs on macrophages and other immune cells systemically release many chemokines and cytokines that may distract neutrophils and prevent them from locating bacteria46. In addition, circulating activated neutrophils become rigid and can be trapped within the small capillaries of the lung and in liver sinusoids and thereby are prevented from finding bacteria in compartments like the peritoneum, from which the bacteria may be emanating. Finally, certain bacterial proteases have been shown to cleave and inactivate CXC chemokines, thereby impeding the chemotaxis of neutrophils to the source of infection, which decreases bacterial clearance and increases systemic dissemination47.
Other mechanisms underlying the disruption of neutrophil chemotaxis during systemic infection have been summarized elsewhere38 and include activation of the low-affinity fMLP receptor, which inactivates the high-affinity fMLP receptor responsible for chemotaxis48. Nitric oxide (NO) is known to have antiadhesive properties that are mediated by downregulating endothelial adhesion molecules49,50, and the induction of vast amounts of NO through inducible NO synthase could also reduce neutrophil chemotaxis38,51. Moreover, therapeutically administered inhaled NO given to septic individuals with pulmonary hypertension could dislodge neutrophils from lungs, which might be beneficial but could also inhibit their chemotaxis to the necessary sites. There are many unanswered questions about the role of NO in sepsis, but general inhibition of NO may actually harm septic individuals52, although specific inhibition of inducible NO synthase could perhaps be of benefit. Finally, peroxisome proliferator–activated receptor-γ (PPAR-γ) seems to be an endogenous inhibitor of chemotaxis. This molecule is activated during sepsis and could also contribute to neutrophil impairment.
In addition to the disruption of chemokine signaling observed in sepsis, neutrophil adhesion molecules may also be altered. Neutrophil recruitment into the liver and lungs seems to use selectin- and integrin-independent adhesive mechanisms in response to bacteremia or systemic LPS administration53,54,55,56. In lungs, the adhesive mechanisms remain unknown, and some investigators have suggested that physical trapping rather than adhesion molecules may be dominant57. Although physical trapping of neutrophil sequestration in the liver was also proposed55, CD44 has recently been shown to account for 60–70% of adhesion in livers from endotoxemia or bacterial sepsis models42,56.
It seems unclear why humans have not evolved mechanisms to counter these pathogen-disrupting pathways. However, it is possible that all these changes in neutrophils may actually be an attempt to help the host rather than simply a dysregulation. How would a crawling, emigrating neutrophil help eradicate a pathogen in blood? Perhaps neutrophil sequestration in the lungs and liver has an important survival benefit, as we discuss later in this review.
Although mouse models have been used to observe these sepsis-related phenomena, the human data are often less clear. There are notable differences between experimental and clinical sepsis in terms of both the concentrations of bacteria and the bacterial components in circulation and disease progression. Rodents are highly resistant to infections as compared to humans, and the LPS doses used to induce sepsis in mice (typically 0.5–25 mg per kg of body weight) are 1,000–10,000 times higher than those that have been determined to induce severe sepsis in humans. Furthermore, organ failure, which is a major and crucial component of sepsis syndrome in humans, cannot be readily studied in mice that have received high doses of LPS or bacteria because they die before developing these secondary complications. Therefore, the results from models such as cecal ligation and puncture in mice, although more variable, may better reflect the human condition. Even though elevated levels of chemokines, cytokines and many other proinflammatory molecules as well as severe chemotactic defects have all been reported in individuals with severe sepsis38,41, the importance of many of the mechanisms discussed here remain to be confirmed in humans.
Neutrophil recruitment in sterile injury
Another form of vascular inflammation is sterile inflammation. Moreover, paradoxically, sterile injury contributes in a major way to infections and sepsis. Aside from potentially killing the host by hyperreactive immunity, many bacteria and viruses cause cell death and cell lysis, which lead to the release of multiple molecules associated with sterile injury. Moreover, as hypotension develops during traumatic or septic shock, tissue ischemia caused by hypoperfusion progresses. This ischemia is followed by reperfusion during aggressive volume resuscitation or rapid blood transfusion, which are procedures used to treat hypotension. Reperfusion injury as a form of sterile injury has been studied for more than 30 years, and a role for neutrophils and their oxidants and proteases has clearly been established58. Depletion of neutrophils, or blockade of neutrophil responses, is effective at reducing pathology not only in ischemia and reperfusion59 but also in trauma, drug-induced hepatotoxicity and other modes of sterile injury60,61.
Injury that causes cell lysis in vivo releases many proinflammatory damage-associated molecular pattern molecules (DAMPs), also called alarmins or danger signals. These are host components released or generated after tissue injury that have inherent proinflammatory activity and thereby alert the innate immune system. A number of comprehensive reviews covering the identities and functions of individual danger signals have recently been published62,63. DAMPs may be proteolytically or oxidatively modified extracellular molecules (for example, digested hyaluronan or precipitated uric acid crystals) or intracellular substances that are actively secreted in response to cell stress (for example, high mobility group protein B1 (HMGB1)) or passively released when the cell membrane integrity is compromised (for example, ATP)64. DAMPs and pathogen-associated molecular patterns function similarly, both stimulating pattern recognition receptors such as TLRs or NOD-like receptors (NLRs)62,63,64. The NLRs comprise a family of 22 proteins that are mostly cytoplasmic and that recognize host-derived 'danger'-associated molecules. Their activation leads to the assembly of complexes of high molecular mass called inflammasomes that induce the generation of active caspase-1 and the production of mature IL-1β. The discovery that the NOD-like receptor family, pyrin domain–containing 3 (NLRP3) inflammasome can be activated by host-derived particulate matter such as uric acid crystals or ATP has led to its implication in a number of inflammatory diseases63. Non–pattern-recognition receptors including receptor for advanced glycation end products (RAGE), CD44 and CD91 can also contribute to the onset of inflammation in several models of sterile injury65,66,67. Despite much DAMP diversity and organ and/or insult specificity, many of these pathways converge on IL-1. IL-1 acts as a central orchestrator of inflammation and cell recruitment through nuclear factor-κB, p38 and other signaling pathways, which then lead to the formation of chemoattractant gradients. Although many of these DAMPs may indirectly recruit neutrophils, none functions as a true chemoattractant for neutrophil recruitment.
Recently, two independent studies reported that mitochondrial-derived formyl-peptide receptor-1 (FPR1) ligands functioned as chemotactic DAMPs37,68. Figure 3a and Supplementary Video 1 show that following thermal injury, many neutrophils enter the injury site within a few hours. Although the injury was sterile, inhibition of the fMLP receptor (Fig. 3b), which is normally associated with responses to bacterial products, was the most efficient way of inhibiting the neutrophils37. Hauser and his colleagues68 showed that neutrophils crawled toward injured mitochondria and that this movement could also be abrogated by fMLP receptor inhibitors. These observations may be explained by the fact that mitochondria originate from ancient bacteria that have become endosymbionts inside the cells of multicellular organisms and that formylated peptides are synthesized from their mitochondrial genomes, suggesting that neutrophils respond to mitochondria in a similar way as they do to bacteria. It is unclear whether neutrophils can distinguish between dead cells and bacteria, whether dead cells unleash the same neutrophil-killing mechanisms as bacteria and whether this response is ultimately beneficial for the host. There is clear overlap between infection and sterile injury in a condition like sepsis. Mitochondrial injury is often observed in sepsis, and inhibition of mitochondrial injury reduces pathology in models of sepsis69. Although this result initially seems counterintuitive, inhibition of formylated peptide receptors may therefore be a potential therapeutic route in profound bacterial infections.
The multiple signals that can be released by just a few dead cells indicate the complexity of the response to a simple sterile injury. Superimposing a multitude of infectious stimuli on top of this injury makes things far more complex. In experiments that attempted to address some of these issues, a filter containing fMLP was placed on the liver surface, and all neutrophils crawled using CD11b through the liver sinusoids toward this site42. In the presence of LPS, the neutrophils adhered in liver sinusoids using CD44 rather than integrins and did not crawl toward fMLP. This change from neutrophil chemotaxis to adhesion was caused by an LPS-mediated downregulation of CD11b on neutrophils through an IL-10 (interleukin 10)-related mechanism, and removal of IL-10 restored the ability of neutrophils to crawl toward fMLP, even in the presence of LPS. Thus, IL-10 is crucial in this mechanism, is greatly elevated in sepsis and is deemed to be responsible for the delayed immunosuppressive phase that follows the initial potent inflammatory phase in this condition70,71 (Box 1). In a mouse model of sepsis induced by Escherichia coli (which expresses both LPS and fMLP), neutrophil recruitment to the liver was CD44 dependent and CD11b independent, and no neutrophil crawling was observed in the liver, showing that LPS overrode the fMLP signal42. A key question is whether this recruitment and adhesion of neutrophils in the liver is a mechanism that allows bacteria to evade the host immune response or, rather, is a beneficial response of the host; this benefit might be from NET-mediated trapping of bacteria, a process further discussed below.
Box 1: Sepsis is a moving clinical target.
Sepsis is a systemic response characterized by two phases. First, an initial hyperinflammatory state occurs, which is also referred to as systemic inflammatory response syndrome, and this is then followed by a pronounced immunosuppression. The ability to recognize severe sepsis earlier has allowed clinical intervention, which consists of early resuscitation and administration of the appropriate antibiotics. However, this intervention has also resulted in a change in the timing of septic mortality from early in the course of illness to later during intensive care unit hospitalization when individuals are in a state of immunosuppression. It is likely that the initial hyperinflammatory phase sets up the immunoparalysis phase.
As both phases are multifaceted, it is not surprising that the majority of clinical trials focusing on an anti-inflammatory strategy targeted at a single inflammatory mediator have been resounding failures. For example, blocking TNF-α, nitric oxide, thrombin, TLR4, TPFI and many other molecules has failed in sepsis trials. Currently, aggressive fluid replacement and the rapid administration of broad spectrum antibiotics are crucial in improving mortality. APC helps in some individuals, however, even for APC, a second trial in individuals with severe sepsis has been initiated to assess the effectiveness and safety of this treatment. One important issue to address in trial design for potential sepsis therapies is that currently all subjects with sepsis are grouped together regardless of the cause of sepsis. Because the type of infection is often not immediately known at the time of study enrollment, recent clinical trials such as one that examined a TLR4 inhibitor (which detects primarily Gram-negative infections) would have tested the inhibitor in all subjects, even those with Gram-positive infections. Finally, microvascular hypercoagulation and disseminated intravascular coagulation are substantial problems in sepsis, and the only current specific therapy, APC, seems to be beneficial. Currently, intravenous heparin is being tested in the treatment of sepsis. Although thrombocytopenia is likely to be a poor prognostic sign in sepsis, no clinical trials to specifically block platelet function in sepsis have been attempted, to our knowledge, and it will be necessary to evaluate whether targeting this cell is of clinical benefit in future studies.
Neutrophils are essential for restitution
An important component of inflammation is the resolution phase. Most studies have focused on how neutrophil recruitment at this stage is turned off, with molecules such as resolvins taking center stage72. However, neutrophils may also contribute to tissue restitution and angiogenesis. Neutrophils produce and store proangiogenic molecules such as vascular endothelial growth factor A (VEGF-A)73 and matrix metalloproteinase-9 (MMP-9)74 within their granules. VEGF is a key player in blood vessel formation and has a direct chemotactic effect on endothelial cells. The proangiogenic function of MMP-9 is attributed to its ability to release VEGF and other growth factors bound to the extracellular matrix and to digest the extracellular matrix to pave the way for new vessels. The neutrophils are the only cells in the body that can release MMP-9 free of its endogenous inhibitor, TIMP, and are therefore capable of delivering highly active MMP-9 to angiogenic sites74. The proangiogenic capacity of neutrophils has been shown in a corneal injury model, where the number of infiltrated neutrophils positively correlated with angiogenesis and VEGF levels75. Neutrophil depletion markedly impaired the release of VEGF and tissue healing in this model. Furthermore, another study showed that isolated pancreatic islets transplanted to muscle did not revascularize in neutropenic mice, whereas intra-islet vasculature was restored after transplantation to wild-type mice, showing that recruited neutrophils are important in the initiation of angiogenesis76. Outside the focus of this review, but worth mentioning, is the growing evidence that neutrophils may also contribute to cancer through their effects on angiogenesis77,78. A final important point is that neutrophils have been purported to recruit monocytes that may help to remove dead and dying neutrophils but that might also assist in angiogenesis and restitution79.
NETs
As discussed previously, neutrophils have a key role in fighting bacterial infections. Under static in vitro conditions, neutrophils can effectively chase after and phagocytose bacteria, but under in vivo flow conditions in blood, this is expected to be a highly inefficient process. However, neutrophils seem to have adapted additional mechanisms for fighting bacteria. Neutrophils have been shown to release their DNA in a net-like fashion in the presence of various bacteria80 to create traps called NETs. These NETs are covered in proteases (including elastase) and other antimicrobial molecules (such as histones), and these toxic molecules may contribute to host cell injury. However, similar in vivo observations of NETs have yet to be published, perhaps because of difficulties in imaging these events in blood.
NET formation was shown to start with the production of oxidants by neutrophils followed by the degradation of the nuclear envelope and DNA release into the cell, culminating with neutrophil lysis 3–4 h after activation by phorbol 12-myristate 13-acetate or bacteria81. In separate studies, eosinophils and neutrophils were found to catapult their DNA out of the cell into the extracellular space, but the DNA source was non-nuclear and, instead, the DNA originated from the mitochondria82,83. However, most other groups have not reported a role for mitochondrial DNA and have documented the presence of histones on the NETs, suggesting a nuclear source for the DNA. In addition to their direct antimicrobial effects, there is also evidence that histones may have a regulatory role during NET formation, as the process of NET formation starts with histone hypercitrullination and decondensation of the associated nuclear chromatin84. During NET formation induced by LPS, IL-8, fMLP or Shigella flexneri, increased histone H3 citrullination occurred through peptidylarginine deiminases. Citrullination is the conversion of positively charged arginine side chains into polar but uncharged citrulline side chains by deimination. Although there are five known peptidylarginine deiminases in humans, only peptidylarginine deiminase 4 (PAD4), which is expressed by neutrophils, has a classical nuclear localization signal, and PAD4 inhibition was shown to prevent histone H3 citrullination and substantially reduce NET release. However, in addition to the beneficial roles of histones, Esmon and his colleagues85 recently showed that histones injure the endothelium. They also found that activated protein C (APC), a therapeutic protein approved to treat sepsis, disables histone toxicity and preserves host cells85.
Interestingly, S. aureus has also been shown to induce NETs within 10 min of neutrophil activation through an oxidant-independent mechanism86,87. However, in these conditions, the neutrophils continued to exclude vital dyes, suggesting they were not lysed, and NETs were found to be released independently of neutrophil lysis through a vesicular mechanism86. Figure 4 summarizes this process, first showing the profound rounding and decondensation of the nucleus followed by the formation of large bleb-like structures between the inner and outer nuclear membrane, with vesicles pinching off the nuclear membrane and ultimately releasing NETs. This process was seen in the first 30–60 min after neutrophil activation, when no neutrophil lysis was observed. Eventually, the nuclear membrane completely broke down, perhaps leading to the release of NETs through a lytic event at a later time point (3–4 h after activation). This same nonlytic rapid release of DNA was observed in response to TLR4-activated platelets binding neutrophils (Fig. 5)88.
Figure 4. Formation of neutrophil extracellular traps.
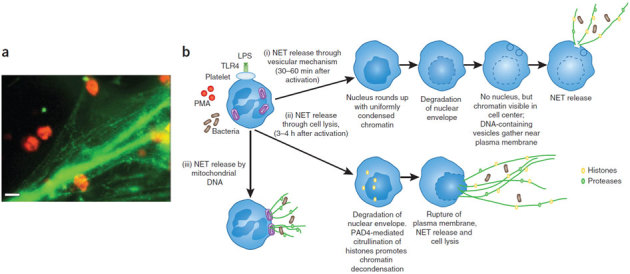
(a) LPS-induced platelet adhesion to neutrophils resulted in NET formation. Representative image of neutrophils visualized by white light through an orange filter using dark-field illumination and fluorescence microscopy with SYTOX Green to stain extracellular DNA green88. Scale bar, 10 μm. (b) Various mechanisms of NET release have been observed. (i) NETs can be released through a vesicular mechanism86,87. Initially, the neutrophils become rounded with uniformly condensed chromatin and then undergo nuclear envelope breakdown. Within these cells, small vesicles containing DNA can be seen in the cytoplasm near the plasma membrane. The DNA-containing vesicles eventually fuse with the plasma membrane, and NETs are released to trap bacteria. (ii) NETs can also be released through cell lysis, and this typically takes longer than the vesicular-mediated mechanism81,82,83,84. The nuclear envelope is degraded, and chromatin decondensation occurs because of PAD4-mediated citrullination of histones. (iii) NET release by mitochondria has also been observed, in one study83, although the steps of this process remain poorly characterized.
Figure 5. Platelet-neutrophil interactions within the liver vasculature during endotoxemia.
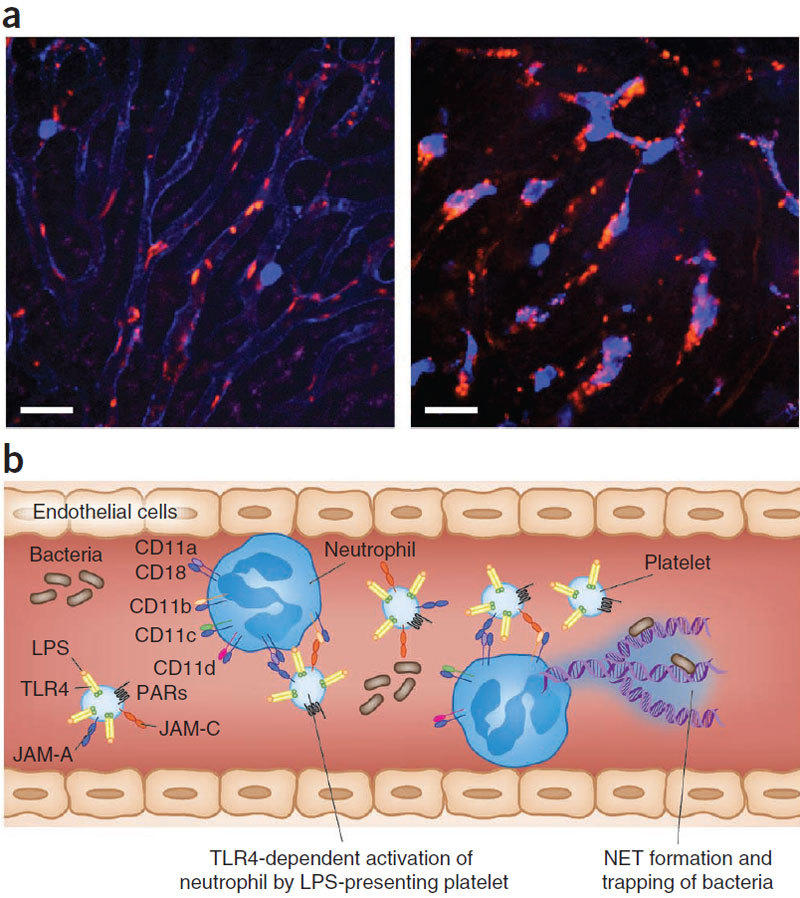
(a) Intravital spinning-disk confocal visualization of neutrophils (blue) and platelets (red) in liver sinusoids in healthy (left) and endotoxemic (right) mice. Although no neutrophil-platelet interactions were detected in healthy mice (left), a notable difference was observed in endotoxemic mice, in which neutrophil-platelet interactions were very common within the liver vasculature (colocalization of red and blue, right). Scale bar, 25 μm. (b) Schematic showing the various cell-surface molecules on platelets and neutrophils. TLR4 expression by the platelet has been shown to be important for neutrophil activation in response to LPS. JAM-A and JAM-C expressed by platelets interact with neutrophil integrins CD11a-CD18 and CD11b-CD18, respectively. Interactions between platelets can also stimulate NET release from neutrophils88.
Many bacteria make DNases that are presumably intended to degrade NETs, allowing for bacterial dissemination89. Indeed, the presence of a DNase increases the virulence of various bacterial strains. This may be of clinical relevance, as DNase is a standard treatment for individuals with cystic fibrosis and helps to clear the lungs of debris87. NETs have been reported in the lungs of individuals with cystic fibrosis, but whether treatment with DNase affects bacterial clearance and susceptibility to infection remains difficult to determine because many other medications, including antibiotics, are concomitantly used in these individuals. Addressing many of the outstanding questions regarding both the beneficial and detrimental aspects of NETs will require a specific NET inhibitor. Currently, only inhibitors that also affect oxidant production and/or additional aspects of neutrophil biology are available, which makes it difficult to elucidate the precise role of NETs.
Platelets are an extension of neutrophils
There is growing interest in the roles of platelets in inflammation and inflammatory diseases90. In arthropods, hemolymph contains hemocytes, which are nucleated circulating cells responsible for immunity as well as for coagulation. In higher order species, these functions have diverged into more specialized cells, but the platelet has retained some essential immune functions, particularly in partnership with neurophils and other phagocytes. Indeed, platelets can bind monocytes and neutrophils, leading to the transcellular synthesis of various inflammatory molecules90,91. Even on its own, the platelet is an important immune cell and outnumbers all other leukocytes tenfold. Platelets express many major innate immune receptors, including most TLRs, thrombin-binding protease-activated receptors, complement receptors and numerous adhesion molecules91. Animal models have indicated that platelets are crucial to sepsis and acute lung injury92. In humans, thrombocytopenia, a prognostic marker for a poor outcome in sepsis, is a major complication in sepsis that often occurs in the sickest individuals (Box 1). Although disseminated intravascular coagulation and bone marrow suppression were thought to be the key reasons for this problem, administration of LPS caused platelets to disappear from the circulation within an hour93, suggesting that bone marrow suppression is an unlikely cause of thromocytopenia in some individuals. Platelets that have been sequestered into organs by binding to the endothelium or to already adherent neutrophils could explain some of the thrombocytopenia that occurs.
In the presence of thrombin, platelets release as many as 300 proteins into the circulation, including proinflammatory, anti-inflammatory and angiogenic factors, as well as mediator-rich microparticles92. Although LPS also activates platelets, LPS-induced platelet responses appear to be far more subtle than those induced by thrombin. In fact, hallmark features of platelet activation, including P-selectin expression and platelet aggregation, are arguably not observed in response to LPS. LPS also induces potent platelet-dependent TNF production93. Following LPS administration, many platelets are sequestered in the lungs93 and bind avidly to adherent neutrophils in the liver88 (Fig. 5). Platelet-neutrophil binding has been shown to depend on TLR4 expressed by the platelet, suggesting that this anuclear cell actively participates in the adhesive interaction. In fact, activated neutrophils alone did not injure the endothelium, but the binding of TLR4-activated platelets to neutrophils induced endothelial cell death88.
Under flow conditions, platelets have been shown to induce neutrophils to release NETs, thereby contributing to the killing of bacteria through this mechanism88. Why might a platelet-dependent NET mechanism be required? As neutrophils are activated at 100-fold lower concentrations of LPS than platelets are, and as LPS induces the non-NET–related functions of neutrophils, the platelet might function as a barometer for detecting substantial levels of bacterial products in blood. Platelets could then bind neutrophils and induce NET production as a last gasp effort to trap and destroy bacteria. This mechanism might occur preferentially in the vast microvasculature of the liver and lung where trapping of blood-borne bacteria may be the most efficient. Indeed, when a neutrophil binds in the very narrow sinusoids of the liver, it does not completely occlude the vessel, allowing plasma and bacteria to be skimmed around the neutrophils and collected in the released NETs. Although concentrations of LPS that are unlikely to ever be seen in infected humans were used in this model system, blood from septic individuals was equally effective at inducing NETs88. The major difference between the LPS and septic blood studies was that TLR4 only accounted for 30% of the NET formation in septic blood, suggesting that additional activators are present in septic blood.
Unresolved issues
There are still many questions regarding the roles and functions of neutrophils in inflammation, and because much of our current knowledge is based on observations in mice, translational studies are needed to evaluate the clinical importance of these observations in humans. Another question that remains to be addressed, the answer to which may also differ in mice and humans, is whether there are different neutrophil subtypes, much like for monocytes. Indeed, mice have been reported to have multiple neutrophil subsets with unique surface receptor phenotypes and effector functions94. During experimental endotoxemia in humans, there is a large mobilization of band-like immature neutrophils that are CD16 low (compared to CD16 high for their mature counterparts). These immature cells tended to be less responsive to stimuli after administration of LPS and showed a smaller oxidative burst and reduced interactions with bacteria95.
Only about 20% of neutrophils make NETs. Whether this is because of maturity levels or subtypes is unclear. The possibility that a population of tissue-resident or perhaps continuously recirculating 'pioneer' neutrophils exists is intriguing96, although there is currently no direct evidence for this hypothesis. Indeed, it is possible that if a group of neutrophils expresses high concentrations of neutrophil chemotactic molecules such as LTB4, this may position it as a unique sentinel population that can recruit further neutrophil subsets97. The continuous expression of cytokines may also alter the neutrophil phenotype by inducing the expression of receptors that have normally been thought to recruit monocytes (such CCR2 and CCR1)98. In fact, the neutrophil phenotype may vary substantially over a typical septic episode that could last weeks. Understanding this moving target will be key to therapeutically modulating neutrophil function and also in the treatment of sepsis. In conclusion, theoretical intervention points affecting neutrophil biology that may be considered when treating sepsis include blocking platelet function or their binding to neutrophils, NET inhibition and targeting formylated peptides while preserving mitochondrial function.
Supplementary information
Supplementary Video 1 (MOV 11923 kb)
Acknowledgements
We wish to express their thanks to the Canadian Foundation for Innovation for the funding of the University of Calgary Live Cell Imaging Facility that enabled this work and to C. Jenne, S. Massena and B. McDonald for helping with the figures. M.P. is supported by the Swedish Medical Research Council (57X-20675) and the Swedish Diabetes Foundation (DIA2010-063), and P.K., an Alberta Heritage Foundation for Medical Research Scientist, Canada Research Chair and the Snyder Chair in Critical Care Medicine, is supported by the Canadian Institutes of Health Research, the Alberta Heritage Foundation for Medical Research and the Heart and Stroke Foundation.
Competing interests
The authors declare no competing financial interests.
References
- 1.Auffray C, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 2.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 3.Petri B, Phillipson M, Kubes P. The physiology of leukocyte recruitment: an in vivo perspective. J. Immunol. 2008;180:6439–6446. doi: 10.4049/jimmunol.180.10.6439. [DOI] [PubMed] [Google Scholar]
- 4.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillipson M, et al. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J. Exp. Med. 2006;203:2569–2575. doi: 10.1084/jem.20060925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Phillipson M, et al. Vav1 is essential for mechanotactic crawling and migration of neutrophils out of the inflamed microvasculature. J. Immunol. 2009;182:6870–6878. doi: 10.4049/jimmunol.0803414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burns AR, et al. Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J. Immunol. 1997;159:2893–2903. [PubMed] [Google Scholar]
- 8.Vaporciyan AA, et al. Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science. 1993;262:1580–1582. doi: 10.1126/science.8248808. [DOI] [PubMed] [Google Scholar]
- 9.Gotsch U, et al. VE-cadherin antibody accelerates neutrophil recruitment in vivo. J. Cell Sci. 1997;110:583–588. doi: 10.1242/jcs.110.5.583. [DOI] [PubMed] [Google Scholar]
- 10.Shaw SK, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J. Exp. Med. 2004;200:1571–1580. doi: 10.1084/jem.20040965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woodfin A, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat. Immunol. 2011;12:761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis RE, Miller RA, Granger HJ. Acute microvascular effects of the chemotactic peptide N-formyl-methionyl-leucyl-phenylalanine: comparisons with leukotriene B4. Microvasc. Res. 1989;37:53–69. doi: 10.1016/0026-2862(89)90072-1. [DOI] [PubMed] [Google Scholar]
- 13.Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J. Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barreiro O, Vicente-Manzanares M, Urzainqui A, Yanez-Mo M, Sanchez-Madrid F. Interactive protrusive structures during leukocyte adhesion and transendothelial migration. Front. Biosci. 2004;9:1849–1863. doi: 10.2741/1285. [DOI] [PubMed] [Google Scholar]
- 15.Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS One. 2008;3:e1649. doi: 10.1371/journal.pone.0001649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petri B, et al. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood. 2011;117:942–952. doi: 10.1182/blood-2010-02-270561. [DOI] [PubMed] [Google Scholar]
- 17.Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Neutrophils emigrate from venules by a transendothelial cell pathway in response to fMLP. J. Exp. Med. 1998;187:903–915. doi: 10.1084/jem.187.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S, et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J. Exp. Med. 2006;203:1519–1532. doi: 10.1084/jem.20051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradfield PF, et al. JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood. 2007;110:2545–2555. doi: 10.1182/blood-2007-03-078733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furze RC, Rankin SM. Neutrophil mobilization and clearance in bone marrow. Immunology. 2008;125:281–288. doi: 10.1111/j.1365-2567.2008.02950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christopher MJ, Link DC. Regulation of neutrophil homeostasis. Curr. Opin. Hematol. 2007;14:3–8. doi: 10.1097/00062752-200701000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Delano MJ, et al. Neutrophil mobilization from the bone marrow during polymicrobial sepsis is dependent on CXCL12 signaling. J. Immunol. 2011;187:911–918. doi: 10.4049/jimmunol.1100588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zimmerman GA, McIntyre TM, Prescott SM. Adhesion and signaling in vascular cell-cell interactions. J. Clin. Invest. 1997;100:S3–S5. [PubMed] [Google Scholar]
- 24.Massena S, et al. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood. 2010;116:1924–1931. doi: 10.1182/blood-2010-01-266072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindahl U. Heparan sulfate-protein interactions—a concept for drug design? Thromb. Haemost. 2007;98:109–115. [PubMed] [Google Scholar]
- 26.Lindahl U, Kjellen L. Heparin or heparan sulfate—what is the difference? Thromb. Haemost. 1991;66:44–48. [PubMed] [Google Scholar]
- 27.Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J. Cell Biol. 1997;139:1349–1360. doi: 10.1083/jcb.139.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funamoto S, Meili R, Lee S, Parry L, Firtel RA. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell. 2002;109:611–623. doi: 10.1016/s0092-8674(02)00755-9. [DOI] [PubMed] [Google Scholar]
- 29.Nishio M, et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat. Cell Biol. 2007;9:36–44. doi: 10.1038/ncb1515. [DOI] [PubMed] [Google Scholar]
- 30.Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J. Cell Biol. 2002;159:91–102. doi: 10.1083/jcb.200202114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nick JA, et al. Common and distinct intracellular signaling pathways in human neutrophils utilized by platelet activating factor and FMLP. J. Clin. Invest. 1997;99:975–986. doi: 10.1172/JCI119263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heit B, et al. PTEN functions to 'prioritize' chemotactic cues and prevent 'distraction' in migrating neutrophils. Nat. Immunol. 2008;9:743–752. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- 33.Gambero A, et al. Signalling pathways regulating human neutrophil migration induced by secretory phospholipases A2. Toxicon. 2004;44:473–481. doi: 10.1016/j.toxicon.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 34.VanUffelen BE, et al. Modulation of neutrophil migration by exogenous gaseous nitric oxide. J. Leukoc. Biol. 1996;60:94–100. doi: 10.1002/jlb.60.1.94. [DOI] [PubMed] [Google Scholar]
- 35.Nishikimi A, et al. Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science. 2009;324:384–387. doi: 10.1126/science.1170179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samanta AK, Oppenheim JJ, Matsushima K. Interleukin 8 (monocyte-derived neutrophil chemotactic factor) dynamically regulates its own receptor expression on human neutrophils. J. Biol. Chem. 1990;265:183–189. [PubMed] [Google Scholar]
- 37.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 38.Reddy RC, Standiford TJ. Effects of sepsis on neutrophil chemotaxis. Curr. Opin. Hematol. 2010;17:18–24. doi: 10.1097/MOH.0b013e32833338f3. [DOI] [PubMed] [Google Scholar]
- 39.Vincent JL. Sepsis and Non-infectious Inflammation. 2009. Definition of sepsis and non infectious SIRS. [Google Scholar]
- 40.Penack O, et al. Management of sepsis in neutropenic patients: guidelines from the infectious diseases working party of the German Society of Hematology and Oncology. Ann. Oncol. 2011;22:1019–1029. doi: 10.1093/annonc/mdq442. [DOI] [PubMed] [Google Scholar]
- 41.Cummings CJ, et al. Expression and function of the chemokine receptors CXCR1 and CXCR2 in sepsis. J. Immunol. 1999;162:2341–2346. [PubMed] [Google Scholar]
- 42.Menezes GB, et al. Selective down-regulation of neutrophil Mac-1 in endotoxemic hepatic microcirculation via IL-10. J. Immunol. 2009;183:7557–7568. doi: 10.4049/jimmunol.0901786. [DOI] [PubMed] [Google Scholar]
- 43.Lokuta MA, Huttenlocher A. TNF-α promotes a stop signal that inhibits neutrophil polarization and migration via a p38 MAPK pathway. J. Leukoc. Biol. 2005;78:210–219. doi: 10.1189/jlb.0205067. [DOI] [PubMed] [Google Scholar]
- 44.Alves-Filho JC, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 2010;16:708–712. doi: 10.1038/nm.2156. [DOI] [PubMed] [Google Scholar]
- 45.Alves-Filho JC, et al. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc. Natl. Acad. Sci. USA. 2009;106:4018–4023. doi: 10.1073/pnas.0900196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andonegui G, et al. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J. Clin. Invest. 2009;119:1921–1930. doi: 10.1172/JCI36411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurupati P, et al. Chemokine-cleaving Streptococcus pyogenes protease SpyCEP is necessary and sufficient for bacterial dissemination within soft tissues and the respiratory tract. Mol. Microbiol. 2010;76:1387–1397. doi: 10.1111/j.1365-2958.2010.07065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrmann JM, et al. Sequential chemotactic and phagocytic activation of human polymorphonuclear neutrophils. Infect. Immun. 2007;75:3989–3998. doi: 10.1128/IAI.00388-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Caterina R, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Invest. 1995;96:60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lefer DJ, et al. Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am. J. Physiol. 1999;276:H1943–H1950. doi: 10.1152/ajpheart.1999.276.6.H1943. [DOI] [PubMed] [Google Scholar]
- 51.Alves-Filho JC, et al. The role of neutrophils in severe sepsis. Shock. 2008;30:3–9. doi: 10.1097/SHK.0b013e3181818466. [DOI] [PubMed] [Google Scholar]
- 52.Hauser B, Bracht H, Matejovic M, Radermacher P, Venkatesh B. Nitric oxide synthase inhibition in sepsis? Lessons learned from large-animal studies. Anesth. Analg. 2005;101:488–498. doi: 10.1213/01.ANE.0000177117.80058.4D. [DOI] [PubMed] [Google Scholar]
- 53.Mizgerd JP, et al. Neutrophil emigration in the skin, lungs, and peritoneum: different requirements for CD11/CD18 revealed by CD18-deficient mice. J. Exp. Med. 1997;186:1357–1364. doi: 10.1084/jem.186.8.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mizgerd JP, et al. Selectins and neutrophil traffic: margination and Streptococcus pneumoniae-induced emigration in murine lungs. J. Exp. Med. 1996;184:639–645. doi: 10.1084/jem.184.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wong J, et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J. Clin. Invest. 1997;99:2782–2790. doi: 10.1172/JCI119468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McDonald B, et al. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 2008;205:915–927. doi: 10.1084/jem.20071765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doerschuk CM. Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation. 2001;8:71–88. [PubMed] [Google Scholar]
- 58.Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am. J. Physiol. 1988;255:H1269–H1275. doi: 10.1152/ajpheart.1988.255.6.H1269. [DOI] [PubMed] [Google Scholar]
- 59.Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23:1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- 60.Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43:1220–1230. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- 61.Hyman MC, et al. Self-regulation of inflammatory cell trafficking in mice by the leukocyte surface apyrase CD39. J. Clin. Invest. 2009;119:1136–1149. doi: 10.1172/JCI36433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu. Rev. Immunol. 2010;28:321–342. doi: 10.1146/annurev-immunol-030409-101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taylor KR, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. J. Biol. Chem. 2007;282:18265–18275. doi: 10.1074/jbc.M606352200. [DOI] [PubMed] [Google Scholar]
- 66.Hofmann MA, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 67.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Larche J, et al. Inhibition of mitochondrial permeability transition prevents sepsis-induced myocardial dysfunction and mortality. J. Am. Coll. Cardiol. 2006;48:377–385. doi: 10.1016/j.jacc.2006.02.069. [DOI] [PubMed] [Google Scholar]
- 70.Brandtzaeg P, et al. Net inflammatory capacity of human septic shock plasma evaluated by a monocyte-based target cell assay: identification of interleukin-10 as a major functional deactivator of human monocytes. J. Exp. Med. 1996;184:51–60. doi: 10.1084/jem.184.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bone RC, Grodzin CJ, Balk RA. Sepsis: a new hypothesis for pathogenesis of the disease process. Chest. 1997;112:235–243. doi: 10.1378/chest.112.1.235. [DOI] [PubMed] [Google Scholar]
- 72.Spite M, et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature. 2009;461:1287–1291. doi: 10.1038/nature08541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gaudry M, et al. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood. 1997;90:4153–4161. [PubMed] [Google Scholar]
- 74.Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA. 2007;104:20262–20267. doi: 10.1073/pnas.0706438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gong Y, Koh DR. Neutrophils promote inflammatory angiogenesis via release of preformed VEGF in an in vivo corneal model. Cell Tissue Res. 2010;339:437–448. doi: 10.1007/s00441-009-0908-5. [DOI] [PubMed] [Google Scholar]
- 76.Christoffersson G, et al. Clinical and experimental pancreatic islet transplantation to striated muscle: establishment of a vascular system similar to that in native islets. Diabetes. 2010;59:2569–2578. doi: 10.2337/db10-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fridlender ZG, et al. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 79.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 80.Brinkmann V, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 81.Fuchs TA, et al. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yousefi S, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat. Med. 2008;14:949–953. doi: 10.1038/nm.1855. [DOI] [PubMed] [Google Scholar]
- 83.Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16:1438–1444. doi: 10.1038/cdd.2009.96. [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009;184:205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu J, et al. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pilsczek FH, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010;185:7413–7425. doi: 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 87.Marcos V, et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 2010;16:1018–1023. doi: 10.1038/nm.2209. [DOI] [PubMed] [Google Scholar]
- 88.Clark SR, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 89.Urban CF, Lourido S, Zychlinsky A. How do microbes evade neutrophil killing? Cell. Microbiol. 2006;8:1687–1696. doi: 10.1111/j.1462-5822.2006.00792.x. [DOI] [PubMed] [Google Scholar]
- 90.Semple JW, Italiano JE, Jr., Freedman J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011;11:264–274. doi: 10.1038/nri2956. [DOI] [PubMed] [Google Scholar]
- 91.Semple JW, Freedman J. Platelets and innate immunity. Cell. Mol. Life Sci. 2010;67:499–511. doi: 10.1007/s00018-009-0205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smyth SS, et al. Platelet functions beyond hemostasis. J. Thromb. Haemost. 2009;7:1759–1766. doi: 10.1111/j.1538-7836.2009.03586.x. [DOI] [PubMed] [Google Scholar]
- 93.Andonegui G, et al. Platelets express functional Toll-like receptor-4. Blood. 2005;106:2417–2423. doi: 10.1182/blood-2005-03-0916. [DOI] [PubMed] [Google Scholar]
- 94.Tsuda Y, et al. Three different neutrophil subsets exhibited in mice with different susceptibilities to infection by methicillin-resistant Staphylococcus aureus. Immunity. 2004;21:215–226. doi: 10.1016/j.immuni.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 95.Pillay J, et al. Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J. Leukoc. Biol. 2010;88:211–220. doi: 10.1189/jlb.1209793. [DOI] [PubMed] [Google Scholar]
- 96.McDonald B, Kubes P. Chemokines: sirens of neutrophil recruitment—but is it just one song? Immunity. 2010;33:148–149. doi: 10.1016/j.immuni.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 97.Chou RC, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33:266–278. doi: 10.1016/j.immuni.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Johnston B, et al. Chronic inflammation upregulates chemokine receptors and induces neutrophil migration to monocyte chemoattractant protein-1. J. Clin. Invest. 1999;103:1269–1276. doi: 10.1172/JCI5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1 (MOV 11923 kb)