

Abstract
Problem
Preeclampsia, a pregnancy-specific hypertensive syndrome, is one of the leading causes of premature births as well as fetal and maternal death. Preeclampsia lacks effective therapies because of the poor understanding of disease pathogenesis. The aim of this paper is to review molecular signaling pathways that could be responsible for the pathogenesis of preeclampsia.
Method of study
This article reviews the English-language literature for pathogenesis and pathophysiological mechanisms of preeclampsia based on genome-wide gene expression profiling and proteomic studies.
Results
We show that the expression of the genes and proteins involved in response to stress, host-pathogen interactions, immune system, inflammation, lipid metabolism, carbohydrate metabolism, growth and tissue remodeling was increased in preeclampsia. Several significant common pathways observed in preeclampsia overlap the datasets identified in TLR (Toll-like receptor)- and RAGE (receptor for advanced glycation end products)-dependent signaling pathways. Placental oxidative stress and subsequent chronic inflammation are considered to be major contributors to the development of preeclampsia.
Conclusion
This review summarizes recent advances in TLR- and RAGE-mediated signaling and the target molecules, and provides new insights into the pathogenesis of preeclampsia.
Keywords: Inflammation, Oxidative stress, Preeclampsia, RAGE, TLR
Introduction
Preeclampsia is the de novo occurrence of pregnancy-specific hypertension and proteinuria after 20 weeks of gestation. It is one of the leading causes of maternal, fetal, and neonatal mortality worldwide. The integration of microarrays and sophisticated bioinformatics can provide specific and efficient high-throughput screening with which to investigate the pathogenesis of preeclampsia. Although the mechanisms have been elusive, new physiologic insights have begun to alter thinking about preeclampsia [1]. Angiogenic factors, including vascular endothelial growth factor (VEGF), have been proposed to play a key role in the maintenance of endothelial cell function [2–5]. Soluble fms-like tyrosine kinase-1 (sFlt-1) and soluble endoglin (sEng) are endogenous anti-angiogenic proteins that neutralize the pro-angiogenic proteins VEGF and placental growth factor (PlGF). Maternal endothelial dysfunction, possibly related to these circulating factors, is a key feature of this syndrome.
Additional factors that may contribute to the pathogenesis of preeclampsia include angiotensin-II receptor autoantibody, infectious ligands, advanced glycation end products (AGEs), Toll-like receptors (TLRs) and receptors for AGEs (RAGE) [1, 6]. It is now increasingly being recognized that abnormal placentation and systemic inflammation represent hallmarks of preeclampsia and infectious agents might increase the risk of this disorder [6]. Invading exogenous pathogens [also known as pathogen-associated molecular pattern (PAMP) molecules] influence immune cell recruitment and pro-inflammatory cytokine secretion. In addition, cellular stress, damage, and inflammation cause release of endogenous damage-associated molecular pattern (DAMP) molecules or endogenous danger signals “alarmins” [7]. The complex bidirectional interplay between angiogenesis and immunity/inflammation can establish a “vicious cycle” that might contribute to the pathogenesis of preeclampsia.
Some parts of the puzzle surrounding pro-inflammatory cytokines and growth factors and their effects on alarmins have begun to unravel. Alarmins activate the repair process through their interaction with pattern recognition receptors (PRRs) such as TLRs. A recent study has shown a crucial link between the development of preeclampsia and defense against both exogenous ligands (foreign pathogens) and endogenously generated alarmins [6]. Women with preeclampsia had increased expressions of TLR2 and TLR4 mRNA and protein, a downstream transcription factor nuclear factor (NF)-kappaB, and interleukin (IL)-1beta mRNA compared with control groups [8]. Thus, TLRs may modulate the innate immune response and appear to be factors contributing to the pathogenesis of preeclampsia [8].
This review focuses on the following six aspects of this emerging field of preeclampsia: an overview of genome-wide gene expression profiling, an overview of microarray analysis and proteomic studies, a history and the molecular basis of promising biomarkers, an overview of specific functions of angiogenic/anti-angiogenic factors, the molecular basis of inflammatory pattern recognition receptors and their ligands, and investigation of TLRs and RAGE and their ligands.
Materials and methods
The present article reviews the literature on biological, pathogenetic and pathophysiological studies on preeclampsia. For studies that reported data on obstetric disorders, only data pertaining to preeclampsia were included. A computerized literature search was performed to identify relevant studies reported in the English language. MEDLINE updates were conducted monthly, and all abstracts were reviewed by two investigators to identify papers for full-text review. We searched PubMed MEDLINE electronic databases (http://www.ncbi.nlm.nih.gov/sites/entrez) for a 10-year period (2000–2010), combining the keywords “gene expression profiling”, “proteomic”, “pathogenesis”, “pathophysiology”, “Toll-like receptor”, “advanced glycation end products”, “receptor for advanced glycation end products”, “inflammation”, “stress”, or “alarmins” with “preeclampsia”. Each gene is also linked to NCBI Entrez Gene pages (http://www.ncbi.nlm.nih.gov/sites/entrez). Additionally, references in each article were searched to identify potentially missed studies for a 20-year period (1990–2010). Target publications are mainly reports on human preeclampsia and mouse models, as well as studies in gene and protein expression systems. A priori, case reports, and abstracts were not included, since abstracts do not undergo a stringent peer-review process. A difficulty in interpreting the literature is that analyses, results and objects (early onset or late onset) are reported differently among the studies. Here, we discuss promising molecular candidates and possible signaling targets for preeclampsia.
Article selection, data extraction and assessment
As the main interest is preeclampsia obtained from human samples, we have not yet included animal preeclampsia models alone in the database. However, we included animal studies performed to support clinical data. Initially, 321 potentially relevant studies were identified by screening electronic databases, and 135 peer-reviewed journal articles were additionally identified from references in each article. The network consists of 92 entities by genome-wide gene expression profiling and proteomic analyses and seven reaction categories by gene ontology analyses. These entities were classified into renin–angiotensin system, angiogenesis, inflammation and cytokines, stress and detoxification, metabolism, adhesion, cell structure, signal, and protease.
Gene profiling and proteomic analysis
Several studies based on genome-wide gene expression profiling analysis and proteomic technology have clarified the specific genes and proteins involved in preeclampsia. We will initially focus on the biology, pathogenesis, and pathophysiology of this disorder. Researchers have investigated candidate markers in samples, including placenta, amniotic membranes, blood, amniotic fluid, and peripheral blood mononuclear cells in severe preeclampsia. There are specific genes that may be of importance for the pathological and pathophysiological changes seen in preeclampsia. Several genes preferentially up-regulated (n = 75) or down-regulated (n = 17) in preeclampsia have been identified (Table 1). These genes have been grouped in clusters or pathways according to their possible influence on certain characteristics of preeclampsia.
Table 1.
Summary of differentially expressed gene and protein profiles in preeclampsia
| Pathways/system | Targets | Refs. |
|---|---|---|
| Up-regulation | ||
| Renin–angiotensin system (RAS) | Angiotensin I converting enzyme (ACE), ACE2, angiotensin II receptor, type 1 (AGTR1a), angiotensin II AT1 receptor, agonistic angiotensin II type 1 (AT1) receptor autoantibodies | [75, 76] |
| Angiogenesis | Fms-like tyrosine kinase-1 (FLT1), vascular endothelial growth factor A (VEGFA), endoglin (ENG) | [43, 55 75, 76, 77, 78, 79] |
| Inflammation and cytokines | NADPH oxidase 4 (NOX4), oncostatin M (OSM), lactotransferrin, interleukin 9 (IL9), Epstein–Barr virus induced 3 (EBI3), interleukin-1 receptor-associated kinase 3 (IRAK3), colony stimulating factor 1 receptor (CSF1R), TNF-alpha, IL-6, TGF-beta | [43, 75, 76, 80] |
| Stress and detoxification | Cytochrome P450, family 11, subfamily A (CYP11A), CYP26A1, heme oxygenase-1 (HMOX1), epoxide hydrolase 2 (EPHX2) | [27, 43, 75, 77] |
| Metabolism | Leptin (LEP), spermine oxidase (SMOX), acetate dehydrogenase A (LDHA), glia maturation factor, beta (GMFB), glycogen synthase kinase 3 beta (GSK3B), transglutaminase 2 (TGM2), cysteine dioxygenase, type I (CDO1), insulin-like 4 (INSL4), phosphomannomutase 2 (PMM2) | [38, 43, 76, 77, 79] |
| Adhesion | Claudin 6 (CLDN6), catenin (CTNNB1), cadherin 5 (CDH5), vitronectin (VTN), protocadherin alpha 3 (PCDHA3), selectin-P (SELP), neural cell adhesion molecule 1 (NCAM1) | [43, 75, 79, 80, 81] |
| Cell structure | Titin (TTN), actinin 4 (ACTN4), fibulin 1 (FBLN1), filamin B (FLNB), talin 1 (TLN1), dystroglycan 1 (DAG1), fibronectin 1 (FN1), adducin 1 (ADD1), integrin, alpha 2 (ITGA2) | [75, 79, 81] |
| Signal | Mitogen-activated protein kinase 1 (MAPK1), MAPK4, protein tyrosine phosphatase, non-receptor type 1 (PTPN1), PTPN6, PTPN11, PTPN12, SHC (Src homology 2 domain containing) transforming protein 1 (SHC1), SMAD4, growth factor receptor-bound protein 2 (GRB2), protein kinase C, zeta (PRKCZ), protein tyrosine kinase 2 (PTK2), v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (SRC), vav 2 guanine nucleotide exchange factor (VAV2), vestigial like 1 (VGLL1), protein C receptor (PROCR), caspase-10 (CASP10), death receptor 3 (DR-3), regulator of G-protein signaling 5 (RGS5), cyclin B1 (CCNB1) | [43, 75, 81, 82] |
| Protease | Laeverin (AQPEP), a disintegrin and metalloproteinase 12, meltrin-alpha (ADAM 12), secretory leukocyte peptidase inhibitor (SLPI), endothelial nitric oxide synthesis (eNOS) | [75, 78, 83] |
| Others | Coagulation factor II (thrombin) receptor (F2R), Fc fragment of IgG, low affinity IIb, receptor (CD32) (FCGR2B), Fc fragment of IgG, high affinity Ia, receptor (CD64) (FCGR1A), inhibin A (INHA), estrogen receptor 1 (ESR1), T-cell immunoglobulin and mucin domain containing 2 (TIMD2), integral membrane protein 2A (ITM2a), immunoglobulin superfamily, member 3 (IGSF3) | [40, 75] |
| Down-regulation | ||
| Angiogenesis | Matrix metallopeptidase 1 (MMP1), MMP10, fibroblast growth factor binding protein 1 (FGFBP1), vasohibin 1 (VASH1), epidermal growth factor receptor (EGFR), epidermal growth factor receptor pathway substrate 15 (EPS15) | [73, 75, 77, 78, 79, 80, 81] |
| Protease | MMP-7, MMP-12, plasminogen activator inhibitor type-1 (SERPINE1, PAI-1) [41] | [80, 84] |
| Metabolism | Aldo–keto reductase family 1, member C3 (AKR1C3), solute carrier family 25, member 13 (SLC25A13) | [85] |
| Growth factors | Platelet derived growth factor D (PDGFD), insulin-like growth factor binding protein-3 (IGFBP-3) | [78, 82] |
| Adhesion | KiSS-1 metastasis-suppressor (KISS-1) [41] | [86] |
| Others | Killer cell immunoglobulin-like receptor, three domains, long cytoplasmic tail, 2 (KIR3DL2), churchill domain containing 1 (CHURC1), nuclear receptor subfamily 4, group A, member 2 (NR4A2) | [81] |
Angiogenesis system
Computational bioinformatic analysis revealed a set of new candidate genes, and highlighted several new pathway systems, such as the vascular endothelial growth factor (VEGF)- and transforming growth factor (TGF)-beta-related pathways and renin–angiotensin system [9]. Preeclamptic placenta is relatively ischemic and produces a variety of factors that might generate profound effects on vascular endothelial cell function [10]. These factors include the soluble VEGF receptor-1 (VEGF-RI, also known as Flt-1), the angiotensin II type-1 receptor autoantibody, and pro-inflammatory cytokines such as tumor necrosis factor (TNF)-alpha and interleukin (IL)-6. The angiogenic activity of VEGF is mainly mediated by VEGF receptor I (Flt-1) and VEGF receptor II [VEGF-RII; also known as Flk-1 (fetal liver kinase 1)/KDR (kinase domain region)]. VEGF expression is primarily regulated by hypoxia [11]. Ischemic placenta also generates sFlt-1, possibly via hypoxia-inducible factor signaling. Circulating mononuclear cells also represent an additional source for circulating sFlt-1 during normal and preeclamptic pregnancies [12]. A recent study has shown decreased production of VEGF by circulating T and natural killer cells in preeclampsia [13]. In the sera of pregnant women with preeclampsia, there was a significant reduction in the levels of free VEGF and placental growth factor (PlGF), whereas the levels of sFlt-1 were significantly higher than those in the sera of normotensive pregnant women [14]. sFlt-1 acts as a negative modulator for the bioavailability of VEGF [15]. These soluble receptors may bind and neutralize VEGF in the maternal circulation, causing endothelial dysfunction [14]. sFlt-1 also induces endothelial cell apoptosis, and decreases nitric oxide (NO) generation in vitro [16].
Furthermore, soluble endoglin (sEng) is another circulating anti-angiogenic protein and may contribute to the pathogenesis of preeclampsia. Endoglin is a transmembrane receptor for TGF-beta that is expressed on proliferating endothelial cells. sEng was found to synergize with a sFlt-1 and induce endothelial cell dysfunction. TGF-beta stimulates production of VEGF and plasminogen activator inhibitor type-1 (PAI-1), that are involved in vascular remodeling [17]. Thus, decreased free VEGF and TGF-beta activities might contribute to the pathogenesis of preeclampsia [14]. Animal experiments demonstrated that co-administration of sFlt-1 and sEng to pregnant rats elicited severe preeclampsia-like symptoms [18].
Renin–angiotensin system (RAS)
Bioinformatic analysis has highlighted the renin-angiotensin pathway [9]. Angiotensin II levels in the placenta are higher in preeclamptic subjects when compared with control pregnant women [19]. Angiotensin II induces gene expression of TGF-beta and matrix metalloproteinase-9 (MMP-9). In addition to elevated angiotensin II levels, angiotensinogen and angiotensin I receptor mRNAs were also increased in preeclamptic placenta [19]. Furthermore, immunoglobulin (Ig) G autoantibody in the serum of preeclamptic women stimulates the angiotensin II type 1 receptor [20]. Angiotensin II type 1 receptor autoantibody can also induce reactive oxygen species (ROS) generation, mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [19]. ROS-induced oxidative stress appears to be both a cause and a consequence of hypertension. This autoantibody also stimulates trophoblast sFlt-1 production [21, 22].
Inflammation, cytokines, stress and detoxification system
VEGF-dependent anti-inflammation and anti-oxidation have been identified, adding a further layer of complexity to the VEGF system [2, 23–26]. The previous studies showed that acute infusions of VEGF cause vasodilation and hypotension [3, 24]. VEGF plays a critical role in blood pressure control and in optimizing blood flow, vascular integrity, and tone by promoting NO activity [2] through VEGFR2-dependent up-regulation of endothelial NO synthase (eNOS) expression [25]. NO has multiple functions in the vasculature, including vasodilatory, anti-inflammatory and anti-thrombotic activities [26]. NO also prevents proliferation of excess endothelial cells and vascular smooth muscle cells, as well as macrophage adhesion and infiltration [26]. Collectively, these studies suggest a protective role for VEGF in the development of preeclampsia, possibly with anti-inflammatory, anti-oxidant and anti-proliferative effects in the vasculature, which may contribute to the prevention of this hypertensive condition.
Considerable evidence implicates the ischemic placenta during early pregnancy, altered proangiogenic and antiangiogenic factor balance, oxidative stress and endothelial dysfunction in the pathophysiology of preeclampsia [18]. Increased expression of some stress-related genes such as heme oxygenase-1 (HMOX1) also supports the role of excessive oxidative stress and maternal inflammatory response [27]. Thus, abnormal cytokine responses and oxidative stress during pregnancy might play a central role in the excessive systemic inflammatory response, as well as in the generalized endothelial dysfunction characteristics of this disorder.
Furthermore, the production of type 1 pro-inflammatory cytokines is dominant in patients with preeclampsia or in individuals with a predisposition to subsequent development of preeclampsia [28]. The production of reactive oxygen intermediates in endothelial cells is induced by Th1 cytokines such as TNF-alpha and IL-6. Chronic subclinical oxidative stresses may also increase maternal and fetal pro-inflammatory cytokines to levels high enough to affect vascular endothelial function. It has recently been reported that healthy pregnant women showed a decrease in IL-17-producing cells, whereas a relative increase in IL-17-producing cells and low T-regulatory cell numbers were observed in preeclamptic women, suggesting that the pathophysiology of preeclampsia might be associated with imbalanced differentiation of T-regulatory cells and Th17 cells [29, 30]. Therefore, systematic immunoactivation might be linked to immuno-maladaptation, chronic inflammation, poor angiogenesis, and endothelial cell dysfunction.
Protease system
A recent study showed that impairment of NO action promotes the genesis of cardiovascular diseases [31]. The synthesis of NO is blocked by inhibition of the NOS active site with guanidino-substituted analogues of l-arginine, such as asymmetric dimethylarginine (ADMA) [29]. Therefore, ADMA acts as an endogenous NOS inhibitor [32, 33]. The administration of ADMA in rats causes an increase in renal vascular resistance and blood pressure [34], confirming its biological action in vivo. Interestingly, a more recent report showed that maternal concentrations of ADMA are higher in mid-pregnancy in women who experience preeclampsia, compared with women with uncomplicated pregnancies [35]. Investigators have studied other promising markers that have been identified up to now, including placental protein 13 (PP13), P-selectin, a disintegrin and metalloprotease-12 (ADAM12), pentraxin 3 (PTX3), pregnancy-associated plasma protein A (PAPP-A), and adrenomedullin[36, 37]. These markers appear to be promising tools, most likely in a combinatorial analysis, to diagnose, predict outcome, and monitor this disorder [36, 37].
Metabolic system
Up-regulation of metabolism-related gene expression such as leptin has been found in several studies [38]. These genes and proteins may be connected to atherosclerosis, diabetes, and Alzheimer’s disease. In addition, Sugathadasa et al. [39] showed that preeclampsia appears to be associated with the AA genotype of −2548 G/A polymorphism of the leptin gene. Genetic factors such as leptin gene polymorphism may increase the susceptibility of pregnant women to develop preeclampsia.
Adhesion system
VEGF is a potent angiogenic and pro-inflammatory cytokine that increases vascular permeability, angiogenesis, vasculogenesis, endothelial cell growth, migration, adhesion, and anti-apoptosis, partly via upregulation of endothelial cell and leukocyte adhesion molecule expression [40]. In contrast to these descriptions of VEGF’s inflammatory actions, several reports describe VEGF neutralization leading to elevated expression of surface adhesion molecules, resulting in increased leukocyte rolling and adhesion [41]. Neutralization of VEGF action also results in endothelial surface expression of P-selectin and impaired peripheral vasodilation [41]. Depending on the local concentrations of VEGF, striking differences in biological functions were found.
Interpretation of microarray and proteomic data sets
We next determined functional categories of coregulated genes and gene pathways. Hypotheses about common regulatory elements or their functional significance were formulated. Genes were classified into nine functional categories according to their expression profile: renin–angiotensin system, angiogenesis, inflammation and cytokines, stress and detoxification, metabolism, adhesion, cell structure, signal transduction, and protease, which are consistent with the oxidative stress of preeclampsia (Table 1 and Fig. 1, Genome-wide gene expression profiling and proteomics). Gene ontology analysis revealed several biological processes which could be associated with the development of preeclampsia [9, 42, 43] (Fig. 1, Gene ontology analysis). A logical and systematic data analysis strategy demonstrated that genes and proteins that participated in response to stress, host–pathogen interactions, immune system, inflammation, lipid metabolism, carbohydrate metabolism, growth, and tissue remodeling pathways were expressed differentially in preeclampsia. The features of the microarray and proteomic data sets, categorized according to gene ontology, indicate that preeclampsia is characterized as “stress” and “immune/inflammatory” responses.
Fig. 1.
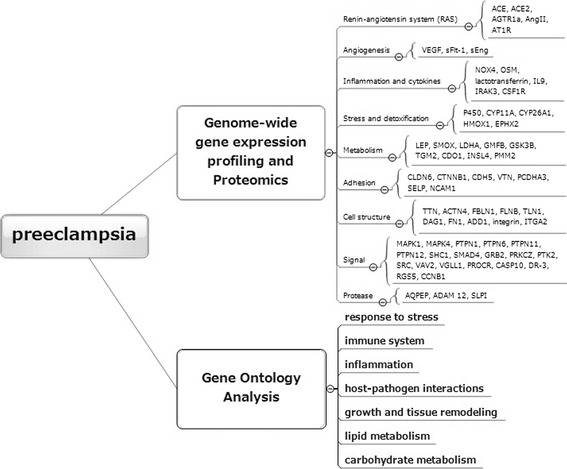
Comparative analysis of candidate markers with altered expression patterns in preeclampsia. We searched PubMed electronic databases, combining the keywords “gene expression profiling”, “proteomic”, “pathogenesis”, “pathophysiology”, “Toll-like receptor”, “advanced glycation end products”, “receptor for advanced glycation end products”, “inflammation”, “stress”, or “alarmins” with “preeclampsia”. This figure provides a summary of differentially expressed gene and protein profiles and a result of gene ontology analysis. ACE angiotensin I converting enzyme (peptidyl-dipeptidase A) 1; ACE2 angiotensin I converting enzyme (peptidyl-dipeptidase A) 2; AGTR1a angiotensin II receptor, type 1a; AngII angiotensin II; AT1R angiotensin II receptor, type 1; VEGF vascular endothelial growth factor; sFlt-1 soluble fms-like tyrosine kinase-1; sEng soluble endoglin; NOX4 nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4; OSM oncostatin M; IL-9 interleukin-9; IRAK3 interleukin-1 receptor-associated kinase 3; CSF1R colony stimulating factor 1 receptor; P450 cytochrome P450; CYP11A cytochrome P450, family 11, subfamily A, polypeptide 1; CYP26A1 cytochrome P450, family 26, subfamily A, polypeptide 1; HMOX1 heme oxygenase (decycling) 1; EPHX2 epoxide hydrolase 2, cytoplasmic; LEP leptin; SMOX spermine oxidase; LDHA lactate dehydrogenase A; GMFB glia maturation factor, beta; GSK3B glycogen synthase kinase 3 beta; TGM2 transglutaminase 2 (C polypeptide, protein-glutamine-gamma-glutamyltransferase), CDO1 cysteine dioxygenase, type I; INSL4 insulin-like 4; PMM2 phosphomannomutase 2; CLDN6 claudin 6; CTNNB1 catenin (cadherin-associated protein), beta; CDH5 cadherin 5, type 2; VTN vitronectin; PCDHA3 protocadherin alpha 3; SELP selectin P; NCAM1 neural cell adhesion molecule 1; TTN transthyretin; ACTN4 actinin, alpha 4; FBLN1 fibulin 1; FLNB filamin B, beta; TLN1 talin 1; DAG1 dystroglycan 1; FN1 fibronectin 1; ADD1 adducin 1; ITGA2 integrin, alpha 2; MAPK1 mitogen-activated protein kinase 1; MAPK4 mitogen-activated protein kinase 4; PTPN1 protein tyrosine phosphatase, non-receptor type; PTPN6 protein tyrosine phosphatase, non-receptor type 6; PTPN11 protein tyrosine phosphatase, non-receptor type 11; PTPN12 protein tyrosine phosphatase, non-receptor type 12; SHC1 SHC (Src homology 2 domain containing) transforming protein 1; SMAD4 SMAD family member 4; GRB2 growth factor receptor-bound protein 2; PRKCZ protein kinase C, zeta; PTK2 protein tyrosine kinase 2; SRC v-src sarcoma (Schmidt–Ruppin A-2) viral oncogene homolog; VAV2 vav 2 guanine nucleotide exchange factor; VGLL1 vestigial like 1; PROCR protein C receptor; CASP10 caspase-10; DR-3 death receptor 3; RGS5 regulator of G-protein signaling; CCNB1 cyclin B1; AQPEP laeverin; ADAM 12 a disintegrin and metalloproteinase 12, meltrin-alpha; SLPI secretory leukocyte peptidase inhibitor
Collectively, preeclampsia is considered to be a disease of at least two stages: the first (subclinical) stage concerns the failure of the proposed sequence of events comprising early trophoblast invasion (Table 1, Angiogenesis and Protease pathways), remodeling (Table 1, Signal pathway), and consequential placental perfusion (Table 1, Inflammation and cytokines pathway), which are believed to play a major role in this disorder through the production of angiogenic factors and immunoregulatory cytokines via oxidative stress (Table 1, Stress and detoxification pathway). Preeclampsia in particular has gene-expression signatures associated with imbalance of oxidative stress generation and detoxification pathways. In a suggested hypothetical event, the link between angiogenesis and cytokine could be provided through oxidative stress by excessive production of ROS. Taken together, placental hypoxia and reoxygenation might induce oxidative stress, which stimulates placental synthesis of cytokines, maternal leukocyte activation, leukocyte/endothelial interaction, immune response regulation and generalized endothelial dysfunction, thereby completing a vicious cycle [44]. These data allow us to speculate that the first subclinical stage has a “stress-resistant” phenotype.
The second (clinical) stage is the overt maternal syndrome, which is characterized by a generalized systemic inflammatory response (Table 1, Inflammation and cytokine pathway) and subsequent generalized endothelial dysfunction involving both leukocytes and endothelial activation (Table 1, Adhesion, Metabolism and Cell structure pathways). Although endothelial cells can cope with a rise in oxidative stress and sFlt-1, the inability to retrieve endothelial function might be a result of oxidative stress imbalance. Maternal inflammatory cells and endothelial cells persistently stimulate the release of pro-inflammatory cytokines, e.g. TNF-alpha [45]. Such environmental stress (the “stress-sensitive” condition) implicitly alters epigenetic and genetic patterns that can lead to the clinical manifestations of preeclampsia. An important role in the second stage is played by endothelial dysfunction, manifested by an imbalance of stress and anti-stress homeostasis.
Promising biomarkers for preeclampsia
Cutting-edge proteomic technologies will be of great help in understanding the heterogeneity of preeclampsia as shown in Table 1 and Fig. 1. The promising biochemical markers have been developed to evaluate the features of placental dysfunction, inflammatory response, or endothelial dysfunction. Potential markers can be classified as “prediction” or “detection” of preeclampsia. Since most of these preeclampsia-specific mediators are increasingly expressed in many different “stress” and “immune/inflammatory response” processes, the following crucial molecules might also be important for the understanding of the pathogenesis of preeclampsia: besides VEGF, they include TLRs, RAGE and RAGE ligands. They are associated with regulatory pathways of inflammation and oxidative stress, along with a number of innate immune systems. The link between “stress” and “immune/inflammatory response” is further supported by the increased levels of inflammatory pattern recognition receptors (PRRs) and their ligands. The relevance of these issues is discussed below.
Inflammatory pattern recognition receptors and ligands
Toll-like receptors (TLRs)
The present review has proposed that hypoxia-induced inflammatory genes and stress-related factors are upstream regulators of preeclampsia [46]. Stress and inflammation are processes by which cells or tissues respond to various insults or stimuli, microbes, and tissue breakdown products. It is characterized by up-regulation of PRRs that sense exogenous (infectious) and endogenous ligands. Two inflammatory PRRs, Toll-like receptors (TLRs) and RAGE, interact with a wide range of common and different ligands, and play a major role in the maternal immune and inflammatory system [47].
There is strong evidence that maternal immune system activation contributes to the development of preeclampsia [6, 8]. TLRs recognize infectious agents (foreign pathogens, exogenous ligands) and other danger signals [endogenously generated inflammatory ligands, also known as alarmins (see above)]. TLR-dependent innate immunity plays a role as the first line of host defense. Activation of TLRs on trophoblast cells influences immune cell recruitment, pro-inflammatory cytokine secretion, and decidual responses to invading pathogens during pregnancy [48]. Importantly, recent data directly implicate signaling by TLRs at multiple maternal–fetal interfaces in the pathogenesis of several pregnancy pathologies, including preeclampsia, intrauterine growth restriction, and preterm labor [48]. Although the participation of the innate immune responses in sustaining the inflammation needs further investigation, preeclampsia is characterized as being a state of persistent inflammation. Animal models for preeclampsia are furthering our understanding of pathophysiology: TLR3 responds to double strand (ds) RNA from viruses, apoptotic cells and necrotic cells. Persistent TLR3 activation during pregnancy causes hypertension, endothelial dysfunction, proteinuria, and intrauterine growth restriction in normal pregnant rats [49]. TLR stimulation of trophoblast cells induces inflammatory mediator production and, in particular, secretion of anti-angiogenic factor sFlt-1 [50]. Exogenous ligand lipopolysaccharide (LPS) itself can also induce sFlt-1 expression in macrophages through protein kinase C signaling [51, 52]. Persistent activation of TLRs might therefore be associated with the development of preeclampsia.
A growing number of defined single nucleotide polymorphisms (SNPs) of TLRs have been described that genetically determine susceptibility to infection and inflammation. Recently, van Rijn et al. [53] observed an association of common TLR4 (D299G and T399I) and nucleotide binding and oligomerization domain (NOD) (R702W, G908R, and L1007fs) gene variants, and pro-inflammatory phenotype with a history of preeclampsia. These findings suggest direct involvement of the maternal innate immune system in this syndrome [53]. On the other hand, functional SNPs of the TLR2 (G2258A) failed to present any significant association with preeclampsia [54]. The impact of SNPs of the TLR molecules therefore remains controversial.
Receptor for advanced glycation end products (RAGE)
Another inflammatory signaling receptor, RAGE, interacts with a wide range of common endogenous ligands [47]. RAGE is an immunoglobulin-like cell surface receptor that is described as a PRR because of the structural heterogeneity of its ligands (Fig. 2). Defined TLR ligands are mainly derived from pathogens, but RAGE ligands do not originate from pathogens [47]. The nonenzymatic glycation and oxidation of proteins and lipids lead to the formation of a broad range of species, called “advanced glycation end products” (AGEs) [55]. Chronic inflammation modifies protein and lipid, leading to production of AGEs. AGEs are proteins, lipids, and polynucleotides, including N-carboxymethyl-lysine, pentosidine, and methylglyoxal derivatives [47] (Fig. 2). AGEs are known to cause oxidative damage in various cells via RAGE signaling.
Fig. 2.
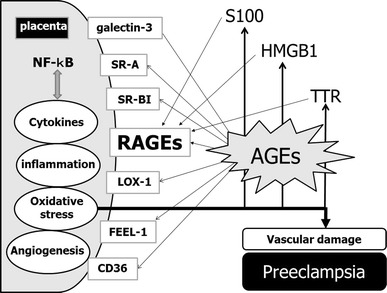
The AGE–RAGE system at the maternal–fetal interface in preeclampsia. RAGEs are pattern recognition receptors. AGEs are proteins, lipids and polynucleotides, including N-carboxymethyl-lysine, pentosidine, and methylglyoxal derivatives, and are the major ligands for RAGEs. Several proteins are known to act as AGE receptors. NF-kB nuclear factor-kappaB, SR-A scavenger receptor 1, SR-BI scavenger receptor BI, RAGE receptor for advanced glycosylation end products, LOX-1 oxidized low density lipoprotein (lectin-like) receptor 1; FEEL-1 stabilin 1, CD36 thrombospondin receptor, S100 S100 calcium binding protein A1, HMGB1 high-mobility group box 1, TTR transthyretin, AGEs advanced glycosylation end products
Not only RAGE but also LOX-1 [oxidized low density lipoprotein (lectin-like) receptor 1], galectin-3 (ectin, galactoside-binding, soluble, 3), SR-A (macrophage scavenger receptor 1), CD36 (thrombospondin receptor), SR-BI (scavenger receptor class B, member 1), FEEL-1 (stabilin 1) and FEEL-2 (stabilin 2) are known to act as AGE receptors (Fig. 2). In addition to AGEs, RAGE binds certain members of the S100/calgranulin family (such as S100A12 and S100B), high-mobility group box 1 (HMGB1), transthyretin (TTR), Mac-1, and amphoterin [56]. RAGE is expressed on multiple cell types, such as endothelial cells, smooth muscle cells, monocytes/macrophages, T lymphocytes, dendritic cells, glomerular epithelial cells, podocytes, cardiomyocytes, neurons of the central and peripheral nervous systems, and transformed cells [56]. RAGE activates nicotinamide adenine dinucleotide phosphate reduced form (NADPH) oxidase, an important source of oxidative stress, and generates ROS [56]. The transcription factor NF-kappaB is a downstream target of RAGE-mediated cellular activation. NF-kappaB activation also leads to increased RAGE expression. Thus, the AGE–RAGE system, which is up-regulated in preeclampsia, is likely to be involved in ROS-induced oxidative stress and might contribute to vascular dysfunction in preeclampsia [57, 58]. Accumulation of markers of oxidative stress such as 4-hydroxy-2-nonenal and 8-hydroxy-2′-deoxyguanosine indicated enhanced oxidative modifications of lipids and DNA in preeclamptic placenta [57]. Cellular stress might result in release of the pro-inflammatory RAGE ligands S100, HMGB1, and transthyretin (TTR) [56]. Taken together, the hypoxia–oxidative stress–persistent inflammation-dependent mechanism during pregnancy may be the triggering stimulus to draw RAGE into preeclampsia pathology.
Other AGE receptors
LOX-1: oxidized low density lipoprotein (lectin-like) receptor 1 (OLR1)
LOX-1 binds, internalizes and degrades oxidized low-density lipoprotein (OxLDL) [59]. This protein may play a role as a scavenger receptor. Investigators have shown overexpression of LOX-1 and arginase, which contribute to endothelial dysfunction and oxidative stress, in the vasculature of women with preeclampsia [59, 60]. Increased arginase expression could contribute to decreased NO and enhanced superoxide formation [59]. LOX-1 also generates superoxide via NADPH oxidase.
Galectin-3: lectin, galactoside-binding, soluble, 3 (LGALS3)
Members of this protein family have an affinity for beta-galactosides. Several findings suggest that the expression of galectin-3 on the extravillous trophoblast is up-regulated in preeclamptic placenta [61]. Galectin-3 was up-regulated under hypoxia [62]. Ligands for galectin-3 include not only AGEs but also other proteins such as laminin, fibronectin, tenascin, integrins, CD98 (solute carrier family 7), cytokeratins, Bcl-2, and Alix/AIP-1 (programmed cell death 6 interacting protein) [63]. Galectin-3 is therefore involved in the inhibition of apoptosis [63].
SR-A: macrophage scavenger receptor 1 (MSR1) and SR-BI: scavenger receptor class B, member 1 (SCARB1)
These proteins mediate the endocytosis of OxLDL and play a role as macrophage scavenger receptors. AGEs induce the gene expression of these OxLDL receptors [64]. These receptors have been implicated in many macrophage-associated physiological and pathological processes including atherosclerosis, Alzheimer’s disease, and host defense. Human extravillous cytotrophoblast cells also express OxLDL receptors [65]. The pathogenesis of preeclampsia appears to overlap with those of atherosclerosis, diabetes, and Alzheimer’s disease.
CD36: thrombospondin receptor, also known as fatty acid translocase (transporter)
CD36 binds to thrombospondin, collagen, anionic phospholipids, and OxLDL. This protein has an important function as a cell adhesion molecule and scavenger receptor. Gene expression of CD36 was decreased in placental tissues from pregnancies complicated by preeclampsia. How CD36 is associated with preeclampsia remains unclear.
FEEL-1: stabilin 1 (STAB 1) and FEEL-2: stabilin 2 (STAB 2)
These receptors have been shown to endocytose ligands such as OxLDL, Gram-positive and Gram-negative bacteria, and AGEs. These molecules may function in angiogenesis, lymphocyte homing, cell adhesion, or receptor scavenging. However, the exact role of FEEL-1 in this disorder and how it mediates stress and inflammation remain unclear.
RAGE ligands
Advanced glycation end products (AGEs)
AGEs are adducts formed by glycoxidation that accumulate in metabolic disorders such as diabetes and atherosclerosis [66]. Interaction of aldoses with proteins initiates a chain of nonenzymatic reactions leading to the covalent addition of AGEs to proteins, known as the Maillard reaction. Attention will be focused on the vascular endothelial cells–AGE interaction. AGEs are heterogeneous in structure, which implies that many products could be measured to estimate formation of AGEs. Of them, pentosidine and carboxymethyl-lysine have been the best characterized [67]. It has been established that AGEs are directly implicated in the development of chronic complications in diabetes [68]. Similar to diabetes, the mean level of serum AGEs in preeclamptic women was higher than those in healthy non-pregnant women or healthy pregnant women [57]. Tsukahara et al. [69] showed that umbilical blood concentrations of pentosidine are high in the maternal preeclamptic condition. Thus, preeclampsia is thought to be complicated with formation and production of AGEs. These data allow us to speculate that AGEs participate in the development of preeclamptic complications.
High-mobility group box 1 (HMGB1)
RAGE was the first receptor identified for extracellular HMGB1. HMGB1 acts as a danger signal “alarmin” to alert the innate immune system for the initiation of host defense or tissue repair [70]. This ligand recognizes not only RAGE but also TLR2 and TLR4, leading to production of pro-inflammatory cytokines via the NF-kappaB signaling cascade. HMGB1 possesses important extranuclear functions as a potent danger signal mediating the late response to infection and inflammation [71]. A higher expression of cytoplasmic HMGB1 was found in the decidua from women with preeclampsia [72]. There are as yet no data on whether HMGB1 levels are correlated with the clinical manifestations of preeclampsia.
S100
Ca2+-binding S100B protein is a RAGE ligand [73]. The concentrations of S100B protein were increased in the placenta under oxidative stress [73]. S100B protein up-regulates sEng release from endothelial cells [73], and thus S100B protein is thought to be associated with preeclampsia.
Transthyretin (TTR)
RAGE has been shown to bind fibrillar transthyretin (TTR), triggering NF-kappaB activation [74]. This protein is responsible for transporting both the thyroid hormone thyroxine and retinol-binding protein. There are several reports that the concentrations of TTR appear to be associated with preeclampsia: TTR was up-regulated in preeclampsia in comparison with normal placentas, and oxidized TTR in amniotic fluid plays a role as an early marker of preeclampsia.
These data allow us to speculate that once RAGE is engaged in the placental tissue, a vicious cycle of ligands–RAGE perturbation ensues, leading to oxidative stress, chronic tissue injury, and vascular damage. This section provides new information regarding PRRs and their ligands that might contribute to pathological processes involved in preeclampsia.
Conclusion
The aim of this review is to summarize current knowledge on the pathogenesis of preeclampsia. We comprehensively analyzed the results of genome-wide gene expression profiling and proteomic studies to delineate the wide array of mediators involved in this disorder. Genes and proteins that participated in response to stress, host–pathogen interactions, immune system, inflammation, lipid metabolism, carbohydrate metabolism, growth, and tissue remodeling pathways were expressed differentially in preeclampsia (Fig. 1). The most important factors are genes and proteins involved in oxidative stress and inflammation.
Several significant common pathways observed in preeclampsia overlap the datasets identified in TLR- and RAGE-dependent signaling pathways. A novel model of the AGEs–RAGE system is proposed in Fig. 2. It is focused on several main properties of TLRs and RAGE and their ligands and signaling pathways along with angiogenic, inflammatory, and stress-related factors. Notably, up-regulation of TLRs and RAGEs, their ligands and their downstream targets are specifically found in preeclampsia. In addition, sFlt-1, sEng, and angiotensin-II receptor autoantibody may contribute to the pathogenesis of preeclampsia. These potential markers have been implicated in the regulation of vascular endothelial function. The persistent inflammatory response plays an important role in coordinating the global transcriptional regulation leading to the functional switch from the “stress-resistant” response to the “stress-sensitive” phenotype: the loss of angiogenic functions and the rise in PRRs. Our review suggests a progressive shift in cellular homeostasis that may underlie stress/inflammation-associated functional alteration in vascular endothelial cells.
In conclusion, the present article reviews the available scientific literature related to preeclampsia. The key challenge is to move from lists of identified factors to obtaining biological information regarding their functions. This work provides new insights into and aids understanding of the pathogenesis of preeclampsia. The strong correlation of specific genes regulated in preeclampsia with the possible TLR- and RAGE-dependent signaling pathways suggests that placental oxidative stress and subsequent chronic inflammation are considered to be major contributors to the development of preeclampsia.
Acknowledgments
This study was supported by KAKENHI (Japan Society for the Promotion of Science (JSPS) Grant-in-Aid No. 20591958). We thank all the study participants for their time and efforts. We thank Mikiko Kita for editorial assistance.
Conflict of interest
No potential conflicts of interest relevant to this article were reported.
References
- 1.Davison JM, Homuth V, Jeyabalan A, Conrad KP, Karumanchi SA, Quaggin S, Dechend R, Luft FC. New aspects in the pathophysiology of preeclampsia. J Am Soc Nephrol. 2004;15:2440–2448. doi: 10.1097/01.ASN.0000135975.90889.60. [DOI] [PubMed] [Google Scholar]
- 2.Mutter WP, Karumanchi SA. Molecular mechanisms of preeclampsia. Microvasc Res. 2008;75(1):1–8. doi: 10.1016/j.mvr.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Facemire CS, Nixon AB, Griffiths R, Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54:652–658. doi: 10.1161/HYPERTENSIONAHA.109.129973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park CW, Kim HW, Lim JH, Yoo KD, Chung S, Shin SJ, Chung HW, Lee SJ, Chae CB, Kim YS, Chang YS. Vascular endothelial growth factor inhibition by dRK6 causes endothelial apoptosis, fibrosis, and inflammation in the heart via the Akt/eNOS axis in db/db mice. Diabetes. 2009;58:2666–2676. doi: 10.2337/db09-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie F, Turvey SE, Williams MA, Mor G, von Dadelszen P. Toll-like receptor signaling and pre-eclampsia. Am J Reprod Immunol. 2010;63:7–16. doi: 10.1111/j.1600-0897.2009.00745.x. [DOI] [PubMed] [Google Scholar]
- 7.Srikrishna G, Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. 2009;11:615–628. doi: 10.1593/neo.09284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie F, Hu Y, Turvey SE, Magee LA, Brunham RM, Choi KC, Krajden M, Leung PC, Money DM, Patrick DM, Thomas E, von Dadelszen P. Toll-like receptors 2 and 4 and the cryopyrin inflammasome in normal pregnancy and pre-eclampsia. BJOG. 2010;117:99–108. doi: 10.1111/j.1471-0528.2009.02428.x. [DOI] [PubMed] [Google Scholar]
- 9.Nishizawa H, Pryor-Koishi K, Kato T, Kowa H, Kurahashi H, Udagawa Y. Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and late onset severe pre-eclampsia. Placenta. 2007;28:487–497. doi: 10.1016/j.placenta.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 10.Royle C, Lim S, Xu B, Tooher J, Ogle R, Hennessy A. Effect of hypoxia and exogenous IL-10 on the pro-inflammatory cytokine TNF-alpha and the anti-angiogenic molecule soluble Flt-1 in placental villous explants. Cytokine. 2009;47:56–60. doi: 10.1016/j.cyto.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Lemmens L, Claes V, Uzzell M. Managing patients with metastatic colorectal cancer on bevacizumab. Br J Nurs. 2008;17:944–949. doi: 10.12968/bjon.2008.17.15.30695. [DOI] [PubMed] [Google Scholar]
- 12.Rajakumar A, Michael HM, Rajakumar PA, Shibata E, Hubel CA, Karumanchi SA, Thadhani R, Wolf M, Harger G, Markovic N. Extra-placental expression of vascular endothelial growth factor receptor-1, (Flt-1) and soluble Flt-1 (sFlt-1), by peripheral blood mononuclear cells (PBMCs) in normotensive and preeclamptic pregnant women. Placenta. 2005;26:563–573. doi: 10.1016/j.placenta.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Molvarec A, Ito M, Shima T, Yoneda S, Toldi G, Stenczer B, Vásárhelyi B, Rigó J, Saito S. Decreased proportion of peripheral blood vascular endothelial growth factor-expressing T and natural killer cells in preeclampsia. Am J Obstet Gynecol. 2010;203:567–8. doi: 10.1016/j.ajog.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 14.Foidart JM, Schaaps JP, Chantraine F, Munaut C, Lorquet S. Dysregulation of anti-angiogenic agents (sFlt-1, PLGF, and sEndoglin) in preeclampsia—a step forward but not the definitive answer. J Reprod Immunol. 2009;82:106–111. doi: 10.1016/j.jri.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Gruemmer R, Motejlek K, Berghaus D, Weich HA, Neulen J. Regulation of soluble vascular endothelial growth factor receptor (sFlt-1/sVEGFR-1) expression and release in endothelial cells by human follicular fluid and granulosa cells. Reprod Biol Endocrinol. 2005;3:57. doi: 10.1186/1477-7827-3-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Marco GS, Reuter S, Hillebrand U, Amler S, König M, Larger E, Oberleithner H, Brand E, Pavenstädt H, Brand M. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J Am Soc Nephrol. 2009;20(10):2235–2245. doi: 10.1681/ASN.2009010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaminska B, Wesolowska A, Danilkiewicz M. TGF beta signalling and its role in tumour pathogenesis. Acta Biochim Pol. 2005;52:329–337. [PubMed] [Google Scholar]
- 18.Ahmed A, Cudmore MJ. Can the biology of VEGF and haem oxygenases help solve pre-eclampsia? Biochem Soc Trans. 2009;37:1237–1242. doi: 10.1042/BST0371237. [DOI] [PubMed] [Google Scholar]
- 19.Anton L, Brosnihan KB. Systemic and uteroplacental renin–angiotensin system in normal and pre-eclamptic pregnancies. Ther Adv Cardiovasc Dis. 2008;2:349–362. doi: 10.1177/1753944708094529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jüpner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–952. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou CC, Ahmad S, Mi T, Abbasi S, Xia L, Day MC, Ramin SM, Ahmed A, Kellems RE, Xia Y. Autoantibody from women with preeclampsia induces soluble Fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of activated T-cells signaling. Hypertension. 2008;51:1010–1019. doi: 10.1161/HYPERTENSIONAHA.107.097790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irani RA, Xia Y. The functional role of the renin-angiotensin system in pregnancy and preeclampsia. Placenta. 2008;29:763–771. doi: 10.1016/j.placenta.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papaioannou AI, Kostikas K, Kollia P, Gourgoulianis KI. Clinical implications for vascular endothelial growth factor in the lung: friend or foe? Respir Res. 2006;7:128. doi: 10.1186/1465-9921-7-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, Shah PK, Willerson JT, Benza RL, Berman DS, Gibson CM, Bajamonde A, Rundle AC, Fine J, McCluskey ER, VIVA Investigators The VIVA trial: vascular endothelial growth factor in ischemia for vascular angiogenesis. Circulation. 2003;107:1359–1365. doi: 10.1161/01.CIR.0000061911.47710.8A. [DOI] [PubMed] [Google Scholar]
- 25.Zachary I, Gliki G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc Res. 2001;49:568–581. doi: 10.1016/S0008-6363(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 26.Nakagawa T, Segal M, Croker B, Johnson RJ. A breakthrough in diabetic nephropathy: the role of endothelial dysfunction. Nephrol Dial Transplant. 2007;22:2775–2777. doi: 10.1093/ndt/gfm380. [DOI] [PubMed] [Google Scholar]
- 27.Eide IP, Isaksen CV, Salvesen KA, Langaas M, Schønberg SA, Austgulen R. Decidual expression and maternal serum levels of heme oxygenase 1 are increased in pre-eclampsia. Acta Obstet Gynecol Scand. 2008;87:272–279. doi: 10.1080/00016340701763015. [DOI] [PubMed] [Google Scholar]
- 28.Saito S, Shiozaki A, Nakashima A, Sakai M, Sasaki Y. The role of the immune system in preeclampsia. Mol Aspects Med. 2007;28:192–209. doi: 10.1016/j.mam.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Santner-Nanan B, Peek MJ, Khanam R, Richarts L, Zhu E, Fazekas de St Groth B, Nanan R. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J Immunol. 2009;183:7023–7030. doi: 10.4049/jimmunol.0901154. [DOI] [PubMed] [Google Scholar]
- 30.Saito S. Th17 cells and regulatory T cells: new light on pathophysiology of preeclampsia. Immunol Cell Biol. 2010;88:615–617. doi: 10.1038/icb.2010.68. [DOI] [PubMed] [Google Scholar]
- 31.Toda N, Ayajiki K. Vascular actions of nitric oxide as affected by exposure to alcohol. Alcohol Alcohol. 2010;45:347–355. doi: 10.1093/alcalc/agq028. [DOI] [PubMed] [Google Scholar]
- 32.Ueda S, Yamagishi S, Okuda S. New pathways to renal damage: role of ADMA in retarding renal disease progression. J Nephrol. 2010;23:377–386. [PubMed] [Google Scholar]
- 33.Anderssohn M, Schwedhelm E, Lüneburg N, Vasan RS, Böger RH. Asymmetric dimethylarginine as a mediator of vascular dysfunction and a marker of cardiovascular disease and mortality: an intriguing interaction with diabetes mellitus. Diab Vasc Dis Res. 2010;7:105–118. doi: 10.1177/1479164110366053. [DOI] [PubMed] [Google Scholar]
- 34.Gardiner SM, Kemp PA, Bennett T, Palmer RM, Moncada S. Regional and cardiac haemodynamic effects of NG,NG,dimethyl-l-arginine and their reversibility by vasodilators in conscious rats. Br J Pharmacol. 1993;110:1457–1464. doi: 10.1111/j.1476-5381.1993.tb13985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Speer PD, Powers RW, Frank MP, Harger G, Markovic N, Roberts JM. Elevated asymmetric dimethylarginine concentrations precede clinical preeclampsia, but not pregnancies with small-for-gestational-age infants. Am J Obstet Gynecol. 2008;198:112.e1–112.e7. doi: 10.1016/j.ajog.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grill S, Rusterholz C, Zanetti-Dällenbach R, Tercanli S, Holzgreve W, Hahn S, Lapaire O. Potential markers of preeclampsia—a review. Reprod Biol Endocrinol. 2009;7:70. doi: 10.1186/1477-7827-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Than NG, Romero R, Hillermann R, Cozzi V, Nie G, Huppertz B. Prediction of preeclampsia—a workshop report. Placenta. 2008;29:S83–S85. doi: 10.1016/j.placenta.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laivuori H. Genetic aspects of preeclampsia. Front Biosci. 2007;12:2372–2382. doi: 10.2741/2239. [DOI] [PubMed] [Google Scholar]
- 39.Sugathadasa BH, Tennekoon KH, Karunanayake EH, Kumarasiri JM, Wijesundere AP. Association of −2548 G/A polymorphism in the leptin gene with preeclampsia/pregnancy-induced hypertension. Hypertens Pregnancy. 2010;29(4):366–374. doi: 10.3109/10641950903214617. [DOI] [PubMed] [Google Scholar]
- 40.Goebel S, Huang M, Davis WC, Jennings M, Siahaan TJ, Alexander JS, Kevil CG. VEGF-A stimulation of leukocyte adhesion to colonic microvascular endothelium: implications for inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2006;290(4):G648–G654. doi: 10.1152/ajpgi.00466.2005. [DOI] [PubMed] [Google Scholar]
- 41.Walshe TE, Dole VS, Maharaj AS, Patten IS, Wagner DD, D’Amore PA. Inhibition of VEGF or TGF-{beta} signaling activates endothelium and increases leukocyte rolling. Arterioscler Thromb Vasc Biol. 2009;29:1185–1192. doi: 10.1161/ATVBAHA.109.186742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Centlow M, Junus K, Nyström H, May K, Larsson I, Olsson MG, Akerström B, Sager R, Schneider H, Hansson SR. Perfusion of the human placenta with red blood cells and xanthine oxidase mimics preeclampsia in vitro. Z Geburtshilfe Neonatol. 2009;213:89–95. doi: 10.1055/s-0029-1224196. [DOI] [PubMed] [Google Scholar]
- 43.Enquobahrie DA, Meller M, Rice K, Psaty BM, Siscovick DS, Williams MA. Differential placental gene expression in preeclampsia. Am J Obstet Gynecol. 2008;199:566.e1–566.e11. doi: 10.1016/j.ajog.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Redman CW, Sargent IL. Pre-eclampsia, the placenta and the maternal systemic inflammatory response—a review. Placenta. 2003;24:S21–S27. doi: 10.1053/plac.2002.0930. [DOI] [PubMed] [Google Scholar]
- 45.Poston L. Endothelial dysfunction in pre-eclampsia. Pharmacol Rep. 2006;58:69–74. [PubMed] [Google Scholar]
- 46.Sharma S, Norris WE, Kalkunte S. Beyond the threshold: an etiological bridge between hypoxia and immunity in preeclampsia. J Reprod Immunol. 2010;85:112–116. doi: 10.1016/j.jri.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin L, Park S, Lakatta EG. RAGE signaling in inflammation and arterial aging. Front Biosci. 2009;14:1403–1413. doi: 10.2741/3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riley JK, Nelson DM. Toll-like receptors in pregnancy disorders and placental dysfunction. Clin Rev Allergy Immunol. 2010;39:185–193. doi: 10.1007/s12016-009-8178-2. [DOI] [PubMed] [Google Scholar]
- 49.Tinsley JH, Chiasson VL, Mahajan A, Young KJ, Mitchell BM. Toll-like receptor 3 activation during pregnancy elicits preeclampsia-like symptoms in rats. Am J Hypertens. 2009;22:1314–1319. doi: 10.1038/ajh.2009.185. [DOI] [PubMed] [Google Scholar]
- 50.Nakada E, Walley KR, Nakada T, Hu Y, von Dadelszen P, Boyd JH. Toll-like receptor-3 stimulation upregulates sFLT-1 production by trophoblast cells. Placenta. 2009;30:774–779. doi: 10.1016/j.placenta.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Mezquita J, Mezquita B, Pau M, Mezquita C. Down-regulation of Flt-1 gene expression by the proteasome inhibitor MG262. J Cell Biochem. 2003;89:1138–1147. doi: 10.1002/jcb.10587. [DOI] [PubMed] [Google Scholar]
- 52.Lee MC, Wei SC, Tsai-Wu JJ, Wu CH, Tsao PN. Novel PKC signaling is required for LPS-induced soluble Flt-1 expression in macrophages. J Leukoc Biol. 2008;84:835–841. doi: 10.1189/jlb.1007691. [DOI] [PubMed] [Google Scholar]
- 53.van Rijn BB, Franx A, Steegers EA, de Groot CJ, Bertina RM, Pasterkamp G, Voorbij HA, Bruinse HW, Roest M. Maternal TLR4 and NOD2 gene variants, pro-inflammatory phenotype and susceptibility to early-onset preeclampsia and HELLP syndrome. PLoS One. 2008;3:e1865. doi: 10.1371/journal.pone.0001865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fraser R, Walker JJ, Ekbote UV, Martin KL, McShane P, Orsi NM. Interleukin-4–590 (C>T), toll-like receptor-2 +2258 (G>A) and matrix metalloproteinase-9–1562 (C>T) polymorphisms in pre-eclampsia. BJOG. 2008;115:1052–1056. doi: 10.1111/j.1471-0528.2008.01771.x. [DOI] [PubMed] [Google Scholar]
- 55.Yan SF, Ramasamy R, Schmidt AM. The receptor for advanced glycation endproducts (RAGE) and cardiovascular disease. Expert Rev Mol Med. 2009;11:e9. doi: 10.1017/S146239940900101X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med. 2009;87:235–247. doi: 10.1007/s00109-009-0439-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chekir C, Nakatsuka M, Noguchi S, Konishi H, Kamada Y, Sasaki A, Hao L, Hiramatsu Y. Accumulation of advanced glycation end products in women with preeclampsia: possible involvement of placental oxidative and nitrative stress. Placenta. 2006;27:225–233. doi: 10.1016/j.placenta.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 58.Cooke CL, Brockelsby JC, Baker PN, Davidge ST. The receptor for advanced glycation end products (RAGE) is elevated in women with preeclampsia. Hypertens Pregnancy. 2003;22:173–184. doi: 10.1081/PRG-120021068. [DOI] [PubMed] [Google Scholar]
- 59.Sankaralingam S, Xu H, Jiang Y, Sawamura T, Davidge ST. Evidence for increased methylglyoxal in the vasculature of women with preeclampsia: role in upregulation of LOX-1 and arginase. Hypertension. 2009;54:897–904. doi: 10.1161/HYPERTENSIONAHA.109.135228. [DOI] [PubMed] [Google Scholar]
- 60.Sankaralingam S, Xu Y, Sawamura T, Davidge ST. Increased lectin-like oxidized low-density lipoprotein receptor-1 expression in the maternal vasculature of women with preeclampsia: role for peroxynitrite. Hypertension. 2009;53:270–277. doi: 10.1161/HYPERTENSIONAHA.108.122630. [DOI] [PubMed] [Google Scholar]
- 61.Jeschke U, Mayr D, Schiessl B, Mylonas I, Schulze S, Kuhn C, Friese K, Walzel H. Expression of galectin-1, -3 (gal-1, gal-3) and the Thomsen-Friedenreich (TF) antigen in normal, IUGR, preeclamptic and HELLP placentas. Placenta. 2007;28:1165–1173. doi: 10.1016/j.placenta.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 62.Hu R, Jin H, Zhou S, Yang P, Li X. Proteomic analysis of hypoxia-induced responses in the syncytialization of human placental cell line BeWo. Placenta. 2007;28:399–407. doi: 10.1016/j.placenta.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 63.Krześlak A, Lipińska A. Galectin-3 as a multifunctional protein. Cell Mol Biol Lett. 2004;9:305–328. [PubMed] [Google Scholar]
- 64.Iwashima Y, Eto M, Hata A, Kaku K, Horiuchi S, Ushikubi F, Sano H. Advanced glycation end products-induced gene expression of scavenger receptors in cultured human monocyte-derived macrophages. Biochem Biophys Res Commun. 2000;277:368–380. doi: 10.1006/bbrc.2000.3685. [DOI] [PubMed] [Google Scholar]
- 65.Wadsack C, Hammer A, Levak-Frank S, Desoye G, Kozarsky KF, Hirschmugl B, Sattler W, Malle E. Selective cholesteryl ester uptake from high density lipoprotein by human first trimester and term villous trophoblast cells. Placenta. 2003;24:131–143. doi: 10.1053/plac.2002.0912. [DOI] [PubMed] [Google Scholar]
- 66.Schmidt AM, Vianna M, Gerlach M, Brett J, Ryan J, Kao J, Esposito C, Hegarty H, Hurley W, Clauss M, Wangll F, Panll Y-CE, Tsang TC, Stern D. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. 1992;267:14987–14997. [PubMed] [Google Scholar]
- 67.Leslie RD, Cohen RM. Biologic variability in plasma glucose, hemoglobin A1c, and advanced glycation end products associated with diabetes complications. J Diabetes Sci Technol. 2009;3:635–643. doi: 10.1177/193229680900300403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Méndez JD, Xie J, Aguilar-Hernández M, Méndez-Valenzuela V. Molecular susceptibility to glycation and its implication in diabetes mellitus and related diseases. Mol Cell Biochem. 2010;344:185–193. doi: 10.1007/s11010-010-0541-3. [DOI] [PubMed] [Google Scholar]
- 69.Tsukahara H, Ohta N, Sato S, Hiraoka M, Shukunami K, Uchiyama M, Kawakami H, Sekine K, Mayumi M. Concentrations of pentosidine, an advanced glycation end-product, in umbilical cord blood. Free Radic Res. 2004;38:691–695. doi: 10.1080/1071576042000220256. [DOI] [PubMed] [Google Scholar]
- 70.Zhang S, Zhong J, Yang P, Gong F, Wang CY. HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. Int J Clin Exp Pathol. 2009;3:24–38. [PMC free article] [PubMed] [Google Scholar]
- 71.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 72.Holmlund U, Wähämaa H, Bachmayer N, Bremme K, Sverremark-Ekström E, Palmblad K. The novel inflammatory cytokine high mobility group box protein 1 (HMGB1) is expressed by human term placenta. Immunology. 2007;122:430–437. doi: 10.1111/j.1365-2567.2007.02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tskitishvili E, Sharentuya N, Temma-Asano K, Mimura K, Kinugasa-Taniguchi Y, Kanagawa T, Fukuda H, Kimura T, Tomimatsu T, Shimoya K. Oxidative stress-induced S100B protein from placenta and amnion affects soluble Endoglin release from endothelial cells. Mol Hum Reprod. 2010;16:188–199. doi: 10.1093/molehr/gap104. [DOI] [PubMed] [Google Scholar]
- 74.Anan I, Kiuru-Enari S, Obayashi K, Ranløv PJ, Ando Y. Investigation of AGE, their receptor and NF-kappaB activation and apoptosis in patients with ATTR and Gelsolin amyloidosis. Histol Histopathol. 2010;25:691–699. doi: 10.14670/HH-25.691. [DOI] [PubMed] [Google Scholar]
- 75.Goyal R, Yellon SM, Longo LD, Mata-Greenwood E. Placental gene expression in a rat ‘model’ of placental insufficiency. Placenta. 2010;31:568–575. doi: 10.1016/j.placenta.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 76.Herse F, Dechend R, Harsem NK, Wallukat G, Janke J, Qadri F, Hering L, Muller DN, Luft FC, Staff AC. Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsia. Hypertension. 2007;49:604–611. doi: 10.1161/01.HYP.0000257797.49289.71. [DOI] [PubMed] [Google Scholar]
- 77.Lee GS, Joe YS, Kim SJ, Shin JC. Cytokine-related genes and oxidation-related genes detected in preeclamptic placentas. Arch Gynecol Obstet. 2010;282:363–369. doi: 10.1007/s00404-009-1222-x. [DOI] [PubMed] [Google Scholar]
- 78.Sitras V, Paulssen RH, Grønaas H, Leirvik J, Hanssen TA, Vårtun A, Acharya G. Differential placental gene expression in severe preeclampsia. Placenta. 2009;30:424–433. doi: 10.1016/j.placenta.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 79.Farina A, Morano D, Arcelli D, De Sanctis P, Sekizawa A, Purwosunu Y, Zucchini C, Simonazzi G, Okai T, Rizzo N. Gene expression in chorionic villous samples at 11 weeks of gestation in women who develop preeclampsia later in pregnancy: implications for screening. Prenat Diagn. 2009;29:1038–1044. doi: 10.1002/pd.2344. [DOI] [PubMed] [Google Scholar]
- 80.Farina A, Sekizawa A, Purwosunu Y, Rizzo N, Banzola I, Concu M, Morano D, Giommi F, Bevini M, Mabrook M, Carinci P, Okai T. Quantitative distribution of a panel of circulating mRNA in preeclampsia versus controls. Prenat Diagn. 2006;26:1115–1120. doi: 10.1002/pd.1562. [DOI] [PubMed] [Google Scholar]
- 81.Cox B, Kotlyar M, Evangelou AI, Ignatchenko V, Ignatchenko A, Whiteley K, Jurisica I, Adamson SL, Rossant J, Kislinger T. Comparative systems biology of human and mouse as a tool to guide the modeling of human placental pathology. Mol Syst Biol. 2009;5:279. doi: 10.1038/msb.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Han JY, Kim YS, Cho GJ, Roh GS, Kim HJ, Choi WJ, Paik WY, Rho GJ, Kang SS, Choi WS. Altered gene expression of caspase-10, death receptor-3 and IGFBP-3 in preeclamptic placentas. Mol Cells. 2006;22:168–174. [PubMed] [Google Scholar]
- 83.Gack S, Marmé A, Marmé F, Wrobel G, Vonderstrass B, Bastert G, Lichter P, Angel P, Schorpp-Kistner M. Preeclampsia: increased expression of soluble ADAM 12. J Mol Med. 2005;83:887–896. doi: 10.1007/s00109-005-0714-9. [DOI] [PubMed] [Google Scholar]
- 84.Lian IA, Toft JH, Olsen GD, Langaas M, Bjørge L, Eide IP, Børdahl PE, Austgulen R. Matrix metalloproteinase 1 in pre-eclampsia and fetal growth restriction: reduced gene expression in decidual tissue and protein expression in extravillous trophoblasts. Placenta. 2010;31:615–620. doi: 10.1016/j.placenta.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 85.Sun CJ, Zhang L, Zhang WY. Gene expression profiling of maternal blood in early onset severe preeclampsia: identification of novel biomarkers. J Perinat Med. 2009;37:609–616. doi: 10.1515/JPM.2009.103. [DOI] [PubMed] [Google Scholar]
- 86.Vascotto C, Salzano AM, D’Ambrosio C, Fruscalzo A, Marchesoni D, di Loreto C, Scaloni A, Tell G, Quadrifoglio F. Oxidized transthyretin in amniotic fluid as an early marker of preeclampsia. J Proteome Res. 2007;6:160–170. doi: 10.1021/pr060315z. [DOI] [PubMed] [Google Scholar]