

Abstract
The fusion glycoproteins of human respiratory syncytial virus (RSV) and human parainfluenza virus type-3 (PIV-3) mediate virus entry and syncytium formation. Interaction between the fusion protein of RSV and RhoA, a small GTPase, facilitates virus-induced syncytium formation. We show here a RhoA-derived peptide inhibits RSV and syncytium formation induced by RSV and PIV-3, both in vitro by inhibition of cell-to-cell fusion and in vivo by reduction of peak titer by 2 log10 in RSV-infected mice. These findings indicate that the interaction between these two paramyxovirus fusion proteins and RhoA is an important target for new antiviral strategies.
Main
Human respiratory syncytial virus (RSV) and parainfluenza virus type-3 (PIV-3) belong to the Paramyxoviridae family. RSV and PIV-3 are the main viral causes of acute lower respiratory tract illness in infants1,2. There is no effective antiviral therapy or vaccine available for either virus.
RSV and PIV-3 each have two main surface envelope glycoproteins: an attachment protein (RSV G or PIV-3 hemagglutinin–neuraminidase) and a fusion protein (F). The G or hemagglutinin–neuraminidase glycoproteins are thought to mediate virus attachment to the cell receptor3,4. The receptor for PIV-3 is sialic acid4, and RSV G may bind to heparan sulfate5,6. The F glycoprotein mediates at least two essential steps in the virus life cycle that require membrane fusion: it promotes fusion of the viral and cellular membranes with subsequent transfer of viral genome material into the cell, and promotes fusion of the infected cell membrane with those of adjacent cell membranes, leading to syncytia formation. The F glycoproteins of RSV and PIV-3 are synthesized as inactive precursors, which are co-translationally modified by the addition of N-linked glycosylation in the endoplasmic reticulum. The F precursor is cleaved by cellular trypsin-like endoproteases into two disulfide-linked subunits, F1 and F2, before reaching the cell surface, and is assembled as a higher-order homooligomer7,8. RSV can infect cells in vitro despite lacking the G glycoprotein9, indicating that F may have additional functions of attachment to a host co-receptor.
The process of membrane fusion is essential for the entry of both cell-free and cell-associated virus. RSV and PIV-3 share the properties of syncytium formation and pH-independent fusion from without. Common host determinants may be involved in the essential processes required for viral entry and syncytium formation. The fusion glycoproteins of these viruses share elements of similar function, structure and, in some cases, sequence, indicating a common membrane fusion mechanism10. In particular, paramyxovirus F proteins share conserved hydrophobic sequences at their amino termini, important for interactions with the lipid bilayer, followed by a region of heptad repeats10.
Although much is known about the properties of membrane fusion in selected cellular compartments or in artificial lipid membranes, the molecular mechanisms for both virus-induced and cellular fusion reactions remain mostly undefined11. Cell-to-cell fusion mediated by some viral envelope proteins involves cellular actin cytoskeleton and cell surface integrins12,13. Host cellular proteins that maintain cell membrane integrity and cell mobility, such as RhoA, might be expected to be involved in virus-induced fusion and syncytium formation; however, there is no direct evidence at present for their involvement.
RhoA binds to RSV F protein and mediates virus-induced syncytium formation14. In addition, RhoA amino acids 67–110 bind to RSV F amino acids 146–155. RhoA, a small GTPase of the Ras superfamily, controls a plethora of biological functions including actin reorganization, gene expression, cell morphology, cell motility and cell proliferation15. We report here a RhoA peptide derived from the domain that binds the RSV F protein has potent antiviral activity against RSV and PIV-3 infection in cell culture and in RSV-infected mice. These data provide additional evidence for conserved structural and functional features between the fusion proteins of RSV and PIV-3 and for the possibility of common mechanisms involving virus-induced membrane fusion. The inhibitory effect of this RhoA77-95 peptide may be useful for the development of new antiviral strategies with broad application.
Effects of RhoA peptides on RSV-induced syncytium
We synthesized three overlapping 19-amino-acid peptides spanning the F binding domain of RhoA (amino acids 67–110) and used these to study the effect of peptides on virus-induced syncytium formation. Pretreatment of virus inoculum with the RhoA77-95 peptide completely blocked plaque formation, whereas pretreatment with RhoA87-105 did not have any effect on the plaque formation relative to media-treated controls (Fig. 1). The RhoA67-85 peptide did not block RSV-induced syncytium formation. Varying the concentration of RhoA77-95 showed that the concentration required to inhibit number of plaques by 50% (IC50) was 0.54 μg/ml, or 0.25 μM. Immunoperoxidase staining did not show any RSV antigen-positive cells treated with RhoA77-95, indicating that RSV replication was inhibited at a step before viral protein synthesis. These data indicate that the RhoA77-95 peptide inhibits RSV at an early step of the replication cycle.
Figure 1. RhoA-derived peptide inhibits RSV infection and syncytium formation.

Addition of RhoA77-95 (left) but not RhoA87-105 (right) to RSV stock18 prevents plaque formation in HEp-2 cells.
We next determined whether the RhoA77-95 peptide could inhibit cell-to-cell spread of RSV after infection. We used a recombinant RSV (rgRSV) expressing a gene for green fluorescent protein, located at the first position in RSV gene order. We added 5 μg/ml RhoA77-95 to cells 0, 4 and 24 hours after RSV adsorption, and evaluated HEp-2 monolayers by fluorescent microscopy 48 hours after infection (Fig. 2). There was inhibition of cell-to-cell spread and syncytia formation in rgRSV-infected cells treated with the RhoA77-95 peptide compared with that of untreated, infected cells. The number of infected cells increased between 4 and 24 hours after infection, indicating the completion of one round of viral replication, production of new virions and infection of new cells before 24 hours. Therefore, the addition of RhoA77-95 seems to prevent subsequent spread of cell-free virus to other cells as well as preventing syncytium formation. These data also support the possibility of a block at an early step in the viral life cycle.
Figure 2. Effect of RhoA77-95 added after rgRSV infection.

Syncytium formation was assessed 48 h after infection. a and b, Peptide added 0 h after infection. b, Phase contrast image of a. c and d, Peptide added 4 h after infection. d, Phase contrast image of c. e, Peptide added 24 h after infection. f, No peptide added.
Next, we tested whether RhoA77-95 peptide inhibition of RSV was reversible. We dialyzed RSV–RhoA77-95 peptide, RSV–RhoA87-105 peptide and RSV–media suspensions overnight and added these to HEp-2 cell monolayers. There were no RSV plaques in wells treated with dialyzed RSV–RhoA77-95 peptide suspension, in contrast to the presence of many plaques in wells treated with dialyzed RSV–RhoA87-105 peptide or RSV–media suspensions (data not shown). This indicates that the RhoA77-95 peptide binds with high avidity to RSV F.
Cell-to-cell fusion assay
To determine whether the RhoA77-95 and RhoA87-105 peptides have biological effects in cell-to-cell fusion induced by RSV F, we used an assay based on the cytoplasmic activation of the reporter gene β-galactosidase. The RSV envelope proteins F, G and SH have been shown in this assay to optimize cell-to-cell fusion16,17. We did this assay as described16 with some modifications. One cell population was infected with the recombinant vaccinia virus vTF7-3 and transfected with three separate plasmids containing RSV F, G and SH genes. The other cell population was infected with a recombinant vaccinia virus expressing β-galactosidase. The two cell populations were mixed and either treated with 5 μg/ml peptides or left untreated. Cell fusion was measured by an in situ assay using X-gal staining of cells (Fig. 3a–c) or a quantitative colorimetric lysate assay for β-galactosidase (Fig. 3d). Wells treated with the RhoA77-95 peptide showed no blue fused cells (Fig. 3b), in contrast to wells treated with the RhoA87-105 peptide (Fig. 3c) or untreated wells (Fig. 3 a). The in situ assay correlated with the quantitative colorimetric assay for the treated and untreated wells (Fig. 3d). Control wells containing one population of cells expressing G and SH and another population of cells infected with vaccinia virus expressing β-galactosidase did not show β-galactosidase activity (data not shown). These data show that the cell-to-cell fusion induced by RSV F was blocked by RhoA77-95 peptide, and indicate that the cell-to-cell spread of virus through syncytium formation may require the binding of F to RhoA.
Figure 3. Effect of RhoA-derived peptides on cell-to-cell fusion.
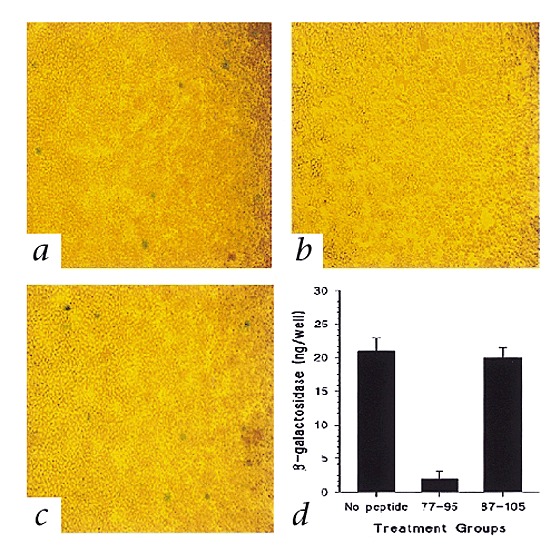
HEp-2 cells expressing RSV F, G, and SH glycoproteins were untreated ( a) or treated with RhoA77-95 (b) or RhoA87-105 (c), then mixed with another cell population infected with recombinant vaccinia virus expressing β-galactosidase, then assessed by in situ X-gal staining (a, b and c) or quantitative colorimetric lysate assay (d). d, Equal volumes of cell lysates and 2× substrate solution were mixed and the rates of substrate hydrolysis was monitored by measuring absorbance at 590 nm with a spectrophotometer. The data are presented as β-galactosidase (ng/well) produced by the cells. Each point represents an average of three experiments ± standard deviation.
RhoA 77-95 peptide inhibits RhoA–RSV F interaction
To determine whether the RhoA77-95 peptide can interfere with the interaction between F and RhoA, we did a competitive enzyme-linked immunosorbent assay (ELISA) (Fig. 4). We coated wells with 25 ng purified F, and after blocking, added 50 ng RhoA and increasing concentrations of peptide (range, 100 ng/ml to 500 μg/ml), then detected bound RhoA with monoclonal antibody against RhoA. We used RhoA without the peptide as a positive control. As a negative control, we used Rac1 protein and RhoA87-105 instead of RhoA and RhoA77-95, respectively. There was a correlation between concentration of RhoA77-95 peptide and inhibition of RhoA interaction with F. The results for all concentrations of RhoA87-105 were similar to data from the untreated positive control, whereas the results for Rac1 binding to F were similar to background values. These data indicate that the biologic effects demonstrated with the peptide in cell culture and in vivo are based on its inhibition of the interaction between RhoA and the viral fusion protein.
Figure 4. RhoA77-95 peptide competitively inhibits RhoA interaction with RSV F in an ELISA.

Wells were coated with purified F protein and blocked, and RhoA was added in the presence of RhoA77-95 (▪) or RhoA87-105 (▴) peptide (horizontal axis, increasing concentrations). Bound RhoA was detected by monoclonal antibodies against RhoA.
Next, we determined whether the binding of RhoA77-95 peptide to F was reversible or nonreversible in the ELISA. We added RhoA protein 1 hour after allowing RhoA77-95 peptide to bind F protein, incubated this for 1 hour, and detected RhoA using monoclonal antibodies against RhoA. There was no binding of RhoA to F (data not shown). Thus, the RhoA77-95 peptide binds F with high avidity.
RhoA 77-95 peptide can block infection in an animal model for RSV
To determine whether the RhoA peptide would alter the course of infection in vivo, we infected BALB/c mice intranasally with 0.1 ml containing 107 plaque-forming units (PFU) live RSV, using a well-established model18. We administered 500 μg RhoA77-95 peptide, RhoA87-105 peptide or PBS intranasally immediately before or 2 hours or 4 days after RSV infection. Mice treated with the RhoA77-95 peptide immediately before or 2 hours after virus infection had no discernable illness or weight loss, whereas PBS-treated control mice experienced a typical illness pattern (Fig. 5a) with a 22% peak weight loss from baseline. Mice treated with the peptide 4 days after virus infection had illness or weight loss similar to that of mice treated with PBS. Plaque assays of lungs on day 4 after RSV challenge showed that peptide treatment immediately before or 2 hours after virus infection reduced RSV titers by 2 log10 compared to the RSV titers in lungs from control mice treated with PBS or the RhoA87-105 peptide (Fig. 5b). These data indicate that the diminished illness in mice treated with the RhoA77-95 peptide before RSV infection could be due to inhibition of virus replication by the peptide.
Figure 5. RhoA77-95 peptide treatment reduces illness and viral titers in RSV-infected mice.

a, Mean clinical illness scores18 in RSV-infected mice treated with peptide (○) or PBS (▪) on day 0 immediately before RSV infection. Error bars represent standard deviations. b, RSV titers in lung on day 4 after challenge. Mice were treated with PBS (left) or the RhoA77-95 peptide (right) immediately before RSV infection on day 0. Data represent geometric means with standard deviations (error bars). These are the combined results of two independent experiments with a total of 10 mice in each group. pfu, plaque-forming unit.
RhoA 77-95 peptide can inhibit infection by PIV-3
As the amino terminus of the RSV fusion glycoprotein is similar in structure and hydrophobicity to that of other enveloped viruses10, we next determined whether the RhoA77-95 peptide could block infection with PIV-3, influenza A virus (H3N2 A/Beijing/1996), herpes simplex virus type I, vaccinia virus or a coronavirus (mouse hepatitis virus strain A59). Syncytium formation was inhibited in PIV-3-infected HEp-2 cells (Fig. 6) by the RhoA77-95 peptide. However, infection with influenza virus, herpes simplex virus type I, vaccinia virus, or coronavirus was not inhibited by the RhoA77-95 peptide (data not shown). Treatment of PIV-3 with the RhoA87-105 peptide had no effect on syncytium formation (Fig. 6). These data indicate that a common mechanism of virus-induced syncytium formation involving RhoA may be shared between these two paramyxoviruses. They also indicate that although a common motif is present at the amino terminus in the fusion proteins of many enveloped viruses, host cell components involved in syncytium formation may differ between virus families.
Figure 6. Inhibition of PIV-3 infection by RhoA77-95 peptide.

a and b, Peptides RhoA77-95 (a) or RhoA87-105 (b) were incubated with PIV-3, then the suspension was added to HEp-2 cells. c, PIV-3-infected HEp-2 cells.
Discussion
The hallmark of the RSV and PIV-3 cytopathic effect in cell culture is extensive syncytium formation. The fusion glycoprotein (F) mediates virus-induced fusion at neutral pH, and for PIV-3, membrane fusion depends on the density of the fusion protein on the infected cell surface19. Cellular proteins contributing to paramyxovirus-induced syncytia formation have not been identified before. However, the lipid composition and other properties of the target cell membrane can influence the ability of a virus to produce syncytia20,21. A host protein, RhoA, has been shown to bind to viral fusion glycoprotein and facilitates syncytium formation14. We have shown here the inhibition of RSV- and PIV-3-induced syncytium formation by a RhoA-derived peptide (RhoA77-95) from the RSV F binding domain. This inhibition indicates that two viruses from the Paramyxoviridae family may use the same host cell protein and same binding site on this protein in the process of inducing cell-to-cell fusion, strengthening the concept of conserved functional and structural features of paramyxovirus fusion proteins. The role of RhoA in virus-induced fusion is not yet defined and might include a direct effect, or an indirect effect through signaling events on cellular cytoskeletal organization, cell shape or cell motility. The F protein interaction with RhoA might also be involved in other aspects of virus replication including assembly, filament formation or budding.
Peptides derived from the heptad repeat region of viral fusion protein22,23,24,25 inhibit syncytium formation by homologous virus by binding to the heptad repeats interfering with fusion protein structure. The inhibitory peptide described here is of a different nature. We have shown here that a peptide derived from a cellular protein that does not have sequence homology to the viral fusion proteins can specifically inhibit virus-induced syncytium formation and possibly virus entry. In a yeast two-hybrid assay, RhoA was shown to not bind a RSV F construct expressing the heptad repeat region of the fusion proteins (FN155 construct encoding F residues 155–550)(ref. 14), indicating that the mechanism of fusion inhibition by the RhoA-derived peptide is different from that for peptides interfering with the heptad repeat interactions.
The complete inhibition of RSV infection when the RhoA77-95 peptide was added before RSV adsorption to cells (Fig. 1) or immediately after rgRSV infection (Fig. 2a) indicates that the peptide interferes with virus-mediated fusion events. Immunohistochemical staining for RSV F (Fig. 1) showed no expression of RSV F in HEp-2 cells. Analysis of the virus growth curve indicated that there was no virus yield (data not shown). Peptide inhibition of green fluorescent protein expression in rgRSV-infected cells indicated that the RhoA77-95 peptide interfered at an early step in the virus replication pathway, possibly at entry. The same could be true with PIV-3 infection ( Fig.6). The inhibitory effect of RhoA77-95 peptide was specific for the amino-acid sequence of the synthetic peptide, as a RhoA77-95 peptide analog with the same amino acid content but random sequence did not inhibit RSV-induced syncytium formation (data not shown). This also indicates that the inhibitory effect of the RhoA77-95 peptide is not based on hydrophobicity or charge.
The role of RhoA in cell-to-cell fusion is supported by the syncytium-inhibiting effect of RhoA77-95 peptide in a RSV-free cell-to-cell fusion assay (Fig. 3). Membrane fusion involves the mixing of both membrane lipid layers, resulting in the mixing of the aqueous contents of donor and recipient cells. In our fusion assay, quantitation of the reporter gene (β-galactosidase) activation demonstrates the content mixing potential of the RSV F protein in the presence or absence of peptides. Cell-to-cell fusion was not inhibited by RhoA87-105 peptide, but RhoA77-95 peptide inhibited content mixing by preventing cell-to-cell fusion. This result is consistent with the data from rgRSV-infected HEp-2 cells in the presence of RhoA peptides (Fig. 2), in which the RhoA77-95 peptide inhibited not only virus–cell fusion but also virus-induced cell-to-cell fusion.
The data from competitive ELISA strongly indicate that the biological effects of the RhoA77-95 peptide are based on interference with the interaction between RhoA and the viral fusion proteins. The high avidity binding of the RhoA77-95 peptide to F supports prior analysis of the RSV F–RhoA interaction by biomolecular interaction analysis based on surface plasmon resonance. This observation was also supported by data obtained from the dialysis experiment, in which the RhoA77-95 peptide irreversibly prevented RSV-induced syncytium formation despite overnight dialysis.
Blockade of RSV and PIV-3 syncytium formation by the same peptide, RhoA77-95 (Figs. 1 and 6, respectively), indicates that the mechanism of virus-induced membrane fusion is similar in both viruses. This provides additional evidence for conserved structural and functional features between the fusion proteins of RSV and PIV-3 indicated by sequence analysis10, and raises the possibility of designing a single antiviral agent against both these paramyxovirus-induced diseases. For example, a RhoA-derived peptide (RhoA77-95), a related peptidomimetic compound or a small molecule capable of interfering with the RhoA-fusion protein interaction could potentially have a clinical effect in RSV treatment similar to that of T-20, a peptide derived from the C-terminal heptad repeat of gp41, in HIV-1 treatment26. The inhibitory effect of RhoA77-95 in vivo on virus replication and potential for diminishing illness in the mouse model of RSV (Fig. 5) indicate that early treatment with a RhoA-derived peptide has therapeutic potential in RSV or PIV-3 infections, particularly in giant cell pneumonia, seen in patients with bone marrow transplantation, lung transplantation or severe combined immunodeficiency. Although the peptide treatment immediately before or 2 hours after RSV infection prevented illness in mice, treatment on day 4 after RSV infection had no effect on the illness (data not shown). This is consistent with other antiviral approaches and reflects the fact that when illness is mediated by the T-cell response and is not direct virus-induced cytopathology, anti-viral therapy must be given early or combined with immunomodulators.
In conclusion, a RhoA-derived peptide (RhoA77-95) was shown here to inhibit RSV and PIV-3 infection and syncytium formation. RhoA77-95 also blocked cell-to-cell-fusion in a virus fusion glycoprotein (F)-induced cell-to-cell fusion assay. Given immediately before infection or 2 hours after RSV infection, the RhoA77-95 peptide reduced RSV titer and prevented illness in mice. Exploiting the interaction between RhoA and F protein may also be useful for designing peptidomimetic or low-molecular-weight antiviral drugs.
Methods
Virus and cells.
The A2 strain of RSV was provided by R. Chanock (National Institutes of Health, Bethesda, Maryland). The rgRSV was provided by M. Peeples (Rush Medical College, Chicago, Illinois) and P. Collins (National Institutes of Health, Bethesda, Maryland). Wild-type parainfluenza virus type 3 (PIV-3), influenza A virus (H3N2 A/Beijing/1996), herpes simplex virus type I and the coronavirus, mouse hepatitis virus strain A59 strains were provided by B. Murphy, P. Wright, P. Spearman and M. Denison, respectively. RSV stocks were prepared as described18. HEp-2 cells were maintained in Eagle's minimal essential media (EMEM) supplemented with glutamine, gentamicin, penicillin G and 10% fetal bovine serum (FBS).
RhoA-derived peptides.
The peptides were synthesized by Research Genetics (Huntsville, Alabama) as the free acid, at approximately 70% purity in the desalted, lyophilized form. The major species in each preparation had the calculated molecular weight of the desired peptide, based on mass spectrometry. The amino-acid sequence of the peptides are: RhoA67-85, DRLRPLSYPDTDVILMCFS; RhoA77-95, TDVILMCFSIDSPDSLENI; RhoA87-105, DSPDSLENIPEKWTPEVKH; and RhoA77-95 Scrambled, DDMSVISELICTSPLDFIN. All the peptides were resuspended in deionized distilled water.
Blockade of RSV infection in cell culture with peptides.
For the assay, the peptides were incubated for 1 h on ice with 1×103 PFU/ml RSV at peptide concentrations of 5 and 10 μg/ml of media, then 100 μl of the suspension was added to HEp-2 cells in 96-well plates. For defining the IC50, concentrations of peptide ranging from 0.1 μg/ml to 5 μg/ml were used. After 3 d, plates were fixed with methanol and RSV-specific immunoperoxidase staining was done27. RSV plaques were counted in each well.
For experiments using rgRSV, HEp-2 cells in 12 well plates were infected with 100 μl rgRSV stock virus. The RhoA77-95 peptide (5 μg/ml) was added at 0, 4 and 24 h after infection. At 48 h after infection, infected cells were viewed by fluorescent microscopy. Cells infected with rgRSV without the peptide were used as a control.
For testing whether the peptide binding to RSV was reversible, 1 ml media containing 1×108 PFU/ml RSV and 100 μg/ml peptide (RhoA77-95 or RhoA87-105) suspension was incubated on ice for 1 h. RSV without the peptide was used as a control. A cellulose ester membrane (Spectrum Medical Industries, Houston, Texas) with a 10,000-dalton molecular weight cutoff was used to dialyze RSV–peptide suspension in PBS at 4°C overnight. After overnight dialysis, the suspension was added to HEp-2 cells in a 12-well plate. After 2 d, plates were fixed and stained as described above. RSV plaques were counted in each well.
Cell fusion assay using vaccinia virus-based expression of RSV envelope glycoproteins.
The ability of the RhoA77-95 peptide to inhibit RSV F-induced cell-to-cell fusion was assessed using a quantitative assay based on the cytoplasmic activation of reporter gene β-galactosidase16. One population of HEp-2 cells was infected with recombinant vaccinia virus vTF7-3, which encodes T7 polymerase, at a multiplicity of infection of 10 PFU per cell, and was transfected with plasmids encoding RSV glycoproteins F, G and SH under control of the T7 promoter (gifts from P. Collins, National Institutes of Health, Bethesda, Maryland) using LipofectAMINE (Life Technologies). At 5 h after transfection, the cells expressing viral envelope proteins were trypsinized, suspended in MEM containing 2.5% FBS to a density of 2×107 cells per ml, and incubated overnight at 32°C. The cells were then washed and suspended in Opti-MEM (Life Technologies) at a concentration of 1×106 cells per ml. A second population of HEp-2 cells was infected with recombinant vaccinia virus expressing β-galactosidase under control of the T7 promoter (provided by E.A. Berger, National Institutes of Health, Bethesda, Maryland). At 5 h after infection, cells were trypsinized and finally suspended at a concentration of 1×106 cells per ml. The cell population expressing the viral glycoproteins was treated with 5 μg/ml RhoA77-95 or RhoA87-105 peptide or left untreated, and incubated for 30 min at 37°C. The two cell populations were mixed in triplicate by adding 100 μl of each cell population to 96-well tissue culture plates, which were then incubated at 37 °C for 4 h. Cell fusion was measured by the quantitative colorimetric lysate assay for β-galactosidase or an in situ assay using X-Gal (5-bromo-4-chloro-3-indolyl β-D-galactopyranoside; Life Technologies) staining of cells. In the colorimetric lysate assay, 5 μl 20% (volume/volume) Nonidet-P40 was added to each well, and the contents were mixed by pipetting. β-galactosidase activity was quantified at ambient temperature in 96-well flat-bottomed plates by mixing 50 μl of each lysate with 50 μl 2X substrate solution (16 mM CPRG (chlorophenol-red-β-D-galactopyranoside; Boehringer), 0.12 M Na2HPO4.7H2O, 0.08 M NaH2PO4.H2O, 0.02 M KCl, 0.002 M MgSO4.7H2O and 0.01 M β-mercaptoethanol). The rate of substrate hydrolysis at ambient temperature was monitored by measuring the absorbance at 590 nm with a spectrophotometer. The quantity of β-galactosidase was calculated by comparing the hydrolysis rates for each sample with that obtained for a standard commercial preparation of Escherichia coli β-galactosidase (600 U/mg; Boehringer). β-galactosidase levels were expressed as nanograms per well. For the in situ assay, 20 μl 10X fixative solution (20% formaldehyde and 2% glutaraldehyde in PBS) was added to each well. The plates were incubated at 4 °C for 5 min. Without disturbing the settled cells, 0.15 ml medium was gently removed and replaced with 0.15 ml of 37 °C-equilibrated staining solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM magnesium chloride and 1 mg/ml X-Gal, freshly diluted from a 40 mg/ml stock solution in dimethyl formamide). The plates were incubated overnight at 37 °C to allow complete staining. Blue-stained syncytia were viewed with an inverted phase-contrast microscope.
Competitive ELISA.
Immunoaffinity-purified RSV F protein (a gift from Wyeth–Lederle–Praxis Biologicals, West Henrietta, New York) was diluted to a concentration of 250 ng/ml in carbonate/bicarbonate buffer, pH 9.6. A suspension (100 μl) containing 25 ng F protein was applied to wells of Immulon II 96-well plates (Nunc, Roskilde, Denmark). Blocking was accomplished overnight with 3% nonfat dry milk and 3% BSA. Blocking buffer (100 μl) containing 50 ng RhoA or Rac1, another Rho family GTPase (CalBiochem, La Jolla, California), and increasing concentrations of either RhoA77-95 or RhoA87-105 peptide (range, 100 ng/ml to 500 μg/ml) was added separately and incubated overnight at 4 °C. RhoA added without the peptide was used as a positive control. As a negative control, Rac1 protein and RhoA87-105 were used instead of RhoA and RhoA77-95, respectively. Monoclonal antibodies against RhoA or Rac1 (1:4,000 dilution; Santa Cruz Biotech, Santa Cruz, California) in blocking buffer were added after wells were washed with PBS and 0.1% Tween 20. After 1 h, plates were washed and goat antibody against mouse IgG conjugated to horseradish peroxidase (1:7,000 dilution) was added. After washing, the substrate 3,3′,5,5′-tetramethylbenzidine (Sigma) was added and the absorbance was measured at 450 nm using a 'microtiter plate reader' (Dynatech, Chantilly, Virginia).
Treatment of RhoA77-95 peptide in RSV-infected mice.
Pathogen-free, 8-week-old BALB/c mice were obtained from Charles Rivers Laboratories (Raleigh, North Carolina) and were housed in a barrier facility. The mice were anesthetized and intranasally infected with 0.1 ml containing 1×107 PFU live RSV, as described18. Immediately before or 2 h or 4 d after RSV infection, 500 μg RhoA77-95 peptide or PBS were given intranasally. Lungs were obtained from five mice from each group on day 4 after infection from RSV-infected mice treated with peptide or PBS immediately before or 2 h after RSV infection. Five RSV-infected mice from each group were weighed for 12 d after infection. Illness was graded daily by an observer 'blinded' to treatment status of mice; clinical features of illness were scored as: 0, no apparent illness; 1, slightly ruffled fur; 2, ruffled fur, but active; 3, ruffled fur and inactive; 4, ruffled, inactive, hunched posture and gaunt; 5, dead.
RSV plaque assay from lung tissue.
Four days after RSV infection, mice were killed by CO2 narcosis and cervical dislocation. The lungs were removed, placed in EMEM with 10% FBS, and were quickly frozen in a bath of alcohol–dry ice. RSV titers in the lungs were measured by standard plaque assays18 using HEp-2 monolayers that were 80% confluent. Lungs were thawed quickly and ground with a mortar and pestle. Serial 10-fold dilutions of lung supernatants were used to infect the monolayers in triplicate, and cultures were grown under 0.75% methylcellulose in EMEM with 10% FBS. Cells were formalin-fixed 5 d after being infected, and were stained with hematoxylin and eosin; plaques were counted using a dissecting microscope. Data are presented as the geometric mean log10 PFU per gram of lung tissue ± standard deviation (s.d.) at the dilution producing more than five plaques per well.
Blockade of PIV-3 infection in cell culture with peptides.
Peptides RhoA77-95 or RhoA87-105 at concentrations of 50 μg/ml were incubated with 100 μl PIV-3 stock for 1 h on ice, then the suspension was added to HEp-2 cells in 12-well plates. PIV-3 plaques were viewed by phase contrast microscopy.
Acknowledgements
Rauf Kuli-Zade assisted with the animal experiments. M. Denison, V. Varthakavi and P. Wright did neutralization assays on coronavirus, herpes simplex virus type I and influenza virus, respectively. J. Exton, S. Aung and T. Johnson contributed through editorial comments and discussions. Fluorescent microscopy analyses were done in part through use of the Vanderbilt University Medical Center Cell Imaging Resource (supported by CA68485 and DK20593). This work was supported by National Institutes of Health grant RO1-AI-33933.
References
- 1.Collins PL, McIntosh K, Chanock RM, et al. Fields Virology. 1996. pp. 1313–1351. [Google Scholar]
- 2.Welliver R, Wong DT, Choi TS, Ogra PL. Natural history of parainfluenza virus infection in childhood. J. Pediatr. 1982; 101:180–187. doi: 10.1016/S0022-3476(82)80113-3. [DOI] [PubMed] [Google Scholar]
- 3.Levine S, Klaiber-Franco R, Paradiso PR. Demonstration that glycoprotein G is the attachment protein of respiratory syncytial virus. J. Gen. Virol. 1987; 68:2521–2524. doi: 10.1099/0022-1317-68-9-2521. [DOI] [PubMed] [Google Scholar]
- 4.Scheid A, Caliguiri LA, Compans RW, Choppin PW. Isolation of paramyxovirus glycoproteins. Association of both hemagglutinating and neuraminidase activities with the larger SV5 glycoprotein. Virology. 1972;50:640–652. doi: 10.1016/0042-6822(72)90418-7. [DOI] [PubMed] [Google Scholar]
- 5.Krusat T, Streckert HJ. Heparin-dependent attachment of respiratory syncytial virus (RSV) to host cells. Arch. Virol. . 1997;142:1247–1254. doi: 10.1007/s007050050156. [DOI] [PubMed] [Google Scholar]
- 6.Feldman SA, Hendry RM, Beeler JA. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J. Virol. 1999;73:6610–6617. doi: 10.1128/jvi.73.8.6610-6617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins PL, Mottet G. Post-translational processing and oligomerization of the fusion glycoprotein of human respiratory syncytial virus. J. Gen. Virol. 1991;72:3095– 3101. doi: 10.1099/0022-1317-72-12-3095. [DOI] [PubMed] [Google Scholar]
- 8.Russell R, Paterson RG, Lamb RA. Studies with cross-linking reagents on the oligomeric form of the paramyxovirus fusion protein. Virology. 1994; 199:160–168. doi: 10.1006/viro.1994.1108. [DOI] [PubMed] [Google Scholar]
- 9.Karron RA. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc. Natl. Acad. Sci. USA. 1997;94:13961– 13966. doi: 10.1073/pnas.94.25.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chambers P, Pringle CR, Easton AJ. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J. Gen. Virol. 1990;71:3075–3080. doi: 10.1099/0022-1317-71-12-3075. [DOI] [PubMed] [Google Scholar]
- 11.Hernandez LD, Hoffman LR, Wolfsberg TG, White JM. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 1996; 12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- 12.Sylwester A. HIV-induced syncytia of a T cell line form single giant pseudopods and are motile. J. Cell Sci. 1993;106:941– 953. doi: 10.1242/jcs.106.3.941. [DOI] [PubMed] [Google Scholar]
- 13.Yang C, Compans RW. Analysis of the murine leukemia virus R peptide: delineation of the molecular determinants which are important for its fusion inhibition activity. J. Virol. 1997;71: 8490–8496. doi: 10.1128/jvi.71.11.8490-8496.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pastey MK, Crowe JE, Graham BS. RhoA interacts with the fusion glycoprotein of respiratory syncytial virus and facilitates virus-induced syncytium formation. J. Virol. 1999;73:7262–7270. doi: 10.1128/jvi.73.9.7262-7270.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narumiya S. The small GTPase Rho: cellular functions and signal transduction. J. Biochem . J. Biol. Chem. 1996;120:215– 228. doi: 10.1093/oxfordjournals.jbchem.a021401. [DOI] [PubMed] [Google Scholar]
- 16.Pastey MK, Samal SK. Analysis of bovine respiratory syncytial virus envelope glycoproteins in cell fusion. J. Gen. Virol. 1997; 78:1885–1889. doi: 10.1099/0022-1317-78-8-1885. [DOI] [PubMed] [Google Scholar]
- 17.Heminway BR. Analysis of respiratory syncytial virus F, G, and SH proteins in cell fusion. Virology. 1994;200:801– 805. doi: 10.1006/viro.1994.1245. [DOI] [PubMed] [Google Scholar]
- 18.Graham BS, Perkins MD, Wright PF , Karzon DT. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 1988;26:153–162. doi: 10.1002/jmv.1890260207. [DOI] [PubMed] [Google Scholar]
- 19.Dutch RE, Joshi SB, Lamb RA . Membrane fusion promoted by increasing surface densities of the paramyxovirus F and HN proteins: comparison of fusion reactions mediated by simian virus 5 F, human parainfluenza virus type 3 F, and influenza virus HA. J. Virol. 1998;72:7745– 7753. doi: 10.1128/jvi.72.10.7745-7753.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aroeti B, Henis YI. Effects of fusion temperature on the lateral mobility of Sendai virus glycoproteins in erythrocyte membranes and on cell fusion indicate that glycoprotein mobilization is required for cell fusion. Biochemistry. 1988;27:5654– 5661. doi: 10.1021/bi00415a039. [DOI] [PubMed] [Google Scholar]
- 21.Daya M, Cervin M, Anderson R. Cholesterol enhances mouse hepatitis virus-mediated cell fusion. Virology. 1988;163:276– 283. doi: 10.1016/0042-6822(88)90267-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rapaport D, Ovadia M, Shai Y . A synthetic peptide corresponding to a conserved heptad repeat domain is a potent inhibitor of Sendai virus-cell fusion: an emerging similarity with functional domains of other viruses. EMBO J. 1995; 14:5524–5531. doi: 10.1002/j.1460-2075.1995.tb00239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA. 1994; 91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lambert DM. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA. 1996;93:2186–2191. doi: 10.1073/pnas.93.5.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao Q, Compans RW. Peptides corresponding to the heptad repeat sequence of human parainfluenza virus fusion protein are potent inhibitors of virus infection. Virology. 1996;223: 103–112. doi: 10.1006/viro.1996.0459. [DOI] [PubMed] [Google Scholar]
- 26.Kilby JM. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nature Med. 1998; 4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 27.Murphy BR. Immunization of cotton rats with the fusion (F) and large (G) glycoproteins of respiratory syncytial virus (RSV) protects against RSV challenge without potentiating RSV disease. Vaccine. 1989; 7:533–540. doi: 10.1016/0264-410X(89)90278-8. [DOI] [PubMed] [Google Scholar]