

Abstract
The successful replication of a viral pathogen in a host is a complex process involving many interactions. These interactions develop from the coevolution of pathogen and host and often lead to a species specificity of the virus that can make interspecies transmissions difficult. Nevertheless, viruses do sporadically cross species barriers into other host populations, including humans. In zoonotic infections, many of these interspecies transfer events are dead end, where transmission is confined only to the animal-to-human route but sometimes viruses adapt to enable spread from human to human. A pathogen must overcome many hurdles to replicate successfully in a foreign host. The viral pathogen must enter the host cell, replicate with the assistance of host factors, evade inhibitory host products, exit the first cell and move on to the next, and possibly leave the initial host and transmit to another. Each of these stages may require adaptive changes in the pathogen. Although the factors that influence each stage of the replication and transmission of most agents have not been resolved, the genomics of both hosts and pathogens are now at hand and we have begun to understand some of the molecular changes that enable some viruses to adapt to a new host.
Main
Emerging and reemerging infectious diseases are a continuing threat to human health and to the domestic animal population of the world. Humans continue to influence global ecology with our ever-growing demands on land use and intensified farming to feed an ever-increasing population which has developed means of transportation permitting rapid global spread of infectious diseases1. Each of these factors—and many others—has implications for the emergence of novel disease agents: severe acute respiratory syndrome (SARS) was the global threat in 2003; will H5N1 influenza be successful in 2004 or 2005?
The increasing number of lethal cases of H5N1 influenza in humans in southeast Asia, coupled with a cluster of probable human-to-human transmissions (World Health Organization, http://www.who.int/csr/don/2004_09_28a/en/), has all of the hallmarks of an incipient pandemic. The summer outbreak of H5N1 influenza in poultry has raised the level of concern from the earlier outbreaks that occurred in winter months2. The cocirculation of human influenza viruses during winter provides an opportunity for reassortment with avian influenza viruses (see below) with unpredictable outcomes. Whether H5N1 will be able to acquire potent human-to-human transmissibility is the pressing issue: exactly what are the constraints remaining to the occurrence of this transmission?
Although we do not know the answer to this or other important questions, we are beginning to understand at least some of the obstacles that pathogens must overcome in order to emerge in the human population. Here, we discuss some of these obstacles by using emerging viral agents as examples. In general terms, for a virus to emerge successfully in a human population it must achieve two feats. The first feat is replication in human cells. This replication requires a virus to accomplish at least five steps: first, contact with a human host; second, entry into the appropriate cell type; third, production of more copies of itself; fourth, overcoming any immediate host response; and fifth, exiting from the cell and transmitting to another. The second feat is human-to-human transmission.
It is obvious from the number of viruses that have achieved the first feat, but not the second, that the adaptive changes necessary for a virus to replicate in a foreign host are independent of, but necessary for, those required for successful transmission between individuals. Of greatest concern are the RNA viruses, which have developed several ways to adapt. RNA viruses such as the Nipah virus, Hendra virus, SARS coronavirus, H5N1 influenza A virus and Ebola virus have all jumped from animals to humans but have yet to achieve the next step of successful establishment. By contrast, the H3N2 and H1N1 influenza A viruses and human immunodeficiency virus (HIV) are examples of animal RNA viruses that have become endemic in humans.
Partly because molecular studies are more easily done at the cellular than at the organismal level, we know very little about the factors that allow successful human-to-human transmission of a virus. We do, however, understand some of the constraints to viral emergence at the other steps. In this Perspective, we briefly summarize some of the salient molecular features of these remaining steps with regard to viral replication in the human population. Although our main focus is the virological side of the equation, we note that emergence is a complex interaction of both host and virus.
Entry into the host cell
Viruses are obligate intracellular parasites. As such, the need to enter the host cell is a prerequisite to replication. Entry of a virus particle into a cell is initially mediated by the interaction of a virus protein with its corresponding host-cell receptors. In many virus systems, this virus-cell interaction is well understood and can be a prime determinant of host range. If the appropriate receptor is present, the virus may replicate; if it is absent, the virus will not. Recent studies on the SARS coronavirus (SARS-CoV) highlight this point. Using SARS-CoV pseudotyped HIV particles, Nie et al.3 have shown that the susceptibility of a given cell type to infection correlates well with levels of the SARS-CoV receptor molecule angiotensin-converting enzyme 2.
Different viruses use various host molecules as receptors, some of which are more globally conserved than others. Consequently, in some virus systems this interaction is very host-specific, whereas in others the virus can attach to numerous cell types of different origin. For example, arenaviruses, the causative agents of several hemorrhagic fevers, use the widely conserved α-dystroglycan protein as a receptor and therefore show a broad host range and cell tropism4. Coronaviruses, by contrast, generally show a restricted host range that is mediated by specific interaction of the viral spike protein with glycoproteins on the host cell surface5. Although this is a species-specific interaction, Thackray and Holmes6 have shown that the alteration of only a few residues in the amino-terminal region of the viral spike protein can extend the host range of a mouse coronavirus to nonmouse cells. The underlying mechanisms remain undefined, but the increase in host range seems to be mediated by the recognition of alternative receptor molecules by the spike protein.
Thackray and Holmes6 also venture that, theoretically, one or more of the ten changes between the spike proteins of the SARS-CoV and similar viruses isolated from the Himalayan palm civet and raccoon dog7 could be responsible for the outbreak of SARS-CoV infection in the human population. The increased contact between animals and humans in the live poultry markets of China8 might be the driving pressure that selected these changes. Similar examples of minor changes in viral proteins that affect receptor usage have been described for other viruses9. The infidelity of the replication machinery of many viruses—RNA viruses in particular—means that the necessary changes can be quickly accumulated on minimal passage in an alternative host.
Similar to the possible situation in coronaviruses, the emergence of influenza A viruses in a new host population seems, at least in part, to be mediated by virus-receptor interactions, although other factors are almost certainly involved. The main reservoir of influenza A viruses is the aquatic birds of the world, from which viruses sporadically transmit to other hosts in whom they can adapt and form stable lineages. Influenza viruses from avian and human sources preferentially bind different forms of the virus receptor on the host cell, namely, sialic acid. Avian influenza viruses have a preference for sialic acid that is linked to the galactose unit in a α2-3 conformation, whereas human viruses preferentially bind those with a α2-6 linkage10,11. This binding preference makes biological sense when one considers the host environment in which these viruses grow. The gastrointestinal tract of avian species contains predominantly α2-3-linked sialic acid, in contrast to the abundance of α2-6 linkages in the human respiratory tract.
Correspondingly, it has been shown that human viruses replicate poorly in avian hosts and avian viruses replicate poorly in humans12,13,14,15 (Fig. 1); however, receptor specificity does not seem to be an absolute barrier to infection. Avian H5N1 viruses were transmitted from infected poultry to humans on at least 18 occasions in 1997 despite their retention of avian virus–like receptor specificity16. Similarly, contemporary H9N2 viruses circulating throughout Asia are confined primarily to avian species, despite a move to a more human virus–like receptor specificity17. Recent studies by Matrosovich et al.18 suggest that the receptor-mediated host range factors for influenza viruses might be more subtle than the simple presence or absence of the appropriate receptor. In an elegant set of experiments, these investigators showed that human viruses with a α2-6 receptor preference almost exclusively infect nonciliated human tracheal cells during the early phase of replication, whereas avian viruses with a α2-3 preference target ciliated cells18 (Fig. 2). Thus, the host range barrier for replication of viruses in humans may not be necessarily at the level of binding, but rather at the level of targeting the appropriate cell type.
Figure 1. Receptor specificities of influenza A viruses affect host range.
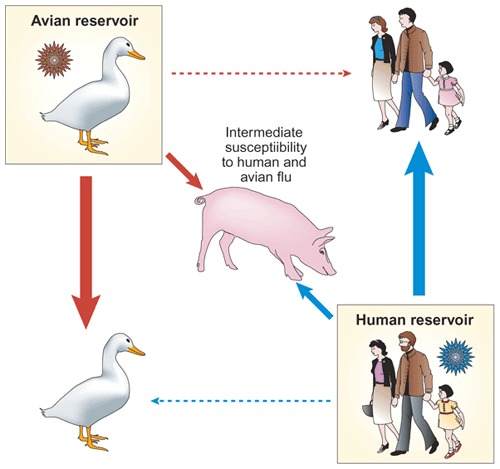
Although the primary reservoir of influenza A viruses is the aquatic birds of the world, stable lineages are present in other hosts such as humans. Analysis of the receptor-binding preferences of viruses from each of these hosts has shown that human and avian viruses have preferences for different conformations of the cellular receptor, sialic acid. Because of this, viruses adapted to humans replicate in and transmit well to other humans, but poorly to aquatic birds. Conversely, viruses adapted to aquatic birds replicate in and transmit well to other aquatic birds, but poorly to humans. Swine seem to be unique in terms of influenza A ecology and show an intermediate susceptibility to both human and avian viruses.
Figure 2. Distinct cellular tropism of influenza viruses in differentiated cultures of human tracheobronchial epithelial cells.

The cultures were infected with either human virus (left) or duck virus (right), fixed 7 h after infection, and immunostained for both virus antigen (brown) and cilia of ciliated cells (gray). Whereas the human virus preferentially infects nonciliated cells, the duck virus infects ciliated cells.
Viral replication
Entry into a cell is only the first replication challenge that a virus faces during an infectious process. Once inside a cell, the virus has to uncoat, transport its genetic material to the appropriate cellular compartment, gather all of the necessary replication machinery, produce copies of its genome and virion components, package these newly produced genomes within virions, exit the cell, move on to the next cell, and finally exit the body and find another susceptible host.
With the exception of some of the larger DNA viruses, the limited coding capacity of viruses means that they have to rely heavily on host functions to navigate all of these steps successfully. As such, this requirement of host factors may exert pressure on a virus to undergo adaptation to optimize interactions with homologous proteins from host cells of a different origin. The number and magnitude of these changes may be the difference between a virus emerging in an alternative population and it being unable to replicate. Of course, if the changes needed are minor in terms of genetic alteration, then there is a high chance that the virus will adapt; however, if the changes needed require substantial genetic change or the host protein is absent, then the chances of adaptation are minimal.
Studies on hepatitis C virus replication in the human hepatoma cell line Huh7 provide an example of how minor changes can improve replication ability. After several independent adaptation experiments, Blight et al.19 identified a mutation 'hot spot' in the nonstructural protein NS5a that accompanied an increase in replication efficiency of the viral genome. Although these mutations occurred in an area known to be associated with resistance to interferon (IFN), the NS5a mutants did not show any changes in IFN susceptibility. A proposed, albeit unproven, explanation is that this area of NS5a interacts with a host protein and changes in this interaction induced by the amino acid substitutions improve the ability of the virus to replicate in Huh7 cells. From these and similar experiments it can be shown that, like the substitutions that can accommodate changes in receptor usage, slight alterations in amino acid composition in viral proteins can substantially alter viral replication. Although for most emerging viruses the exact nature of these interactions between host and viral proteins is poorly understood, a comparison of nonhuman isolates with the corresponding human isolates of emerging viruses may help to identify protein regions for further study.
Antagonizing the innate antiviral response
When faced with the challenge of replication in a foreign host, a virus not only has to propagate its own genome physically, but must do so in the presence of an immune response from the host organism. Once a host recognizes an invading organism, specific and nonspecific immune responses are initiated that are charged with protecting the body from serious threat. As far as viral replication in humans is concerned, one of the first nonspecific lines of defense that must be overcome by a virus is the IFN response. IFN was first described by Isaacs and Lindenmann20 in chicken chorioallantoic membranes infected with heat-inactivated influenza A virus. These landmark studies showed that a substance (IFN) produced from infected cells had the capacity to protect surrounding cells from viral infection. Experiments expanding on these original observations showed that, although viral replication was necessary for the expression of IFN, partially inactivated viruses were more potent IFN inducers21. These results strongly suggested that components produced during viral replication were actively repressing the expression and/or action of IFN. It is now known that this is indeed true, and many viruses have evolved mechanisms with which to antagonize the host IFN response.
Type I IFNs are produced by most cell types in response to viral replication and are crucial components of the host innate response to viral attack. The production of IFN leads to the activation of numerous gene products, some of which have direct antiviral activity22. The ability of a virus to replicate successfully in a given host therefore requires the virus to overcome this potent antiviral response. As a direct consequence of this challenge, viruses have developed various mechanisms by which to counteract the host IFN response. Krug et al.23 have referred to this phenomenon as 'intracellular warfare.' In this war, if the virus has an effective mechanism with which to combat the IFN response of the host, the virus wins the battle and its replication ensues. If, however, the virus does not have the appropriate IFN inhibiting abilities, the battle is won by the cell and viral replication is aborted.
Mechanisms to dampen the host IFN response have been described for many emerging viruses, such as the Nipah virus24,25, Hendra virus26, Ebola virus27, hepatitis C virus28 and influenza viruses29 to name a few. As varied as these viruses are, so are the mechanisms by which IFN circumvention is achieved. Some viruses suppress the production of IFN, others interfere with signaling downstream of IFN production, and yet others target the function of IFN-induced proteins. Owing to the varied nature of the different viral mechanisms, the IFN inhibitory effects of some viral proteins can be species-specific, whereas others are broadly active.
Mechanisms of IFN inhibition such as binding of the NS1 protein of influenza A to double-stranded (dsRNA) would seem to be able to act in cell types of any origin. In this interaction, the IFN antagonistic activity has been suggested to result from the binding and sequestering of dsRNA by NS1, followed by the subsequent inhibition of dsRNA-mediated activation of the protein kinase PKR30. Other interactions, such as Hendra virus V protein binding to complexes of STAT1 and STAT2 (ref. 26), proteins involved in downstream IFN signal transduction, or the proposed binding of influenza A NS1 protein to cellular factors involved in post-transcriptional modifications31,32, require a more specific context. It thus follows that in cases where specific interactions are required, the ability of an IFN antagonist to adapt to specific host factors must be an essential step in the successful emergence of a pathogen in an alternative host. Indeed, it has been shown that the host range of paramyxovirus can be controlled by the viral V protein and that this host range is determined by the interaction of V with STAT2 in a species-dependent manner33.
Overcoming the hurdles
Given the above obstacles to successful emergence, one wonders how viruses ever enter the human population at all. Fortunately for the human population, the number of emerging diseases is small considering the numerous viruses that exist in animal reservoirs. Nevertheless, history and experience tell us that successful adaptations of animal viruses to the human host, albeit relatively rare, do occur. Many of the emerging diseases that threaten humans are caused by RNA viruses and this is not by chance. One of the continuing themes in the above discussions is that viruses need to adapt in perhaps numerous ways to cross the species barrier successfully. Of all pathogens, RNA viruses are most, although not exclusively, able to achieve this.
RNA viruses are extremely mutable and use very efficient strategies for generating viral diversity during evolution (Fig. 3). RNA viruses have few or no proofreading mechanisms and many mutations are introduced during replication. As such, RNA viruses exist as a quasi-species comprising viruses of slightly different genetic composition. Thus, when selective conditions arise, such as the conditions present after jumping to a new host, variants with an advantage are quickly amplified. It can therefore take only limited replication under suboptimal conditions before an adapted virus emerges. Unfortunately, the evolution of viruses is not only confined to point mutations inserted during replication. Viruses can also undergo leaps in evolution through the processes of recombination and reassortment. These processes achieve population heterogeneity in viruses through the acquisition of large sections of genomic material from other viruses.
Figure 3. Molecular mechanisms for generating viral diversity.
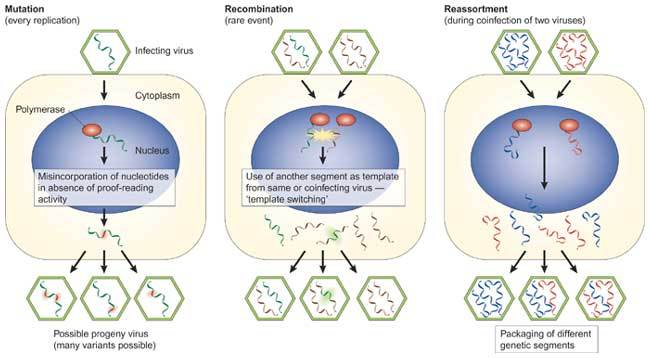
Viruses have evolved three main mechanisms for generating diversity on replication. (a) During replication, single point mutations are incorporated into one or more genomic positions as a result of a lack of proofreading activity of the viral polymerase. (b) During recombination, foreign genetic material is incorporated into the viral genome through mechanisms such as template switching during replication. (c) During reassortment, which occurs on dual infection of a cell with segmented genome viruses, whole gene segments can be swapped. All three mechanisms, which are not exclusive, may result in viruses that have new biological properties, such as new host range and pathogenic potential.
Reassortment occurs in viruses with segmented genomes when sections of different viruses are exchanged on dual infection. Both the Asian (1957) and Hong Kong (1968) human influenza pandemics arose through reassortment of human and avian strains34,35. Global health authorities are concerned that the same may happen with the H5N1 viruses that are currently circulating in birds in southeastern Asia, with the result being a virus that is transmissible in humans. Unfortunately, we know little about the conditions for successful reassortment and it is difficult to assess the possibility of this happening. The available evidence from influenza A viruses is that there is a selective rather than random mechanism for packaging RNA segments into infectious virions36 and not all combinations of viral genes are compatible. Recent findings from vaccine development have shown that the surface proteins of the H5N1 influenza viruses are at least compatible with older human strains37, suggesting that reassortants of H5N1 and human influenza virus may well be viable. Arenaviruses are also capable of reassortment, although reassortment again does not seem to be completely random and some combinations of genes do not seem to be compatible38. Nevertheless, there has been some speculation that reassortment may have contributed to the emergence of new arenaviruses with disease associations38.
Recombination leads to the same end as reassortment—that is, the acquisition of foreign genetic material—but through a different route. Rather than through the acquisition of gene segments, recombination occurs through 'splicing' of the foreign component into the virus genome. Recombination is often observed in positive-sense RNA viruses such as picornaviruses and coronaviruses39,40, although it also occurs to a limited extent in negative-sense viruses such as influenza A41. As with reassortment, recombination has the capacity to change the phenotype of a virus markedly and has the potential to aid in the transmission of viruses between species.
Concluding remarks
Despite advances in our understanding of some viruses, for the most part we know very little about the specific molecular changes that enable many viruses to overcome the known barriers. A significant omission from this review is any discussion on HIV—perhaps the single most important viral agent to emerge in recent times. But the fact is that we know very little about the molecular changes that led to the emergence of HIV. Phylogenetic analysis indicates that several interspecies transmissions from simian species introduced two genetically distinct types of HIV into the human population—HIV-1, which is closely related to simian immunodeficiency virus (SIV) from chimpanzees, and HIV-2, which is closely related to SIV from sooty mangabeys (reviewed in ref. 43); however, the molecular changes that enabled the virus to adapt to its new host are unknown. Indeed, data from the HIV field is instead revealing several new cellular proteins that create intracellular barriers to virus replication and restrict virus host range. Some of these so-called 'restriction factors' seem to have evolved as a result of an evolutionary association with endogenous retroviruses (reviewed in refs. 44,45).
Although we are very far from a complete understanding of the molecular mechanisms that lead to the interspecies transmission of most pathogens, the achievements in the past decade have been considerable. The number of new molecular tools available to the virologist has lead to a greater resolution of factors that restrict viruses to certain hosts. In addition to numerous tools with which to dissect these cellular factors, the development of reverse genetics systems for many RNA viruses has facilitated the manipulation of the viral genome to an extent that was not previously possible. For example, the resurrection of several pieces of the 1918 Spanish influenza genome46,47 provides the possibility that a genomic analysis of virus and host will explain the extreme pathogenicity of the 1918 outbreak of Spanish influenza that killed up to 50 million people worldwide. Perhaps the greatest advances in the next decade will come from a genomic and proteomic analysis in which all of the interactions between a parasite and a host can be resolved. These areas are the engines that will drive our understanding of both host range and the avenues that are available to a pathogen to overcome host range barriers. This knowledge will identify targets and intervention strategies to manipulate the different anti-infective host responses and to develop new drugs and vaccines.
Our increased understanding of the molecular properties of viruses underpins the development of control strategies and techniques that hold future promise both for the rapid development of vaccines and for resolving the issues concerning pathogenesis and transmissibility. In this Perspective, we have discussed interspecies transmission only from the perspective of the viral pathogen and have not considered the key issues of susceptibility and the immune status of the host. It must be remembered that the molecular basis of interspecies transmission constitutes a complex interaction between virus and host and between the genomics of both.
Acknowledgements
We thank M. Matrosovich for supplying data for this review and C. Walsh for editorial assistance. The preparation of this report was supported by contract AI95357 from the National Institute of Allergy and Infectious Diseases of the United States National Institutes of Health and from the American Lebanese Syrian Associated Charities (ALSAC).
Competing interests
The authors declare no competing financial interests.
References
- 1.Smolinski M, Hamburg M, Lederberg J. Microbial Threats to Health: Emergence, Detection, and Response. 2003. [PubMed] [Google Scholar]
- 2.Li KS, et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature. 2004;430:209–213. doi: 10.1038/nature02746. [DOI] [PubMed] [Google Scholar]
- 3.Nie Y, et al. Highly infectious SARS-CoV pseudotyped virus reveals the cell tropism and its correlation with receptor expression. Biochem. Biophys. Res. Commun. 2004;321:994–1000. doi: 10.1016/j.bbrc.2004.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer BJ, de la Torre JC, Southern PJ. Arenaviruses: genomic RNAs, transcription, and replication. Curr. Top. Microbiol. Immunol. 2002;262:139–157. doi: 10.1007/978-3-642-56029-3_6. [DOI] [PubMed] [Google Scholar]
- 5.Holmes KV, Zelus BD, Schickli JH, Weiss SR. Receptor specificity and receptor-induced conformational changes in mouse hepatitis virus spike glycoprotein. Adv. Exp. Med. Biol. 2001;494:173–181. doi: 10.1007/978-1-4615-1325-4_29. [DOI] [PubMed] [Google Scholar]
- 6.Thackray LB, Holmes KV. Amino acid substitutions and an insertion in the spike glycoprotein extend the host range of the murine coronavirus MHV-A59. Virology. 2004;324:510–524. doi: 10.1016/j.virol.2004.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guan Y, et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276–278. doi: 10.1126/science.1087139. [DOI] [PubMed] [Google Scholar]
- 8.Webster RG. Wet markets—a continuing source of severe acute respiratory syndrome and influenza? Lancet. 2004;363:234–236. doi: 10.1016/S0140-6736(03)15329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baranowski E, Ruiz-Jarabo CM, Domingo E. Evolution of cell recognition by viruses. Science. 2001;292:1102–1105. doi: 10.1126/science.1058613. [DOI] [PubMed] [Google Scholar]
- 10.Couceiro JN, Paulson JC, Baum LG. Influenza virus strains selectively recognize sialyloligosaccharides on human respiratory epithelium; the role of the host cell in selection of hemagglutinin receptor specificity. Virus Res. 1993;29:155–165. doi: 10.1016/0168-1702(93)90056-S. [DOI] [PubMed] [Google Scholar]
- 11.Ito T, et al. Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J. Virol. 1998;72:7367–7373. doi: 10.1128/jvi.72.9.7367-7373.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beare AS, Webster RG. Replication of avian influenza viruses in humans. Arch. Virol. 1991;119:37–42. doi: 10.1007/BF01314321. [DOI] [PubMed] [Google Scholar]
- 13.Hinshaw VS, Webster RG, Easterday BC, Bean WJ. Replication of avian influenza A viruses in mammals. Infect. Immun. 1981;34:354–361. doi: 10.1128/iai.34.2.354-361.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinshaw VS, Webster RG, Naeve CW, Murphy BR. Altered tissue tropism of human-avian reassortant influenza viruses. Virology. 1983;128:260–263. doi: 10.1016/0042-6822(83)90337-9. [DOI] [PubMed] [Google Scholar]
- 15.Murphy BR, et al. Virulence of avian influenza A viruses for squirrel monkeys. Infect. Immun. 1982;37:1119–1126. doi: 10.1128/iai.37.3.1119-1126.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matrosovich M, Zhou N, Kawaoka Y, Webster R. The surface glycoproteins of H5 influenza viruses isolated from humans, chickens, and wild aquatic birds have distinguishable properties. J. Virol. 1999;73:1146–1155. doi: 10.1128/jvi.73.2.1146-1155.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matrosovich MN, Krauss S, Webster RG. H9N2 influenza A viruses from poultry in Asia have human virus-like receptor specificity. Virology. 2001;281:156–162. doi: 10.1006/viro.2000.0799. [DOI] [PubMed] [Google Scholar]
- 18.Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl. Acad. Sci. USA. 2004;101:4620–4624. doi: 10.1073/pnas.0308001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 20.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B. 1957;147:258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 21.Isaacs A, Burke DC. Mode of action of interferon. Nature. 1958;182:1073–1074. doi: 10.1038/1821073a0. [DOI] [PubMed] [Google Scholar]
- 22.Katze MG, He Y, Gale M., Jr. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- 23.Krug RM, Yuan W, Noah DL, Latham AG. Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology. 2003;309:181–189. doi: 10.1016/S0042-6822(03)00119-3. [DOI] [PubMed] [Google Scholar]
- 24.Park MS, et al. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol. 2003;77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez JJ, Parisien JP, Horvath CM. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 2002;76:11476–11483. doi: 10.1128/JVI.76.22.11476-11483.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez JJ, Wang LF, Horvath CM. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J. Virol. 2003;77:11842–11845. doi: 10.1128/JVI.77.21.11842-11845.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harcourt BH, Sanchez A, Offermann MK. Ebola virus inhibits induction of genes by double-stranded RNA in endothelial cells. Virology. 1998;252:179–188. doi: 10.1006/viro.1998.9446. [DOI] [PubMed] [Google Scholar]
- 28.Enomoto N, et al. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N. Engl. J. Med. 1996;334:77–81. doi: 10.1056/NEJM199601113340203. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Sastre A, et al. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 30.Lu Y, Wambach M, Katze MG, Krug RM. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology. 1995;214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z, Li Y, Krug RM. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 1999;18:2273–2283. doi: 10.1093/emboj/18.8.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemeroff ME, Barabino SM, Li Y, Keller W, Krug RM. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′ end formation of cellular pre-mRNAs. Mol. Cell. 1998;1:991–1000. doi: 10.1016/S1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- 33.Parisien JP, Lau JF, Horvath CM. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J. Virol. 2002;76:6435–6441. doi: 10.1128/JVI.76.13.6435-6441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawaoka Y, Krauss S, Webster RG. Avian-to-human transmission of the PB1 gene of influenza A viruses in the 1957 and 1968 pandemics. J. Virol. 1989;63:4603–4608. doi: 10.1128/jvi.63.11.4603-4608.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scholtissek C, Rohde W, von HV, Rott R. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology. 1978;87:13–20. doi: 10.1016/0042-6822(78)90153-8. [DOI] [PubMed] [Google Scholar]
- 36.Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA. 2003;100:2002–2007. doi: 10.1073/pnas.0437772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Webby RJ, et al. Responsiveness to a pandemic alert: use of reverse genetics for rapid development of influenza vaccines. Lancet. 2004;363:1099–1103. doi: 10.1016/S0140-6736(04)15892-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sevilla N, Domingo E, de la Torre JC. Contribution of LCMV towards deciphering biology of quasispecies in vivo. Curr. Top. Microbiol. Immunol. 2002;263:197–220. doi: 10.1007/978-3-642-56055-2_10. [DOI] [PubMed] [Google Scholar]
- 39.Lai MM. Genetic recombination in RNA viruses. Curr. Top. Microbiol. Immunol. 1992;176:21–32. doi: 10.1007/978-3-642-77011-1_2. [DOI] [PubMed] [Google Scholar]
- 40.Nagy PD, Simon AE. New insights into the mechanisms of RNA recombination. Virology. 1997;235:1–9. doi: 10.1006/viro.1997.8681. [DOI] [PubMed] [Google Scholar]
- 41.Suarez DL, et al. Recombination resulting in virulence shift in avian influenza outbreak, Chile. Emerg. Infect. Dis. 2004;10:693–699. doi: 10.3201/eid1004.030396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hahn BH, Shaw GM, De Cock KM, Sharp PM. AIDS as a zoonosis: scientific and public health implications. Science. 2000;287:607–614. doi: 10.1126/science.287.5453.607. [DOI] [PubMed] [Google Scholar]
- 43.Lemey P, et al. Tracing the origin and history of the HIV-2 epidemic. Proc. Natl. Acad. Sci. USA. 2003;100:6588–6592. doi: 10.1073/pnas.0936469100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bieniasz PD. Restriction factors: a defense against retroviral infection. Trends Microbiol. 2003;11:286–291. doi: 10.1016/S0966-842X(03)00123-9. [DOI] [PubMed] [Google Scholar]
- 45.Goff, S.P. Genetic control of retrovirus susceptibility in mammalian cells. Annu. Rev. Genet. advance online publication, 26 May 2004 (doi:10.1146/annurev.genet.38.072902.094136). [DOI] [PubMed]
- 46.Kobasa D, et al. Enhanced virulence of influenza A viruses with the haemagglutinin of the 1918 pandemic virus. Nature. 2004;431:703–707. doi: 10.1038/nature02951. [DOI] [PubMed] [Google Scholar]
- 47.Tumpey TM, et al. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc. Natl. Acad. Sci. USA. 2004;101:3166–3171. doi: 10.1073/pnas.0308391100. [DOI] [PMC free article] [PubMed] [Google Scholar]