

Abstract
Chronic HIV infection in the era of anti-retroviral therapy is associated with dramatically increased risk of developing severe cardio pulmonary disease. Common to these diseases is increased oxidative burden and chronic inflammation despite low viremia and restoration of CD4+ T-cell levels. Soluble viral factors are heavily implicated in these disease processes, including the HIV Transactivator of Transcription (Tat). Tat is produced in high levels during infection and secreted from infected cells into circulation where it is internalized by bystander cells and is known to regulate inflammatory pathways and elicit a pro-oxidant environment. We have examined the effects of Tat on the anti-oxidant regulatory network driven by the transcription factor Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) in primary human pulmonary arterial endothelial cells, which are heavily involved in pathogenesis of HIV associated lung diseases including pulmonary arterial hypertension and COPD. Co-expression of Tat and a luciferase reporter construct driven by the Nrf2 activated anti-oxidant response element (ARE) demonstrated markedly reduced Nrf2/ARE activity, even when stimulated by the potent Nrf2 activating compound PB125. Additionally, Heme-oxygenase-1 (HO-1) transcription was potently repressed by Tat in a cell line as well as primary endothelial cells, and treatment with PB125 failed to restore transcriptional activity. Other anti-oxidant Nrf2 genes examined included NADPH Dehydrogenase Quinone 1 (NQO1) and Sulfiredoxin-1 (SRXN1). NQO1 was repressed basally by Tat, while SRXN1 transcription was refractory to activation by PB125 in the presence of Tat. Lastly, we demonstrated that Tat expressing cells have increased indicators of oxidative stress including elevated production of reactive oxygen species, measured by electron paramagnetic resonance spectroscopy, and increased levels of nitrotyrosine content. These observations suggest a novel mechanism by which HIV Tat increases oxidative burden by dysregulation of the Nrf2/ARE pathway.
Graphical Abstract
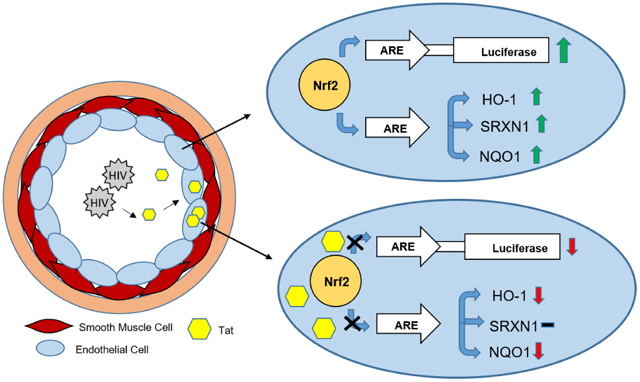
Introduction
HIV infection remains a major global health concern, with an estimated 36.7 million individuals currently infected worldwide [1]. The implementation of combinatorial anti-retroviral therapy (ART) has been widely successful in controlling viral load in HIV infected patients, thus mitigating morbidity and mortality associated with AIDS [2]. However, upon cessation of therapy, patients with very low and even undetectable viral titers experience a prolific rebound in viremia, thus rendering infection with HIV a chronic medical condition. Despite the longevity associated with ART, a significant gap in life expectancy, as well as diminished quality of life, still exists between HIV patients receiving therapy and the general population, and can be attributed largely to non-AIDS associated morbidities of chronic HIV infection [3]. Concomitant with the advent of ART has been a dramatically increased epidemiological incidence of inflammatory non-infectious pulmonary diseases including pulmonary arterial hypertension, COPD, and lung cancer [4-6]. Despite apparent virologic control and improvement of CD4+ T cell counts by ART, biomarkers of immune activation, oxidative stress and inflammation do not completely normalize [7,8]. These persistent phenotypes correlate with, and indeed may directly contribute to these non-AIDS complications. With over 17 million HIV infected individuals receiving ART worldwide, there is an ever growing need to understand the mechanisms driving the development of these diseases at the host/pathogen interface.
The incidence of inflammation-mediated diseases in the HIV infected population is significantly increased despite low viral titers and apparent lack of productive infection of vascular cells. However, HIV transgenic animal models exhibit exacerbated systemic vascular inflammation, increased biomarkers of oxidative stress, and vascular remodeling [9-12]. The development of these pathological phenotypes in the absence of replication competent virus strongly supports a role for exposure to viral proteins in the etiology of HIV associated inflammatory disease, of which the HIV Transactivator of Transcription (Tat) is a particularly likely culprit. Tat is an early HIV trans-acting regulatory element that is required for initiation of transcription of integrated pro-viral DNA as well as elongation of nascent viral transcript [13, 14]. During HIV infection, Tat secretion is highly active, and Tat circulates diffusely through the bloodstream where it is indiscriminately internalized by bystander cells via its basic core domain containing the cell penetrating peptide (PTD) [15, 16]. Once internalized, Tat is capable of altering cellular physiology by regulating both transcriptional and post-translational processes, and is implicated in a number of physiological responses in line with the Janus like nature of inflammation [17, 18]. As such, Tat is uniquely poised to contribute to the pathophysiological characteristics of uninfected cells, such as pulmonary vascular cells, which exist in close proximity to infected immune cells.
The transcription factor Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is integral in maintaining cellular redox homeostasis, controlling many redox-sensitive cellular functions, and resolving inflammation [19]. Under physiological conditions, Nrf2 resides in the cytoplasm bound to its repressor, Keap1 [20]. The Nrf2 regulatory network is largely dictated by the presence of the cis-acting anti-oxidant response element (ARE) in the promoter regions of various anti-oxidant and anti-inflammatory genes. In response to oxidative stress, Nrf2 dissociates from Keap1 and translocates to the nucleus where it binds to the ARE, resulting in the activation of cytoprotective enzymes including heme oxygenase 1 (HO-1), sulfiredoxin-1 (SRXN-1), NADPH quinone dehydrogenase 1 (NQO1) and glutamate-cysteine ligase catalytic subunit (GCLC) [21,22]. As such, Nrf2 activation elicits cytoprotective effects to acute and chronic oxidative insult including hypoxia, ischemic reperfusion, and xenobiotic exposure. HO-1 is of particular importance to the maintenance of pulmonary redox homeostasis and resolution of vascular inflammation. Indeed, HO-1 deficiency is associated with increased severity of hypoxic injury and pulmonary vascular inflammation [23]. Additionally, selective overexpression of HO-1 in the lungs of rodent models of pulmonary arterial hypertension attenuates pulmonary inflammation, mitigates hyper-proliferation of vascular smooth muscle cells, and prevents the formation of complex obstructive vascular lesions [24.25]. While the pro-oxidant and pro-inflammatory properties of Tat are well established, the driving mechanisms remain elusive. Here, we demonstrate repression of Nrf2/ARE activity by Tat and repression of Nrf2 activated genes. In particular, we observe the potent repression of HO-1 by Tat both basally as well as in the presence of the potent Nrf2 activating compound PB125.
Material and Methods
Cells and Generation of Stable Tat Transfectants
HeLa cells were transfected (Superfect reagent, Qiagen LLC) with the FLAG tagged Tat expressing vector pCMV-Tat-86-FLAG (Sino Biologicals). This plasmid, hereafter referred to as pTat-FLAG, expresses the 86 amino acid isoform of Tat from the HXB2 HIV strain. After overnight incubation, standard media was supplanted with media containing 200 mg/L Hygromycin-B (Thermo Fisher Scientific). Cells were passaged in Hygromycin-B containing media and serially diluted to obtain isolated molecular clones. To confirm the presence and expression of the Tat gene, total genomic DNA, RNA and protein were extracted and analyzed via PCR, RT-qPCR and immunoblotting. To determine if Tat was biologically active in the stable Tat transfectant, hereafter referred to as HeLa-Tat-FLAG, the cells were co-transfected with a luciferase reporter driven by the HIV LTR and a control plasmid with Renilla luciferase expression driven from the cytomegalovirus (CMV) promoter and a dual luciferase assay (Promega Corp.) was performed (S1). Human Primary Arterial Endothelial Cells (HPAEC) were procured from Lonza (catalog # CC-2530) and cultured in Endothelial Basal Media-2 (Lonza catalog #: CC-3516) supplemented with endothelial growth factors optimized for aortic and pulmonary arterial endothelial cells (Lonza catalog # CC-3162). HPAEC expansion was limited to six passages in order to prevent senescence and de-differentiation.
PB125
The novel Nrf2 activating compound PB125 was acquired from collaborators at the University of Colorado for use in this study [26].
Transfections, PB125 Treatment, and Dual Luciferase Assays
Transfections were performed using the Qiagen Superfect® system according to manufacturer’s protocol for cell lines or primary cells. To asses Nrf2/ARE activity, cells were transfected with an ARE driven luciferase reporter plasmid/Renilla luciferase plasmid mix (Cignal Antioxidant Response Reporter kit, Qiagen catalog #: 336481) according to manufacturer’s protocol. To assess the effect of Tat on Nrf2/ARE activity, cells were co-transfected with the ARE-driven luciferase reporter and pTat-FLAG. At the time of washing, transfection media was supplanted with media containing PB125 or vehicle (0.016% ethanol) and incubated overnight, after which a Dual-Luciferase® assay was carried out (Dual-Luciferase® Reporter Assay System, Promega catalog #: E1910) following manufacturer’s protocol. For this study, 8 mg/L was the standard PB125 dose. For the dose response curve, cells were treated with PB125 concentrations ranging from 1 – 10 mg/L.
Nrf2 Target Gene Transcriptional Analyses, Western Blots and ELISA
Total cellular RNA was extracted from cells using Qiagen’s RNeasy Mini Kit (Qiagen, Catalog #: 74106) according to manufacturer spin method protocol. Reactions were carried out as technical duplicates from experimental replicates of each condition (mock transfected or Tat transfected, un-treated, vehicle treated, or PB125 treated, n = 6). RT-qPCR was carried out (50 ng total RNA/reaction) using Biorad iSCript™ Reverse Transcription Supermix (Catalog #: 1708841). For relative expression analysis, the average Ct values were calculated from technical replicates and analyzed utilizing the delta-delta Ct method, where all samples were normalized to GAPDH (first delta) and then compared to un-treated controls (second delta). To calculate absolute copy numbers, a standard curve was generated using plasmids containing the target sequences and normalized to copies of gapdh. Primer sequences are summarized in supplementary table 1. For immunoblot and ELISA assays total protein was extracted from cells using Mammalian Protein Extraction Reagent (Thermo Fisher, Catalog #: 78501) supplemented with 1 mM DTT and 1XProtease/Phosphatase inhibitor cocktail (HALT, Thermo Fisher catalog #: 78440).For immunoblot analysis 0.01 mg of protein per lane were resolved by Sodium dodecyl sulfate (SDS) poly-acrylamide gel electrophoresis and transferred to Polyvinylidene fluoride (PVDF). Proteins were detected with antibodies from commercial sources, Santa-Cruz Biotechnology (HO-1) and Thermo Fisher (FLAG, β-actin and secondary HRP-conjugated anti-mouse or anti-rabbit). Immunoblots were imaged by chemiluminescence using Supersignal West Femto Maximum Sensitivity substrate (Thermo Fisher, catalog #: 34095). Densitometry values are expressed as net intensity of the target protein (HO-1) over net intensity of the loading control, (β-actin) from biological replicates (n = 3). 3-nitrotyrosine-ELISA was carried out on total protein extracts using the Cell Biolabs Nitrotyrosine ELISA kit (catalog #: STA-305) according to manufacturer’s protocol. Tat-FLAG levels were quantified from pTat-FLAG transfected HPAEC (n = 6) total protein extract by competitive anti-FLAG ELISA using the DYKDDDDK-Tag Detection ELISA Kit (Cayman Chemicals, catalog # 501560).
Electron Paramagnetic Resonance Spectroscopy
Total ROS production was measured by EPR using the superoxide sensitive spin probe 1-hydroxy-3-methoxycarbonyl-2, 2, 5, 5-tetramethylpyrrolidine (CMH) while mitochondrial ROS production was measured using the mitochondrial spin probe 1-hydroxy-4-[2-triphenylphosphonio)-acetamido]-2,2,6,6-tetramethyl-piperidine,1-hydroxy-2,2,6,6-tetramethyl-4-[2-(triphenylphosphonio)acetamido] piperidinium dichloride (mito-TEMPO-H. Cells were seeded 18 hours prior to the EPR measurements. CMH and mito-TEMPO-H probes were prepared in deoxygenated 50 mM phosphate buffer. Cells were washed and treated with CMH and mito-TEMPO-H 0.25 mM in Krebs-HEPES buffer (KHB) containing 100 μM of a metal chelator DTPA to avoid direct oxidation with metal ion or hydroxyl radical generation by Fenton reaction. Cells were incubated for 50 min at 37° C then gently scraped and transferred to ice. 5 0 μl of cell suspension was loaded in an EPR capillary tube and EPR measurements were performed at room temperature using Bruker EMXnano X-band spectrometer. EPR acquisition parameters are: microwave frequency = 9.6 GHz; center field = 3432 G; modulation amplitude = 2.0 G; sweep width = 80 G; microwave power = 19.9 mW; total number of scans = 5; sweep time = 12.11 s; and time constant = 20.48 ms. CM· Or mito-TEMPO· nitroxide radicals concentration was obtained by simulating the spectra using the SpinFit module incorporated in the Xenon software of the bench-top EMXnano EPR spectrometer followed by the SpinCount module (Bruker) [27]. Total protein was extracted from analyzed samples and quantified via a Bradford protein assay and nitroxide concentrations were normalized to total protein.
Results
Nrf2/ARE Activity and Transcriptional Regulation in Tat Expressing Cells
Nrf2 is a transcription factor that is vital to mitigating cellular oxidative burden through activation of cytoprotective enzymes, which contain the cis-acting anti-oxidant response element (ARE) sequence in their promoter regions. As such, we surmised that dysregulation of the Nrf2 signaling pathway may be involved in the increase in oxidative burden witnessed in cells exposed to Tat. To asses Nrf2/ARE activity, an ARE driven luciferase reporter was transfected into HeLa-Tat-FLAG and HeLa wild type (HeLa-WT) and the cells were either un-treated or treated with the potent Nrf2 activating compound PB125 or vehicle (0.016 ethanol) for 18-hours. Basal ARE activity, without PB125 treatment, was significantly increased in HeLa-Tat-FLAG cells compared to HeLa-WT (fig. 1A), and this effect was further increased by treatment with PB125 (fig. 1B). We next sought to observe the transcriptional activity of two well characterized Nrf2/ARE driven antioxidant genes, HO-1 and SRXN1, via RT-qPCR. As expected, basal levels of SRXN1 in HeLa-Tat-FLAG cells were significantly increased, correlating with the observed increased ARE activity (fig. 1D). Perplexingly, HO-1 levels were consistently decreased in HeLa-Tat-FLAG cells, suggesting negative transcriptional regulation in the presence of Tat (fig. 1C). We next sought to determine if exogenous activation of Nrf2, by PB125 treatment, could restore HO-1 transcriptional levels in HeLa-Tat-FLAG. While SRXN1 and HO-1 transcription were dramatically increased by PB125 treatment in HeLa-WT cells (fig. 2A and B), and while SRXN1 transcript levels were increased by PB125 treatment in HeLa-Tat-FLAG cells, HO-1 transcript levels remained unchanged (fig 2-A and B). Western blot analysis confirmed that induction of HO-1 by PB125 was attenuated in Tat expressing cells (fig. 2C and D). The surprising observations that HO-1 levels in Tat expressing cells are basally repressed and remain repressed even in the presence of a potent Nrf2 activator suggest a mechanism by which Tat may elicit pro-oxidant properties through transcriptional repression of select anti-oxidant genes.
Figure 1. ARE Activity and HO-1/SRXN1 Transcriptional Profile in WT and Tat-FLAG Cells.
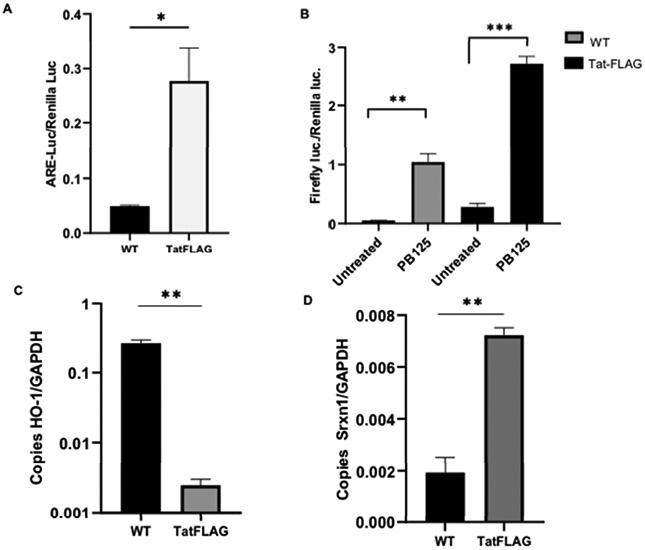
(A)Basal ARE activity is significantly higher in Tat-FLAG cells. Values shown are normalized to renilla luciferase. Mean ± SEM of biological replicates (WT n = 3, Tat-FLAG n = 4)(**p ≤ 0.01 by student’s T-test).
(B)ARE activity is enhanced in PB125 treated WT and Tat-FLAG cells. Cells were left un-treated in culture media or treated with culture media containing 8 mg/L of PB125 for 18 hours. Mean ± SEM of biological replicates (WT n = 3, TatFLAG n = 4) (**p ≤ 0.01, ***p ≤ 0.001 by student’s T-test)
(C) HO-1 Transcription is basally repressed in Tat-FLAG cells compared to WT. (Values shown as absolute copy number. Mean ± SEM of biological replicates (n = 3) (**p ≤ 0.01 by student’s T-test).
(D) SRXN1 Transcription is basally enhanced in Tat-FLAG cells compared to WT. (Values shown as absolute copy number. Mean ± SEM of biological replicates (n = 3) (**p ≤ 0.01 by student’s T-test).
Figure 2. Effects of Tat Expression on HO-1 and SRXN1 in Response to PB125.

(A) HO-1 transcription is induced by PB125 in HeLa-WT cells but remains repressed in HeLa-Tat-FLAG cells. HeLa-WT or HeLa-Tat-FLAG cells were incubated overnight with 8 mg/LPB125 or left un-treated, after which total RNA was extracted and RT-qPCR performed. Values shown as fold change relative to un-treated controls. Mean ± SEM of biological replicates (n = 3). (B) SRXN1 transcription is induced by PB125 in both WT and Tat-FLAG cells. HeLa-WT or HeLa-Tat-FLAG cells were incubated overnight with 8 mg/LPB125 or left un-treated, after which total RNA was extracted and RT-qPCR performed. Values shown as fold change relative to un-treated controls. Mean ± SEM of biological replicates (n = 3). (C-D) HO-1 protein levels in response to PB125 in WT and Tat-FLAG cells. HeLa-WT or HeLa-Tat-FLAG cells were incubated overnight with 8 mg/L PB125 or treated with vehicle (0.016% ethanol), after which total protein extract was harvested for SDS-PAGE and western blot analysis. Densitometry values shown as relative to actin loading controls. Mean ± SEM of biological replicates (n = 3) (**p ≤ 0.01 by unpaired student’s T-test)
Tat Expressing Cells Exhibit Increased Oxidative Burden and Protein Modification
Tat is known to induce oxidative and pro-inflammatory pathways in-vitro, and the pro oxidant properties of Tat have been implicated in the pathological processes of chronic HIV associated co-morbidities including HIV associated pulmonary arterial hypertension(HIV-PAH). To assess whether the cellular oxidative burden was increased by Tat, levels of cellular superoxide were quantified via spin trapping and Electron Paramagnetic Resonance (EPR) spectroscopy. HeLa-Tat-FLAG and HeLa-WT cells were incubated for 50 minutes with spin trapping probes CMH or Mito-TEMPO to detect levels of total cellular superoxide or mitochondrial superoxide respectively. Nitroxide levels were then quantified via electron paramagnetic resonance (EPR) spectroscopy and normalized to mg of protein. HeLa-Tat-FLAG cells demonstrated increased total cellular oxidative burden compared to their wild type counterparts (fig. 3A). Interestingly, while mitochondrial superoxide levels were significantly increased (fig. 3B), it accounts for only a fraction of the total cellular increase in superoxide, suggesting important contributions of cellular sources of oxygen radicals other than mitochondrial respiration. In addition to increased levels of reactive oxygen species, HeLa-Tat-FLAG cells exhibited increased oxidative protein modification as measured by 3-nitrotyrosine ELISA of total protein extract (fig. 3C). Treatment with PB125 slightly reduced 3-nitrotyrosine levels in Tat-FLAG cells, although levels were still markedly increased compared to un-treated WT cells (fig. 4).
Figure 3. Tat Expressing Cells Exhibit Increased Oxidative Burden and Protein Modification.

.(A) Total ROS is increased in HeLa-Tat-FLAG cells. Total ROS were measured by incubating cells with 1-Hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine-HCl (CMH) and nitroxide was measured via EPR spectroscopy. Values shown as nmol of nitroxide per gram of protein.
(WT n = 6, TatFLAG n = 9) (***p ≤ 0.001 by un-paired student’s T-test)
(B) Mitochondrial ROS is increased in HeLa-Tat-FLAG Cells. Mitochondrial ROS were measured by incubating cells with (2-(2,2,6,6-Tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl)triphenylphosphonium chloride (Mito-TEMPO-H) and nitroxide was measured via EPR spectroscopy. Values shown as nmol of nitroxide per gram protein. (n = 6) (*p ≤ 0.05 by un-paired student’s T-test)
(C) HeLa-Tat-FLAG cells demonstrate increased protein nitrosylation as measured by ELISA. Total 3-nitrotyrosine was measured by ELISA in HeLa-WT and HeLa-Tat-FLAG cells from total protein extract. (n = 6) (**p ≤ 0.01 by unpaired student’s T-test)
Figure 4. HeLa-Tat-FLAG cells showed increased nitrotyrosine levels which were partially attenuated by PB125.
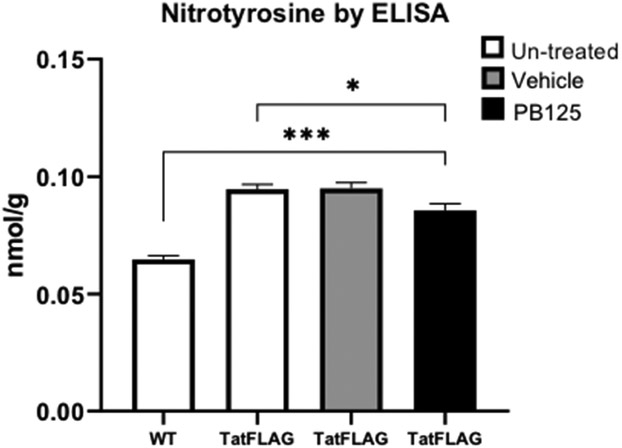
Mean ± SEM of biological replicates (n = 6),) (*p ≤ 0.05, ***p ≤ 0.001 by un-paired student’s T-test)
Nrf2/ARE Activity is Repressed in HPAEC Expressing Tat
The pulmonary vascular endothelium is a large endocrine organ that is profoundly involved in the pathogenesis of pulmonary arterial hypertension (PAH) through various mechanisms. Endothelial dysfunction, pro-inflammatory signaling, abnormal proliferation, and the aberrant production of ROS are all heavily implicated in the PAH disease process. The observation that HO-1 was repressed in HeLa-Tat-FLAG cells led us to surmise that Nrf2 signaling may also be dysregulated by Tat in primary Human Pulmonary Arterial Endothelial Cells (HPAEC). To test whether ARE activity is sensitive to Tat in primary endothelial cells, HPAEC were co-transfected with an ARE driven luciferase vector (ARE-luc) pTat-FLAG, and a dual luciferase assay was carried out after overnight incubation. In contrast to HeLa-Tat-FLAG cells, basal ARE activity was decreased ~6 fold in Tat expressing HPAEC (fig. 5A). To test whether different concentrations of PB125 could restore ARE-luc levels in Tat(+) cells, a dose curve analysis was performed, with concentrations ranging from 1 to 10 mg/L. At every concentration, ARE-luc activity was heavily repressed in Tat(+) HPAEC (fig. 5B). While the signal in Tat(−) HPAEC plateaued at 6 and 8 mg/L, maximum ARE-luc activity was not achieved in Tat(+) cells even at 10 mg/L. In fact, the signal in Tat(+) cells at 10 mg/L was equivalent to those in Tat(−) cells at 1 mg/L, further demonstrating a potent predilection of Tat to inhibit the activation of Nrf2.
Figure 5. The Effect of Tat on Nrf2/ARE Activity in Human Pulmonary Arterial Endothelial Cells.

(A) Nrf2 activation is attenuated by Tat basally as well as in the presence of PB125. HPAEC were transfected with ARE-luc or co-transfected with ARE-luc and a plasmid expressing Tat-FLAG (pTatFLAG) and media containing 8 mg/L PB125 was added at the time of washing. A dual luciferase assay was carried out after overnight incubation. Mean ± SEM of biological replicates (Tat(−)PB125(+) n = 3, Tat(+) PB125(+) n = 3, Tat(−) PB125(−) n = 13, Tat(+) PB125(−) n = 15). (***p ≤ 0.001 by unpaired student’s T-test). (B) PB125 Dose Response in Tat+/− HPAEC. HPAEC were transfected with ARE-luc alone or co-transfected with ARE-luc and pTat-FLAG and media containing 1 to 10 mg/L PB125 was added at the time of washing. A dual luciferase assay was carried out after overnight incubation. Values graphed as individual biological replicates. (n = 3)
Nrf2 Regulated Anti-oxidant and Anti-inflammatory Genes Are Repressed by Tat in HPAEC
Next, to assess activation of Nrf2/ARE driven genes, Tat-FLAG transfected (Tat(+)) and mock transfected HPAEC (Tat(−)) were treated with either PB125 (8 mg/L), vehicle (0.016% ethanol), or left untreated, and relative expression of target genes was assessed via RT-qPCR from total cellular RNA. As seen in HeLa-Tat-FLAG cells, HO-1 transcription was significantly decreased in un-treated Tat(+) cells (fig. 6. A, C-D). Although PB125 led to an ~14 fold increase in HO-1 transcription in Tat(+) HPAEC, compared to un-treated Tat(+) HPAEC, these levels were still well below basal HO-1 levels in un-treated Tat(−) HPAEC. While SRXN1 levels were increased ~4 fold in un-treated HeLa-Tat-FLAG compared to un-treated HeLa-WT, they were increased <1 fold in un-treated Tat(+) HPAEC. Furthermore, SRXN1 was not significantly increased by PB125 treatment in Tat(+) HPAEC compared to controls whereas it was increased ~2.5 fold in PB125 treated Tat(−) HPAEC (fig 6B). We further characterized the effect of Tat on Nrf2/ARE target gene transcriptional activation by assessing transcript levels of NAD(P)H Dehydrogenase, Quinone 1 (NQO1) and glutamate-cysteine ligase, catalytic subunit (GCLC), both of which contain the ARE in their promoter regions. Compared to untreated cells, NQO1 levels were increased ~2.6 and ~3.6 fold by PB125 in Tat(−) and Tat(+) HPAECS respectively. However, basal levels of NQO1 transcript were significantly decreased in un-treated Tat(+) when compared to un-treated Tat(−) cells (fig. 7, A). While GCLC transcription was not significantly increased by PB125 in Tat(+) or Tat(−) HPAEC, basal levels of GCLC were slightly elevated across all treatments in Tat(+) compared to Tat(−) cells (fig. 7B). We hypothesized that, due to Tat’s transcriptional modulatory abilities, Nrf2 transcription itself may be repressed by Tat. However, Nrf2 transcription was unaffected under all conditions between Tat(−) and Tat(+) HPAEC, indicating post-transcriptional mechanisms of Nrf2 inhibition by Tat (fig. 7C). Importantly, Tat concentrations observed in transfected HPAEC ranged from 4.39 to 97.62 ng/mL, well within the physiological range observed in HIV patients.
Figure 6. Effects of Tat on HO-1 and SRXN1 Expression in HPAEC.

(A) Tat represses HO-1 transcription in HPAEC. Cells were mock transfected or transfected with pTat-FLAG and incubated overnight with either regular growth medium or medium supplemented with vehicle (0.016% ethanol) or PB125 (8 mg/L). Total RNA was collected after overnight incubation and RT-qPCR was performed. Values shown as fold change relative to Tat(−) un-treated controls. Mean ± SEM of biological replicates (n = 6). (B) SRXN1 transcriptional induction is refractory to PB125 treatment in Tat expressing HPAEC. Experiment carried out as in A. Mean ± SEM of biological replicates (n = 6). (C-D) Induction of HO-1 Expression by PB125 is attenuated by Tat in HPAEC. Transfections and PB125 treatments were performed as in A. Total protein extracts were collected after overnight incubation for immunoblot analysis. Densitometry values expressed as relative intensity normalized to beta-actin. Mean ± SEM of biological replicates (n = 6). (**p ≤ 0.01 by unpaired student’s T-test) (UT = Un-treated, V = Vehicle, P/PB = PB125).
Figure 7. NQO1, GCLC, and Nrf2 Transcript levels in HPAEC.

(A) Tat represses NQO1 basally (B) GCLC transcription is unaffected by PB125 in HPAEC but is enhanced by Tat. (C) Nrf2 transcription is unaffected by PB125 and Tat in HPAEC. Cells were mock transfected or transfected with pTat-FLAG and either left untreated or were treated with vehicle (0.016% ethanol) or PB125 (8 mg/L) for 18 hours. RT-qPCR was performed from total RNA and normalized to GAPDH. Values are shown as fold change relative to Tat(−) un-treated controls. Mean ± SEM of biological replicates (n = 6) (*p ≤ 0.05, **p ≤ 0.01 by un-paired student’s T-test). (UT = Untreated, V = Vehicle, PB = PB125).
Discussion
The pulmonary disease burden in the HIV infected population in developed countries has undergone a shift from predominately infectious to non-infectious disease, and this shift can be largely ascribed to improvements in the development and implementation of anti-retroviral therapies (ART). Such diseases are associated with poor prognosis and clinical outcome due largely to poor screening/detection and limited therapeutic options, and include pulmonary arterial hypertension (PAH), COPD, and lung cancer. Common to these pathologies are chronic inflammation and increased oxidative burden in patients who often present with low viral load as a result of ART, implicating these phenotypes in the development of HIV associated non-infectious lung disease. Though there is an apparent lack of evidence for productive infection of the tissues and cell types involved in these pathologies, HIV transgenic animal models exhibit exacerbated systemic vascular inflammation, increased biomarkers of pulmonary oxidative stress, and vascular remodeling. The development of these pathological phenotypes in the absence of replication competent virus strongly support a role for exposure to viral proteins in the etiology of HIV associated pulmonary inflammatory disease, though the mechanisms are poorly understood. Identification of specific viral/host molecular interactions and pathophysiological consequences will facilitate a better understanding of how host cytoprotective responses are affected and which targeted therapeutic interventions might best serve this specific population.
The Nrf2/ARE pathway has been the target of a large body of research centered on understanding redox regulation under normal physiological conditions as well as in response to exogenous stressors and various disease states. These efforts have led to the discovery of several anti-oxidant and anti-inflammatory genes which are regulated by Nrf2 via a cis-acting regulatory element located in the promoter region known as the Anti-oxidant Response Element (ARE). Targeted activation of these genes through Nrf2 have demonstrated profound cytoprotective effects in animal models of lung disease, including PAH [28,29]. As such, the induction of these genes has been of intense therapeutic interest, and dysregulation of the Nrf2/ARE regulatory system continues to be implicated in the mechanisms driving such diseases [21,30]. Thus, the Nrf2 pathway provided an enticing target of research in regards to mechanisms by which soluble HIV factors may contribute to the pro-oxidant and pro-inflammatory environment of the pulmonary system of HIV patients. The HIV Transactivator of transcription (Tat) is an early HIV gene required for successful infection and is a critical viral transcriptional regulatory element. During infection, large amounts of Tat are secreted into circulation and are consequently internalized from the extracellular milieu by bystander cells due to the presence of a transducing peptide sequence in Tat’s basic domain. The pulmonary endothelium represents a target of particular interest due to its heavy involvement in inflammatory pulmonary diseases such as PAH as well as its direct proximity to potentially HIV infected cells and circulating Tat protein. As Tat is known to regulate host cellular transcriptional and post transcriptional processes, we sought to determine if Nrf2 activity is sensitive to Tat in primary Human Pulmonary Arterial Endothelial Cells (HPAEC). Here, we have demonstrated a profound effect of Tat on Nrf2/ARE regulated genes including HO-1, SRXN1, and NQO1 as well as an increase in cellular oxidative burden.
We found that in a constitutively Tat expressing cell line, Nrf2/ARE activity was actually enhanced compared to wild-type controls, and treatment with PB125 further augmented this effect. Accordingly, SRXN1 transcript levels were increased in Tat expressing cells, and this was further stimulated by PB125. Confoundingly, HO-1 expression was severely attenuated in Tat expressing cells both basally and in the presence of PB125, suggesting differential regulation of HO-1 and SRXN1 by Nrf2. In line with this observation, total oxidative burden was greater in Tat expressing cells and while treatment with PB125 slightly reduced 3-nitrotyrosine levels, it did not normalize them back to those of un-treated wild type controls.
As the pulmonary endothelium is heavily involved in non-infectious HIV associated lung disease, we sought to characterize the effect of Tat on the Nrf2/ARE regulatory network in primary Human Arterial Endothelial Cells (HPAEC). In HPAEC, Tat expression resulted in markedly lower Nrf2/ARE activity and exogenous activation of Nrf2 by PB125 failed to restore Nrf2 activity. Corresponding with repression of Nrf2/ARE activity, HO-1 transcription was strongly repressed by Tat in HPAEC, and while treatment with PB125 enhanced HO-1 expression, these levels were still significantly below those seen in Tat(−) HPAEC treated with PB125. While SRXN1 transcription was not basally attenuated, transcriptional induction by PB125 was repressed by Tat. NQO1 transcription was basally repressed by Tat although treatment with PB125 induced transcription to similar levels in both Tat(+) and Tat(−) HPAEC. Unexpectedly, GCLC transcription was unaffected by PB125 in all conditions, but was slightly enhanced in Tat(+) expressing cells. Taken together, these results suggest a predilection of Tat to interfere with global Nrf2 activity, with a particular predisposition for repression of HO-1.
Chronically infected HIV patients are at a dramatically increased risk of developing a number of lung complications, in particular HIV associated Pulmonary Arterial Hypertension (HIV-PAH) [31]. Once considered a relatively static selectively permeable barrier, the vascular endothelium has emerged as a dynamic and adaptive endocrine organ with sensory and effector capabilities, and is a critical regulator of vascular contractility, immune cell recruitment/extravasation, angiogenesis, and initiation and resolution of inflammatory responses [32,33]. Endothelial dysfunction is an early event in PAH and excess proliferation and impaired apoptotic response of pulmonary endothelial cells contributes to the obliteration of the pulmonary vasculature seen in HIV-PAH patients, resulting in eventual right ventricular failure due to dramatically increased pulmonary arterial resistance. In this study, we noted a marked repression of Nrf2/ARE activity and its target genes by the HIV Tat protein in human pulmonary arterial endothelial cells.
While we observed that various Nrf2/ARE driven genes were transcriptionally repressed or refractory to exogenous activation by PB125, the most dramatic response was seen in HO-1. HO-1 functions as both an antioxidant and mediator of inflammation and is induced by a plethora of exogenous stimuli including oxidative and xenobiotic stress. Interestingly, HO-1 knockout mice demonstrate increased systemic inflammation, hyper-susceptibility to oxidative stress, and right heart failure [34, 35]. Furthermore, it was demonstrated that the vascular protective effects of rampamycin are mediated by HO-1 in a monocrotaline rat model of PAH by eliciting anti-proliferative effects and attenuation of vascular remodeling [25]. Given that, in HPAEC, ARE activity was potently repressed by Tat as measured by an ARE driven luciferase construct, it is apparent that these genes are differentially regulated in the larger context of the Nrf2/ARE pathway. Transcriptional activation requires a coordinated response by various transcription factors and repressors. Small Maf proteins (sMafs) are a family of transcriptional co-factors that form heterodimers with “cap n’ collar” proteins including Nrf2, which cannot bind DNA as monomers [36]. sMafs also form heterodimers with the transcriptional regulator Bach1 [37]. Bach1 is primarily a transcriptional repressor of ARE driven genes and is a critical component in the maintenance of cellular redox homeostasis [38]. Nrf2 dimerization with different sMaf family members appears to confer specific activity. For instance, Nrf2/MafK dimerization confers transcriptional activation to the ARE driven gene NQO1, while other ARE genes such as NAPDH generating enzymes are sensitive to Nrf2/MafG dimerization [39]. It has also been demonstrated that Bach1/MafK heterodimers act to repress transcription of HO-1 in a feedback loop designed to maintain normal heme levels [40]. Tat is known to regulate a number of host transcription factors during HIV infection, and it is possible that Tat modulates the dimerization of various sMafs to different transcription factors. Tat my also occupy elements proximal to the ARE in the promoter such as enhancer elements and proximal AP-1 sequences that are unique to different ARE driven genes. sMAFs may also contribute to the discrepancy in responses seen between HeLa cells and HPAEC, as HeLa cells are a transformed cancerous cell line and aberrant expression of sMAFs is observed in a plethora of malignant cancers.
HIV utilizes an arsenal of virulence factors to modulate its host micro and macro environments, avoid immune detection and neutralization, and influence cellular function. Repression of Nrf2/ARE activity has been previously noted in alveolar macrophages infected with HIV or exogenously exposed to Tat and Gp120 by way of decreased Nrf2 expression [43]. Our findings support the observation that Tat is involved in impairment of the Nrf2/ARE transcriptional network. However, our results suggest an alternative/additional mechanism as we demonstrated that Nrf2 levels are unchanged by Tat in HPAEC though its downstream effectors are affected. The increased epidemiological incidence of PAH in the HIV infected population has been well established, and rates may in fact be currently under-represented due to poor screening practices. While the exact mechanisms driving these pathologies are not entirely understood, soluble HIV factors such as Tat, Nef, and Gp120 have been heavily implicated [41,42]. While Tat has been demonstrated to be a pro-oxidant and pro-inflammatory factor in-vitro and in-vivo, the mechanisms driving these effects remain elusive. Our observations that Tat represses Nrf2/ARE activity, as well is dysregulates a number of ARE driven genes including HO-1, SRXN1, and NQO1 sheds light on a novel mechanism by which Tat may contribute to pro-oxidant and pro-inflammatory states in the pulmonary system of chronically HIV infected patients.
Supplementary Material
Highlights.
Total oxidative burden is increased in cells expressing the HIV Transactivator of transcription (Tat) protein as quantified by electron paramagnetic resonance spectroscopy.
Nrf2/ARE activity is repressed by Tat in primary Human Pulmonary Arterial Endothelial Cells (HPAEC).
Key Nrf2/ARE driven anti-oxidant genes including HO-1 and NQO1 are repressed by Tat in HPAEC.
Nrf2/ARE activity is refractory to exogenous stimulation in HPAEC expressing Tat.
Acknowledgements:
This work was supported by the National Institutes of Health grants R01HL125050, R25HL103286, 1R35HL139726-01 (ENG), and the UCD CFRet fellowship award (HE).
Footnotes
Declarations of interest: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Unaids Global, AIDS Update 2016, (2016).
- [2].Triplette M, Crothers K, Attia EF, Non-infectious pulmonary diseases and HIV, Curr. HIV AIDS Rep. 13 (2016) 140–148. [DOI] [PubMed] [Google Scholar]
- [3].Langebeek N, et al. , Impact of comorbidity and ageing on health-related quality of life in HIV-positive and HIV-negative individuals, AIDS 31 (2017) 1471–1481. [DOI] [PubMed] [Google Scholar]
- [4].Presti RM, et al. , Mechanisms underlying HIV-associated noninfectious lung disease, Chest 152 (2017) 1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Almodovar S, Cicalini S, Petrosillo N, Flores SC, Pulmonary hypertension associated with HIV infection, Chest 137 (2010) 6S–12S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sitbon O, et al. , Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era, Am. J. Respir. Crit. Care Med 177 (2008) 108–113. [DOI] [PubMed] [Google Scholar]
- [7].Ivanov AV, et al. , Oxidative stress during HIV infection: mechanisms and consequences, Oxid. Med. Cell. Longev 2016 (2016) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Masiá M, et al. , Oxidative stress predicts all-cause mortality in HIV-infected patients, PLoS One 11 (2016) e0153456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cota-Gomez A, Flores AC, Ling XF, Varella-Garcia M, Flores SC, HIV-1 Tat increases oxidant burden in the lungs of transgenic mice, Free Radic. Biol. Med 51 (2011) 1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Reid W, et al. , An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction, Proc. Natl. Acad. Sci. Unit. States Am 98 (2001) 9271–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fiume G, et al. , Impairment of T cell development and acute inflammatory response in HIV-1 Tat transgenic mice, Sci. Rep 5 (2015) 13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Porter KM, et al. , Human immunodeficiency virus-1 transgene expression increases pulmonary vascular resistance and exacerbates hypoxia-induced pulmonary hypertension development, Pulm. Circ 3 (2013) 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Marciniak RA, Sharp PA, HIV-1 Tat protein promotes formation of more-processive elongation complexes, EMBO J. 1013 (1991) 4189–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Marciniak RA, Calnan BJ, Frankel AD, Sharp PA, HIV-1 Tat protein tram-activates transcription in vitro, Cell 63 (1990) 791–602. [DOI] [PubMed] [Google Scholar]
- [15].Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B, HIV-1 Tat Protein Exits from Cells via a Leaderless Secretory Pathway and Binds to Extracellular Matrix-Associated Heparan Sulfate Proteoglycans through its Basic Region, (1997). [DOI] [PubMed] [Google Scholar]
- [16].Debaisieux S, Rayne F, Yezid H, Beaumelle B, The ins and outs of HIV-1 Tat, Traffic 13 (2012) 355–363. [DOI] [PubMed] [Google Scholar]
- [17].Cota-Gomez A, et al. , The human immunodeficiency virus-1 Tat protein activates human umbilical vein endothelial cell E-selectin expression via an NF-κB-dependent mechanism, J. Biol. Chem 277 (2002) 14390–14399. [DOI] [PubMed] [Google Scholar]
- [18].Jiang Y, Chai L, Fasae MB, Bai Y, The role of HIV Tat protein in HIV-related cardiovascular diseases, J. Transl. Med 16 (2018) 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tebay LE, et al. , Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease, Free Radic. Biol. Med 88 (2015) 108–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Velichkova M, Hasson T, Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism, Mol. Cell. Biol 25 (2005) 4501–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Reddy SP, The antioxidant response element and oxidative stress modifiers in airway diseases, Curr. Mol. Med 8 (2008) 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McMahon M, Itoh K, Yamamoto M, Hayes JD, Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression, J. Biol. Chem 278 (2003) 21592–21600. [DOI] [PubMed] [Google Scholar]
- [23].Kapturczak MH, et al. , Heme oxygenase-1 modulates early inflammatory responses: evidence from the heme oxygenase-1-deficient mouse, Am. J. Pathol 165 (2004) 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Minamino T, et al. , Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia, Proc. Natl. Acad. Sci. U.S.A 98 (2001) 8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou H, et al. , Heme oxygenase-1 mediates the protective effects of rapamycin in monocrotaline-induced pulmonary hypertension, Lab. Investig 86 (2006) 62–71. [DOI] [PubMed] [Google Scholar]
- [26].Hybertson Brooks, Bifeng Gao, Bose Swapan, McCord J, Phytochemical Combination PB125 Activates the Nrf2 Pathway and Induces Cellular Protection against Oxidative Injury. Antioxidants in Submiss, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Elajaili HB, Hernandez-Lagunas L, Ranguelova K, Dikalov S, Nozik-Grayck E, Use of electron paramagnetic resonance in biological samples at ambient temperature and 77 K, J. Vis. Exp (2019), 10.3791/58461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cho H-Y, Reddy SP, Kleeberger SR, Nrf2 defends the lung from oxidative stress, Antioxidants Redox Signal. 8 (2006) 76–87. [DOI] [PubMed] [Google Scholar]
- [29].Chen Y, et al. , Activation of Nrf2 attenuates pulmonary vascular remodeling via inhibiting endothelial-to-mesenchymal transition: an insight from a plant polyphenol, Int. J. Biol. Sci 13 (2017) 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Singh S, Vrishni S, Singh BK, Rahman I, Kakkar P, Nrf2-ARE stress response mechanism: a control point in oxidative stress-mediated dysfunctions and chronic inflammatory diseases, Free Radic. Res 44 (2010) 1267–1288. [DOI] [PubMed] [Google Scholar]
- [31].Pulmonary hypertension in HIV, Can. J. Cardiol 35 (2019) 288–298. [DOI] [PubMed] [Google Scholar]
- [32].Durand MJ, Gutterman DD, Diversity in mechanisms of endothelium-dependent vasodilation in health and disease, Microcirculation 20 (2013) 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Norton K-A, Popel AS, Effects of endothelial cell proliferation and migration rates in a computational model of sprouting angiogenesis, Sci. Rep 6 (2016) 36992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yet SF, et al. , Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice, J. Clin. Investig 103 (1999) R23–R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Poss KD, Tonegawa S, Reduced stress defense in heme oxygenase 1-deficient cells, Proc. Natl. Acad. Sci. U.S.A 94 (1997) 10925–10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Katsuoka F, Yamamoto M, Small maf proteins (MafF, MafG, MafK): history, structure and function, Gene 586 (2016) 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Igarashi K, et al. , Multivalent DNA binding complex generated by small Maf and Bach1 as a possible biochemical basis for beta-globin locus control region complex, J. Biol. Chem 273 (1998) 11783–11790. [DOI] [PubMed] [Google Scholar]
- [38].Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK, Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants, J. Biol. Chem 280 (2005) 16891–16900. [DOI] [PubMed] [Google Scholar]
- [39].Hirotsu Y, et al. , Nrf2–MafG heterodimers contribute globally to antioxidant and metabolic networks, Nucleic Acids Res. 40 (2012) 10228–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sun J, et al. , Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network, Proc. Natl. Acad. Sci. Unit. States Am 101 (2004) 1461–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Marecki JC, et al. , HIV-1 Nef is associated with complex pulmonary vascular lesions in SHIV- nef –infected macaques, Am. J. Respir. Crit. Care Med 174 (2006) 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kanmogne GD, Primeaux C, Grammas P, Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins, Biochem. Biophys. Res. Commun 333 (2005) 1107–1115. [DOI] [PubMed] [Google Scholar]
- [43].Staitieh BS, Ding L, Neveu WA, Spearman P, Guidot DM, Fan X, HIV-1 decreases Nrf2/ARE activity and phagocytic function in alveolar macrophages, J. Leukoc. Biol 102 (2) (2017) 517–525, 10.1189/jlb.4A0616-282RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.