

Oligophrenin-1 (Ophn1) encodes a Rho GTPase activating protein whose mutations cause X-linked intellectual disability (XLID) in humans. Loss of function of Ophn1 leads to impairments in the maturation and function of excitatory and inhibitory synapses, causing deficits in synaptic structure, function and plasticity. Epilepsy is a frequent comorbidity in patients with Ophn1-dependent XLID, but the cellular bases of hyperexcitability are poorly understood.
Keywords: epilepsy, electrographic seizures, fasudil, GABAergic interneurons, inhibitory synapses
Abstract
Oligophrenin-1 (Ophn1) encodes a Rho GTPase activating protein whose mutations cause X-linked intellectual disability (XLID) in humans. Loss of function of Ophn1 leads to impairments in the maturation and function of excitatory and inhibitory synapses, causing deficits in synaptic structure, function and plasticity. Epilepsy is a frequent comorbidity in patients with Ophn1-dependent XLID, but the cellular bases of hyperexcitability are poorly understood. Here we report that male mice knock-out (KO) for Ophn1 display hippocampal epileptiform alterations, which are associated with changes in parvalbumin-, somatostatin- and neuropeptide Y-positive interneurons. Because loss of function of Ophn1 is related to enhanced activity of Rho-associated protein kinase (ROCK) and protein kinase A (PKA), we attempted to rescue Ophn1-dependent pathological phenotypes by treatment with the ROCK/PKA inhibitor fasudil. While acute administration of fasudil had no impact on seizure activity, seven weeks of treatment in adulthood were able to correct electrographic, neuroanatomical and synaptic alterations of Ophn1 deficient mice. These data demonstrate that hyperexcitability and the associated changes in GABAergic markers can be rescued at the adult stage in Ophn1-dependent XLID through ROCK/PKA inhibition.
SIGNIFICANCE STATEMENT In this study we demonstrate enhanced seizure propensity and impairments in hippocampal GABAergic circuitry in Ophn1 mouse model of X-linked intellectual disability (XLID). Importantly, the enhanced susceptibility to seizures, accompanied by an alteration of GABAergic markers were rescued by Rho-associated protein kinase (ROCK)/protein kinase A (PKA) inhibitor fasudil, a drug already tested on humans. Because seizures can significantly impact the quality of life of XLID patients, the present data suggest a potential therapeutic pathway to correct alterations in GABAergic networks and dampen pathological hyperexcitability in adults with XLID.
Introduction
Oligophrenin-1 (Ophn1) is a gene whose mutations cause X-linked intellectual disability (XLID) in humans. Ophn1 encodes for a Rho GTPase activating protein (RhoGAP) which negatively regulates Rac, RhoA, and Cdc42 (Billuart et al., 1998; Fauchereau et al., 2003; Khelfaoui et al., 2007). Ophn1 is expressed in several brain regions, including the cerebral cortex and the hippocampus, where it contributes to synapse maturation and plasticity (Govek et al., 2004; Khelfaoui et al., 2007; Powell et al., 2012, 2014). Ophn1 knock-out (KO) mice represent an excellent model of Ophn1 mutations in humans (Khelfaoui et al., 2007). These mice exhibit impairments in spatial memory and social behavior, alterations in adult neurogenesis, and defects in dendritic spines associated with altered synaptic plasticity (Khelfaoui et al., 2013; Meziane et al., 2016; Redolfi et al., 2016; Allegra et al., 2017; Zhang et al., 2017).
At the electrophysiological level, the loss of function of Ophn1 leads to alterations of both excitatory and inhibitory synaptic transmission. Patch-clamp recordings from the hippocampus of Ophn1 KO mice have shown reductions in evoked and spontaneous EPSCs and IPSCs (Powell et al., 2012, 2014). By contrast, the Ophn1 deficiency leads to an increased spontaneous activity in the medial prefrontal cortical (mPFC) neurons, where Zhang et al. (2017) found a higher frequency of EPSPs. Synaptic deficits were rapidly rescued by inhibition of Rho-associated protein kinase (ROCK)/protein kinase A (PKA), which are overactivated after loss of Ophn1 (Compagnucci et al., 2016; Meziane et al., 2016; Zhang et al., 2017). Gamma oscillations were also found to be reduced in Ophn1 deficient hippocampal slices, pointing to deficits in synaptic inhibition (Powell et al., 2014). However, a detailed analysis of inhibitory circuits in Ophn1-dependent XLID is still lacking. A growing body of evidence has highlighted the crucial role of GABAergic interneurons in the pathophysiology of XLID (Papale et al., 2017; Zapata et al., 2017). In this context, distinct subsets of GABAergic interneurons such as parvalbumin (PV)-positive basket cells, somatostatin (SOM)-positive cells and neuropeptide Y (NPY)-positive interneurons play distinct roles in fine-tuning and synchronization of the hippocampal network (Pelkey et al., 2017).
Epilepsy is a frequent comorbidity of Ophn1-dependent XLID and may significantly impact quality of life in the patients (Bergmann et al., 2003). The electrographic analysis has demonstrated seizure episodes as well as interictal epileptic activity in Ophn1-mutated subjects (Bergmann et al., 2003; des Portes et al., 2004; Santos-Rebouças et al., 2014). However, the mechanisms by which mutations in Ophn1 affect the balance between excitation and inhibition, with consequent cognitive impairment and network hyperexcitability remain still incompletely understood.
In subjects with refractory epilepsy and intellectual disability, the treatment of epileptic symptoms is normally performed by conventional anti-epileptic drugs (AEDs). However, these treatments can lead to adverse events and, importantly, a proportion of the patients remains pharmacoresistant (Jackson et al., 2015). This prompts the need for alternative treatments to reduce seizure burden and epileptic activity in subjects with XLID.
Lack of Ophn1 leads to high level of ROCK/PKA activity, and several pathological deficits of Ophn1 KO mice are at least partially rescued by treatment with ROCK/PKA inhibitor fasudil (Khelfaoui et al., 2013; Meziane et al., 2016; Redolfi et al., 2016; Allegra et al., 2017), an isoquinoline derivative drug approved for use in humans in China and Japan, and currently tested in multiple clinical trials in the United States and Europe.
In this manuscript, we have used recordings of local field potentials (LFPs) to describe the electrographic alterations of Ophn1 KO mice and their association with alterations in GABAergic hippocampal networks. We have also explored the possibility of rescuing the pathological hyperexcitability and GABAergic network defects induced by loss of function of Ophn1 via treatment with fasudil in adulthood.
Materials and Methods
Animals and treatment.
All experiments were performed in compliance with ARRIVE guidelines and the EU Council Directive 2010/63/EU on the protection of animals used for scientific purposes and were approved by the Italian Ministry of Health (authorization #738/2017-PR). All experiments and analyses were performed blind to the genotype and treatment. The animals used for all experiments were of the C57BL/6-J strain. Mice were housed in cages in a controlled environment (21°C and 60% of humidity) with 12 h/12 h light/dark cycle, with food and water available ad libitum. All experiments were performed using Ophn1−/y KO mice and Ophn1+/y wild-type (WT) littermates of 2 months of age, generated by breeding heterozygote females (Ophn1+/−) with WT males (Ophn1+/y). Because the Ophn1 gene is located on X chromosome, only male mice were used for our experiments because they develop X-linked ID, while females are not-affected carriers. The genotype was revealed through PCR analysis on small samples of tail tissue taken from pups at postnatal stage P10, as described by Khelfaoui et al. (2007) and Allegra et al. (2017).
The rescue experiments were performed by using the clinically approved drug fasudil, an inhibitor of ROCK/PKA signaling (Khelfaoui et al., 2013; Compagnucci et al., 2016; Meziane et al., 2016; Redolfi et al., 2016; Allegra et al., 2017). For chronic treatment, fasudil was administered for 7 weeks in drinking water (0.65 mg/ml; Allegra et al., 2017) to WT (n = 5) and KO animals (n = 8) at 2 months of age. Control animals received only water (WT, n = 5; KO, n = 7). These animals were recorded at the end of the treatment, and their brains used for countings of interneurons in the hilus. A second cohort of animals with chronic treatment (WT+water, n = 4; WT+fasudil, n = 4; KO+water, n = 4; KO+fasudil, n = 4) were used for interneuron counts in CA1 and for the analysis of synaptic boutons (see below). For acute treatment, after a baseline recording of epileptiform activity, KO mice (n = 7) were intraperitoneally injected with saline as control and the following day with fasudil (10 mg/kg). After 1 week (time necessary for wash out), mice were injected with a higher fasudil dose (25 mg/kg). The same batch of fasudil was proven to be effective in rescuing the neuroanatomical synaptic impairments of Ophn1 KO mice.
Kainic acid injection and behavioral analysis of seizures.
Seizures in adult male mice were evoked by intraperitoneal administration of kainic acid (KA) (stock solution, 2 mg/ml in PBS; administered at 10 mg/kg; Coremans et al., 2010; Corradini et al., 2014). We used a total of 15 mice for these experiments (WT, n = 8; KO, n = 7). Seizure severity was quantified by an observer blind to the mouse genotype using the following scale: stage 0, normal behavior; stage 1, immobility; stage 2, forelimb and/or tail extension, rigid posture; stage 3, repetitive movements, head bobbing; stage 4, sporadic clonus of forelimbs with rearing and falling (LMS, Limbic Motor Seizures); stage 5, continuous rearing and falling (status epilepticus); stage 6, severe whole-body convulsions; and stage 7, death. For each animal, behavior was scored every 10 min over a period of 2 h after KA administration. The maximum score reached by each animal over the entire observation period was used to calculate the maximum seizure score for each treatment group.
Placement of electrodes for LFP recordings.
For electrodes implants, mice were anesthetized by i.p. injection of avertin (20 ml/kg, 2,2,2-tribromoethanol 1.25%; Sigma-Aldrich) and mounted on a stereotaxic apparatus. A burr hole was drilled at stereotaxic coordinates corresponding to the hippocampus (anteroposterior −2.00 mm, mediolateral 1.50 mm to bregma). With the aid of a micromanipulator, a twisted steel wire bipolar electrode was lowered to a depth of 1.70 mm to reach the hippocampus. The wires were cut to have two end points with a distance of 0.5 mm that allows to integrate the electrical signal coming from two nearby regions of the hippocampus: the longer end from the dorsal blade of dentate gyrus while the shorter from the CA1 region. The voltage difference between the two ends was measured and compared with the ground electrode, i.e., a screw placed on the bone over the cerebellum. The electrodes and the reference were soldered to an electrical connector and the whole implant secured with dental acrylic cement.
LFP recordings.
LFP recordings were performed in freely moving mice for 2 h for three consecutive days between 10:00 A.M. and 6:00 P.M. and care was taken to record each animal at the same time of the day. The animals were placed in a recording chamber, where, after a 1 h habituation, LFP recording sessions were performed. For the acute treatment experiments, 2 daily sessions of 1 h baseline recordings were performed to habituate the animals to the recording chamber and to verify the stability of the signal. On the third day, after another 1 h of baseline signal acquisition, all animals received saline injection without being removed from the apparatus. Subsequent 2 h of recordings were acquired. The day after, after 1 h of baseline recording, animals were injected with fasudil (10 mg/kg in saline solution) and the effect was followed for 2 h. Finally, we left animals untreated for a week to allow drug washout and we repeated the same protocol with a higher dose of fasudil (25 mg/kg in saline solution).
Signals were acquired by a miniature head stage (NPI) connected to an amplifier (EXT-02F; NPI). Signals were amplified (10,000-fold), filtered (low pass, 100 Hz), digitized (National Instruments card), and conveyed to a computer for a storage and analysis. Detection of seizures was performed with custom software written in LabView (Antonucci et al., 2009; Mainardi et al., 2012; Cerri et al., 2016; Vannini et al., 2016). Data analysis was performed blind to experimental condition. The program first identified epileptiform alterations in the LFP using a voltage threshold. This voltage threshold was set to 4 times the SD of the signal. Every crossing of this threshold corresponded to a “spike” and spikes were considered to be clustered when separated by <1 s. Electrographic seizures were defined as repetitive spiking activity lasting for at least 4 s (Antonucci et al., 2009; Cerri et al., 2016). Spike clusters lasting <4 s and isolated spikes were considered as interictal events. Visual inspection revealed no obvious behavioral abnormalities during the electrographic seizure activity. For each recording session, we determined the frequency of electrographic seizures and interictal events, as well as the total time spent in electrographic seizures (calculated by adding together the duration of all paroxysmal episodes).
Immunohistochemistry.
Animals were deeply anesthetized with an overdose of chloral hydrate (10.5%, in saline) and then perfused through the heart with PBS, followed by paraformaldehyde (PFA) diluted at 4% in 0.1 m phosphate buffer, pH 7.4.
Brains were postfixed for 2 h (except for synaptic terminals analysis, in which postfixation was 30 min) and then cryoprotected in sucrose (30% in phosphate buffer). Brain coronal sections (50 micrometers) were obtained using a freezing microtome. Sections were cut in anteroposterior way, in a serial order, and kept in culture wells, in PBS solution at 4°C. Serial sections (one of six) were selected to perform immunohistochemical stainings.
Immunohistochemistry for counting of PV-positive and NPY-positive cells was performed on the same slices. Free floating sections were blocked for 1 h at room temperature (RT) with 10% normal goat serum (NGS), 0.3% Triton and PBS. Slices were then incubated overnight at 4°C with anti-PV and anti-NPY primary antibodies (respectively guinea pig, 1:1000, Synaptic Systems, #195004 and rabbit, 1:1000, Peninsula Laboratories, #T-4070), in a solution containing 2% NGS, 0.2% Triton and PBS. The primary antibodies were revealed by incubation for 2 h at RT with secondary antibodies (respectively anti-guinea pig Alexa Fluor 488, 1:500, Jackson ImmunoResearch, #706545148 and anti-rabbit Rhodamine Red X, 1:500, Jackson ImmunoResearch, #711025152) diluted in 1% NGS and 0.1% Triton X-100 and PBS. SOM somas were counted in the slices labeled for presynaptic terminals (see below).
For a further analysis of presynaptic terminals, double immunohistochemistry were performed with SOM and the vesicular GABA transporter (VGAT), or PV and synaptotagmin-2 (SYNT2). Free-floating sections were first blocked for 2 h at RT with 10% donkey serum (DS), 0.3% Triton in PBS. Then sections were incubated overnight at RT with primary antibodies (guinea pig anti-SOM, 1:500, Synaptic Systems, #366004; rabbit anti-VGAT, 1:500, Synaptic Systems, #131003; guinea pig anti-PV, 1:500, Synaptic Systems, #195004; rabbit anti-SYT2, 1:500, Synaptic Systems, #105223), 1% DS, 0.1% Triton X-100, and PBS. We then incubated the slices with the appropriate secondary antibodies, i.e., anti-rabbit rhodamine red X (1:500, Jackson ImmunoResearch, #711025152) and anti-guinea pig Alexa Fluor 488 (1:500, Jackson ImmunoResearch, #706545148) with 1% DS, 0.1% Triton X-100 and PBS.
Postsynaptic terminals were analyzed by performing double immunohistochemistry with SOM or PV and gephyrin (Geph). For Geph labeling (rabbit, 1:500, Synaptic Systems, #147018), the slices were preincubated in a citrate solution (10 mm, pH = 6) at 80°C for 30 min for antigen retrieval. After cooling down and washing the slices with PBS, immunohistochemistry was performed as explained above.
Stereological and cell density quantification.
Stereology was used to analyze the labeled cells in the hilus of dentate gyrus. As previously described in Allegra et al. (2017), no difference between WT and KO was found in the volume of the dentate gyrus. Cell countings were done blind to genotype and treatment. A fluorescence microscope (Zeiss Axioskop) with a 20× objective (air, 0.50 numerical aperture [NA]) and Stereoinvestigator software (MicroBrightField) were used to count the stained cells. For each staining, the analysis was performed in serial sections (one of six). All cells in the hilus/dentate gyrus of every sixth section were counted, and the resulting number of cells was multiplied by six to give an estimate of the total number of labeled cells. The analysis was performed in the same animals used for LFP recordings, in the hemisphere contralateral to the electrode implant. Three KO animals (one treated with water and two with fasudil) were excluded from the neuroanatomical analyses due to poor histology, which precluded stereological quantification.
Cell density analysis was performed to identify possible differences between interneurons in the hippocampal CA1 subregion. Cell counting was done blinded to genotype and treatment. Similarly to stereology, a fluorescence microscope (Zeiss Axioskop) with a 20× objective (air, 0.50 NA) and Stereoinvestigator software (MicroBrightField) were used to count the stained cells. For each staining (PV, NPY, SOM), the analysis was performed in serial coronal sections (one of six) as follows. Cells were counted within defined regions of interest (ROIs), drawn across the dorsal CA1 region (including the pyramidal layer, stratum oriens and stratum radiatum). The area of each ROI was multiplied by the thickness of the slice (∼50 μm) to obtain its volume, and the density was then calculated as the total number of-positive cells divided by the volume of each ROI.
Analysis of synaptic boutons formed by PV- and SOM-positive interneurons.
Representative images of PV- and SOM-positive boutons within the dentate gyrus and CA1 region were acquired with an epifluorescence microscope (Axio Imager.Z2, Zeiss) equipped with Apotome.2 (Zeiss), using a 63× objective (oil, 1.40 NA). For each experimental animal (n = 3–4 mice per treatment), 3–4 different sections were acquired with ZENpro software (Zeiss). By using this software, 3–4 z-series spaced by 1 μm intervals were imaged. Synaptic boutons were counted using the program Punta Analyzer plugin and the Fiji ImageJ software (Ippolito and Eroglu, 2010; Caleo et al., 2018), which allows the quantification of positive puncta for each channel (red and green channels) and their colocalization. Different synaptic markers (VGAT, SYNT2, Geph) were used to identify the presynaptic and postsynaptic component of PV- and SOM-positive synapses and the colocalization between them was analyzed. Images from both channels were thresholded to remove background levels and only the brightest puncta were retained. Analyses were performed blind to the experimental treatment. The size of the puncta to be analyzed was set at 15–500 square pixels. The number of double-labeled puncta was then calculated. The percentage of colocalization between the inhibitory synapse markers (green channel) and the presynaptic or postsynaptic markers (red channel) was determined by calculating the ratio between the number of the colocalised puncta and the total number of inhibitory synaptic boutons (green puncta).
Statistical analysis.
Statistical analysis was performed using SigmaPlot 12.0 software (Systat Software), setting the significance level at p < 0.05. We performed a pilot experiment to quantify LFP alterations in an initial cohort of KO animals with respect to WT (see Fig. 2). From the data of Figure 2C, we calculated an effect size of 1.22 and we used this value to extrapolate the minimum number of animals required to detect an improvement in the epileptiform alterations following fasudil treatment. We found that that a number of 4 animals per group was enough to have a power of > 80% in two-way ANOVA with four experimental groups and three degrees of freedom (df). We thus considered a minimum of 4 animals per group for electrophysiological characterization of the treatment. Power calculations were performed with G Power Software (version 3.1.5). To compare two groups, we used either Student's t test or Mann–Whitney rank-sum test, depending on whether the data were normally or not normally distributed and had or not equal variances. The trajectories in behavioral seizure scores of WT and KO animals (see Fig. 1A) were compared using two-way repeated-measures (RM) ANOVA, with genotype and time after KA as relevant factors. The same statistical test was also used to compare data obtained in the acute fasudil treatment (vehicle, fasudil 10 mg/kg and fasudil 25 mg/kg) (see Fig. 4). Two-way ANOVA (followed by appropriate post hoc tests) was used to compare electrographic and neuroanatomical alterations in WT+water, WT+fasudil, KO+water, KO+fasudil groups. All statistical analyses were performed on raw data (α value 0.05), if not differently indicated. Graphs were obtained using GraphPad Prism 6 software.
Figure 2.

Electrographic alterations in the hippocampus of Ophn1 KO mice. A, Representative LFP traces in WT and KO animals. Note high-amplitude spiking activity in the KO mouse. B, Spectrogram showing the increase of LFP oscillatory activity in a wide bandwidth during paroxysmal discharges in the KO mouse. The analysis has been performed on the LFP trace shown in A (bottom). C, Number of seizures per 10 min of recording in WT (n = 6) and KO (n = 8) animals. Seizure frequency is only detectable in KO animals (Ophn1+/y: 0.05 ± 0.03; Ophn1−/y: 0.92 ± 0.29, Mann–Whitney rank-sum test, p = 0.008). D, Time spent in ictal activity per 10 min of recording (Ophn1+/y: 0.23 ± 0.12; Ophn1−/y: 5.00 ± 1.55, Mann–Whitney rank-sum test, p = 0.008). E, Number of interictal events per 10 min of recording. Frequency of hippocampal short discharges (< 4 s) is significantly higher in KO animals compared with WT (Ophn1+/y: 16.09 ± 1.77; Ophn1−/y: 41.72 ± 7.82; Mann–Whitney rank-sum test, p = 0.003). Each point represents one animal. Histograms indicate mean ± SEM. **p < 0.01.
Figure 1.

Behavioral analysis of seizures in KA-injected Ophn1+/y and Ophn1−/y mice. A, Progression of behavioral changes after systemic KA injection (10 mg/kg i.p.) in WT (n = 8) and KO mice (n = 7) over a 2 h observation period. Data show an increased susceptibility to KA-induced seizures in KO mice (two-way RM ANOVA, p < 0.0001, followed by Tukey's test). All data are shown as mean seizure scores ± SEM. B, Maximum seizure score reached by each animal during 2 h of observation. Data show that there is a significant difference between WT and KO (Ophn1+/y: 3.00 ± 0; Ophn1−/y: 4.42 ± 0.52; Mann–Whitney rank-sum test, p = 0.02). Typical limbic motor convulsions (stages 4–5) are only detectable in KO mice. Horizontal lines indicate the mean of the group. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 4.
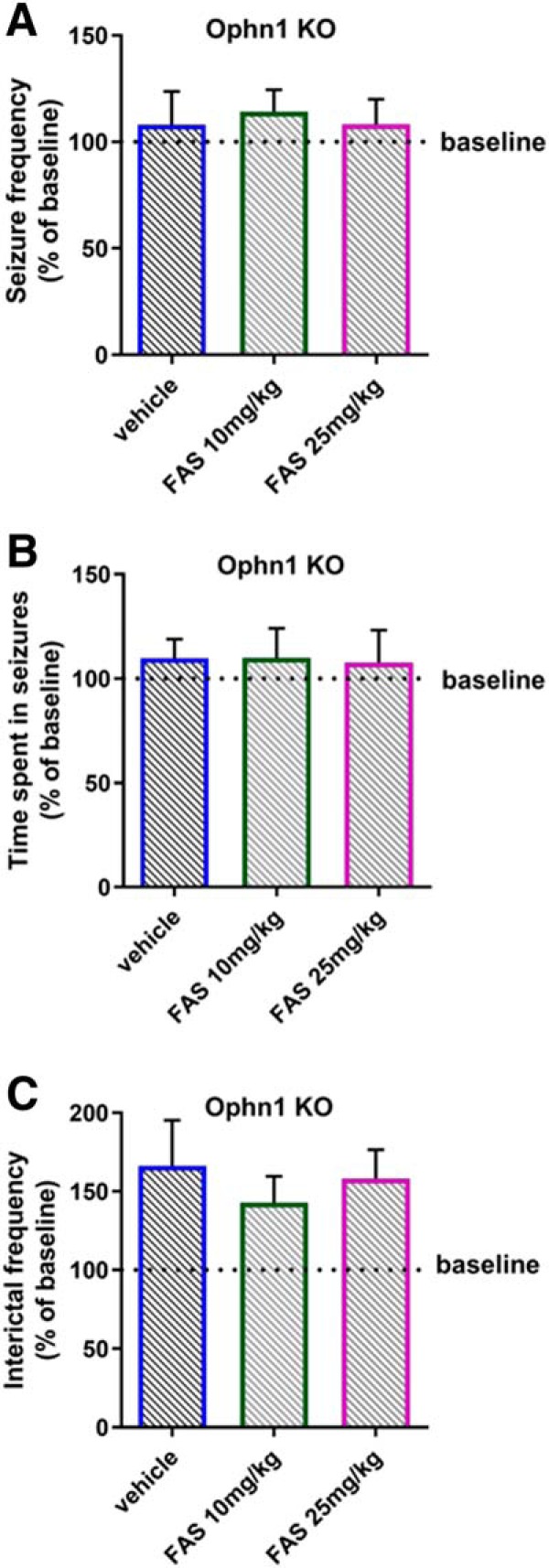
A single fasudil administration is not effective in rescuing the electrographic alterations in Ophn1 KO mice. Fasudil (FAS) was intraperitoneally administered in a group of KO mice (n = 7) at two different doses, 10 mg/kg and 25 mg/kg. Intraperitoneal injection of vehicle (saline solution) was used as control. No significant changes were found for the electrophysiological parameters: A, Number of seizures per 10 min of recording (two-way RM ANOVA, p = 0.203), B, Time spent in ictal activity per 10 min of recording (two-way RM ANOVA, p = 0.139), and C, Number of interictal events per 10 min of recording (two-way RM ANOVA, p = 0.404). Data are normalized on their baseline value (pretreatment condition) which is indicated in the plot by the dotted line. Histograms indicate mean ± SEM.
Results
Higher susceptibility to kainic acid-induced seizures in Ophn1 KO mice
We initially compared the susceptibility to evoked seizures in Ophn1−/y (KO) and Ophn1+/y (WT) mice. Ophn1+/y and Ophn1−/y animals were intraperitoneally injected with kainic acid (KA), a glutamate agonist, at a dose (10 mg/kg) that is normally not sufficient to induce generalized convulsions in WT C57BL6 mice (Coremans et al., 2010; Corradini et al., 2014; Testa et al., 2019). Mice behavior was observed for a period of 2 h by an experimenter blind to genotype. Both WT and KO mice showed preconvulsive behaviors during the first hour of observation, exhibiting immobility (stage 1), sharing and tail extension (stage 2), repetitive movements and head bobbing (stage 3).
Seizure progression was significantly different in Ophn1 KO animals with respect to controls, particularly during the second hour after KA. Indeed, five of seven KO mice exhibited limbic motor seizures with rearing and falling, forelimb clonus and praying posture (stage 4–5). Overall, the trajectory of behavioral score was shifted upward in KO mice (Fig. 1A; two-way RM ANOVA followed by Tukey's test, p < 0.0001).
The maximum seizure score reached by each animal during the 2 h of observations after KA administration is reported in Figure 1B. Such scores were significantly higher in the Ophn1 KO mice with respect to the control group, which showed only preconvulsive behaviors (stage 3) (Fig. 1B; Mann–Whitney rank-sum test, p = 0.02). Thus, Ophn1−/y animals exhibit an enhanced propensity to KA-induced behavioral seizures.
Spontaneous epileptiform spiking in the hippocampus of Ophn1 KO mice
We next checked spontaneous epileptiform activity in Ophn1−/y and Ophn1+/y mice. We performed in vivo recordings of LFPs from the hippocampus of freely moving animals (WT, n = 6; KO, n = 8), which were implanted with a chronic bipolar electrode. Representative recordings are shown in Figure 2A, and the power spectrogram for the LFP of the Ophn1−/y animal is reported in Figure 2B. We found an overall alteration of hippocampal LFPs in KO mice, which were characterized by high-amplitude spikes and sharp waves. Repetitive spiking activity was classified as either electrographic seizures (> 4 s) or short interictal discharges based on duration. The majority of KO animals displayed electrographic seizures (Fig. 2C), whereas no seizures could be detected in control animals. The difference in seizure frequency between WT and KO mice was highly significant (Fig. 2C; Mann–Whitney rank-sum Test, p = 0.008). Similarly, Ophn1−/y mice differed in the total time spent in electrographic seizures (Fig. 2D; Mann–Whitney rank-sum test, p = 0.008). We also analyzed the frequency of interictal events, and we found a higher frequency of interictal discharges in KO mice with respect to WT (Fig. 2E; Mann–Whitney rank-sum test, p = 0.003). Together with the higher susceptibility to KA-induced seizures, these findings indicate an overall hippocampal hyperexcitability in Ophn1 KO mice.
Pharmacological rescue of hippocampal network hyperexcitability in Ophn1 KO mice
Previous data have shown that several alterations caused by Ophn1 deficiency can be rescued by treatment with the ROCK/PKA inhibitor fasudil (Powell et al., 2012, 2014; Compagnucci et al., 2016; Meziane et al., 2016; Allegra et al., 2017). Based on these findings, we tested whether treatment with fasudil in drinking water for 7 weeks can rescue the pathological phenotypes of Ophn1 KO mice.
We examined four experimental groups of animals: WT mice treated with water (n = 5), WT mice treated with fasudil (n = 5), KO mice treated with water (n = 7) and KO mice treated with fasudil (rescue group, n = 8). Overall, we found that the epileptiform alterations were substantially reduced by treating KO mice with fasudil (Fig. 3A).
Figure 3.

Chronic fasudil treatment rescues hippocampal hyperexcitability in Ophn1 KO mice. Fasudil (FAS) was administered for 7 weeks in drinking water. Control animals received only water. At the end of this period, LFP recordings were performed. A, Representative traces showing LFP recordings obtained from the hippocampus of KO mice treated either with water (top) or fasudil (bottom). B, Number of seizures per 10 min of recording in WT (n = 5), WT with fasudil (n = 5), KO (n = 7), KO with fasudil (n = 8) animals. The presence of hippocampal discharges was consistently confirmed in this group of KO mice. Importantly, fasudil significantly reduced seizure frequency in KO animals (Ophn1−/y treated with water: 1.91 ± 0.29; Ophn1−/y treated with fasudil: 0.61 ± 0.28; two-way ANOVA followed by Tukey's test, p = 0.004). C, Time spent in ictal activity per 10 min of recording. KO animals treated with fasudil spent significantly less time in seizures than KO animals treated with water (Ophn1−/y treated with water: 11.03 ± 1.85; Ophn1−/y treated with fasudil: 3.40 ± 1.73; two-way ANOVA followed by Tukey's test, p = 0.007). D, Number of interictal events per 10 min of recording. Interictal discharges are significantly prevented in KO animals administered with fasudil (Ophn1−/y treated with water: 26.22 ± 2.59; Ophn1−/y treated with fasudil: 15.91 ± 2.43; two-way ANOVA followed by Tukey's test, p = 0.008). Each point represents one animal. Histograms indicate mean ± SEM. **p < 0.01; ***p < 0.001.
Quantification of epileptiform activity is reported in Figure 3B–D for the four groups of mice. The results obtained in the KO+water group were statistically coherent with the data reported in Figure 2 (t test, p = 0.124), confirming the reproducibility of the phenotype. We found that the frequency of spontaneous electrographic seizures was significantly decreased in KO+fasudil animals compared with KO+water group (Fig. 3B; two-way ANOVA followed by Tukey's test p = 0.004). A robust rescue effect of fasudil was also detected for the total time spent in seizures (Fig. 3C; two-way ANOVA followed by Tukey's test, p = 0.007). Fasudil treatment also dampened the frequency of interictal events (Fig. 3D; two-way ANOVA followed by Tukey's test, p = 0.008).
In the WT animals, fasudil treatment had no significant impact on the electrophysiological activity (Fig. 3B–D; WT+water vs WT+fasudil, two-way ANOVA p > 0.378 for all parameters). Altogether these results indicate that a prolonged inhibition of ROCK/PKA signaling via fasudil administration is effective in counteracting the hippocampal electrographic alterations caused by Ophn1 loss of function.
Acute fasudil delivery has no impact on electrographic alterations
Previous reports (Powell et al., 2012, 2014), have found that brief treatment with a ROCK inhibitor is effective in reversing electrophysiological deficits in hippocampal slices from Ophn1 KO mice, suggesting a direct effect on synaptic transmission. We therefore wondered whether seizure activity may be impacted by a single administration of fasudil. To this aim, we recorded hippocampal LFP in a subset of Ophn1 KO animals (n = 7) before and after acute delivery of either fasudil or saline as control. Fasudil was administered by intraperitoneal administration at two different doses (10 mg/kg and 25 mg/kg). Similar doses have been previously shown to reverse behavioral deficits during working memory (Swanson et al., 2017), and goal-directed decision making tasks (Zimmermann et al., 2017). We found that acute fasudil treatment was not effective to rescue the altered electrophysiological phenotype (Fig. 4). Indeed, number of seizures, total time spent in seizures and interictal activity were not different among the groups (two-way RM ANOVA p > 0.139 for all the parameters; the exact p-value is indicated in the legend). Overall, these results underline the importance of a long-term fasudil treatment to rescue the epileptiform alterations.
Pharmacological rescue of the number of GABAergic hippocampal interneurons in Ophn1 KO mice
Network hyperexcitability is typically associated with an alteration of the excitatory/inhibitory balance (Casalia et al., 2017; Di Cristo et al., 2018). Therefore, we performed a systematic analysis of the major GABAergic interneuron populations in the dentate gyrus and CA1 region, from which LFPs were sampled. We also assessed the impact of a prolonged fasudil treatment in both Ophn1−/y and Ophn1+/y mice. Counts of the total number of inhibitory interneurons was conducted in the same groups of animals used for the LFP recordings (see above), and the quantifications were performed within the hilus and CA1 region of the hippocampus contralateral to the electrode implant.
First, we analyzed the stereological number of NPY-, SOM-, and PV-positive cells in the hilus of dentate gyrus. For NPY, we found a statistically consistent decrease in the number of NPY interneurons in the hilus of KO+vehicle mice with respect to the control group (Fig. 5A,B; two-way ANOVA, followed by Tukey's test, p = 0.013). Notably, treatment with fasudil was effective in normalizing completely the total number of NPY-positive cells (Fig. 5A,B; KO+water vs KO+fasudil: two-way ANOVA followed by Tukey's test, p = 0.02). Treatment of WT mice with fasudil had no impact on the NPY-positive cell population (Fig. 5B; WT+water vs WT+fasudil: two-way ANOVA followed by Tukey's test p = 0.740).
Figure 5.

Reduction of NPY-positive interneurons in KO mice and rescue by fasudil administration in the hilus of hippocampus. A, Representative images showing examples of NPY labeling in the four group of animals in the hilus of hippocampus. Dotted lines define the region where interneurons were counted. DG: dentate gyrus. Scale bar, 100 μm. B, Number of NPY-positive cells in WT (n = 5), WT with fasudil (n = 5), KO (n = 6), and KO with fasudil (n = 6) animals. The data show a significant impairment in the number of NPY-positive cells in KO mice compared with WT animals (Ophn1+/y: 1408.80 ± 108.95; Ophn1−/y: 1000.00 ± 99.46; two-way ANOVA, followed by Tukey's test, *p = 0.013). This reduction was counteracted by 7 weeks of fasudil treatment in KO mice (Ophn1−/y treated with fasudil: 1361.67 ± 99.46; two-way ANOVA, followed by Tukey's test, *p = 0.02). All data are shown as mean ± SEM. C, Number of SOM-positive cells in WT (n = 5), WT with fasudil (n = 5), KO (n = 6) and KO with fasudil (n = 6). No significant difference was found between WT and KO animals (Ophn1+/y: 1107.60 ± 106.68; Ophn1−/y: 994.00 ± 97.39; two-way ANOVA, p = 0.413). Seven weeks of fasudil administration had no impact on the number of SOM-positive cells (Ophn1−/y treated with fasudil: 1213.00 ± 97.39; two-way ANOVA, p = 0.084). D, Number of PV-positive cells in WT (n = 5), WT with fasudil (n = 5), KO (n = 6), and KO with fasudil (n = 6). No significant difference was found between WT and KO groups of animals but only a tendency to decrease in KO animals (two-way ANOVA, p = 0.127). All data are shown as mean ± SEM.
We performed the same anatomical analysis on another class of hippocampal interneurons, namely SOM-positive cells. The results of immunohistochemical labeling with anti-SOM antibody demonstrated that the number of SOM-positive cells remained unaltered in both WT and KO mice, treated either with water or fasudil (Fig. 5C; two-way ANOVA, p = 0.413 and p = 0.084).
Next, we measured the density of PV-positive cells and the stereological counts revealed no significant alterations in the total number of such interneurons, although there was a tendency to reduction in Ophn1 KO mice (Fig. 5D; two-way ANOVA, p = 0.127).
We next focused on the hippocampal CA1 subregion and we found a significant decrease of PV interneurons in Ophn1−/y mice treated with water (Fig. 6A,B; WT+water vs KO+water: two-way ANOVA followed by Tukey's test p = 0.01). Fasudil administration for 7 weeks was able to restore a normal density of PV-positive cells. Indeed, fasudil-treated KO mice displayed a significant enhanced number of PV-labeled interneurons, compared with the KO mice treated with water (Fig. 6A,B; KO+water vs KO+fasudil: two-way ANOVA followed by Tukey's test, p = 0.007). Fasudil treatment in WT mice had no effect (Fig. 6A,B; WT+water vs WT+fasudil: two-way ANOVA followed by Tukey's test, p = 0.403).
Figure 6.

Decrease of PV-positive interneurons in KO mice and rescue by fasudil administration in the CA1 region of hippocampus. A, Representative images showing examples of PV labeling in the four groups of animals in the hippocampal CA1 subregion. Dotted lines define the region where cell counts were performed. s.o., Stratum oriens; s.r., stratum radiatum. Scale bar, 50 μm. B, Density of PV-positive interneurons in WT (n = 4), WT with fasudil (n = 4), KO (n = 4) and KO with fasudil (n = 4) (number of cells × mm3). A significant decrease of PV-positive cells was found in KO animals with respect to WT mice (Ophn1+/y: 1976.29 ± 233.32; Ophn1−/y: 1344.02 ± 46.06; two-way ANOVA, followed by Tukey's test, **p = 0.01). fasudil treatment significantly enhanced the density of PV-positive interneurons in KO animals (Ophn1−/y treated with fasudil: 2014.89 ± 116.62; two-way ANOVA, followed by Tukey's test, **p = 0.007). All data are shown as mean ± SEM. C, Density of NPY interneurons in the different groups. No significant difference was found between WT and KO animals (Ophn1+/y: 1969.76 ± 185.30; Ophn1−/y: 2160.32 ± 247.103; two-way ANOVA, p = 0.352). Seven weeks of fasudil administration had no impact on the number of NPY-positive cells (Ophn1−/y treated with fasudil: 1947.77 ± 293.66; two-way ANOVA p = 0.238). D, Density of SOM interneurons in the different groups. No significant difference was found between WT and KO groups of animals (two-way ANOVA, p = 0.830). All data are shown as mean ± SEM.
Considering the other two classes of interneurons, we found that their density remained unaltered in the CA1 region. In particular, the analysis of NPY cell density revealed no significant differences between WT and KO animals treated with water (Fig. 6C; two-way ANOVA followed by Tukey's test, p = 0.352). In the same way, fasudil administration was ineffective to determine any robust changes (Fig. 6C; two-way ANOVA followed by Tukey's test, p = 0.238).
Similarly, we demonstrated that SOM cells density in the CA1 region of the hippocampus was not affected by either genotype or drug treatment (Fig. 6D; two-way ANOVA followed by Tukey's test, p = 0.830).
Altogether, these data indicate that GABAergic interneurons display region-specific alterations in Ophn1 deficient mice. In particular, lack of Ophn1 causes a significant downregulation in NPY-positive cells in hippocampal hilus and a consistent decrease of PV-positive cells in the CA1 region. The population of SOM interneurons was instead not affected. Remarkably, administration of fasudil rescues the neuroanatomical impairments in GABAergic circuitry, normalizing the number of NPY- and PV-positive interneurons.
Pharmacological rescue of GABAergic synaptic impairments in Ophn1 KO mice
The anatomical alterations of GABAergic interneurons may be associated with impairments in the synaptic function of the circuit, leading to the hippocampal hyperexcitability observed in Ophn1 KO mice. Notably, rearrangements of inhibitory synapses have been demonstrated in human samples and animal models of neurodevelopmental disorders, including intellectual disability (Coghlan et al., 2012; Mircsof et al., 2015). To test this hypothesis in Ophn1−/y, we dissected the presynaptic and postsynaptic sites of the GABAergic inputs to granule and pyramidal cells in the dentate gyrus and CA1, respectively. In particular, we focused on the SOM- and PV-positive boutons because of their powerful inhibitory control on the excitatory neurons via dendritic (SOM) or perisomatic/axonal (PV) synapses (Klausberger and Somogyi, 2008).
We performed an initial analysis of the postsynaptic compartment in the dentate gyrus and CA1 of WT and Ophn1 KO mice, by costaining for either PV or SOM and gephyrin (Geph), a protein which anchors GABA receptors to the postsynaptic cytoskeleton (Schneider Gasser et al., 2006). Representative images of colocalization between PV/Geph and SOM/Geph in the dentate gyrus and CA1 region of hippocampus for both WT and KO mice are shown in Figure 7A and C. We found no significant differences in the colocalization of both PV and SOM boutons with postsynaptic Geph clusters (Fig. 7B,D, t test p > 0.325; the exact p-value is indicated in the legend), indicating no change at the postsynaptic level in these synapses.
Figure 7.

Normal postsynaptic phenotype of PV- and SOM-positive clusters in the dentate gyrus and CA1 of Ophn1 KO mice. A, Representative images of PV (green)-Geph (red) immunolabeling in the dentate gyrus (DG) and CA1 region of WT and KO animals. The dotted lines define the regions where analyses have been performed (for DG, s.m. refers to stratum moleculare, g.c,l. to granule cell layer, h. to hilus; for CA1, s.o. refers to stratum oriens; s.r. to stratum radiatum; p.l. to pyramidal layer). Scale bar, 25 μm. B, Colocalization between PV and Geph clusters in the dentate gyrus and CA1. The analysis revealed no substantial changes in KO animals compared with controls (DG, t test, p = 0.526; CA1, t test, p = 0.325). C, Representative images of SOM (green)-Geph (red) immunolabeling in the dentate gyrus (DG) and CA1 region of WT and KO animals. Abbreviations as above. Scale bar, 25 μm. D, Colocalization between SOM and Geph in the dentate gyrus and CA1. No significant difference was found between WT and KO animals (DG, t test, p = 0.893; CA1, t test, p = 0.776). All data are shown as mean ± SEM.
In contrast, robust changes were observed in the presynaptic compartment of both PV- and SOM-positive inputs to principal cells. Brain coronal sections were costained with a presynaptic marker to label the putative functional boutons. For PV, we used synaptotagmin 2 (SYNT2), a specific marker of PV terminals (Sommeijer and Levelt, 2012; Cameron et al., 2019) (Fig. 8A,B). We found an overall reduction of the double positive terminals in the dentate gyrus of KO animals treated with water (Fig. 8C; two-way ANOVA, followed by Tukey's test, p = 0.002). The loss of PV boutons was completely counteracted by long-term fasudil treatment (Fig. 8C; two-way ANOVA, followed by Tukey's test p < 0.001). It is noteworthy that the same rescue effects were detected in CA1 (Fig. 8D; two-way ANOVA, followed by Tukey's test, p < 0.001). WT mice treated treated with fasudil showed an enhancement of the PV-SYNT2 puncta, only in the CA1 region of the hippocampus (Fig. 8D; two-way ANOVA, followed by Tukey's test, p = 0.012).
Figure 8.

Alterations in presynaptic terminals of PV interneurons are rescued after 7 weeks of fasudil treatment. A, B, Representative images showing examples of PV (green)-SYNT2 (red) double labeling in the four groups of animals in the dentate gyrus (DG, A) and CA1 (B) region of hippocampus. Yellow puncta represent the sites of colocalization. The dotted lines define the regions where analyses have been performed. Abbreviations as in Figure 7. Scale bar, 25 μm. C, Percentage of colocalization between PV and SYNT2 in the four groups of animals in the dentate gyrus of hippocampus. A robust decrease of double positive boutons (PV-SYNT2) was observed in KO mice with respect to controls (Ophn1+/y: 48.85 ± 1.66; Ophn1−/y: 36.66 ± 4.75; two-way ANOVA, followed by Tukey's test, **p = 0.002). The data also indicated that fasudil treatment significantly rescued this impairment, by enhancing the percentage of colocalization (Ophn1−/y treated with fasudil: 52.11 ± 0.95; two-way ANOVA, followed by Tukey's test, ***p < 0.001). D, Percentage of colocalization between PV and Synt2 in the four groups of animals in CA1. A consistent reduction of double positive PV-SYNT2 boutons was also found in the CA1 region of KO animals compared with WT mice (Ophn1+/y: 53.30 ± 1.99; Ophn1−/y: 46.27 ± 3.73; two-way ANOVA, followed by Tukey's test, *p = 0.038). Administration of fasudil recovered a normal PV-SYNT2 colocalization (Ophn1−/y treated with fasudil: 59.68 ± 0.98; two-way ANOVA, followed by Tukey's test, ***p < 0.001). WT mice treated with fasudil also showed enhanced PV-SYNT2 double staining (62.04 ± 1.62; two-way ANOVA, followed by Tukey's test, *p = 0.012). All data are shown as mean ± SEM.
We also costained for SOM and VGAT (i.e., the vesicular GABA transporter) and surprisingly, we found that the density of SOM boutons was very significantly increased in both DG and CA1 of KO mice treated with water (Fig. 9A,C, DG: two-way ANOVA, followed by Tukey's test, p < 0.001; Fig. 9B,D, CA1: two-way ANOVA, followed by Tukey's test, p = 0.008). fasudil treatment was ineffective in WT but reduced to normal levels the density of SOM terminals in KO (Fig. 9C, DG: two-way ANOVA, followed by Tukey's test, p < 0.001; Fig. 9D, CA1: two-way ANOVA, followed by Tukey's test, p < 0.001).
Figure 9.

Alterations in presynaptic terminals of SOM interneurons are rescued after 7 weeks of fasudil treatment. A, B, Representative images showing examples of SOM (green)-VGAT(red) double labeling in the four groups of animals in the dentate gyrus (DG, A) and CA1 (B) region of hippocampus. The yellow puncta represent the sites of colocalization. The dotted lines define the regions where analyses have been performed. Abbreviations are as in Figures 7 and 8. Scale bar, 25 μm. C, Percentage of colocalization between SOM and VGAT in the four groups of animals in the dentate gyrus of hippocampus. We observed a significant increase of SOM-VGAT double positive boutons in KO animals compared with WT mice (Ophn1+/y: 15.93 ± 1.80; Ophn1−/y: 34.96 ± 4.77; two-way ANOVA, followed by Tukey's test, ***p < 0.001). Seven weeks of fasudil treatment had a robust impact in reducing the percentage of SOM-VGAT-positive terminals (Ophn1−/y treated with fasudil: 17.54 ± 0.79; two-way ANOVA, followed by Tukey's test, ***p < 0.001). D, Percentage of colocalization between SOM and VGAT in the four groups of animals in the CA1 region of hippocampus. KO animals showed an increased percentage of SOM-VGAT colocalization compared with controls (Ophn1+/y: 28.93 ± 3.85; Ophn1−/y: 42.14 ± 5.14; two-way ANOVA, followed by Tukey's test, **p = 0.008). This phenotype was recovered after fasudil treatment (Ophn1−/y treated with fasudil: 21.41 ± 1.53; two-way ANOVA, followed by Tukey's test, ***p < 0.001). All data are shown as mean ± SEM.
In summary, Ophn1 deficiency leads to a bidirectional regulation of presynaptic inhibitory terminals (i.e., a net loss of PV- and an increase in SOM-positive boutons) in the DG and CA1 which is rescued by chronic interference with ROCK/PKA signaling.
Discussion
In this manuscript, we investigated how Ophn1 deficiency affects the function and synaptic structure of the adult hippocampal circuit. In vivo LFP recordings from the hippocampus revealed high-amplitude spiking activity and spontaneous electrographic seizures in Ophn1−/y mice. Hyperexcitability was accompanied by alterations in specific subtypes of GABAergic interneurons and their synaptic inputs onto excitatory cells, in both the dentate gyrus and CA1 of Ophn1−/y animals. To the best of our knowledge, this is the first demonstration of circuit hyperexcitability in Ophn1-dependent XLID, highlighting its essential role in maintaining a correct excitatory-inhibitory balance. Importantly, a prolonged ROCK/PKA inhibition via fasudil treatment was effective in rescuing the pathological hyperexcitability and GABAergic network defects induced by loss of function of Ophn1, indicating a therapeutic strategy applicable in adulthood.
Patients with Ophn1 mutations often display epileptiform discharges (Bergmann et al., 2003; des Portes et al., 2004; Santos-Rebouças et al., 2014), but the underlying cellular and molecular mechanisms remain still incompletely understood. In animal models, loss of function of Ophn1 is known to lead to impairments in synaptic development and transmission, which may result in alterations in the excitatory-inhibitory ratio and consequent hyperexcitability. In the olfactory bulb, Ophn1 deficiency perturbs maturation and synaptogenesis of adult-born inhibitory granule cells acting via ROCK/PKA (Redolfi et al., 2016). Ophn1 KO mice have disrupted dendritic spines (Khelfaoui et al., 2007; Allegra et al., 2017), which may contribute to seizure propensity. The KO mice also display reduced synaptic vesicle availability, consistent with a role for Ophn1 and its interaction with endophilin A1 in clathrin-mediated endocytosis (Nakano-Kobayashi et al., 2009).
Here, we first showed a higher propensity to KA-induced seizures (Ben-Ari, 1985; Ben-Ari and Cossart, 2000; Bozzi et al., 2000; Costantin et al., 2005), in Ophn1−/y KO mice compared with WT. Moreover, by LFP recordings, we also revealed hippocampal hyperexcitability in Ophn1−/y mice, highlighted by the presence of high-amplitude spiking activity and electrographic seizures (Fig. 2). Quantification of electrographic seizures indicated some variability in the epileptic phenotype. This may be due to the short periods of LFP recordings that were analyzed (i.e., 1 h per day, during the light period) which preclude a complete description of the pathological phenotype. Despite this limitation, hippocampal hyperexcitability was consistently detected in distinct cohorts of Ophn1-deficient mice (Figs. 2, 3).
Only few electrophysiological reports are available concerning synaptic transmission in slices from Ophn1−/y mice (Powell et al., 2012, 2014; Zhang et al., 2017). These data can be useful to understand the possible causes underlying the epileptic phenotype. Using repetitive high-frequency stimuli, a facilitation of inhibitory currents has been observed in Ophn1+/y neurons but not Ophn1−/y cells. The origin of this presynaptic malfunction results from a difference in the readily releasable pool (RPP) of synaptic vesicles that is significantly smaller in KO animals compared with WT, suggesting a possible causal mechanism to our findings on epileptiform activity. Indeed the deficits in GABAergic synapses observed in Ophn1−/y slices may make more difficult to buffer the build-up of excitation during high-frequency activity, thus leading to hyperexcitability and seizures. Interestingly, it has been recently shown that the Ophn1 loss of function leads to a network hyperexcitability in other brain regions. For instance, the mPFC neurons displayed a slightly more depolarized membrane potential and an increase of spontaneous EPSP frequency, producing an “increased synaptic noise” responsible for the spatial working memory deficits in Ophn1 KO mice (Zhang et al., 2017).
Hippocampal hyperexcitability in Ophn1−/y mice was accompanied by an alteration of GABAergic markers. GABAergic interneurons are strongly implicated in different forms of intellectual disabilities and epilepsy (Marín, 2012; Allegra et al., 2014; Ledri et al., 2014; Stamboulian-Platel et al., 2016). Indeed, structural and functional abnormalities of GABAergic interneurons might represent the neuroanatomical substrate of an unbalanced excitation/inhibition ratio leading to seizure onset (Avoli and de Curtis, 2011; Gu et al., 2017). Here, the neuroanatomical analysis demonstrated that the Ophn1 deficiency produces region specific impairments. In particular, we observed a decrease in the total number of NPY-positive interneurons in the hilus of the hippocampus and a reduction of PV cell density in CA1 (Figs. 5, 6). Such changes are likely due to protein downregulation, or phenotypic changes (rather than interneuron cell death), as they can be fully rescued by fasudil administration. Specifically, the reduction of NPY expression may be causally related to hippocampal hyperexcitability in Ophn1-deficient mice, as NPY is a powerful anti-convulsant peptide. Its overexpression in the rat hippocampus results in significant reduction of seizures (Noè et al., 2008). Decreased numbers of NPY-positive interneurons have been described in different epilepsy models (e.g., Huusko et al., 2015).
We detected rearrangements at the level of GABAergic presynaptic inputs to excitatory cells in both the DG and CA1, while no differences were found at the postsynaptic sites. Analysis of distinct hippocampal subregions (DG and CA1) was consistent in indicating a differential regulation of the PV- and SOM-positive boutons on granule and CA1 pyramidal neurons. Specifically, the data were clear in indicating a reduction of the PV-SYNT2 double positive terminals, while the increase of SOM-VGAT colabeling may appear counterintuitive based on the electrographic phenotype of Ophn1 KO mice. Synapses formed by SOM- and PV-positive interneurons may be differentially regulated by Ophn1 deficiency because of their different molecular composition (Horn and Nicoll, 2018). Recent studies indicate a differential regulation of dendritic and somatic inhibition in the hippocampus of a mouse model of Down syndrome (Schulz et al., 2019). The loss of functional PV boutons may be crucial for predisposing to spontaneous seizures, as fast-spiking interneurons contact the soma and axon initial segment of pyramidal neurons, thus potently impacting on their firing frequency (Contreras et al., 2019). In this context, an enhancement of the density of functional synapses made by SOM interneurons on the dendrites might represent a compensatory change in the inhibitory network designed to buffer baseline hyperexcitability.
Altogether, our findings concur with electrophysiological evidence showing no alterations of the function of postsynaptic GABA receptors in Ophn1−/y mice (Powell et al., 2012), strengthening the hypothesis that the structural and functional presynaptic deficits in Ophn1 KO mice play a key role in dysregulating the excitation-inhibition balance in the hippocampal network.
ROCK/PKA inhibitors such as Y-27632 and fasudil have already been successfully used in Ophn1-dependent form of intellectual disability (Compagnucci et al., 2016; Meziane et al., 2016; Redolfi et al., 2016; Allegra et al., 2017). Moreover, these compounds have also shown anticonvulsant effects in specific seizure models (Inan and Büyükafşar, 2008; Kourdougli et al., 2015; Çarçak et al., 2018), suggesting that they could be used as an adjuvant or alternative therapy for refractory epilepsy. Here, based on in vitro results (Powell et al., 2012), we first investigated whether a single pharmacological treatment could abolish the enhanced excitability in Ophn1 KO mice in vivo, and we found that such treatment was not effective (Fig. 4). This evidence prompted us to evaluate the effectiveness of a prolonged fasudil treatment administered in the drinking water, over the course of 7 weeks (Meziane et al., 2016; Redolfi et al., 2016; Allegra et al., 2017). Strikingly, all the quantitative measures of epileptiform activity (electrographic seizure frequency, time spent in seizure and frequency of interictal discharges), as well as the neuroanatomical and synaptic GABAergic changes were completely normalized by this pharmacological treatment (Figs. 5, 6, 8, 9, and see summary in Table 1). The rescue of the aberrant phenotype in Ophn1 KO mice upon the prolonged fasudil administration (but not the single treatment) indicates that hyperexcitability is likely due to structural rearrangements in interneurons which take place across a well defined temporal window.
Table 1.
Summary of neuroanatomical changes in interneuron populations in Ophn1 KO mice, which are rescued by chronic Fasudil treatment
| Markers | Region | H2O | Fasudil |
|---|---|---|---|
| NPY-positive cells | DG | Decreased number | Rescue |
| PV-positive cells | CA1 | Decreased number | Rescue |
| PV-SYNT2 boutons | DG, CA1 | Decreased colocalization | Rescue |
| SOM-VGAT boutons | DG, CA1 | Increased colocalization | Rescue |
In this context, it remains to be determined whether the hyperexcitability phenotype is developmental in origin, as we have not performed LFP recordings at an early developmental stage. Precocious seizure onset may indeed alter hippocampal wiring during “critical periods” (Zhou et al., 2009) and disturb cognitive development of the affected individuals. Developmental defects lead to neuronal circuit alterations resulting in circuit hyperexcitability and susceptibility to seizures (Bozzi et al., 2012). In the case of Ophn1 loss of function, developmental changes may alter the contribution of various interneurons to the control of the excitation/inhibition ratio. ROCK/PKA inhibitors such as fasudil, which acts on RhoGTPase signaling, have a general plastic effect on cell cytoskeleton/morphology of various cells, including neurons and astrocytes. The finding that fasudil treatment in adulthood reduces hyperexcitability is of hope in therapeutic terms, as it indicates that at least some of the developmental changes can be reversed by drug therapy at a symptomatic stage. In Meziane et al. (2016), ROCK/PKA inhibition could rescue some but not all of the behavioral deficits, suggesting that an early therapeutic treatment may be more effective.
In summary, we have demonstrated hippocampal hyperexcitability and associated defects in specific GABAergic networks in the Ophn1 mouse model of XLID. These alterations can be significantly rescued by prolonged inhibition of ROCK/PKA signaling in adulthood. We obtained this rescue with ROCK/PKA inhibitor fasudil, a promising drug already tested on humans. Because seizures can significantly impact the quality of life of XLID patients, the present data suggest a potential therapeutic pathway to dampen pathological hyperexcitability in adults with XLID.
Footnotes
The authors declare no competing financial interests.
This work was funded by Telethon Foundation (project #GGP11116 to M.C.) and by Fondazione Cassa di Risparmio di Padova e Rovigo (Foundation Cariparo, project #52000 to M.C.). We thank Francesca Biondi (CNR Pisa) for animal care and Elena Novelli (CNR Pisa) for microscopy technical support.
References
- Allegra M, Genovesi S, Maggia M, Cenni MC, Zunino G, Sgadò P, Caleo M, Bozzi Y (2014) Altered GABAergic markers, increased binocularity and reduced plasticity in the visual cortex of engrailed-2 knockout mice. Front Cell Neurosci 8:163. 10.3389/fncel.2014.00163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegra M, Spalletti C, Vignoli B, Azzimondi S, Busti I, Billuart P, Canossa M, Caleo M (2017) Pharmacological rescue of adult hippocampal neurogenesis in a mouse model of X-linked intellectual disability. Neurobiol Dis 100:75–86. 10.1016/j.nbd.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonucci F, Bozzi Y, Caleo M (2009) Intrahippocampal infusion of botulinum neurotoxin E (BoNT/E) reduces spontaneous recurrent seizures in a mouse model of mesial temporal lobe epilepsy. Epilepsia 50:963–966. 10.1111/j.1528-1167.2008.01983.x [DOI] [PubMed] [Google Scholar]
- Avoli M, de Curtis M (2011) GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog Neurobiol 95:104–132. 10.1016/j.pneurobio.2011.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y. (1985) Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14:375–403. 10.1016/0306-4522(85)90299-4 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R (2000) Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci 23:580–587. 10.1016/S0166-2236(00)01659-3 [DOI] [PubMed] [Google Scholar]
- Bergmann C, Zerres K, Senderek J, Rudnik-Schoneborn S, Eggermann T, Häusler M, Mull M, Ramaekers VT (2003) Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain 126:1537–1544. 10.1093/brain/awg173 [DOI] [PubMed] [Google Scholar]
- Billuart P, Bienvenu T, Ronce N, des Portes V, Vinet MC, Zemni R, Roest Crollius H, Carrié A, Fauchereau F, Cherry M, Briault S, Hamel B, Fryns JP, Beldjord C, Kahn A, Moraine C, Chelly J (1998) Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 392:923–926. 10.1038/31940 [DOI] [PubMed] [Google Scholar]
- Bozzi Y, Vallone D, Borrelli E (2000) Neuroprotective role of dopamine against hippocampal cell death. J Neurosci 20:8643–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi Y, Casarosa S, Caleo M (2012) Epilepsy as a neurodevelopmental disorder. Front Psychiatry 3:19. 10.3389/fpsyt.2012.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caleo M, Spinelli M, Colosimo F, Matak I, Rossetto O, Lackovic Z, Restani L (2018) Transsynaptic action of botulinum neurotoxin type A at central cholinergic boutons. J Neurosci 38:10329–10337. 10.1523/JNEUROSCI.0294-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron S, Lopez A, Glabman R, Abrams E, Johnson S, Field C, Gulland FMD, Buckmaster PS (2019) Proportional loss of parvalbumin-immunoreactive synaptic boutons and granule cells from the hippocampus of sea lions with temporal lobe epilepsy. J Comp Neurol 527:2341–2355. 10.1002/cne.24680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Çarçak N, Yavuz M, Eryiğit Karamahmutoğlu T, Kurt AH, Urhan Küçük M, Onat FY, Büyükafsar K (2018) Suppressive effect of rho-kinase inhibitors Y-27632 and fasudil on spike-and-wave discharges in genetic absence epilepsy rats from Strasbourg (GAERS). Naunyn Schmiedebergs Arch Pharmacol 391:1275–1283. 10.1007/s00210-018-1546-9 [DOI] [PubMed] [Google Scholar]
- Casalia ML, Howard MA, Baraban SC (2017) Persistent seizure control in epileptic mice transplanted with gamma-aminobutyric acid progenitors. Ann Neurol 82:530–542. 10.1002/ana.25021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerri C, Genovesi S, Allegra M, Pistillo F, Püntener U, Guglielmotti A, Perry VH, Bozzi Y, Caleo M (2016) The chemokine CCL2 mediates the seizure-enhancing effects of systemic inflammation. J Neurosci 36:3777–3788. 10.1523/JNEUROSCI.0451-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan S, Horder J, Inkster B, Mendez MA, Murphy DG, Nutt DJ (2012) GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci Biobehav Rev 36:2044–2055. 10.1016/j.neubiorev.2012.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnucci C, Barresi S, Petrini S, Billuart P, Piccini G, Chiurazzi P, Alfieri P, Bertini E, Zanni G (2016) Rho kinase inhibition is essential during in vitro neurogenesis and promotes phenotypic rescue of human induced pluripotent stem cell-derived neurons with oligophrenin-1 loss of function. Stem Cells Transl Med 5:860–869. 10.5966/sctm.2015-0303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras A, Hines DJ, Hines RM (2019) Molecular specialization of GABAergic synapses on the soma and axon in cortical and hippocampal circuit function and dysfunction. Front Mol Neurosci 12:154. 10.3389/fnmol.2019.00154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coremans V, Ahmed T, Balschun D, D'Hooge R, DeVriese A, Cremer J, Antonucci F, Moons M, Baekelandt V, Reumers V, Cremer H, Eisch A, Lagace D, Janssens T, Bozzi Y, Caleo M, Conway EM (2010) Impaired neurogenesis, learning and memory and low seizure threshold associated with loss of neural precursor cell survivin. BMC Neurosci 11:2. 10.1186/1471-2202-11-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini I, Donzelli A, Antonucci F, Welzl H, Loos M, Martucci R, De Astis S, Pattini L, Inverardi F, Wolfer D, Caleo M, Bozzi Y, Verderio C, Frassoni C, Braida D, Clerici M, Lipp HP, Sala M, Matteoli M (2014) Epileptiform activity and cognitive deficits in SNAP-25+/- mice are normalized by antiepileptic drugs. Cereb Cortex 24:364–376. 10.1093/cercor/bhs316 [DOI] [PubMed] [Google Scholar]
- Costantin L, Bozzi Y, Richichi C, Viegi A, Antonucci F, Funicello M, Gobbi M, Mennini T, Rossetto O, Montecucco C, Maffei L, Vezzani A, Caleo M (2005) Antiepileptic effects of botulinum neurotoxin E. J Neurosci 25:1943–1951. 10.1523/JNEUROSCI.4402-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- des Portes V, Boddaert N, Sacco S, Briault S, Maincent K, Bahi N, Gomot M, Ronce N, Bursztyn J, Adamsbaum C, Zilbovicius M, Chelly J, Moraine C (2004) Specific clinical and brain MRI features in mentally retarded patients with mutations in the oligophrenin-1 gene. Am J Med Genet 124A:364–371. [DOI] [PubMed] [Google Scholar]
- Di Cristo G, Awad PN, Hamidi S, Avoli M (2018) KCC2, epileptiform synchronization, and epileptic disorders. Prog Neurobiol 162:1–16. 10.1016/j.pneurobio.2017.11.002 [DOI] [PubMed] [Google Scholar]
- Fauchereau F, Herbrand U, Chafey P, Eberth A, Koulakoff A, Vinet MC, Ahmadian MR, Chelly J, Billuart P (2003) The RhoGAP activity of OPHN1, a new F-actin-binding protein, is negatively controlled by its amino-terminal domain. Mol Cell Neurosci 23:574–586. 10.1016/S1044-7431(03)00078-2 [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Akerman CJ, Cross JR, Van der Veken L, Van Aelst L (2004) The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci 7:364–372. 10.1038/nn1210 [DOI] [PubMed] [Google Scholar]
- Gu F, Parada I, Shen F, Li J, Bacci A, Graber K, Taghavi RM, Scalise K, Schwartzkroin P, Wenzel J, Prince DA (2017) Structural alterations in fast-spiking GABAergic interneurons in a model of posttraumatic neocortical epileptogenesis. Neurobiol Dis 108:100–114. 10.1016/j.nbd.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn ME, Nicoll RA (2018) Somatostatin and parvalbumin inhibitory synapses onto hippocampal pyramidal neurons are regulated by distinct mechanisms. Proc Natl Acad Sci U S A 115:589–594. 10.1073/pnas.1719523115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huusko N, Römer C, Ndode-Ekane XE, Lukasiuk K, Pitkänen A (2015) Loss of hippocampal interneurons and epileptogenesis: a comparison of two animal models of acquired epilepsy. Brain Struct Funct 220:153–191. 10.1007/s00429-013-0644-1 [DOI] [PubMed] [Google Scholar]
- Inan S, Büyükafşar K (2008) Antiepileptic effects of two rho-kinase inhibitors, Y-27632 and fasudil, in mice. Br J Pharmacol 155:44–51. 10.1038/bjp.2008.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C (2010) Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp 45:2270. 10.3791/2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson CF, Makin SM, Marson AG, Kerr M (2015) Pharmacological interventions for epilepsy in people with intellectual disabilities. Cochrane Database Syst Rev 9:CD005399. 10.1002/14651858.CD005399.pub3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelfaoui M, Denis C, van Galen E, de Bock F, Schmitt A, Houbron C, Morice E, Giros B, Ramakers G, Fagni L, Chelly J, Nosten-Bertrand M, Billuart P (2007) Loss of X-linked mental retardation gene Oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J Neurosci 27:9439–9450. 10.1523/JNEUROSCI.2029-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelfaoui M, Gambino F, Houbaert X, Ragazzon B, Muller C, Carta M, Lanore F, Srikumar BN, Gastrein P, Lepleux M, Zhang CL, Kneib M, Poulain B, Reibel-Foisset S, Vitale N, Chelly J, Billuart P, Lüthi A, Humeau Y (2013) Lack of the presynaptic RhoGAP protein oligophrenin1 leads to cognitive disabilities through dysregulation of the cAMP/PKA signalling pathway. Philos Trans R Soc Lond B Biol Sci 369:20130160. 10.1098/rstb.2013.0160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P (2008) Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321:53–57. 10.1126/science.1149381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourdougli N, Varpula S, Chazal G, Rivera C (2015) Detrimental effect of post status epilepticus treatment with ROCK inhibitor Y-27632 in a pilocarpine model of temporal lobe epilepsy. Front Cell Neurosci 9:413. 10.3389/fncel.2015.00413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledri M, Madsen MG, Nikitidou L, Kirik D, Kokaia M (2014) Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci 34:3364–3377. 10.1523/JNEUROSCI.2734-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi M, Pietrasanta M, Vannini E, Rossetto O, Caleo M (2012) Tetanus neurotoxin-induced epilepsy in mouse visual cortex. Epilepsia 53:e132–e136. 10.1111/j.1528-1167.2012.03510.x [DOI] [PubMed] [Google Scholar]
- Marín O. (2012) Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci 13:107–120. 10.1038/nrn3155 [DOI] [PubMed] [Google Scholar]
- Meziane H, Khelfaoui M, Morello N, Hiba B, Calcagno E, Reibel-Foisset S, Selloum M, Chelly J, Humeau Y, Riet F, Zanni G, Herault Y, Bienvenu T, Giustetto M, Billuart P (2016) fasudil treatment in adult reverses behavioural changes and brain ventricular enlargement in oligophrenin-1 mouse model of intellectual disability. Hum Mol Genet 25:2314–2323. 10.1093/hmg/ddw102 [DOI] [PubMed] [Google Scholar]
- Mircsof D, Langouët M, Rio M, Moutton S, Siquier-Pernet K, Bole-Feysot C, Cagnard N, Nitschke P, Gaspar L, Žnidarič M, Alibeu O, Fritz A-K, Wolfer DP, Schröter A, Bosshard G, Rudin M, Koester C, Crestani F, Seebeck P, Boddaert N, et al. (2015) Mutations in NONO lead to syndromic intellectual disability and inhibitory synaptic defects. Nat Neurosci 18:1731–1736. 10.1038/nn.4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano-Kobayashi A, Kasri NN, Newey SE, Van Aelst L (2009) The rho-linked mental retardation protein OPHN1 controls synaptic vesicle endocytosis via endophilin A1. Curr Biol 19:1133–1139. 10.1016/j.cub.2009.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noè F, Pool AH, Nissinen J, Gobbi M, Bland R, Rizzi M, Balducci C, Ferraguti F, Sperk G, During MJ, Pitkänen A, Vezzani A (2008) Neuropeptide Y gene therapy decreases chronic spontaneous seizures in a rat model of temporal lobe epilepsy. Brain 131:1506–1515. 10.1093/brain/awn079 [DOI] [PubMed] [Google Scholar]
- Papale A, d'Isa R, Menna E, Cerovic M, Solari N, Hardingham N, Cambiaghi M, Cursi M, Barbacid M, Leocani L, Fasano S, Matteoli M, Brambilla R (2017) Severe intellectual disability and enhanced gamma-aminobutyric acidergic synaptogenesis in a novel model of rare RASopathies. Biol Psychiatry 81:179–192. 10.1016/j.biopsych.2016.06.016 [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ (2017) Hippocampal GABAergic inhibitory interneurons. Physiol Rev 97:1619–1747. 10.1152/physrev.00007.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AD, Gill KK, Saintot PP, Jiruska P, Chelly J, Billuart P, Jefferys JG (2012) Rapid reversal of impaired inhibitory and excitatory transmission but not spine dysgenesis in a mouse model of mental retardation. J Physiol 590:763–776. 10.1113/jphysiol.2011.219907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AD, Saintot PP, Gill KK, Bharathan A, Buck SC, Morris G, Jiruska P, Jefferys JG (2014) Reduced gamma oscillations in a mouse model of intellectual disability: a role for impaired repetitive neurotransmission? PLoS One 9:e95871. 10.1371/journal.pone.0095871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redolfi N, Galla L, Maset A, Murru L, Savoia E, Zamparo I, Gritti A, Billuart P, Passafaro M, Lodovichi C (2016) Oligophrenin-1 regulates number, morphology and synaptic properties of adult-born inhibitory interneurons in the olfactory bulb. Hum Mol Genet 25:5198–5211. 10.1093/hmg/ddw340 [DOI] [PubMed] [Google Scholar]
- Santos-Rebouças CB, Belet S, Guedes de Almeida L, Ribeiro MG, Medina-Acosta E, Bahia PR, Alves da Silva AF, Lima dos Santos F, Borges de Lacerda GC, Pimentel MM, Froyen G (2014) A novel in-frame deletion affecting the BAR domain of OPHN1 in a family with intellectual disability and hippocampal alterations. Eur J Hum Genet 22:644–651. 10.1038/ejhg.2013.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider Gasser EM, Straub CJ, Panzanelli P, Weinmann O, Sassoè-Pognetto M, Fritschy JM (2006) Immunofluorescence in brain sections: simultaneous detection of presynaptic and postsynaptic proteins in identified neurons. Nat Protoc 1:1887–1897. 10.1038/nprot.2006.265 [DOI] [PubMed] [Google Scholar]
- Schulz JM, Knoflach F, Hernandez MC, Bischofberger J (2019) Enhanced dendritic inhibition and impaired NMDAR activation in a mouse model of down syndrome. J Neurosci 39:5210–5221. 10.1523/JNEUROSCI.2723-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommeijer JP, Levelt CN (2012) Synaptotagmin-2 is a reliable marker for parvalbumin positive inhibitory boutons in the mouse visual cortex. PLoS One 7:e35323. 10.1371/journal.pone.0035323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamboulian-Platel S, Legendre A, Chabrol T, Platel JC, Pernot F, Duveau V, Roucard C, Baudry M, Depaulis A (2016) Activation of GABA A receptors controls mesiotemporal lobe epilepsy despite changes in chloride transporters expression: in vivo and in silico approach. Exp Neurol 284:11–28. 10.1016/j.expneurol.2016.07.009 [DOI] [PubMed] [Google Scholar]
- Swanson AM, DePoy LM, Gourley SL (2017) Inhibiting rho kinase promotes goal-directed decision making and blocks habitual responding for cocaine. Nat Commun 8:1861. 10.1038/s41467-017-01915-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa G, Mainardi M, Olimpico F, Pancrazi L, Cattaneo A, Caleo M, Costa M (2019) A triheptanoin-supplemented diet rescues hippocampal hyperexcitability and seizure susceptibility in FoxG1+/- mice. Neuropharmacology 148:305–310. 10.1016/j.neuropharm.2019.01.005 [DOI] [PubMed] [Google Scholar]
- Vannini E, Restani L, Pietrasanta M, Panarese A, Mazzoni A, Rossetto O, Middei S, Micera S, Caleo M (2016) Altered sensory processing and dendritic remodeling in hyperexcitable visual cortical networks. Brain Struct Funct 221:2919–2936. 10.1007/s00429-015-1080-1 [DOI] [PubMed] [Google Scholar]
- Zapata J, Moretto E, Hannan S, Murru L, Longatti A, Mazza D, Benedetti L, Fossati M, Heise C, Ponzoni L, Valnegri P, Braida D, Sala M, Francolini M, Hildebrand J, Kalscheuer V, Fanelli F, Sala C, Bettler B, Bassani S, et al. (2017) Epilepsy and intellectual disability linked protein Shrm4 interaction with GABABRs shapes inhibitory neurotransmission. Nat Commun 8:14536. 10.1038/ncomms14536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Aime M, Laheranne E, Houbaert X, El Oussini H, Martin C, Lepleux M, Normand E, Chelly J, Herzog E, Billuart P, Humeau Y (2017) Protein kinase A deregulation in the medial prefrontal cortex impairs working memory in murine oligophrenin-1 deficiency. J Neurosci 37:11114–11126. 10.1523/JNEUROSCI.0351-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP (2009) Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med 15:1208–1214. 10.1038/nm.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann KS, Yamin JA, Rainnie DG, Ressler KJ, Gourley SL (2017) Connections of the mouse orbitofrontal cortex and regulation of goal-directed action selection by brain-derived neurotrophic factor. Biol Psychiatry 81:366–377. 10.1016/j.biopsych.2015.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]