

The KMT2-Cre1-Hyd4 regulatory pathway modulates infection-related morphogenesis and pathogenicity in an insecticidal fungus.
Abstract
Entomopathogenic fungi can overcome insecticide resistance and represent promising tools for the control of mosquitoes. Better understanding of fungus-mosquito interactions is critical for improvement of fungal efficacy. Upon insect cuticle induction, pathogenic fungi undergo marked infection-related morphological differentiation. However, regulatory mechanisms of fungal infection–related morphogenesis are poorly understood. Here, we show that a histone lysine methyltransferase KMT2 in Metarhizium robertsii (MrKMT2) is up-regulated upon cuticle induction. MrKMT2 plays crucial roles in regulating infection-related morphogenesis and pathogenicity by up-regulating the transcription factor gene Mrcre1 via H3K4 trimethylation during mosquito cuticle infection. MrCre1 further regulates the cuticle-induced gene Mrhyd4 to modulate infection structure (appressorium) formation and virulence. Overall, the MrKMT2-MrCre1-MrHyd4 regulatory pathway regulates infection-related morphogenesis and pathogenicity in M. robertsii. These findings reveal that the epigenetic regulatory mechanism plays a pivotal role in regulating fungal pathogenesis in insects, and provide new insights into molecular interactions between pathogenic fungi and insect hosts.
INTRODUCTION
The increasing global threat of emerging and reemerging mosquito-borne diseases poses a serious human health problem. Malaria is the leading cause of human morbidity and mortality worldwide among mosquito-borne diseases, and the steady reduction in global malaria cases has stalled in the past 4 years (1). Because of the lack of vaccines or effective treatments against these human pathogens, vector control using chemical insecticides remains the major tool for combating mosquito-borne diseases (2). However, the growing threat of mosquito insecticide resistance requires the development of more sustainable vector control tools that can overcome insecticide resistance (3).
Fungal biopesticides are a promising environmentally friendly alternative to chemical insecticides. Entomopathogenic fungi such as Metarhizium robertsii and Beauveria bassiana are equally effective at killing insecticide-resistant and insecticide-susceptible mosquitoes and are considered the next-generation control agent for mosquito vectors (4, 5). However, the relatively slow action of fungal pathogens in comparison with that of chemical insecticides has hampered their widespread application (4). Development of approaches to improve fungal efficacy requires a better understanding of the molecular interactions between the fungi and mosquitoes (6, 7).
Entomopathogenic fungi are the only pathogens that can infect the insect host directly through the exoskeletons. During the course of infection in insects, the fungi undergo marked infection-related morphological differentiation and form a series of infection structures (8). One such infection structure, called appressorium, forms at the beginning of the infection by expansion of germ tube tips, which are produced during germination of conidia adhered to the hydrophobic surface of the insect. The appressoria promote the localized secretion of enzymes that degrade insect cuticle and build up turgor pressure to allow cuticle penetration by mechanical force. Then, the appressoria germinate into penetration pegs that break the host cuticle. After entering the insect hemocoel, the penetration pegs grow into the fungal hyphae, which further switch into hyphal bodies through yeast-type budding, for rapid proliferation and evasion of the host immune responses. Once the fungus depletes the hemocoelic nutrients, the fungal hyphae erupt through the insect cadaver and produce asexual conidia for the next infection cycle (8, 9).
The complex infection-related morphological transitions of the fungus require rapid and fine-tuning regulation of gene expression programs (9, 10). Epigenetic modifications allow rapid, reversible, and readily available phenotypic plasticity that can be directly shaped by both the pathogen and host selection pressures (11, 12). However, the epigenetic regulatory mechanism of fungal pathogenesis remains poorly understood. Histone lysine methylation is widely accepted as a critically important epigenetic mechanism, which controls the intricate regulatory networks of gene expression, and has been implicated in developmental switches and stress responses (13, 14). During an examination of B. bassiana transcriptome profiles over the course of infection in mosquitoes, we found a putative histone lysine methyltransferase gene, kmt2, whose expression substantially increased (about threefold) during cuticle infection. KMT2 is a homolog of Saccharomyces cerevisiae SET1 [Su(var)3-9, Enhancer-of-zeste, Trithorax], which specifically catalyzes methylation at lysine 4 of histone H3 (H3K4) (15). This indicates the potential role of KMT2-mediated H3K4 methylation in the regulation of fungal pathogenesis.
In the current study, we reveal that the KMT2-Cre1-Hyd4 regulatory pathway plays a crucial role in coordinated regulation of the infection-related morphogenesis and fungal pathogenicity against mosquitoes. We show that a histone lysine methyltransferase KMT2 in M. robertsii (MrKMT2) is up-regulated upon cuticle induction. MrKMT2 regulates infection structure (appressorium) formation and fungal virulence by targeting a transcription factor gene Mrcre1 via H3K4me3 modification. The up-regulated MrCre1 further regulates the expression of cuticle-induced gene hydrophobin 4 (hyd4) to modulate appressorium formation and fungal virulence. This regulatory pathway provides new insights into the epigenetic mechanism of fungal pathogenesis.
RESULTS
MrKMT2 is a histone methyltransferase whose expression increases in response to host cuticle
To assess whether MrKMT2 is likely to be a methyltransferase, we performed a phylogenetic analysis. We found that MrKMT2 is highly similar to homologs from another entomopathogenic fungus B. bassiana (EJP63881.1, 64.25% identity) and the phytopathogenic fungus Fusarium graminearum (SET1, ESU13710.1, 64.20% identity; Fig. 1A). Similar to other fungal KMT2-type histone methyltransferases, MrKMT2 contains five domains, including the catalytic SET domain, the RNA recognition motif, SET-associated domain (found only in fungi), N-SET, and post-SET domains (Fig. 1A). These findings indicate that MrKMT2 is likely a histone methyltransferase.
Fig. 1. Characterization of the H3K4 methyltransferase gene Mrkmt2 in M. robertsii ARSEF 2575.
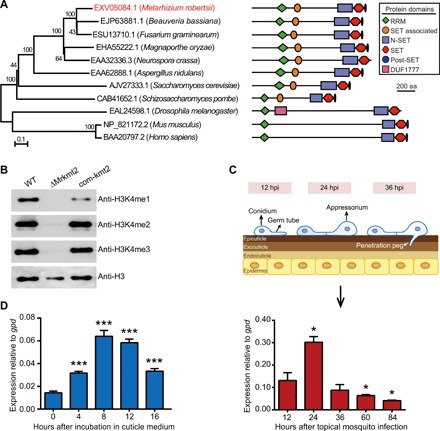
(A) Phylogenetic relationships and domain structures of MrKMT2 and its homologs. The amino acid sequences were aligned with ClustalW, and a neighbor-joining tree was generated with 1000 bootstrap replicates using the program MEGA v7.0. Protein domain structure is shown on the right. aa, amino acid. (B) Western blotting analysis of histone modifications in the WT, deletion mutant (ΔMrkmt2), and complemented strain (com-kmt2). Histones extracted from M. robertsii mycelium grown in the MM medium were resolved by 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and probed with specific antibodies against H3K4me1, H3K4me2, H3K4me3, and a C-terminal peptide of histone H3. Representative blot from three independent experiments is shown. (C) Top: Schematic diagram of the fungal appressorium differentiation on the host cuticle. Bottom: Real-time quantitative PCR (qPCR) analysis of Mrkmt2 expression during topical infection of A. stephensi with WT fungus. (D) qPCR analysis of Mrkmt2 expression during incubation in MM medium supplemented with 0.8% locust cuticle. Data are shown as means ± SD of three technical replicates. One and triple asterisks represent significant differences compared with that at 12 hours after topical mosquito infection (C) and 0 hour after incubation in cuticle medium (D) determined by Student’s t test at P < 0.05 and P < 0.001, respectively. gpd was used as a reference gene.
To determine the catalytic function of MrKMT2, we constructed an Mrkmt2 deletion mutant (ΔMrkmt2; fig. S1). Western blotting analysis indicated that H3K4me1 and H3K4me3 were present in wild-type (WT) fungus but were not detected in the ΔMrkmt2 strain (Fig. 1B). In addition, H3K4me2 levels were drastically reduced in ΔMrkmt2 compared with the WT strain. The defects in H3K4 methylation in ΔMrkmt2 were restored in the complemented strain com-kmt2 (Fig. 1B). These observations indicated that MrKMT2 is involved in H3K4 mono-, di-, and trimethylation in M. robertsii.
Next, we studied the expression of Mrkmt2 during M. robertsii topical infection of Anopheles stephensi. Mrkmt2 expression significantly increased during appressoria formation [24 hours post infection (hpi)] compared with 12 hpi (germination), followed by a marked decrease in gene expression at 36 hpi when the fungus penetrates the cuticle (Fig. 1C). Furthermore, we use the minimal medium (MM) supplemented with 0.8% insect cuticle, which mimics the nutrient-poor insect epicuticle, to activate the fungal infection system (16, 17). We found that Mrkmt2 was highly induced at 8 hours after inoculation (Fig. 1D). These results indicated that Mrkmt2 is up-regulated in response to the host cuticle.
Mrkmt2 is required for appressorium formation and fungal pathogenicity
To determine the biological functions of Mrkmt2, we analyzed fungal growth and pathogenicity of the ΔMrkmt2 strain. Deletion of Mrkmt2 resulted in a significant reduction in radial growth on potato dextrose agar (PDA) and conidia production relative to WT (fig. S2, A to C), indicating that MrKMT2 is required for asexual development in M. robertsii. However, conidia germination was not affected by Mrkmt2 deletion (fig. S2, D and E). An insect bioassay revealed that mosquitoes died significantly more slowly after topical inoculation with the ΔMrkmt2 strain [median lethal time, LT50 = 5.8 ± 0.3 days] than after topical inoculation with the same amount of conidia of WT strain (LT50 = 3.2 ± 0.1 days; P < 0.01, t test; Fig. 2, A and B), indicating that fungal virulence was significantly reduced in the ΔMrkmt2 strain. The germination rate of the ΔMrkmt2 strain on cicada hindwings was similar to that of the WT, but the mutant formed far fewer typical swollen appressoria than the WT (Fig. 2, C and D). Furthermore, the ΔMrkmt2 strain failed to produce hyphal bodies in the insect hemocoel within 72 hpi (Fig. 2, E and F), which might be attributed to cuticle penetration defects triggered by fewer appressoria numbers. These phenotypic defects were restored in the complemented strain com-kmt2. Collectively, these observations indicated that MrKMT2 plays a pivotal role in infection-related morphogenesis in M. robertsii.
Fig. 2. ΔMrkmt2 exhibits severe defects in fungal pathogenicity.
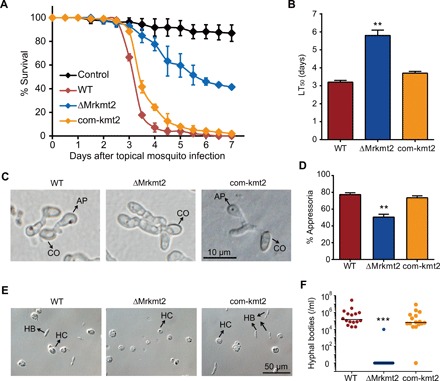
(A) Survival of A. stephensi female adults following topical application of suspensions of 1 × 107 conidia/ml of the WT, ΔMrkmt2, and com-kmt2 strains. Control mosquitoes were treated with 0.01% Triton X-100. Each treatment was replicated three times, with 50 mosquitoes per replicate. (B) LT50 values of WT, ΔMrkmt2, and com-kmt2 strains against mosquitoes. (C) Microscopic observation of fungal appressoria induced by the cicada hindwings. CO, conidium; AP, appressorium. (D) Percentage of fungal conidia forming appressoria relative to total conidia induced in MM2-Gly on hydrophobic surfaces in plastic plates. One hundred conidia per plate were counted. Microscopic observation (E) and quantification (F) of fungal hyphal bodies in the silkworm hemocoel 72 hours after topical infection with 1 × 107 conidia/ml of the WT, ΔMrkmt2, and com-kmt2 strains. Sixteen silkworms were detected for each treatment. HB, hyphal body; HC, hemocyte. Data are shown as means ± SD of three technical replicates. Double and triple asterisks represent significant differences in mutants compared with that in WT determined by Student’s t test at P < 0.01 and P < 0.001, respectively. The experiments were repeated three times with similar results.
MrKMT2 regulates expression of M. robertsii genes that are induced by host cuticle
Since we had observed ΔMrkmt2 defect in appressorium formation during host cuticle infection, and expression of the genes involved in this process is known to be induced upon insect cuticle (17, 18), we decided to investigate the role of MrKMT2 in fungal transcriptional regulation upon exposure to the cuticle. We analyzed global gene expression in the WT and ΔMrkmt2 strains grown in Sabouraud dextrose broth (SDB; before cuticle induction) and further incubation in MM supplemented with 0.8% locust cuticle, mimicking cuticle induction, for 8 hours, i.e., the time point at which Mrkmt2 exhibited the highest expression (Fig. 1D). We expected that MrKMT2 would perform the most pronounced regulation of cuticle-induced genes at the time point at which it is most active.
Upon cuticle induction, 2425 (19.4%) and 2954 (23.7%) genes showed at least 1.5-fold up-regulation and down-regulation in the WT grown in MM with cuticle compared with expression in the WT grown in SDB, respectively (Fig. 3A). Comparison of the fold induction and repression of the differentially expressed genes (DEGs) in the ΔMrkmt2 and WT strains showed that, in the mutant strain, 770 and 728 genes were induced or suppressed less than 50% of the WT level; these genes were considered MrKMT2 dependent (Fig. 3A). Therefore, approximately 27% of the transcriptional changes in cuticle-induced genes were directly or indirectly dependent on MrKMT2 regulation.
Fig. 3. Global transcriptional changes and their correlation with histone modifications.
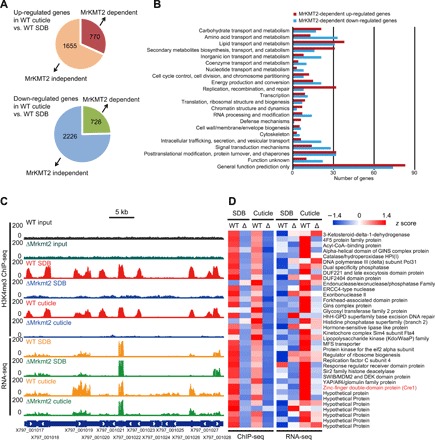
The WT and ΔMrkmt2 strains grown in the SDB and MM medium supplemented with 0.8% locust cuticle were analyzed by a combined RNA-seq and H3K4me3 ChIP-seq analysis. (A) Schematic diagram showing the number of differentially expressed genes (DEGs) after cuticle induction in an MrKMT2-dependent manner. On the basis of the comparison of fold change values (fold change values of gene expression level in the cuticle-induced culture relative to the SDB culture) between the WT and ΔMrkmt2 strains, MrKMT2-dependent genes were identified when the rate of fold change increase or decrease in the ΔMrkmt2 strains was less than 50% that of the WT; the remaining genes were classified as MrKMT2 independent. (B) KOG functional classification of MrKMT2-dependent up-regulated and down-regulated genes. (C) Representative genome browser view of the enrichment of H3K4me3 and mRNA signals in WT and ΔMrkmt2 strains grown in the SDB and MM media supplemented with 0.8% locust cuticle. (D) Heat map showing the H3K4me3 enrichment at transcription start site ±1 kb of 37 directly H3K4me3-modified genes whose up-regulated expression was dependent on MrKMT2 (left) and their corresponding transcription patterns (right). The zinc-finger double-domain–containing transcription factor Cre1 is labeled in red.
Functional analysis using eukaryotic orthologous group (KOG) database revealed that some of the MrKMT2-dependent up-regulated genes were overrepresented in the KOG functional categories of “lipid transport and metabolism,” “carbohydrate transport and metabolism,” and “secondary metabolite biosynthesis, transport, and metabolism” (Fig. 3B). These MrKMT2-dependent up-regulated genes may be involved in the degradation of the insect epicuticle, which consists of complex mixtures of nonpolar lipids, including hydrocarbons, fatty acids, and wax esters (19). By contrast, the KOG functional categories overrepresented among the MrKMT2-dependent down-regulated genes were involved in “amino acid transport and metabolism,” “inorganic ion transport and metabolism,” “intracellular trafficking, secretion, and vesicular transport,” “signal transduction mechanisms,” and “posttranslational modification, protein turnover, and chaperones” (Fig. 3B). Collectively, the RNA sequencing (RNA-seq) analysis revealed that almost a third of cuticle-induced genes are regulated by MrKMT2, highlighting the role of this gene in regulation of fungal gene expression upon cuticle induction.
MrKMT2-dependent transcriptional changes are associated with H3K4me3 modification
Next, to examine the global MrKMT2-mediated H3K4me3 modification, we performed chromatin immunoprecipitation (ChIP) using H3K4me3 antibody followed by DNA sequencing [ChIP sequencing (ChIP-seq)], and profiled the chromatin landscape of H3K4me3 in the WT and ΔMrkmt2 strains grown in the SDB and cuticle induction media. Genome-wide analysis revealed that H3K4me3 was predominantly localized in the 5′ region of the genes (Fig. 3C and fig. S3A).
We then compared the ChIP-seq results to those of the RNA-seq analysis to identify target genes whose changes in H3K4me3 levels are associated with changes in gene expression levels. Globally, H3K4me3 levels were higher in genes with high expression levels than in genes with medium or lower expression levels in both SDB and cuticle-induced cultures (fig. S3B), indicating that H3K4me3 is associated with active gene expression in M. robertsii. When we compared the H3K4me3 and gene expression levels in the ΔMrkmt2 and WT strains grown in SDB, we detected 2722 genes with notably reduced methylation in ΔMrkmt2. Among these, the expression of 795 and 77 genes was down-regulated and up-regulated, respectively, in the ΔMrkmt2 strain (1.5-fold change; fig. S4). Similarly, among 2987 genes with notably reduced H3K4me3 modification in the ΔMrkmt2 cuticle-induced cultures compared with H3K4me3 levels in the WT cuticle-induced cultures, 405 and 97 genes were down- and up-regulated, respectively, compared with their expression in the WT cuticle-induced cultures (1.5-fold change; fig. S4). In total, 1150 genes were identified as MrKMT2 targets whose transcription was regulated by H3K4me3 (fig. S4).
To assess whether MrKMT2-dependent transcriptional changes upon cuticle induction occur via H3K4me3 modification, we compared the MrKMT2-dependent cuticle-induced genes with MrKMT2 target genes. We found that among the MrKMT2-dependent genes, only a small number were MrKMT2 target genes (37 of 770 MrKMT2-dependent up-regulated genes and 184 of 728 MrKMT2-dependent down-regulated genes; Fig. 3D and fig. S5). The ChIP-seq and RNA-seq data for six randomly selected target genes (fig. S6A) were validated by ChIP with quantitative polymerase chain reaction (ChIP-qPCR) (fig. S6B) and reverse transcription (RT)–qPCR analysis (fig. S6C). These results indicated that the majority of MrKMT2-dependent genes are not directly regulated by H3K4me3.
MrKMT2 regulates infection-related morphogenesis and fungal pathogenicity via MrCre1
Because H3K4me3 is mainly associated with active transcription, we speculated that MrKMT2-mediated H3K4me3 might directly activate other important regulatory factors to indirectly regulate the transcription of a large number of downstream cuticle-induced genes. Among the 37 MrKMT2-dependent cuticle-induced genes that are targets of H3K4me3 (Fig. 3D), we identified the transcription factor Cre1 (named here MrCre1), a zinc-finger double-domain DNA binding protein (EXV01725.1; fig. S7). In addition to functioning as a carbon catabolite repressor, Cre1 plays important roles in nutrient utilization, cell homeostasis, and virulence in B. bassiana (20).
By ChIP-seq analysis, we found that pronounced H3K4me3 peaks existed in the cre1 gene in the WT strain and lost in the ΔMrkmt2 strain (Fig. 4A). Consistent with the ChIP-seq data, ChIP-qPCR revealed no H3K4me3 modification in Mrcre1 in SDB and cuticle-induced cultures of the ΔMrkmt2 strain in comparison with that in WT strain (Fig. 4B). The expression of the Mrcre1 gene was notably up-regulated in both WT and ΔMrkmt2 fungal cells exposed to the insect cuticle, but the induction was lower in the ΔMrkmt2 strain confirming MrKMT2-dependent expression (Fig. 4A). Similarly, RT-qPCR confirmed that the expression of Mrcre1 was significantly decreased in the ΔMrkmt2 strain grown in the SDB and cuticle induction media compared with that in the WT strain, as observed by RNA-seq analysis (Fig. 4C). These results confirmed that Mrcre1 is induced in cuticle-supplemented medium and that this induction is dependent on MrKMT2-mediated H3K4me3. Then, we analyzed the expression patterns of Mrcre1 during M. robertsii topical infection of A. stephensi and culture in cuticle-supplemented medium. Mrcre1 was markedly up-regulated at 24 hours after topical inoculation of mosquitoes (Fig. 4D) and 8 hours after inoculation into the cuticle induction medium (Fig. 4E), which was highly consistent with the expression patterns of Mrkmt2 (Fig. 1, C and D). Together, these data indicated that upon cuticle exposure, MrKMT2 up-regulates the expression of the transcription factor gene Mrcre1 via H3K4me3.
Fig. 4. Mrcre1 is a direct downstream target of MrKMT2-mediated H3K4me3.
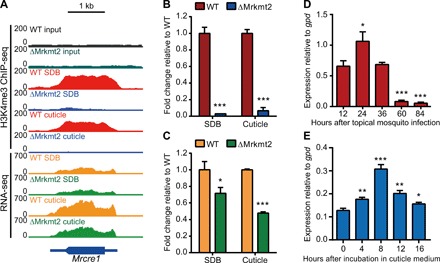
(A) A genome browser view showing H3K4me3 signals at Mrcre1, and Mrcre1 expression in the WT and ΔMrkmt2 strains grown in the SDB and MM medium supplemented with 0.8% locust cuticle. (B) ChIP-qPCR validation of ChIP-seq shown as the fold change of the percentage of the signal from immunoprecipitation over the input in ΔMrkmt2 relative to that in WT. (C) qPCR validation of RNA-seq shown as the fold change of Mrcre1 expression in ΔMrkmt2 relative to that in WT. Actin was used as a reference gene. (D) qPCR analysis of Mrcre1 expression during topical infection of A. stephensi by the WT strain. (E) qPCR analysis of Mrcre1 expression during incubation in MM medium supplemented with 0.8% locust cuticle. gpd was used as a reference gene. Data are shown as means ± SD of three technical replicates. Single, double, and triple asterisks represent significant differences compared with that in WT (B and C), that in 12 hours after topical mosquito infection (D), and that in 0 hour after incubation in cuticle medium (E) determined by Student’s t test at P < 0.05, P < 0.01, and P < 0.001, respectively. The experiments were repeated three times with similar results.
Next, to evaluate whether Mrcre1 is also necessary for MrKMT2-mediated regulation of fungal virulence, we deleted this gene from the M. robertsii genome. While conidiation of the ΔMrcre1 strain was significantly reduced compared with the WT strain (fig. S8), the deletion did not affect conidial germination on hydrophobic surfaces (fig. S2, D and E). Further, the deletion markedly reduced fungal virulence in A. stephensi mosquito (Fig. 5A) and resulted in the formation of far fewer appressoria (Fig. 5, B and C) compared with the WT strain. These results indicated that MrCre1 positively regulates the appressorium formation and fungal virulence. The phenotypic defects of the ΔMrcre1 strain resembled those of the ΔMrkmt2 strain. Furthermore, overexpression of Mrcre1 largely rescued the defects in fungal conidiation (fig. S8), fungal pathogenicity in mosquito (Fig. 5A), and appressorium formation on the cicada hindwings (Fig. 5, B and C) in the ΔMrkmt2 strain. These observations confirmed that, at a genetic level, MrKMT2 regulates infection-related morphogenesis and fungal pathogenicity via the downstream transcription factor MrCre1.
Fig. 5. Overexpression of Mrcre1 in ΔMrkmt2 largely rescues the phenotypic defects in fungal virulence and appressorium formation.
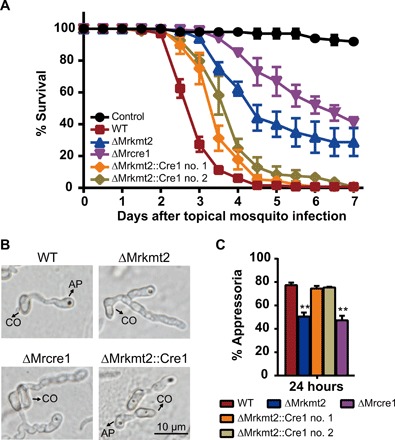
(A) Survival of A. stephensi female adults following topical application of conidial suspensions (1 × 107 conidia/ml) of the WT, ΔMrkmt2, ΔMrcre1, and ΔMrkmt2::Cre1 no. 1 and 2 strains. Control mosquitoes were treated with 0.01% Triton X-100. Each treatment was replicated three times, with 50 mosquitoes per replicate. Data are shown as means ± SEM of two biological replicates. (B) Microscopic observation of fungal appressoria induced on the cicada hindwings. (C) Percentage of fungal conidia forming appressoria relative to total conidia induced in MM2-Gly on hydrophobic surfaces in plastic plates. One hundred conidia per plate were counted. Data are shown as means ± SD of three technical replicates. The experiments were repeated three times with similar results. Double asterisks represent a significant difference in mutants compared with that in WT determined by Student’s t test at P < 0.01.
MrCre1 regulates appressorium formation and fungal pathogenicity by targeting a hydrophobin gene Mrhyd4
To gain a better understanding of how the transcription factor MrCre1 modulates fungal pathogenicity, we used RNA-seq to compare transcriptional changes in the ΔMrcre1 and WT strains upon cuticle induction. Similar to what we observed for MrKMT2, MrCre1-dependent up-regulated genes were highly associated with the transport and metabolism of lipids, carbohydrates, and amino acids, as well as secondary metabolism (fig. S9).
Next, we used ChIP-seq to identify the direct target genes of MrCre1. We constructed a Cre1:5myc fusion by inserting a 5×myc tag at the 3′ end of cre1 in the genome of the WT strain (fig. S10, A and B). We used anti-Myc antibody to isolate Cre1-binding DNA and identified 298 genes that have the MrCre1-binding signal (a representative genome browser view was shown in fig. S10C). Among them, 17 and 21 MrCre1-binding genes were down- and up-regulated in ΔMrcre1 cuticle cultures compared with that in WT (1.5-fold change), respectively. Further, we selected six genes for detailed investigation, based on their functional annotation associated with carbohydrate and lipid metabolism as well as functions on cell surface, which are presumably involved in appressorium formation and fungal virulence (Fig. 6A and fig. S11A). These genes included two genes encoding cell surface proteins [hydrophobin 4, hyd4, and glycosylphosphatidylinositol (GPI)–anchored protein, gpi], two genes involved in carbohydrate metabolism [major intrinsic protein (MIP) family transporter, mip, and GH88 glycosidase, gh88], and two genes involved in lipid metabolism [acyl–coenzyme A (CoA) dehydrogenase, acdh, and catalase C, catC]. ChIP-qPCR analysis validated MrCre1 binding to these selected genes (Fig. 6A and fig. S11A). Further, RT-qPCR analysis revealed that the transcript levels of these six genes were significantly reduced in the ΔMrkmt2 and ΔMrcre1 strains compared with the WT strain (Fig. 6B and fig. S11B), indicating that both MrKMT2 and MrCre1 positively regulate these genes after exposure to the host cuticle. In addition, overexpression of Mrcre1 in ΔMrkmt2 largely restored expression of these six genes (Fig. 6B and fig. S11B), suggesting that MrKMT2 regulates these genes largely in an MrCre1-dependent manner.
Fig. 6. Mrhyd4 is the direct target of the transcription factor MrCre1.
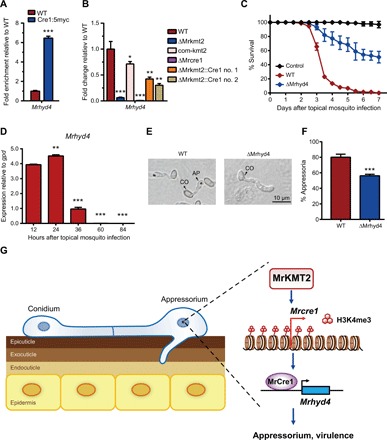
(A) ChIP-qPCR validation of MrCre1 binding to Mrhyd4, shown as fold enrichment of the percentage of the signal from immunoprecipitation over the relative input DNA in the Cre1:5myc strain relative to that in WT. (B) qPCR analysis of Mrhyd4 expression shown as the fold change in different strains cultured in the MM medium supplemented with 0.8% locust cuticle relative to that in WT; actin was used as a reference gene. (C) Survival of A. stephensi female adults following topical application of conidial suspensions (1 × 107 conidia/ml) of WT and ΔMrhyd4 strains. Control mosquitoes were treated with 0.01% Triton X-100. Each treatment was replicated three times, with 50 mosquitoes per replicate. (D) qPCR analysis of Mrhyd4 expression during the infection of A. stephensi by WT strain; gpd was used as a reference gene. Significant differences were compared with that at 12 hours after topical mosquito infection (E) Microscopic observation of fungal appressorium induced on the cicada hindwings. (F) Percentage of fungal conidia forming appressoria relative to total conidia induced in MM2-Gly on hydrophobic surfaces in plastic plates. One hundred conidia per plate were counted. Data are shown as means ± SD of three technical replicates. Experiments were repeated three times with similar results. Single, double, and triple asterisks represent significant differences determined by Student’s t test at P < 0.05, P < 0.01, and P < 0.001, respectively. (G) A schematic model of the MrKMT2-MrCre1-MrHyd4 regulatory pathway required for infection-related morphogenesis and fungal pathogenicity. During cuticle infection, up-regulated MrKMT2 up-regulates the transcription of the transcription factor gene Mrcre1 via H3K4me3 modification. MrCre1 further directly activates downstream target genes, including Mrhyd4, to regulate appressorium formation and fungal pathogenicity of M. robertsii.
To determine whether the six MrCre1-regulated genes identified above are involved in infection-related morphogenesis and fungal virulence, we next constructed individual deletion mutants of these genes in M. robertsii. Similar to the Mrkmt2 and Mrcre1 deletions, Mrhyd4 deletion resulted in a significant reduction in conidia production compared with the WT strain (fig. S12, A and B). Further, fungal virulence against A. stephensi mosquito was severely compromised in the ΔMrhyd4 strain (Fig. 6C). During topical infection of A. stephensi mosquito, hyd4 was highly expressed at the early stages of cuticle infection (Fig. 6D), consistent with the expression patterns of Mrkmt2 and Mrcre1. Similarly, the ΔMrhyd4 strain formed notably fewer appressoria on both cicada hindwings (Fig. 6E) and hydrophobic surfaces (Fig. 6F) than the WT strain. By contrast, the conidiation and pathogenicity of the other five deletion mutants (ΔMrgpi, ΔMrmip, ΔMrgh88, ΔMracdh, and ΔMrcatC) was unaffected (fig. S12). Collectively, these observations indicated that hyd4, but not gpi, mip, gh88, acdh, and catC, is involved in infection-related morphogenesis and plays a pivotal role in fungal virulence.
DISCUSSION
The fine-tuning transcriptional regulation of fungal morphological differentiation during insect infection is poorly understood. In the current study, we demonstrated that MrKMT2-mediated H3K4me3 modification plays a crucial role in regulating infection-related morphogenesis and pathogenicity of M. robertsii. MrKMT2 up-regulates the transcription factor gene Mrcre1 via H3K4me3 modification. The up-regulated MrCre1 further induces the downstream target gene Mrhyd4 to regulate appressorium formation and fungal pathogenicity (Fig. 6G).
MrKMT2 is the single ortholog of S. cerevisiae SET1, which belongs to the KMT2 family responsible for methylation at H3K4. In yeast, SET1 is required for normal cell growth and ribosomal DNA (rDNA) silencing (21). Expression of the Mrkmt2 gene is markedly up-regulated during appressorium formation on the host cuticle, and deletion of the gene results in severe defects in appressorium formation and pathogenicity in the host mosquito. Deletion of set1 in Magnaporthe oryzae and F. graminearum also affects their pathogenicity (22, 23). But the molecular mechanism of how SET1 regulates pathogenicity remains elusive in pathogenic fungi.
Entomopathogenic fungi infect insect hosts directly through the exoskeleton. Upon recognition of the physicochemical and nutritional cues on the host surface, M. robertsii initiates morphological differentiation, including conidial germination and appressorium formation on the insect cuticle (19). The epicuticle, or the outermost layer, provides a hydrophobic barrier rich in lipids, covering the procuticle that contains chitin and sclerotized protein (24). The fungus must tightly regulate gene transcription to assimilate various hydrocarbon and lipid components of the insect cuticle and to properly induce infection-related morphogenesis (19). Accordingly, 2425 and 2954 genes were up-regulated and down-regulated upon insect cuticle induction, and one-third were regulated in an MrKMT2-dependent manner. MrKMT2-dependent genes were mainly involved in lipid and carbohydrate transport and metabolism, indicating that MrKMT2 plays an important role in transcriptional regulation of the fungal genes involved in hydrolysis and assimilation of the cuticular hydrocarbon and lipid components.
MrKMT2-mediated H3K4 methylation is an evolutionarily conserved epigenetic marker of gene activation (13). However, only a small number of MrKMT2-dependent cuticle-induced genes were directly regulated by MrKMT2 via H3K4me3 as observed by ChIP-seq and RNA-seq combined analysis, indicating that MrKMT2 may positively regulate a transcription activator to control cuticle-induced genes. We identify the transcription factor gene Mrcre1 as the MrKMT2 direct key target. We found that MrKMT2-mediated H3K4me3 up-regulates Mrcre1 transcription and that the expression of one-third of the MrKMT2-dependent genes was MrCre1 dependent, suggesting that MrKMT2 regulates the expression of these cuticle-induced genes via MrCre1.
Cre1 is conserved in microorganisms and is considered to play an important role in “glucose-first” assimilation by repressing genes required for the utilization of less favored carbon sources (25). Although the transcription factor Cre1 has been therefore termed a carbon catabolite repressor, accumulating evidence points to Cre1 function as an activator of certain genes involved in virulence in addition to glucose/carbon repression (10, 20, 26). In the current study, we found that the expression patterns of Mrcre1 during host cuticle infection and upon cuticle induction were consistent with those of Mrkmt2. Further, the pathogenicity of the ΔMrcre1 strain in the mosquito model was also severely compromised, with significantly reduced appressorium formation, i.e., phenotypic defects similar to those of the ΔMrkmt2 strain. Furthermore, overexpression of Mrcre1 in the ΔMrkmt2 background largely rescued these phenotypic defects, demonstrating that MrKMT2 regulates infection-related morphogenesis and fungal pathogenicity via MrCre1-mediated gene regulation.
Using ChIP-seq and RNA-seq analyses, we identified several direct gene targets of MrCre1 that are involved in lipid and carbohydrate transport and metabolism, as well as cuticle surface interactions. We showed that one of these genes, the hydrophobin 4 gene (Mrhyd4), is required for appressorium formation and fungal pathogenicity. Hydrophobins are generally found on the outer surface of conidia and of the hyphal wall in ascomycetes and basidiomycetes and may be involved in mediating contact and communication between the fungus and its environment (27). In plant pathogenic and entomopathogenic fungi, hydrophobins were described as pathogenicity factors (27). The hydrophobin MPG1 in M. oryzae (homolog of MrHyd4) functioned as a developmental sensor for appressorium formation (28). In B. bassiana, the nonspecific hydrophobic interaction between the conidial coat hydrophobins and the insect epicuticle is involved in establishing pathogenicity (29).
In conclusion, here we revealed that epigenetic regulation via MrKMT2-mediated H3K4me3 modification plays a crucial role in the regulation of infection-related morphogenesis and fungal pathogenicity in M. robertsii. The identified MrKMT2-MrCre1-MrHyd4 regulatory pathway allows the fungus to reprogram its gene expression for the appressorium differentiation during cuticle infection. Understanding the regulatory mechanism of MrKMT2-mediated H3K4me3 modification not only provides new insights into the entomopathogenic fungus-insect interactions but may also open a new avenue to improve fungal virulence to control insect pests and mosquito-borne diseases.
MATERIALS AND METHODS
Fungal strains and culture conditions
The WT and mutant strains of M. robertsii ARSEF 2575 [obtained from the U.S. Department of Agriculture Entomopathogenic Fungus Collection (ARSEF) in Ithaca, NY] were routinely grown on PDA (BD Difco) at 27°C. For liquid cultures, fungi were grown in SDB (BD Difco) at 27°C in a rotary shaker. For insect cuticle induction, they were grown in a cuticle-containing medium to activate the fungal infection system (16, 17). Fungal conidia (1 × 106 conidia/ml) were first cultured for 40 hours in the nutrient-rich SDB medium. The mycelia were then harvested and washed with phosphate-buffered saline (pH 7.4). Set mycelial mass (1 g wet weight) was transferred to 100 ml of the MM medium (0.1% w/v KH2PO4, 0.1% w/v MgSO4·7H2O, and 50% v/v tap water) supplemented with 0.8% (w/v) adult locust (Schistocerca gregaria) cuticles for 4 to 16 hours (18).
Mosquito rearing
A. stephensi (Dutch strain) mosquitoes were maintained on 10% (w/v) sucrose at 26° ± 1°C and 80 ± 5% relative humidity, with a 12-hour day/12-hour night cycle. The larvae were fed cat food pellets and ground fish food supplement (30).
Construction of M. robertsii gene deletion, complementation, and overexpression strains
For targeted deletion of Mrkmt2 (EXV05084.1), Mrcre1 (EXV01725.1), Mrmip (EXU98391.1), Mrhyd4 (EXU97165.1), MrcatC (EXU95657.1), Mracdh (EXU99334.1), Mrgpi (EXU97885.1), or Mrgh88 (EXV01871.1) genes, the 5′ and 3′ flanking regions of each open reading frame were PCR amplified from the ARSEF 2575 genomic DNA as a template and then subcloned into the Xba I and Eco RV sites of the binary vector pBarGFP, respectively (31). The gene disruption constructs were then used to separately transform Agrobacterium tumefaciens AGL-1 for targeted gene disruption by the split-marker homologous recombination (32). Replacement-specific PCR amplifications of the gene locus were performed using specific primer pairs (listed in table S1) to amplify either the WT or mutated gene locus.
To complement the ΔMrkmt2 and ΔMrcre1 strains, the full-length target genes and their native promoter and terminator sequences were PCR amplified and cloned into the Eco RV site of pSurGFP, to generate the complementation vectors pSurGFP-Mrkmt2 and pSurGFP-Mrcre1, respectively. The complementary strain com-kmt2 was generated by introducing pSurGFP-Mrkmt2 into the deletion mutants ΔMrkmt2, using A. tumefaciens–mediated transformation.
In addition, the ΔMrkmt2::Cre1 strain was obtained by overexpression of the Mrcre1 gene in the ΔMrkmt2 strain using the expression vector pSurGFPOE-MrCre1. To construct the vector, the coding region of Mrcre1 lacking the 3′ TGA (thymidine guanine adenine) stop codon was PCR amplified from the ARSEF 2575 complementary DNA (cDNA) and inserted into the Spe I site of the overexpression plasmid pSurGFPOE downstream of the constitutive B. bassiana gpd promoter and upstream of a myc tag sequence followed by a trpC terminator. The resultant overexpression plasmid pSurGFPOE-MrCre1 was used for A. tumefaciens–mediated transformation of strain ΔMrkmt2 to generate the overexpression strain ΔMrkmt2::Cre1.
To tag Cre1 with 5×myc, the strain Cre1:5myc was constructed by inserting a sequence encoding 5×myc tag at the native genomic locus of the cre1 gene in the WT strain. Mrcre1 open reading frame without a stop codon (as a 5′ homologous arm) and its 3′ flanking region were PCR amplified from ARSEF 2575 genomic DNA and then subcloned into the Xba I and Eco RV sites of the binary vector pBarGFP-5×myc. The 5×myc tag sequence was, hence, fused at the 3′-Mrcre1 under the control of the native promoter and the trpC terminator and used to transform the WT strain to generate cre1:5myc fusion in situ, using the split-marker homologous recombination method. Replacement-specific PCR amplifications of the Mrcre1 locus were performed using specific primer pairs (listed in table S1) to amplify either the WT cre1 or the cre1:5myc locus.
Insect bioassays
The virulence of the WT, deletion mutants, complemented strains, and overexpression strain ΔMrkmt2::Cre1 was assayed using A. stephensi female adults (7). Conidial suspensions in 0.01% (v/v) Triton X-100 were prepared from 14-day-old fungal PDA cultures. For fungal infection, adult female A. stephensi mosquitoes were sprayed with a fungal conidial suspension (1 × 107 conidia/ml). The treated mosquitoes were maintained at 26° ± 1°C and 80 ± 5% relative humidity, with a 12-hour day/12-hour night cycle, until they died. Mosquitoes sprayed with sterile 0.01% Triton X-100 were used as controls. Each treatment was replicated three times, with 50 mosquitoes per replicate, and the infection bioassays were repeated three times. Mortality was recorded every 12 hours. The LT50 value was calculated using the Kaplan-Meier analysis in the SPSS program (version 16.0).
Conidiation, conidial germination, appressorium induction, and hyphal body monitoring
For the conidiation assay, conidia were harvested in 0.01% (v/v) Triton X-100 from 10-day-old fungal PDA cultures. Conidial concentration was determined by microscopic observation using a hemocytometer. The germination rate of conidia was determined by inoculating 20 μl of conidial suspension (2 × 107 conidia/ml) in 5.5-cm polystyrene petri dishes containing 2 ml of SDB and incubating at 27°C for up to 12 hours.
Appressorium induction assays were conducted using the cicada hindwings or an MM2 [containing NaNO3 (6 g/liter), KCl (0.52 g/liter), MgSO4·7H2O (0.52 g/liter), and KH2PO4 (0.25 g/liter)] supplemented with 1% (v/v) glycerol as the sole carbon source (MM2-Gly) on plastic hydrophobic surfaces (33). The cicada hindwings were collected, surface sterilized in 37% H2O2 for 1 min, washed three times with sterile water, and dipped into conidial suspensions (2 × 107 conidia/ml) for 1 min. The inoculated wings were then placed on 0.7% water agar at 27°C for 16 to 24 hours for appressorium induction. Conidial suspension (20 μl; 1 × 108 conidia/ml) was added into 2 ml of MM2-Gly in Thermoplastic plates (Φ = 6 cm) and incubated at 27°C for 16 to 24 hours for appressorium induction.
To monitor hyphal body differentiation, the fifth instar larvae of Bombyx mori (Nistari) were dipped into the fungal conidial suspension (1 × 107 conidia/ml) for 20 s. The hemolymph was collected 72 hours after topical infection. The hyphal bodies induced in the hemolymph were determined by microscopic observation using a hemocytometer. Each treatment involved 15 silkworm larvae. The experiment was repeated three times.
Western blot analysis
The mycelia for histone extraction were obtained by inoculating fungal conidia in SDB (1 × 106 conidia/ml) and incubating at 200 rpm at 27°C for 40 hours. The mycelia were then transferred into MM medium for 24 hours. They were next harvested by filtration, frozen in liquid nitrogen, and grounded to a fine powder using a mortar and pestle. Histones were acid extracted as previously described (34). Approximately 10 to 20 μg of total nuclear protein per lane was analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Proteins were then transferred to polyvinylidene fluoride membranes and blotted using standard procedures. Primary antibodies specific against the following proteins were used for Western blotting: H3K4me1 (Abcam, ab8895), H3K4me2 (Active Motif, 39141), H3K4me3 (Abcam, ab8580), and H3 (Abcam, ab1791). The secondary antibodies used were peroxidase-conjugated AffiniPure goat anti-rabbit (Jackson ImmunoResearch Inc., 111-035-003) or peroxidase-conjugated AffiniPure goat anti-mouse antibodies (Jackson ImmunoResearch Inc., 115-035-003).
ChIP and high-throughput ChIP-seq
ChIP was performed as described previously with some modifications (22, 35). Briefly, mycelia were generated by growing fungal conidia in 100 ml of SDB at 200 rpm, at 27°C for 40 hours, and then transferring them to the MM medium supplemented with adult locust (S. gregaria) cuticle [0.8% (w/v)] for an 8-hour induction. For chromatin fixation, fungal cultures were treated with 1% formaldehyde with gentle shaking (125 rpm) for 15 min at 27°C. After cell lysis, the chromatin was sheared into 0.1- to 0.3-kb fragments. The soluble chromatin fraction was immunoprecipitated using antibodies specific for H3K4me3 (Abcam, ab8580) or myc tag (Abmart, M20002). DNA fragments were recovered from immunoprecipitated chromatin or total chromatin (input DNA) by treatment with proteinase K. Indexed ChIP-seq libraries were prepared using the NEBNext UltraDNA library prep kit for Illumina (New England Biolabs) according to the manufacturer’s instructions. Selected DNA fragments (0.2 to 0.5 kb) were purified and enriched by PCR. Then, 50–base pair (bp) single-end sequencing was performed using an Illumina HiSeq 2500 genome analyzer (Guangzhou RiboBio Co. Ltd).
ChIP quantitative PCR
ChIP input and IP samples were diluted 10-fold and 100-fold for PCR, respectively. Then, qPCR was performed using an AceQ qPCR SYBR Green Master Mix kit (Vazyme Biotech Co. Ltd, Q111-02) and a PikoReal instrument (Thermo Fisher Scientific, N11471), under the following conditions: denaturation at 95°C for 7 min, followed by 40 cycles of denaturation at 95°C for 10 s, and annealing and extension at 60°C for 30 s. The primers used for the target genes are listed in table S1.
RNA isolation and RNA-seq
Aliquots of cultures used for ChIP were lyophilized and grounded in liquid nitrogen. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) with RNase-free DNase (Qiagen) to remove genomic DNA. RNA-seq libraries were constructed using an Illumina TruSeq RNA Sample Preparation kit, and cDNA was sequenced on an Illumina HiSeq 2000 genome analyzer (BGI, Shenzhen).
Reverse transcription quantitative polymerase chain reaction
To monitor target gene expression under different growth conditions, the WT conidia were first incubated in fresh SDB medium for 40 hours. The mycelia were then harvested and transferred to the MM medium supplemented with adult locust (S. gregaria) cuticle for 4- to 16-hour induction. Samples were also prepared during fungal topical infection of A. stephensi female adults. The mosquitoes were sprayed with conidial suspension (1 × 108 conidia/ml) and collected 24, 36, 60, and 84 hours after the topical infection. Samples were lyophilized and ground in liquid nitrogen. Total RNA was extracted using the RNAiso Plus Kit (Takara, D9108A), and cDNA was synthesized using the PrimeScript RT Reagent Kit with genomic DNA Eraser (Takara, DRR047A) according to the manufacturer’s instructions. Then, qPCR was performed using an AceQ qPCR SYBR Green Master Mix Kit (Vazyme Biotech Co. Ltd, Q111-02) and a PikoReal instrument (Thermo Fisher Scientific, N11471) under the following conditions: denaturation at 95°C for 7 min, followed by 40 cycles of denaturation at 95°C for 10 s, and annealing and extension at 60°C for 30 s. The primers used for target genes and the reference gene actin are listed in table S1.
Next-generation sequencing data analysis
The ChIP-seq reads (50 bp) were mapped to the M. robertsii ARSEF 2575 genome (http://ncbi.nlm.nih.gov/bioproject/PRJNA230500) using bwa (36). Peaks were called using Model-based Analysis for ChIP-seq (MACS2) (37). RNA-seq clean reads were also aligned with the M. robertsii ARSEF 2575 genome using TopHat2 and assembled using Cufflinks with the default settings (38). KOG was used to identify the statistically significantly enriched functional KOG categories, according to the annotation provided at genome.jgi.doe.gov/Trire2/Trire2.home.html (39). The ChIP-seq and RNA-seq data were visualized using the Integrative Genomics Viewer genome browser (40).
Statistical analysis
The statistical significance for survival data from the fungal bioassays was analyzed using the log-rank (Mantel-Cox) test. Statistical significance of other variables was determined using Student’s t test for unpaired comparisons between two treatments. A value of P < 0.05 was regarded as statistically significant. All statistical analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software).
Supplementary Material
Acknowledgments
We thank Fang Li for rearing mosquitoes. Funding: This work was supported by the National Key R&D Program of China (grants 2017YFD0200400, 2017YFD0201202, and 2018YFA0900502), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB11010500), the National Natural Science Foundation of China (grant nos. 31501703, 31772534, 31701841, and 31771431), and the Key Research Program of the Chinese Academy of Sciences (grant no. KFZD-SW-219). Author contributions: S.W. and Y.L. conceived and designed the experiments. Y.L., J.C., and L.W. performed the experiments and analyzed the data. X.C. and G.W. analyzed the NGS data. Y.L. and S.W. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. KMT2 ChIP-seq and RNA-seq data, and Cre1 ChIP-seq and RNA-seq data have been deposited in the National Center for Biotechnology Information Sequence Read Archive with accession numbers PRJNA541058 and PRJNA541070, respectively. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaaz1659/DC1
Fig. S1. Disruption of the Mrkmt2 gene in M. robertsii ARSEF 2575.
Fig. S2. Growth and conidial germination assay.
Fig. S3. MrKMT2-mediated H3K4me3 is associated with gene activation.
Fig. S4. Characterization of genes transcriptionally regulated by MrKMT2-mediated H3K4me3.
Fig. S5. Characterization of genes directly modulated by H3K4me3 in an MrKMT2-dependent manner.
Fig. S6. Six direct gene targets of MrKMT2-mediated H3K4me3.
Fig. S7. Characterization of MrCre1 (EXV01725.1).
Fig. S8. Overexpression of Mrcre1 in ΔMrkmt2 rescues sporulation defects.
Fig. S9. Global transcriptional analysis of WT and ΔMrcre1 strains grown in the SDB and MM medium supplemented with 0.8% locust cuticle.
Fig. S10. Gene tagging and ChIP-seq of Cre1:5myc.
Fig. S11. The other five targets of the transcription factor MrCre1.
Fig. S12. Effects of deletion of each Cre1 direct target gene on fungal growth, conidiation, and virulence.
Table S1. Primers used in this study.
REFERENCES AND NOTES
- 1.World Health Organization, World Malaria Report (World Health Organization, 2018).
- 2.Chanda E., Ameneshewa B., Bagayoko M., Govere J. M., Macdonald M. B., Harnessing integrated vector management for enhanced disease prevention. Trends Parasitol. 33, 30–41 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Ranson H., Lissenden N., Insecticide resistance in African Anopheles mosquitoes: A worsening situation that needs urgent action to maintain malaria control. Trends Parasitol. 32, 187–196 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Knols B. G. J., Bukhari T., Farenhorst M., Entomopathogenic fungi as the next-generation control agents against malaria mosquitoes. Future Microbiol. 5, 339–341 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Lovett B., Bilgo E., Millogo S. A., Ouattarra A. K., Sare I., Gnambani E. J., Dabire R. K., Diabate A., Leger R. J. S., Transgenic Metarhizium rapidly kills mosquitoes in a malaria-endemic region of Burkina Faso. Science 364, 894–897 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Thomas M. B., Read A. F., Can fungal biopesticides control malaria? Nat. Rev. Microbiol. 5, 377–383 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Wei G., Lai Y., Wang G., Chen H., Li F., Wang S., Insect pathogenic fungus interacts with the gut microbiota to accelerate mosquito mortality. Proc. Natl. Acad. Sci. U.S.A. 114, 5994–5999 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang C., Wang S., Insect pathogenic fungi: Genomics, molecular interactions, and genetic improvements. Annu. Rev. Entomol. 62, 73–90 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Lai Y., Chen H., Wei G., Wang G., Li F., Wang S., In vivo gene expression profiling of the entomopathogenic fungus Beauveria bassiana elucidates its infection stratagems in Anopheles mosquito. Sci. China Life Sci. 60, 839–851 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Beattie S. R., Mark K. M. K., Thammahong A., Ries L. N. A., Dhingra S., Caffrey-Carr A. K., Cheng C., Black C. C., Bowyer P., Bromley M. J., Obar J. J., Goldman G. H., Cramer R. A., Filamentous fungal carbon catabolite repression supports metabolic plasticity and stress responses essential for disease progression. PLOS Pathog. 13, e1006340 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonduriansky R., Day T., Nongenetic inheritance and its evolutionary implications. Annu. Rev. Ecol. Evol. Syst. 40, 103–125 (2009). [Google Scholar]
- 12.Gómez-Díaz E., Jordà M., Peinado M. A., Rivero A., Epigenetics of host-pathogen interactions: The road ahead and the road behind. PLOS Pathog. 8, e1003007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bannister A. J., Kouzarides T., Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kouzarides T., Chromatin modifications and their function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Roguev A., Schaft D., Shevchenko A., Pijnappel W. W., Wilm M., Aasland R., Stewart A. F., The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 20, 7137–7148 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santi L., Silva W. O. B., Pinto A. F. M., Schrank A., Vainstein M. H., Metarhizium anisopliae host-pathogen interaction: Differential immunoproteomics reveals proteins involved in the infection process of arthropods. Fungal Biol. 114, 312–319 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Wang C., Hu G., St. Leger R. J., Differential gene expression by Metarhizium anisopliae growing in root exudate and host (Manduca sexta) cuticle or hemolymph reveals mechanisms of physiological adaptation. Fungal Genet. Biol. 42, 704–718 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Wang C., St. Leger R. J., Developmental and transcriptional responses to host and nonhost cuticles by the specific locust pathogen Metarhizium anisopliae var. acridum. Eukaryot. Cell 4, 937–947 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pedrini N., Ortiz-Urquiza A., Huarte-Bonnet C., Zhang S., Keyhani N. O., Targeting of insect epicuticular lipids by the entomopathogenic fungus Beauveria bassiana: Hydrocarbon oxidation within the context of a host-pathogen interaction. Front. Microbiol. 4, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo Z., Qin Y., Pei Y., Keyhani N. O., Ablation of the creA regulator results in amino acid toxicity, temperature sensitivity, pleiotropic effects on cellular development and loss of virulence in the filamentous fungus Beauveria bassiana. Environ. Microbiol. 16, 1122–1136 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Briggs S. D., Bryk M., Strahl B. D., Cheung W. L., Davie J. K., Dent S. Y., Winston F., Allis C. D., Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev. 15, 3286–3295 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pham K. T. M., Inoue Y., Vu B. V., Nguyen H. H., Nakayashiki T., Ikeda K.-I., Nakayashiki H., MoSET1 (histone H3K4 methyltransferase in Magnaporthe oryzae) regulates global gene expression during infection-related morphogenesis. PLOS Genet. 11, e1005385 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y., Liu N., Yin Y., Chen Y., Jiang J., Ma Z., Histone H3K4 methylation regulates hyphal growth, secondary metabolism and multiple stress responses in Fusarium graminearum. Environ. Microbiol. 17, 4615–4630 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Ortiz-Urquiza A., Keyhani N. O., Action on the surface: Entomopathogenic fungi versus the insect cuticle. Insects 4, 357–374 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ries L. N. A., Beattie S. R., Espeso E. A., Cramer R. A., Goldman G. H., Diverse regulation of the CreA carbon catabolite repressor in Aspergillus nidulans. Genetics 203, 335–352 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Görke B., Stülke J., Carbon catabolite repression in bacteria: Many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Bayry J., Aimanianda V., Guijarro J. I., Sunde M., Latgé J.-P., Hydrophobins--unique fungal proteins. PLOS Pathog. 8, e1002700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talbot N. J., Kershaw M. J., Wakley G. E., DeVries O. M. H., Wessels J. G. H., Hamer J. E., MPG1 encodes a fungal hydrophobin involved in surface interactions during infection-related development of Magnaporthe grisea. Plant Cell 8, 985–999 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang S., Xia Y. X., Kim B., Keyhani N. O., Two hydrophobins are involved in fungal spore coat rodlet layer assembly and each play distinct roles in surface interactions, development and pathogenesis in the entomopathogenic fungus Beauveria bassiana. Mol. Microbiol. 80, 811–826 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Wang S., Ghosh A. K., Bongio N., Stebbings K. A., Lampe D. J., Jacobs-Lorena M., Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc. Natl. Acad. Sci. U.S.A. 109, 12734–12739 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S., Fang W., Wang C., St. Leger R. J., Insertion of an esterase gene into a specific locust pathogen (Metarhizium acridum) enables it to infect caterpillars. PLOS Pathog. 7, e1002097 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang W., Pei Y., Bidochka M. J., Transformation of Metarhizium anisopliae mediated by Agrobacterium tumefaciens. Can. J. Microbiol. 52, 623–626 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Gao Q., Shang Y., Huang W., Wang C., Glycerol-3-phosphate acyltransferase contributes to triacylglycerol biosynthesis, lipid droplet formation, and host invasion in Metarhizium robertsii. Appl. Environ. Microbiol. 79, 7646–7653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Honda S., Selker E. U., Direct interaction between DNA methyltransferase DIM-2 and HP1 is required for DNA methylation in Neurospora crassa. Mol. Cell. Biol. 28, 6044–6055 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamaru H., Zhang X., McMillen D., Singh P. B., Nakayama J.-I., Grewal S. I., Allis C. D., Cheng X., Selker E. U., Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat. Genet. 34, 75–79 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L., TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Derntl C., Kluger B., Bueschl C., Schuhmacher R., Mach R. L., Mach-Aigner A. R., Transcription factor Xpp1 is a switch between primary and secondary fungal metabolism. Proc. Natl. Acad. Sci. U.S.A. 114, E560–E569 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., Mesirov J. P., Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaaz1659/DC1
Fig. S1. Disruption of the Mrkmt2 gene in M. robertsii ARSEF 2575.
Fig. S2. Growth and conidial germination assay.
Fig. S3. MrKMT2-mediated H3K4me3 is associated with gene activation.
Fig. S4. Characterization of genes transcriptionally regulated by MrKMT2-mediated H3K4me3.
Fig. S5. Characterization of genes directly modulated by H3K4me3 in an MrKMT2-dependent manner.
Fig. S6. Six direct gene targets of MrKMT2-mediated H3K4me3.
Fig. S7. Characterization of MrCre1 (EXV01725.1).
Fig. S8. Overexpression of Mrcre1 in ΔMrkmt2 rescues sporulation defects.
Fig. S9. Global transcriptional analysis of WT and ΔMrcre1 strains grown in the SDB and MM medium supplemented with 0.8% locust cuticle.
Fig. S10. Gene tagging and ChIP-seq of Cre1:5myc.
Fig. S11. The other five targets of the transcription factor MrCre1.
Fig. S12. Effects of deletion of each Cre1 direct target gene on fungal growth, conidiation, and virulence.
Table S1. Primers used in this study.