

By mimicking the RIBE machinery, we provided an indirect radiotherapy approach based on RT-MP for eradication of MPE in mice.
Abstract
Radiotherapy (RT) is routinely used in cancer treatment, but expansion of its clinical indications remains challenging. The mechanism underlying the radiation-induced bystander effect (RIBE) is not understood and not therapeutically exploited. We suggest that the RIBE is predominantly mediated by irradiated tumor cell–released microparticles (RT-MPs), which induce broad antitumor effects and cause immunogenic death mainly through ferroptosis. Using a mouse model of malignant pleural effusion (MPE), we demonstrated that RT-MPs polarized microenvironmental M2 tumor-associated macrophages (M2-TAMs) to M1-TAMs and modulated antitumor interactions between TAMs and tumor cells. Following internalization of RT-MPs, TAMs displayed increased programmed cell death ligand 1 (PD-L1) expression, enhancing follow-up combined anti–PD-1 therapy that confers an ablative effect against MPE and cisplatin-resistant MPE mouse models. Immunological memory effects were induced.
INTRODUCTION
Radiotherapy (RT) serves as frontline therapy to treat up to 50% of patients with cancer and generally contributes the most to curing cancers when compared with many other treatment modalities (excluding surgery), such as molecular-targeted treatment and traditional chemotherapy (1, 2). The achievement of recent therapeutic breakthroughs in the field of cancer immunotherapy and recognition of the central importance of tumor microenvironments (TMEs) are affecting the standard care of RT for many tumor types (3–6); however, further expansion of the indications of RT for other tumors remains challenging. For example, malignant pleural effusion (MPE), a complication of lung cancer, has seldom been cured in mouse MPE models using current strategies; it is not suited for RT due to its high mobility that causes failure in identifying effective and safe radiation target areas and doses (7–9).
With the aid of radiation energy, radiation causes oxidative stress and DNA damage in cancer cells and in neighboring unirradiated cells or tissues—the so-called “radiation-induced bystander effect (RIBE)”—but underlying mechanisms remain poorly understood (10, 11). Early studies have been mainly focused on gap junctional intercellular communication (GJIC), as bystander signaling can occur through GJIC for cells in direct contact (12, 13). However, GJIC is active in normal cells and is generally down-regulated in tumor cells, indicating that the GJIC is unlikely the main route responsible for RIBE in cancer RT (11). The second possible route by which RIBE is mediated comprises the release of soluble factors, either in the form of extracellular vesicles or soluble signaling molecules from cells that have been irradiated. A recent report found that cysteine protease cathepsin B mediated the bystander effect (14). Despite these fundamental mechanistic studies, RIBE is often regarded as an indicator of the need to limit the dose to surrounding normal tissues but not therapeutically exploited (15, 16).
In this study, we aimed to probe and leverage the RIBE machinery to treat tumors that cannot receive RT by re-engineering biological processes in TMEs. We show that the RIBE is mainly mediated through radiated tumor cell–released microparticles (RT-MPs), which exhibited unique and broad therapeutic antitumor effects, phenotypically different from those of previous reported tumor cell–released microparticles (T-MPs) (e.g., chemotherapy and hypoxia derived) that had prometastatic potential (17–20). Exogenously injected RT-MPs were avidly endocytosed by tumor-associated macrophages (TAMs) and caused proinflammatory M1-TAM polarization both in vitro and in vivo, while the hypoxia-treated tumor cell–derived microparticles reportedly polarized M2-TAM for tumor progression (19). Moreover, tumor cells taking up RT-MPs were prone to be sequestered by activated TAMs, thus promoting immunosuppressive TMEs to perform antitumor functions during treatment of MPE. The internalization of RT-MPs caused remarkably enhanced expression of programmed cell death ligand 1 (PD-L1) in TAMs, which suggests effectiveness of RT-MPs in combination with anti–PD-1 in terms of curing MPE. Mechanistically, CD8+ T cells, macrophages, and CD4+ T cells were shown to play critical roles in regulating the potent in vivo antitumor efficacy of RT-MPs and anti–PD-1 combination treatment. Collectively, our results show that, by learning and mimicking the RIBE machinery, we have generated a translational and potent chemotherapy-free approach for indirect cancer RT.
RESULTS
RT-MPs can mediate the RIBE and have therapeutic antitumor potential
RIBE is a unique phenomenon that has been found in vitro and is sometimes observed in clinical settings (21). To directly visualize the RIBE, we established cell mixing and coculture models: Irradiated cells were directly mixed with unirradiated cells or medium from irradiated cells was cocultured with unirradiated cells passed through different pore size membranes (0.4 and 1 μm), as shown in fig. S1A. In the mixing model, fluorescence microscopy showed fewer green fluorescent protein (GFP) stably transfected Lewis lung carcinoma cell (LLC) cells (LLC-GFP) when mixed with red fluorescent protein (RFP) stably transfected LLC cells that had been irradiated (Fig. 1, A and B); this indicates that the irradiated cells could influence the unirradiated cells. In the coculture model, clone formation analysis showed that fewer cells survived in the 1-μm coculture condition than in the 0.4-μm coculture condition (Fig. 1C and fig. S1B), which suggests that certain nanoscale substances might play a role in RIBE. These findings led us to investigate the function of microvesicles in RIBE. We assessed the production of microparticles and exosomes in murine LLC (lung), human A549 (lung), and other cancer cells; microparticles and exosomes were isolated from the medium of irradiated cells by gradient centrifugation (Fig. 1D). Colony formation analysis showed that RT-MPs played the main role in killing tumor cells, as removal of microparticles from the medium showed that the rest could not effectively kill tumor cells (Fig. 1, E and F, and fig. S1C). Consistent with the in vitro results, we found that the irradiated LLC-RFP cells released greater numbers of red microparticles than unirradiated LLC-RFP cells in vivo using the dorsal skinfold window chamber model (Fig. 1G, fig. S1D, and movies S1 and S2).
Fig. 1. RIBE is mainly mediated by RT-MPs.
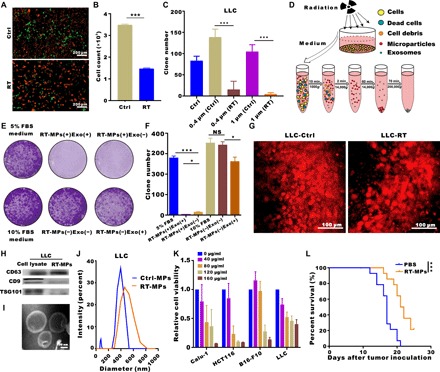
(A) Representative fluorescence microscope images of cell mixtures of unirradiated LLC-RFP and unirradiated LLC-GFP (top) and irradiated LLC-RFP and unirradiated LLC-GFP (bottom). Scale bars, 200 μm. (B) Flow cytometry analysis of green LLC-GFP cells in (A). (C) Quantification of clone formation after coculture with medium from irradiated cells using 0.4- and 1-μm pores. (D) Experimental outline for the production of microparticles and exosomes. (E and F) Representative images showing the colony numbers of LLC cells in the presence of microparticles or exosomes. NS, not significant. (G) Titanium chambers were surgically implanted in C57BL/6 mice, and irradiated or control tumor cells were then subcutaneously injected (2 × 104) into the window. Irradiated tumor cells released more microparticles in the window chamber. Representative images are shown. Scale bars, 100 μm. (H) Western blots of CD9, CD63, and TSG101 expression in LLC whole-cell lysates (positive control) and RT-MPs pellets. (I) TEM images of RT-MPs. Scale bars, 100 nm. (J) Representative size and particle distribution plots of LLC-derived MPs and RT-MPs. (K) Calu-1, HCT116, B16-F10, and LLC cells were treated with various concentrations of RT-MPs for 48 hours, and cell viability was estimated using Cell Counting Kit-8 (CCK-8) assays. (L) Survival analysis in all two groups (n = 14 per group). *P < 0.05 and ***P < 0.001.
Because the amount of RT-MPs differed with variations in radiation dose and plateaued at 20 gray (Gy), we chose this dose for subsequent studies (fig. S1E). RT-MPs were characterized on the basis of protein content, morphology, and size. Western blot analysis revealed the presence of extracellular vesicle–associated proteins, such as CD63, CD9, and tumor susceptibility gene 101 protein (TSG101) (Fig. 1H). We also analyzed the proteins that RT-MPs contain through a proteomic approach and show the top 30 proteins in fig. S1F. Transmission electron microscopy (TEM) imaging showed that RT-MPs had a regular spherical morphology (Fig. 1I and fig. S1G). Nanoparticle tracking analysis revealed that A549- and LLC-derived RT-MPs had mean diameters of 381.8 and 480.1 nm, respectively (Fig. 1J and fig. S1H). To further evaluate the therapeutic effect of RT-MPs on tumor cells, we performed cell toxicity studies involving various cell lines: human Calu-1 (lung), murine B16-F10 (skin), human HCT116 (colon), and murine LLC (lung). We found that RT-MPs could efficiently inhibit the growth of homologous tumor cells in a dose-dependent manner (Fig. 1K and fig. S2A). We observed that A549-derived RT-MPs damaged other types of tumor cells, indicating an indiscriminately therapeutic in vitro antitumor effect (fig. S2B). To evaluate the toxic effect to normal cells, we used fibroblasts and macrophages. The concentration that can kill tumor cells had no obvious killing effect on fibroblasts and promoted proliferation in macrophages (fig. S2C). We next investigated the therapeutic effect of RT-MPs in an MPE mouse model. As shown in Fig. 1L and fig. S2D, we observed increased survival upon treatment with RT-MPs, superior to that of control treatment. To explore the universal therapeutic effect of RT-MPs in vivo, we established a subcutaneous transplanted model by using B16-F10 cells and used Lewis-derived RT-MPs and B16-F10–derived RT-MPs for the treatment, and both of them could induce a delay in tumor growth (fig.S2F). Together, these findings suggest that RT-MPs exhibit broad killing effects with respect to tumor cells and that they mediate in vitro RIBE.
RT-MPs kill tumor cells by causing ferroptosis
To explore the mechanism of RT-MP–induced cell death, we used inhibitors of caspase (Z-VAD-FMK), RIPK1 (necrostatin-1), or autophagy (3-methyladenine); these compounds are known to inhibit forms of apoptosis, necrosis, and autophagic cell death, respectively (22). We observed that RT-MP–induced death was not modulated by these inhibitors. We then tested ferrostatin-1 and l-glutathione (GSH), inhibitors of ferroptosis, and found that RT-MP–induced death in A549 cells was effectively rescued by both GSH and ferrostatin-1 (Fig. 2A and fig. S3, A and B), which was also verified in B16-F10 cells (fig. S3C). To further elucidate the underlying mechanism of cell death caused by RT-MPs, label-free quantitative proteomics analysis was performed on RT-MP–treated and untreated cells. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed that the ferroptosis pathway was enriched; moreover, prostaglandin G/H synthase 2 (ptgs2) and ferritin (ftl1), which were involved in ferroptosis (23), exhibited significantly higher expression in the RT-MP–treated group (Fig. 2, B and C). In RT-MP–treated A549 and LLC, TEM images showed shrunken mitochondria (Fig. 2D and fig. S3D), similar to those observed upon treatment with erastin, a typical ferroptosis-inducing compound (24). To further confirm that ferroptosis was caused by RT-MPs, cytosolic and lipid reactive oxygen species (ROS) were respectively detected by fluorescent probes 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and 4,4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diaza-s-indacene-3-undecanoic acid (BODIPY® 581/591 C11). We observed a time-dependent increase beginning at 4 hours, and both could be partially rescued by ferrostatin-1 (fig. S3, E to G). This mechanism could also be applied in B16-F10 cells (fig. S3, H and I). To further explore the reason why RT-MPs could kill tumor cells compared to normal cancer cell MPs, we used flow cytometry and found RT-MPs contained ROS, and this may be one of the reasons for the enhanced killing (fig. S3J). We further traced the subcellular distribution of RT-MPs and found the colocalization between RT-MPs membranes and lysosomes, mitochondria (fig. S3K).
Fig. 2. RT-MPs induce tumor cell death by causing ferroptosis.
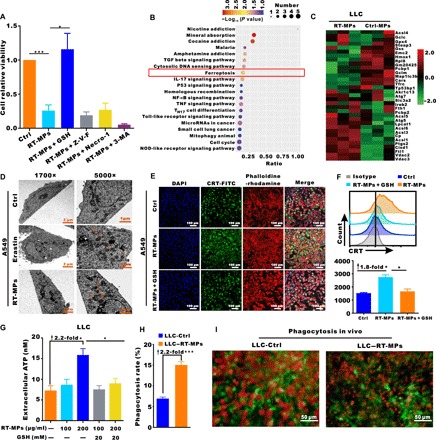
(A) Modulatory profiling of known small-molecule cell death inhibitors in A549 cells treated with RT-MPs (100 μg/ml, 48 hours). (B) KEGG bubble map of differentially enriched proteins. The x axis indicates the ratio of the number of differential proteins in the corresponding pathway to the number of total proteins identified. The colors of the points represent the P values of the hypergeometric test. The sizes of the dots represent the numbers of differential proteins in the corresponding pathway. (C) Heat map of differentially expressed proteins in the ferroptosis pathways of RT-MP–treated and untreated cells. (D) TEM images of A549 cells treated with phosphate-buffered saline (PBS) (24 hours), erastin (2 μM, 24 hours), and RT-MPs (100 μg/ml, 24 hours). Single orange arrowheads indicate shrunken mitochondria. At least 10 cells were examined in each treatment condition. (E) Immunofluorescence staining of calreticulin (CRT) expression (green) on the surface of A549 cells after various treatments. Phalloidine-rhodamine (red) represents the cytoskeleton. Scale bars, 100 μm. (F) Flow cytometric analysis of CRT expression. (G) ATP levels in LLC cells treated as shown with the indicated compounds. (H) BMDMs from C57BL/6 mice showed a higher rate of phagocytosis for RT-MP–treated LLC cells. BMDMs were stained with antibodies to F4/80, and LLC cells were stained with red fluorescence dye PKH26. Flow cytometric analysis was performed to evaluate the rate of phagocytosis. (I) In vivo visualization of phagocytosis of RT-MP–treated cells. LLC-RFP cells were subcutaneously injected (2 × 104) into the window. Macrophages were recruited and phagocytosed more RT-MP–treated LLC-RFP cells in the CX3CR1+/GFP window chamber. Scale bars, 50 μm. *P < 0.05 and ***P < 0.001.
It is known that ferroptosis is immunogenic (22, 25), characterized by the release of tumor-associated antigen and immunologic danger signals from dying tumor cells; thus, we examined danger-related molecular patterns of tumor cells after treatment with RT-MPs. The results showed that levels of extracellular calreticulin (CRT) expression and adenosine triphosphate (ATP) release in the medium were 1.8-fold and 2.2-fold of those in the control group, respectively. Confocal microscopy also showed increased CRT expression after treatment with RT-MPs (Fig. 2, E to G, and fig. S4, A to D). Analysis by flow cytometry revealed that macrophages phagocytosed more RT-MP–treated than untreated LLC (Fig. 2H). In the CX3CR1+/GFP mouse window chamber model, RT-MP–treated LLC-RFP cells could recruit more macrophages (green); these macrophages had greater capacity to capture tumor cells (Fig. 2I and movies S3 and S4). Together, these results indicate that RT-MPs mediate RIBE by causing ferroptosis in tumor cells, which further promotes phagocytosis by macrophages.
RT-MPs promote macrophage polarization and activate the Jak-STAT and MAPK pathways
To elucidate the in vivo effects of RT-MPs on TMEs, we first examined the cellular internalization of PKH26 (PKH26 Red Fluorescent Cell Linker Mini Kit for General Cell Membrane Labeling)–labeled RT-MPs in MPE mice after intrapleural administration. We collected pleural effusion cells and analyzed them by flow cytometry; PKH26-labeled RT-MPs were mainly taken up by CD11b+F4/80+ cells (representing TAMs), followed by Gr-1+ cells (representing myeloid-derived suppressor cells) and CD45− cells (representing tumor cells), whereas few RT-MPs were taken up by CD3+ T cells (Fig. 3A and fig. S5A). In the CX3CR1+/GFP dorsal skinfold window chamber model, following injection of RT-MPs, we observed that large numbers of macrophages migrated to the injection site in a time-dependent manner and avidly phagocytosed red PKH26-labeled RT-MPs (Fig. 3B and movies S5 and S6). On the basis of these findings, we further explored the effect of RT-MPs on phenotypic changes of macrophages due to their robust phagocytosis of RT-MPs (movie S7). Flow cytometry analysis was performed on macrophages incubated with PKH26-labeled RT-MPs for 2, 6, 12, and 24 hours. The results showed time-dependent uptake of RT-MPs by macrophages (Fig. 3C and fig. S5B), which was consistent with in vitro confocal images (fig. S5C). To determine mRNA changes of RT-MP–treated macrophages, subsequent RNA sequencing (RNA-seq) was performed by using bone marrow–derived macrophages (BMDM-M2 cells). RNA-seq results showed that M1-related mRNAs were highly expressed (Fig. 3D) and real-time fluorescent quantitative polymerase chain reaction (RT-qPCR) showed consistently significantly increased mRNA expression levels of inducible nitric oxide synthase (iNOS), interleukin-1 (IL-1), IL-6, IL-12, tumor necrosis factor–α, and interferon-γ (IFN-γ) in RT-MP–treated BMDM-M2 cells (Fig. 3E). KEGG analysis showed that the Janus kinase/signal transducers and activators of transcription (Jak-STAT) signaling pathway and mitogen-activated protein kinase (MAPK) signaling pathway (M1-related pathway) were enriched (Fig. 3F). The protein expression levels of p-STAT1, p-p38, p-ERK (extracellular signal–regulated kinase), p-JNK, and β-actin in BMDM-M2 cells treated with RT-MPs at indicated time points were analyzed by Western blotting. As shown in Fig. 3G, the members of MAPK, ERK1/2, c-Jun N-terminal kinase (JNK), p38, and STAT1 pathways were phosphorylated (26). Following phagocytosis of RT-MPs, macrophages showed M1 polarization concomitant with threefold increased expression of CD86, 5.2-fold increased expression of major histocompatibility complex II, and 46.5% reduction in CD206 expression. Moreover, this was supported by the significant increase in the protein expression level of iNOS (fig. S5D). This change could not be rescued by GSH incubation (Fig. 3H), indicating that ROS did not contribute to macrophage polarization. In MPE mice, injection of RT-MPs reduced CD206 expression in macrophages by 86.8% (Fig. 3, I and J). As expected, RT-MP–treated macrophages showed much stronger ability to phagocytose tumor cells, which was 1.5-fold greater than that of untreated macrophages (Fig. 3K). Furthermore, this effect was visualized in the CX3CR1+/GFP dorsal skinfold window chamber model (Fig. 3L and movies S3 and S8). Together, these results indicated that RT-MPs can modify macrophages from a tumor-promoting to a tumor-inhibiting phenotype by activation of the Jak-STAT and MAPK pathways.
Fig. 3. RT-MPs reprogram macrophages through Jak-STAT and MAPK pathways.
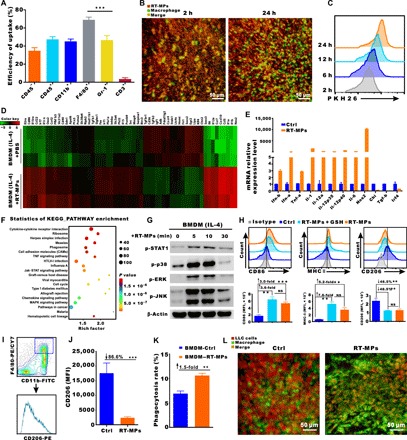
(A) Quantification of the accumulation of PKH26-labeled RT-MPs in various cell types in MPE mice (means ± SEM, n = 5 mice). (B) Representative images of macrophages (green) that were recruited and phagocytosed red RT-MPs over time in the CX3CR1+/GFP window chamber. RT-MPs were stained with red fluorescence dye PKH26 and subcutaneously injected into the window. Scale bar, 50 μm. (C) Flow cytometric analysis of RT-MPs internalization by macrophages at multiple time points (n = 3). (D) Heat map of RNA-seq on RT-MP–treated and untreated BMDM-M2 cells. (E) RT-qPCR analysis of the expression levels of M1- and M2-associated mRNAs in RT-MP–treated BMDM-M2 cells. The results indicate up-regulation of M1-associated mRNAs and down-regulation of M2-associated mRNAs. (F) The top 20 up-regulated functional pathways in RT-MP–treated and untreated groups, as determined by KEGG analysis. (G) Protein expression levels of p-STAT1, p-p38, p-ERK, p-JNK, and β-actin in BMDM-M2 cells treated with RT-MPs at the indicated time points were analyzed by Western blotting. (H) Flow cytometric analysis of CD86, major histocompatibility complex II (MHC II), and CD206 expression in BMDM-M2 cells treated with RT-MPs and GSH-incubated RT-MPs. MFI, mean fluorescence intensity. (I) Flow cytometry gating strategy for measurement of macrophages from MPE mice. (J) Quantification of CD206 expression in MPE mice after treatment with RT-MPs. (K) RT-MP–treated BMDM-M2 cells showed increased phagocytosis of LLC cells compared to that of control BMDM-M2 cells. Flow cytometric analysis was performed to evaluate the phagocytosis rate. FITC, fluorescein isothiocyanate; PE, phycoerythrin. (L) In the CX3CR1+/GFP mouse window chamber, RT-MP–treated macrophages were recruited and phagocytosed a greater number of LLC-RFP cells. Representative images were shown here. Scale bars, 50 μm. **P < 0.01 and ***P < 0.001.
Treatment comprising RT-MPs combined with anti–PD-1 confers antitumor effects and generates memory immune responses
As noted above, RT-MPs induced ferroptosis in tumor cells, causing immunogenic cell death and activated macrophages; RT-MPs also caused 3.2-fold increase in PD-L1 expression on the macrophage surface (Fig. 4A), which prompted a follow-up experiment to investigate combined therapy with anti–PD-1 monoclonal antibody (mAb). The combined therapy for MPE treatment resulted in a significantly prolonged survival time and cured 1 of 15 mice with no recurrence (fig. S6, A and B); these effects were superior to those of treatment with anti–PD-1 or RT-MPs alone (Fig. 4, B and C). By increasing the number of RT-MPs injections to six, the cure rate reached 38.5% (Fig. 4D and fig. S6C). We also established a subcutaneous transplanted model by using B16-F10 cells stably transfected with luciferase. Combined treatment with RT-MPs and anti–PD-1 induced an obvious delay in tumor growth and prolonged survival (Fig. 4, E and F, and fig. S6D).
Fig. 4. Combination treatment with RT-MPs plus anti–PD-1–cured MPE mice and generated strong memory responses.
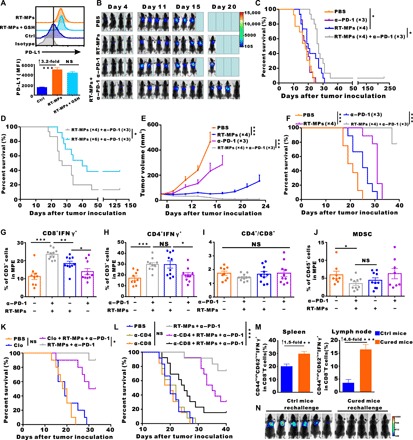
(A) Flow cytometry analysis of PD-L1 expression on the surface of BMDMs treated with RT-MPs. (B) Representative in vivo bioluminescence images of the growth of mice MPE under various treatment conditions. (C) Kaplan-Meier survival plot of MPE mice in the corresponding treatment groups described in (B) (n = 13 to 15 per group). (D) Comparison of the therapeutic efficacy of different frequencies of RT-MPs injection (n = 13 to 15 per group). (E) Tumor growth curves of B16-F10–LUC subcutaneous transplant model in corresponding treatment groups (n = 8 to 9 per group). (F) Kaplan-Meier survival plot of B16-F10–LUC melanoma-bearing mice in the corresponding treatment groups described in (E) (n = 8 to 9 per group). (G to J) Flow cytometry analysis of changes in the immune cells in the pleural lavage fluid of MPE mice that underwent different treatments (n = 8 to 11 per group). (K) Kaplan-Meier survival plot of LLC-LUC MPE-bearing C57BL/6 mice (n = 9 to 10 per group) that were treated with clodronate liposomes and/or RT-MPs plus anti–PD-1. (L) Kaplan-Meier survival plot of LLC-LUC MPE-bearing C57BL/6 mice treated with RT-MPs plus anti–PD-1, concomitant with anti-CD4 or anti-CD8 neutralizing antibody treatment (n = 12 to 13 per group). (M) Proportions of effector memory T cells (Tem and CD44highCD62low IFN-γ+) in the lymph node and spleen analyzed by flow cytometry (gated on CD3+CD8+ T cells) at day 60 (n = 5 to 6 per group). (N) In vivo bioluminescence images to monitor the growth of rechallenged thorax-injected LLC-LUC tumors (n = 5 to 6 per group). *P < 0.05, **P < 0.01, and ***P < 0.001.
To clarify the immune microenvironment changes after injection of RT-MPs in vivo, immune cells were measured from the pleural lavage of MPE mice. We observed a concurrent increase in CD3+CD4+IFN-γ+ and CD3+CD8+IFN-γ+ T cells in the combination group (Fig. 4, G and H). Changes in the CD4+/CD8+ T cell ratio and the frequency of Foxp3+ T regulatory cells (Fig. 4I and fig. S6E) were not statistically different between treated groups. Myeloid-derived suppressor cells (MDSCs) (CD45+CD11b+Gr-1+) decreased significantly in RT-MP–treated and combination treatment groups (Fig. 4J). Combination treatment also led to an increased proportion of CD3+ T cells in pleural tumor nodules (fig. S6F). Collectively, these findings of increased T cell populations (including CD3+CD4+IFN-γ+ and CD3+CD8+IFN-γ+ T cells) and decreased MDSCs in MPE strongly suggested reshaping of TMEs upon combination treatment with RT-MPs and anti–PD-1.
We further explored the roles of macrophages, natural killer (NK) cells, CD4+ T cells, and CD8+ T cells in the antitumor activity of combination treatment with RT-MPs and anti–PD-1; this was achieved by effectively depleting macrophages, NK cells, CD4+ T cells, and CD8+ T cells by clodronate liposomes, NK1.1 antibody, anti-CD4 antibody, and anti-CD8 antibody, respectively (fig. S6, G to I). We observed that macrophage depletion significantly attenuated the inhibitory effect of combination treatment with RT-MPs and anti–PD-1 on MPE (P = 0.0427; Fig. 4K). The inhibitory effect was mostly lost in mice that underwent CD8+ T cell depletion and partially lost in mice that underwent CD4+ T cell depletion (Fig. 4L), suggesting that the anticancer effect of combination treatment with RT-MPs and anti–PD-1 is largely dependent on CD8+ T cells and partially dependent on CD4+ T cells. However, NK cell depletion had no significant effect on the therapeutic effect of combination treatment with RT-MPs and anti–PD-1 (fig. S6J). Together, these findings suggested that combination treatment with RT-MPs and anti–PD-1 initiates a macrophage-, CD4+ T cell–, and CD8+ T cell–dependent pathway that enhances antitumor immunity.
Because combination treatment with RT-MPs and anti–PD-1 triggered innate and adaptive immune responses, we assessed whether the cured mice formed memory responses. For this experiment, six cured mice (completely cured for at least 30 days, confirmed by in vivo bioluminescence images) were assessed for the presence of memory T cells. CD3+CD8+CD44highCD62LlowIFN-γ+ cells were regarded as T effector memory (Tem) cells (27, 28); the numbers of Tem in cured mice increased by 1.5-fold and 4.6-fold in the spleen and lymph nodes, respectively (Fig. 4M). Another five cured mice were rechallenged with LLC-LUC cells, and the same aged naïve mice were challenged in parallel. No tumor growth occurred in the combination treatment–cured groups, indicating a memory response upon tumor antigen recognition. In contrast, in vivo imaging showed that all naïve mice developed MPE (Fig. 4N). Collectively, these results demonstrated that the combination treatment with RT-MPs and anti–PD-1 is favorable for the generation of memory immune responses after eradication of primary MPE.
Combination treatment with RT-MPs and anti–PD-1 demonstrates an ablative effect in the cisplatin (DDP)–resistant MPE model
Diamminedichloroplatinuin (DDP) a classical drug for MPE patients in clinical treatment, but it has limited therapeutic effect because most patients develop resistance to DDP after repeated cycles of administration (29–31). Therefore, we explored whether combination treatment with RT-MPs and anti–PD-1 could contribute to treatment of DDP-resistant (DDR) MPE. DDR LLC-LUC cells, LLC cells, and A549 cells were established by low-concentration DDP induction with a period of approximately 2 months. When DDP (2 μg/ml) was added, the percentages of apoptotic cells, including early and late apoptotic cells, were 35.2 and 6.4% in LLC and DDR-LLC cells, respectively (Fig. 5, A and B). Dose-response curves showed that the IC50 (median inhibitory concentration) values of DDR-LLC and DDR-A549 cells were nearly threefold greater than those of their corresponding wild-type cells (Fig. 5C and fig. S7A). Moreover, we found that DDR-LLC cells had a poor response to DDP treatment in the MPE model (Fig. 5, D and E). Despite resistance to DDP treatment, both DDR-LLC and DDR-A549 cells were indiscriminately killed by RT-MPs (Fig. 5F and fig. S7B). Consistent with this phenomenon, we found that combination treatment with RT-MPs and anti–PD-1 had a good therapeutic effect on the DDR MPE model and cured approximately 20% of MPE mice, whereas DDP did not show this effect (Fig. 5G and fig. S7C). Representative 18F-FDG positron emission tomography–computed tomography images of normal mice, combination treatment–cured MPE mice, and DDP-treated mice are shown in Fig. 5H.
Fig. 5. Combination treatment with RT-MPs plus anti–PD-1 overcomes cisplatin resistance.
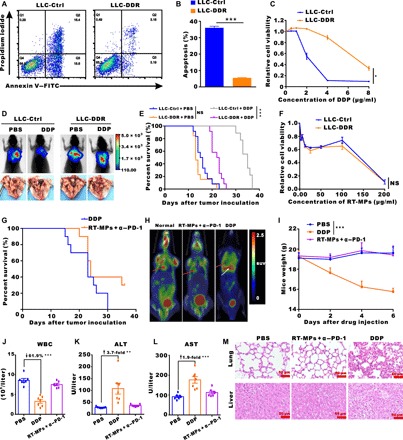
(A and B) Flow cytometry measurement of LLC-Ctrl and LLC-DDR cell apoptosis induced by cisplatin (DDP). (C) Sensitivity of LLC-Ctrl and LLC-DDR cells to DDP treatment. Relative cell viability was measured by CCK-8 assay. (D) Representative in vivo bioluminescence images to monitor the growth of thorax-injected LLC-Ctrl or LLC-DDR cells in different groups. LLC-DDR MPE was more resistant to DDP treatment. Photo credit: Chao Wan, Cancer Center, Wuhan Union Hospital. (E) Kaplan-Meier survival plot of LLC-Ctrl or LLC-DDR MPE-bearing mice in the corresponding treatment groups described in (D) (n = 9 to 13 per group). (F) Sensitivity of LLC-Ctrl and LLC-DDR cells to treatment with RT-MPs. Cell viability was measured by CCK-8 assay. (G) Kaplan-Meier survival plot of MPE-bearing mice treated with DDP or RT-MPs plus anti–PD-1. (H) Representative 18F-FDG PET-CT images of normal mice, RT-MPs plus anti–PD-1–cured MPE mice, and DDP-treated mice, respectively. (I) Weight changes after treatment with PBS, DDP, or RT-MPs plus anti–PD-1. (J to L) Hemanalysis was performed on blood withdrawn from mice on day 3 after treatment. White blood cell (WBC), alanine transaminase (ALT), and aspartate transaminase (AST) are presented as the means ± SEM (n = 7). (M) Representative histological examinations of the main organs with hematoxylin and eosin staining. Images of main organs from mice injected with PBS, DDP, or RT-MPs plus anti–PD-1. Scale bars, 50 μm. *P < 0.05, **P < 0.01, and ***P < 0.001.
To evaluate the biocompatibility of RT-MPs, we recorded weight changes in the mice; hemanalysis and biochemical analysis were performed on blood withdrawn from mice at 3 days after treatment, and normal tissues (such as the heart, liver, spleen, lung, and kidney) were dissected for histopathological analysis. During the 11-day study period, mice that received combination treatment with RT-MPs and anti–PD-1 remained normal, as indicated by the absence of weight loss (Fig. 5I). At a DDP concentration of 3 mg/kg, DDP-treated mice showed significant weight loss compared to the phosphate-buffered saline (PBS)–treated control mice. All treated mice showed similar levels of red blood cell (RBC) number and renal function parameters (such as blood urea nitrogen and creatinine) (fig. S8, A to C). We observed that the DDP group had abnormal levels of white blood cells, alanine transaminase, and aspartate transaminase, while these parameters remained normal in PBS-treated and combination-treated mice (Fig. 5, J to L). Postmortem histopathological examination of the heart, liver, spleen, lung, and kidney demonstrated that these organs remained in normal condition and were not affected by combination treatment with RT-MPs and anti–PD-1; in contrast, hepatocytes showed balloon-like degeneration, and pulmonary congestion were observed in the lung tissues in the DDP group (Fig. 5M and fig. S8D).
DISCUSSION
Although RT plays a central role in treating cancer, it is effective for approximately one-half of newly diagnosed patients with cancer and patients with persistent or recurrent tumors. Currently, RT-related clinical studies are mainly focused on how to combine RT and other therapies (e.g., immunotherapy and molecular-targeted treatment) to maximize therapeutic benefits (32–34); however, few studies have been undertaken to develop strategies that could expand its clinical indications. In classic radiobiology studies, irradiated cells were shown to influence neighboring unirradiated cells in vitro (RIBE), but the mechanism is poorly understood (35). Here, we identified that the action of in vitro RIBE largely depends on release of RT-MPs, which exhibit both direct antitumor effects and indirect immune-stimulating capabilities; these can be used to treat cancers where RT cannot be applied, thus comprising indirect cancer RT.
MPE is nearly incurable with current standard therapy and is poorly responsive to chemotherapy; furthermore, RT cannot be performed as treatment of MPE. In mouse models of MPE, few studies have attempted to establish strategies for treatment of MPE that result in prolonged survival; however, MPE mice were seldom cured (7, 36). Here, by using combination treatment with RT-MPs and anti–PD-1, a cure rate of approximately 20 to 40% was achieved for regular MPE and for cisplatin-resistant MPE. Using RT-MPs for cancer treatment may provide several advantages: (i) Unique antitumor effect: Most of previously reported T-MPs (e.g., chemotherapy and hypoxia derived) often show prometastatic potential, whereas RT-MPs mimic the RIBE and show direct in vitro antitumor capability. When they are taken up by tumor cells, RT-MPs can cause immunogenic cell death mainly through ferroptosis, thus making tumor cells more vulnerable to attack by macrophages (Figs. 1L and 2, D to I). (ii) Immune-stimulating ability: Exogenously injected RT-MPs were more efficiently captured by TAMs in the TMEs, compared to Gr-1+ MDSCs and tumor cells, resulting in proinflammatory M1-TAMs polarization, which is beneficial for eliciting antitumor immune responses (37, 38) (Fig. 3). (iii) Combination treatment with anti–PD-1: The internalization of RT-MPs also increases the expression level of PD-L1 in TAMs, which provides a good rationale for combination therapy involving RT-MPs and anti–PD-1. This combination showed a cure rate of up to 40% and was favorable for the generation of memory immune responses when used for MPE treatment. Moreover, it showed potent therapeutic efficacy in a subcutaneous B16-F10 melanoma model (Fig. 4). (iv) Chemotherapy-free: Another type of tumor cell–derived microparticle containing the chemotherapeutic drug methotrexate (TMP-MTX) reportedly caused marked restriction of MPE growth and provided a survival benefit in MPE models; however, it could not cure the MPE mice (17, 39). TMP-MTX treatment was applied in 11 patients with advanced lung cancer by intrapleural delivery, and chemotherapy-related adverse events (including dizziness, fever, nausea, vomiting, and decreased RBCs) still occurred (17). (v) Biocompatible and safe: Preclinical and clinical evidence suggests that irradiated tumor cells could be used as effective and safe vaccines to generate host adaptive immune responses for prevention of tumor recurrence after resection of lesions (Fig. 5) (40). RT-MPs are derived from irradiated tumor cells and could be obtained from autologous dissected primary tumor cells in clinical applications of this approach, thus providing potential nontoxic and personalized cancer treatment. In our study, we confirmed excellent biocompatibility of combination treatment with RT-MPs and anti–PD-1, as revealed by the absence of obvious side effects. We also compare RT-MPs with ultraviolet-treated tumor cell–derived microparticles (UV-MPs; the most commonly mentioned preparation method for MPs) in vitro and in vivo; we found that the killing ability and also the capacity for reprogramming macrophages of UV-MPs were weaker than that of RT-MPs (figs. S2E and S5E). The effect of RT-MPs was superior to that of UV-MPs in MPE model (fig. S6K). In conclusion, this study demonstrates a chemotherapy-free strategy for indirect cancer RT by putatively mimicking the RIBE machinery.
MATERIALS AND METHODS
Chemical reagents
Erastin (S7242), deferiprone (S4067), ferrostatin-1 (S7243), liproxstatin-1 (S7699), Z-VAD-FMK (Z-VAD fluoromethylketone) (S7023), 3-methyladenine (3-MA) (S2767), and necrostatin-1 (S8037) were purchased from Selleck. β-Mercaptoethanol (07604) and reduced l-glutathione (G4251) were purchased from Sigma-Aldrich. H2DCFDA (D399) and s 4,4-difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diaza-s-indacene-3-undecanoic acid (BODIPY 581/591 C11) (Lipid Peroxidation Sensor) (D3861) were purchased from Invitrogen.
Cell lines and cell culture
All cell lines were purchased from the American Tissue Culture Collection, including human lung carcinoma cell lines (A549, Calu-1, and H1975), the human colon adenocarcinoma cell line (HCT116), the human primary glioblastoma cell line (U87), murine B16F10 melanoma cells, and murine LLC. For in vivo studies, luciferase stably transfected cell lines (LLC-LUC and B16F10-LUC) and RFP stably transfected cell lines (LLC-RFP and A549-RFP) were established. Cisplatin-resistant cells (LLC-DDR, LLC-LUC-DDR, and A549-DDR) were generated by long-term (at least 2 months) culturing with cisplatin (2 μg/ml). All cells were cultured in an incubator at 37°C with 5% CO2 and using complete medium [supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 U/ml)]. LLC, LLC-LUC, HCT116, and U87 cells were maintained in Dulbecco’s modified Eagle medium (DMEM); other cells were maintained in RPMI 1640 medium. All cell lines were routinely tested for mycoplasma infection and were confirmed to be negative.
Isolation of RT-MPs and RT exosomes
A total of 5 × 106 cells that were plated into 10-cm cell culture dishes were irradiated with a single dose of 20 Gy by 6-MV x-rays (600 MU/min, Trilogy System Linear Accelerator, Varian Medical Systems). The medium was then replaced with 20 ml of complete medium (DMEM or RPMI 1640, based on the needs of each cell line). After 72 hours, the medium was collected and centrifuged at 1000g for 10 min and then 14,000g for 2 min to remove tumor cells and debris. Then, the supernatant was centrifuged at 14,000g for 1 hour at 4°C to isolate RT-MPs and further centrifuged for 70 min at 120,000g at 4°C to isolate RT exosomes. The precipitate (containing exosomes or MPs) was washed with sterile 1× PBS twice and resuspended in sterile 1× PBS for animal experiments or resuspended in completed medium for cell experiments.
Quantification of RT-MPs
We quantified RT-MPs by measuring the protein concentration. After being washed, RT-MPs were lysed with radioimmunoprecipitation assay (RIPA) buffer at 4°C for 30 min and then centrifuged for 30 min at 12,000g at 4°C; the supernatant that contained the total protein was switched to a new centrifuge tube. Protein was quantified with the BCA Protein Assay Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s protocol.
Transmission electron microscopy
RT-MPs and cells treated with RT-MPs (100 μg/ml) or erastin (2 μM) were observed by TEM. RT-MPs in suspension were stained with 2% phosphotungstic acid solution for 5 min and then deposited on copper mesh; size and morphology were observed by TEM. Cells were washed in 0.1 M PBS and then fixed with 2.5% glutaraldehyde in 0.1 M Sorenson’s buffer overnight at 4°C; cells were then post-fixed with 1% osmium tetroxide in PBS for 1.5 hours. After dehydration through a graded ethanol series, cells were embedded in Lx-112 (Ladd Research Industries) and Embed-812 (EMS) and then cut on an MT-7000 ultramicrotome. Ultrathin sections were stained with 1% uranyl acetate and 0.4% lead citrate, and then morphology was observed by TEM (HT7700-SS/FEI Tecnai G20 TWIN).
Cell viability study
To measure cell viability, cells were plated in 96-well plates (5000 cells per well) and allowed to grow for 24 hours before treatment. Cells were then treated with different concentrations of RT-MPs or other chemical reagents. After incubation for 48 hours, cell viability was evaluated by using a CCK-8 assay kit (BS350B, Biosharp).
Colony formation assay
Cells were seeded into 48-well plates (10,000 cells per well) or six-well plates (500 cells per well). To test the ability of RT-MPs to inhibit tumor cells, cells in 48-well plates were treated with different doses of RT-MPs and then cultured with complete medium for 3 days. To directly visualize the RIBE, cells were seeded into Transwell chambers with different pore sizes (0.4 and 1 μm) (200,000 cells per chamber) and six-well plates (500 cells per well). After 24 hours, the cells in Transwell chambers were irradiated with a single dose of 20 Gy by 6 MV x-rays and then were cocultured with cells in six-well plates for 5 days. Subsequently, the chambers were moved, and the plates were replaced with 3 ml of complete medium for 10 days. When colonies had more than 50 cells, the cells were fixed with 4% formaldehyde for 30 min, stained with 2% crystal violet (no. HT90132, Sigma-Aldrich) overnight, and washed with water. The stained cells were photographed, and the numbers of clones were counted.
Analysis of ROS production
Cells were plated in 12-well dishes (50,000 cells per well) and then treated with RT-MPs and other chemical reagents for 4, 12, and 24 hours. Cells were subsequently stained with H2DCFDA (10 μM) or C11-BODIPY (581/591) (2 μM) in 1 ml of medium without FBS for 30 min at 37°C in a cell culture incubator. After three washes in PBS, cells were harvested and resuspended in 150 μl of fresh PBS and then analyzed using a flow cytometer (Beckman CytoFLEX S). Each treatment condition included at least 10,000 cells for analysis.
Generation of BMDMs
BMDMs were collected from femurs of 6- to 12-week-old C57BL/6 male mice. RBCs were depleted using RBC lysis buffer and then differentiated in RPMI 1640 medium supplemented with 10% FBS and macrophage colony-stimulating factor (20 ng/ml; PeproTech). The medium was replaced every 2 days. On the sixth day, naïve macrophages (BMDMs) were stimulated with IL-4 (20 ng/ml; PeproTech) for 24 hours to generate BMDM-M2 macrophages. BMDM-M2 cells were harvested for stimulation assay on the seventh day.
Western blotting
RT-MPs and cells were lysed with RIPA buffer supplemented with protease inhibitors and phosphatase inhibitor cocktails II and III at 4°C for 30 min and then centrifuged for 30 min at 12,000g at 4°C; the supernatant contained total protein was switched to a new centrifuge tube. Protein was quantified with the BCA Protein Assay Kit (Thermo Scientific). Samples were mixed in 1× SDS–polyacrylamide gel electrophoresis sample loading buffer (P0015, Beyotime Biotechnology) and denatured by heating to 100°C for 10 min; they were then separated in an SDS-polyacrylamide gel and transferred to a polyvinylidene difluoride (PVDF) membrane. The PVDF membrane was blocked with 5% skim milk powder dissolved in tris-buffered saline with 0.05% Tween 20 (TBST) at room temperature for 1 hour with gentle shaking and then incubated overnight at 4°C with primary antibody. Next, the membrane was washed three times with TBST at room temperature for 15 min each time, followed by incubation at room temperature for 1 hour with diluted secondary antibody in TBST. Following additional washes in TBST, NcmECL Ultra (P10100, NCM Biotech) was used for chemiluminescent exposure of the blot. The antibodies used are listed in table S2.
Real-time quantitative polymerase chain reaction
Total RNA was isolated from cells using the Total RNA Kit I R6834 (Omega), and RNA was measured using the NanoDrop ND-1000 (Thermo Fisher Scientific). Purified RNA was reverse-transcribed into complementary DNA by HiScript III RT SuperMix (+gDNA wiper) (Vazyme, #R323) in accordance with the manufacturer’s instructions. The RT-qPCR reaction was performed in a Step One system using AceQ Universal SYBR qPCR Master Mix (Vazyme). Gene expression was calculated by using the comparative threshold cycle method and then normalized to the reference gene, glyceraldehyde 3-phosphate dehydrogenase. The primers used were synthesized by Sangon Biotech (Shanghai) Co Ltd.; these are listed in table S1.
In vitro cellular uptake assay
For detecting colocalization of cell and RT-MPs, M2-BMDMs were seeded in a glass-bottom cell culture dish (NEST, catalog no. 801001; 1 × 105 per well) and incubated with PKH-26–labeled RT-MPs for 2, 6, 12, and 24 hours. At the end, the cells were washed by PBS for three times and then stained with carboxyfluorescein diacetate succinimidyl ester (10 μM) for 10 min. After that, cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min and then washed by PBS again. Cells were imaged using confocal laser scanning microscopy (Leica TSC SP8). For quantifying assessment of cellular uptake, cells seeded in six-well cell culture dishes and treated as above, then washed by PBS for three times, collected, fixed, and resuspended in PBS (150 μl) for flow cytometry detection.
ATP release assays in vitro
To measure extracellular ATP levels, cell culture supernatants were collected, and ATP concentrations were measured using luciferin-based ENLITEN ATP Assay (Promega) kits in accordance with the manufacturer’s protocol.
CRT assay
Cells were seeded in 24-well cell culture dished with 13-mm round glass coverslips and incubated with RT-MPs for 24 hours. Cells were fixed in formaldehyde (4% in PBS) and permeabilized with 0.1% Triton X-100 in PBS. Subsequently, cells were incubated with anti-CRT antibodies for 30 min, followed by incubation with Alexa Fluor 488 goat anti-rabbit immunoglobulin G (IgG) for 30 min. Then, cells on coverslips were labeled with DY-554 phalloidin, washed with PBS, and stained with DAPI (4′,6-diamidino-2-phenylindole). The stained cells were observed by laser confocal microscopy (Leica TCS SP8). For flow cytometer analysis, the cells were fixed in formaldehyde (4% in PBS) and incubated with anti-CRT antibodies for 30 min, followed by incubation with fluorescein-conjugated AffiniPure goat anti-rabbit IgG for 30 min.
Label-free quantitative proteomics
Label-free quantitative proteomics analysis was performed by Novogene Bioinformatics Technology Co Ltd. (Beijing, China). Label-free quantitative proteomics analysis was used to identify differentially expressed proteins between RT-MP–treated LLC and control cells. Analyses mainly included raw data filtering and quality control; protein function annotation including gene ontology (GO), Clusters of Orthologous Group, KEGG, and InterPro (IPR); protein expression quantification; differential expression analysis; GO enrichment; KEGG enrichment; IPR enrichment; and protein-protein interaction.
RNA sequencing
Total RNA was extracted from BMDMs that were treated with or without RT-MPs. RNA-seq was performed by CapitalBio Technology Inc. (Beijing, China). Basic analysis mainly included raw data filtering and quality control, reference genome comparison, alignment homogeneity analysis, saturation analysis, sample correlation analysis, expression analysis, and differential gene analysis. Advanced analysis mainly included new transcript prediction, splice junction analysis, cSNP/InDel (single nucleotide polymorphism/Insertion/Deletion) analysis, differentially expressed gene function annotation, GO and pathway/disease enrichment, differential gene coexpression network analysis, and differential gene protein interaction analysis.
Animal studies
Male C57BL/6 J wild-type (WT) mice were purchased from Liaoning Changsheng Biotechnology Co Ltd. All animals were maintained in the specific pathogen–free barrier facility in the Animal Center of Huazhong University of Science and Technology (HUST; Wuhan, China). All animal studies were performed in compliance with protocols that had been approved by the Hubei Provincial Animal Care and Use Committee in accordance with the experimental guidelines of the Animal Experimentation Ethics Committee of the HUST (Wuhan, China).
Model animal experiments and evaluation of therapeutic effects
Mice used for experiments were age (6 weeks)–, weight (18 to 20 g)–, and sex-matched. To establish the MPE model, mice were anesthetized by administration of 1% pentobarbital sodium before all operations. LLC-LUC cells (200,000 cells suspended in 50 μl of PBS) were injected into the right pleural cavity through the 10th or 11th intercostal space at the midaxillary line. Four days after inoculation with LLC-LUC cells, each mouse was observed by bioluminescence imaging to ensure that MPE models were established successfully and uniformly; mice were then randomized to four groups—including control, RT-MPs, PD-1 mAb, and RT-MPs and anti–PD-1—and were given treatment. Mice were treated by intrapleural injections with 50 μl of liquid (PBS or RT-MPs suspension) under isoflurane anesthesia, four times at 2-day intervals, or by intraperitoneal injection with PD-1 mAb (10 mg/kg), three times at 2-day intervals. To evaluate MPE growth, five mice in each group were imaged on the day when all treatments were completed and then five times at 3-day intervals under 1% pentobarbital sodium anesthesia using the Bruker In Vivo MS FX PRO Imager. The remaining mice were observed until death. To establish the subcutaneous transplanted B16-F10–LUC tumor model, 5 × 105 B16-F10–LUC cells in 100 μl of PBS were subcutaneously injected in the right flank of each mouse. These animals were then observed until they regained consciousness. Seven days after B16-F10–LUC inoculation, mice were randomized to four groups as above. When the tumor volume reached about 50 mm3, mice were given treatment. The mice were treated by intratumoral injections with 50 μl of liquid (PBS or RT-MPs suspension), four times at 2-day intervals, or intraperitoneal injection with PD-1 mAb (10 mg/kg), three times at 2-day intervals. To evaluate the therapeutic efficacy of the treatments, subcutaneous tumor growth was recorded with the length (L) and width (W) of tumors by vernier calipers, and the tumor size (V) was assessed at 2-day intervals with the formula V = (L × W2) / 2. Mice were sacrificed when the tumor volume reached 1000 mm3.
Bioluminescence imaging
After MPE mice had been anesthetized with 1% pentobarbital sodium, they were intraperitoneally injected with firefly luciferin (150 mg/kg; Sigma-Aldrich; CAS: 103404-75-7). After 15 min, mice were imaged by the Bruker In Vivo MS FX PRO Imager. The luminescent images were acquired with 3-min exposure time, and x-ray photographs were taken with 30-s exposure time.
Positron emission tomography–computed tomography
Before positron emission tomography (PET) imaging, the mice were fasted for 12 hours, and then approximately 200 ± 10 μCi of 18-fluoro-6-deoxy-glucose (FDG) was injected by intravenous injection. After 60 min of FDG uptake, mice were anesthetized with 2% isoflurane and placed on scanning bed. PET/computed tomography (CT) images were obtained with the static mode for 10 min followed by CT scan of normal mode by the TransPET Discoverist 180 system (Raycan Technology Co Ltd., Suzhou, China). The PET images were reconstructed using the three-dimensional ordered subset expectation maximization method with a voxel size of 0.5 mm by 0.5 mm by 0.5 mm. CT images were reconstructed using Feldkamp algorithm with 256 × 256 × 256 matrix. Images were displayed with software Carimas (Turku PET Center, Turku, Finland).
In vivo cellular internalization
To verify the in vivo uptake of RT-MPs, MPE mice received intrapleural injections of 50 μl of PKH26-labeled RT-MPs. Four hours later, mice were sacrificed by anesthetic overdose. Pleural lavages of MPE mice were collected, and RBCs were removed with lysis buffer to prepare single-cell suspensions. Cells were stained with a Zombie Violet Viability kit (BioLegend) according to the manufacturer’s instructions. Flow cytometry immunophenotyping was performed on single-cell suspensions using antibodies including anti-CD45 (clone 30-F11), anti-CD11b (clone M1/70), anti-F4/80 (clone BM8), and anti-LY6G (clone 1A8), as well as the PKH26 label [PE (phycoerythrin) channel, Sigma-Aldrich]. Cell acquisition was performed on a flow cytometer (Beckman CytoFLEX S). Only live cells were gated for analysis.
Dorsal skinfold window chamber model
Titanium chambers were surgically implanted under the dorsal skin of WT C56BL/6 mice or CX3CR1+/GFP mice. One day later, irradiated LLC-RFP cells, control LLC-RFP cells, or RT-MPs were stained with red fluorescent PKH26 and then subcutaneously injected into the window. At specified time points, videos were acquired by double femtosecond laser multiphoton microscope (LSM780, Zeiss). CX3CR1+/GFP mice were provided by the Optical Bioimaging Core Facility of HUST. All above experiments were undertaken in the laboratory too.
Immunomonitoring of MPE mice
To analyze changes of immune cells in TME, the pleural lavages of MPE mice were collected in anticoagulant conditions. Samples were centrifuged at 500g for 5 min, followed by lysis of RBCs, two washes with PBS, and resuspension in PBS. For flow cytometry, single-cell suspensions were stained with a Zombie Violet Viability kit (BioLegend) according to the manufacturer’s instructions to exclude dead cells. Surface antigens were stained with antibodies for 30 min at 4°C; all antibodies (listed below) were against mouse antigens and were purchased from BioLegend. For analysis of TAMs, cell suspensions from tumor tissues were stained with the following surface antibodies: anti-CD45 (clone 30-F11), anti-CD11b (clone M1/70), anti-F4/80 (clone BM8), and anti-CD86 (clone GL-1). After fixation and permeabilization, cells were stained with anti-CD206 (clone C068C2) antibody. For analysis of T cells, a portion of purified cell suspensions from MPE mice was stimulated with ionomycin (1 mg/ml; Abcam) and phorbol 12-myristate 13-acetate (50 ng/ml; Abcam) in the presence of bleomycin (1 mg/ml; Abcam) for 4 hours at 37°C with 5% CO2. Subsequently, single-cell suspensions were stained with anti-CD3ε (clone 145-2C11), anti-CD4 (clone GK1.5), and anti-CD8a (clone 53-6.7) antibodies. Cells were then fixed and stained with anti–IFN-γ (clone XMG1.2) and anti-Foxp3 (clone MF-14) antibodies. To analyze MDSCs, cell suspensions made from tumor tissues were stained with the following surface antibodies: anti-CD45 (clone 30-F11), anti-CD11b (clone M1/70), and anti–Gr-1 (clone RB6-8C5). To analyze memory effector T cells, cell suspensions made from the spleen and lymph nodes were stained with the following surface antibodies: anti-CD3ε (clone 145-2C11), anti-CD8a (clone 53-6.7), anti-CD44 (clone IM7), and anti-CD62L (clone MEL-14). Cells were then fixed and stained with anti–IFN-γ (clone XMG1.2) antibody.
T cell depletion
For the T cell depletion study, 200 μg per mouse of mouse anti-CD4 (clone GK1.5) and mouse anti-CD8 (clone 2.43) antibodies were diluted in PBS and intraperitoneally injected every 4 days, starting on the day before treatment.
Macrophage depletion
For the macrophage depletion study, clodronate liposomes (F70101C-AC) were purchased from FormuMax. Clodronate liposomes (200 μl of each) were injected intraperitoneally every 4 days, starting at the day before treatment.
NK cell depletion
For the NK cell depletion study, 200 μg per mouse of mouse anti-NK1.1 (clone 108760) were diluted in PBS and intraperitoneally injected every 4 days, starting on the day before treatment.
Quantification and statistical analysis
Group sizes, numbers of replicates, and explanations of mean and error bars are provided in figure legends. Statistical analysis was performed with Prism software (GraphPad Prism 6.0 software). Survival curves were compared with the log-rank (Mantel-Cox) test. Tumor growth was assessed by Kaplan-Meier analysis. For comparisons of three or more groups, one-way analysis of variance (ANOVA) was performed with Tukey’s multiple comparisons test. For comparisons of two groups, a two-tailed unpaired t test or the Mann-Whitney U test was performed. P values of <0.05 were considered to be statistically significant. Data are presented as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; and NS, not significant.
Supplementary Material
Acknowledgments
We thank Y. Wang (Cancer Center, Union Hospital, Wuhan, China) for providing us with professional radiation technology, J. Zhu and D. Li (Union Hospital, Wuhan, China) for in vivo bioluminescence imaging, Q. Yuan (Nanfang Hospital, Southern Medical University, Guangzhou, China) for biochemical analyses, F. Zhou from the Analysis and Testing Center of Institute of Hydrobiology, and CAS for the technical support of confocal imaging. Funding: This study was conducted with the support by National Natural Science Foundation of China (grant nos. 81874233, 81874222, 81672978, and 31770983) and the National Key Research and Development Program of China (project no. 2016YFC0105300). Author contributions: K.Y. and H.J. designed the research. C.W., Y.S., and Y.T. performed experiments and analyzed data. L.L. assisted with the experiments about dorsal window chamber model. X.D., J.M., J.H., and Q.H. assisted with mouse experiments and flow cytometry. B.W., Z.Z., K.J., D.H., G.W., and J.F.L. provided resources or conducted experiments. Y.S., C.W., H.J., and Y.T. wrote the manuscript. All authors have given approval to the final version of the manuscript. Competing interests: K.Y., H.J., Y.S., C.W., and Y.T. are inventors on the patent related to this work filed by Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (no. 2019100311342, 14 January 2019). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaay9789/DC1
Fig. S1. Examination of RIBE in irradiated A549 cells.
Fig. S2. Therapeutic effect of RT-MPs.
Fig. S3. RT-MPs induce ferroptosis in tumor cells.
Fig. S4. RT-MPs promote immunogenic cell death.
Fig. S5. Macrophages show robust phagocytosis of RT-MPs.
Fig. S6. Therapeutic effect of combination treatment with RT-MPs and anti–PD-1 depends on immune system activation.
Fig. S7. Therapeutic effect of RT-MPs on A549-DDR cells.
Fig. S8. Hemanalysis and biochemical analyses.
Table S1. Sequences of primers for RT-qPCR analysis.
Table S2. Antibodies used in this article.
Movie S1. Radiated LLC-RFP released more microparticles.
Movie S2. Unradiated LLC-RFP released less microparticles.
Movie S3. Macrophages and LLC-RFP cells.
Movie S4. Macrophages and RT-MP–treated LLC-RFP cells.
Movie S5. Two hours after RT-MPs’ injection.
Movie S6. Twenty-four hours after RT-MPs’ injection.
Movie S7. Macrophages phagocytose PKH26-labeled RT-MPs in vitro.
Movie S8. RT-MP–treated macrophages and LLC-RFP cells.
REFERENCES AND NOTES
- 1.Ethun C. G., Bilen M. A., Jani A. B., Maithel S. K., Ogan K., Master V. A., Frailty and cancer: Implications for oncology surgery, medical oncology, and radiation oncology. CA Cancer J. Clin. 67, 362–377 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Herrera F. G., Bourhis J., Coukos G., Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J. Clin. 67, 65–85 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Joyce J. A., Fearon D. T., T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Sanmamed M. F., Chen L., A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell 175, 313–326 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khalil D. N., Smith E. L., Brentjens R. J., Wolchok J. D., The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 13, 273–290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Q., Chen J., Yang Z., Xu J., Xu L., Liang C., Han X., Liu Z., Nanoparticle-enhanced radiotherapy to trigger robust cancer immunotherapy. Adv. Mater. 31, e1802228 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Fang F., Gong C., Qian Z., Zhang X., Gou M., You C., Zhou L., Liu J., Zhang Y., Guo G., Gu Y., Luo F., Chen L., Zhao X., Wei Y., Honokiol nanoparticles in thermosensitive hydrogel: Therapeutic effects on malignant pleural effusion. ACS Nano 3, 4080–4088 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Taghizadeh N., Fortin M., Tremblay A., US hospitalizations for malignant pleural effusions: Data from the 2012 national inpatient sample. Chest 151, 845–854 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Wu S. G., Yu C. J., Tsai M. F., Liao W. Y., Yang C. H., Jan I. S., Yang P. C., Shih J. Y., Survival of lung adenocarcinoma patients with malignant pleural effusion. Eur. Respir. J. 41, 1409–1418 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Mothersill C., Seymour C. B., Radiation-induced bystander effects—Implications for cancer. Nat. Rev. Cancer 4, 158–164 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Prise K. M., O’Sullivan J. M., Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 9, 351–360 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mesnil M., Yamasaki H., Bystander effect in herpes simplex virus-thymidine kinase/ganciclovir cancer gene therapy: Role of gap-junctional intercellular communication. Cancer Res. 60, 3989–3999 (2000). [PubMed] [Google Scholar]
- 13.Yang X., Xu S., Su Y., Chen B., Yuan H., Xu A., Wu L., Autophagy-Src regulates Connexin43-mediated gap junction intercellular communication in irradiated HepG2 cells. Radiat. Res. 190, 494–503 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Peng Y., Zhang M., Zheng L., Liang Q., Li H., Chen J.-T., Guo H., Yoshina S., Chen Y.-Z., Zhao X., Wu X., Liu B., Mitani S., Yu J.-S., Xue D., Cysteine protease cathepsin B mediates radiation-induced bystander effects. Nature 547, 458–462 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirolikar S., Prasannan P., Raghuram G. V., Pancholi N., Saha T., Tidke P., Chaudhari P., Shaikh A., Rane B., Pandey R., Wani H., Khare N. K., Siddiqui S., D’Souza J., Prasad R., Shinde S., Parab S., Nair N. K., Pal K., Mittra I., Prevention of radiation-induced bystander effects by agents that inactivate cell-free chromatin released from irradiated dying cells. Cell Death Dis. 9, 1142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan W. F., Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat. Res. 159, 581–596 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Guo M., Wu F., Hu G., Chen L., Xu J., Xu P., Wang X., Li Y., Liu S., Zhang S., Huang Q., Fan J., Lv Z., Zhou M., Duan L., Liao T., Yang G., Tang K., Liu B., Liao X., Tao X., Jin Y., Autologous tumor cell-derived microparticle-based targeted chemotherapy in lung cancer patients with malignant pleural effusion. Sci. Transl. Med. 11, eaat5690 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Keklikoglou I., Cianciaruso C., Güç E., Squadrito M. L., Spring L. M., Tazzyman S., Lambein L., Poissonnier A., Ferraro G. B., Baer C., Cassará A., Guichard A., Iruela-Arispe M. L., Lewis C. E., Coussens L. M., Bardia A., Jain R. K., Pollard J. W., De Palma M., Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 21, 190–202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma R., Ji T., Chen D., Dong W., Zhang H., Yin X., Ma J., Liang X., Zhang Y., Shen G., Qin X., Huang B., Tumor cell-derived microparticles polarize M2 tumor-associated macrophages for tumor progression. Oncoimmunology 5, e1118599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Yu Y., Zhou L., Ma J., Tang K., Xu P., Ji T., Liang X., Lv J., Dong W., Zhang T., Chen D., Xie J., Liu Y., Huang B., Circulating tumor microparticles promote lung metastasis by reprogramming inflammatory and mechanical niches via a macrophage-dependent pathway. Cancer Immunol. Res. 6, 1046–1056 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Lorimore S. A., Coates P. J., Scobie G. E., Milne G., Wright E. G., Inflammatory-type responses after exposure to ionizing radiation in vivo: A mechanism for radiation-induced bystander effects? Oncogene 20, 7085–7095 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Tang D., Kang R., Berghe T. V., Vandenabeele P., Kroemer G., The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stockwell B. R., Friedmann Angeli J. P., Bayir H., Bush A. I., Conrad M., Dixon S. J., Fulda S., Gascón S., Hatzios S. K., Kagan V. E., Noel K., Jiang X., Linkermann A., Murphy M. E., Overholtzer M., Oyagi A., Pagnussat G. C., Park J., Ran Q., Rosenfeld C. S., Salnikow K., Tang D., Torti F. M., Torti S. V., Toyokuni S., Woerpel K. A., Zhang D. D., Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., Patel D. N., Bauer A. J., Cantley A. M., Yang W. S., Morrison B. III, Stockwell B. R., Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen Q., Liu J., Kang R., Zhou B., Tang D., The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 510, 278–283 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Lawrence T., Natoli G., Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Jin H., Wan C., Zou Z., Zhao G., Zhang L., Geng Y., Chen T., Huang A., Jiang F., Feng J.-P., Lovell J. F., Chen J., Wu G., Yang K., Tumor ablation and therapeutic immunity induction by an injectable peptide hydrogel. ACS Nano 12, 3295–3310 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Mathios D., Kim J. E., Mangraviti A., Phallen J., Park C.-K., Jackson C. M., Garzon-Muvdi T., Kim E., Theodros D., Polanczyk M., Martin A. M., Suk I., Ye X., Tyler B., Bettegowda C., Brem H., Pardoll D. M., Lim M., Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 8, 370ra180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stathopoulos G. T., Kalomenidis I., Malignant pleural effusion: Tumor-host interactions unleashed. Am. J. Respir. Crit. Care Med. 186, 487–492 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelland L., The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 7, 573–584 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Kodama K., Doi O., Higashiyama M., Yokouchi H., Tatsuta M., Long-term results of postoperative intrathoracic chemo-thermotherapy for lung cancer with pleural dissemination. Cancer 72, 426–431 (1993). [DOI] [PubMed] [Google Scholar]
- 32.Atun R., Jaffray D. A., Barton M. B., Bray F., Baumann M., Vikram B., Hanna T. P., Knaul F. M., Lievens Y., Lui T. Y. M., Milosevic M., O’Sullivan B., Rodin D. L., Rosenblatt E., Van Dyk J., Yap M. L., Zubizarreta E., Gospodarowicz M., Expanding global access to radiotherapy. Lancet Oncol. 16, 1153–1186 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Bernstein M. B., Krishnan S., Hodge J. W., Chang J. Y., Immunotherapy and stereotactic ablative radiotherapy (ISABR): A curative approach? Nat. Rev. Clin. Oncol. 13, 516–524 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ngwa W., Irabor O. C., Schoenfeld J. D., Hesser J., Demaria S., Formenti S. C., Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 18, 313–322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mothersill C., Seymour C., Medium from irradiated human epithelial cells but not human fibroblasts reduces the clonogenic survival of unirradiated cells. Int. J. Radiat. Biol. 71, 421–427 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Stathopoulos G. T., Moschos C., Loutrari H., Kollintza A., Psallidas I., Karabela S., Magkouta S., Zhou Z., Papiris S. A., Roussos C., Kalomenidis I., Zoledronic acid is effective against experimental malignant pleural effusion. Am. J. Respir. Crit. Care Med. 178, 50–59 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Cassetta L., Pollard J. W., Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Singhal S., Stadanlick J., Annunziata M. J., Rao A. S., Bhojnagarwala P. S., O’Brien S., Moon E. K., Cantu E., Danet-Desnoyers G., Ra H.-J., Litzky L., Akimova T., Beier U. H., Hancock W. W., Albelda S. M., Eruslanov E. B., Human tumor-associated monocytes/macrophages and their regulation of T cell responses in early-stage lung cancer. Sci. Transl. Med. 11, eaat1500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang K., Zhang Y., Zhang H., Xu P., Liu J., Ma J., Lv M., Li D., Katirai F., Shen G.-X., Zhang G., Feng Z.-H., Ye D., Huang B., Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat. Commun. 3, 1282 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Soiffer R., Lynch T., Mihm M., Jung K., Rhuda C., Schmollinger J. C., Hodi F. S., Liebster L., Lam P., Mentzer S., Singer S., Tanabe K. K., Cosimi A. B., Duda R., Sober A., Bhan A., Daley J., Neuberg D., Parry G., Rokovich J., Richards L., Drayer J., Berns A., Clift S., Cohen L. K., Mulligan R. C., Dranoff G., Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte–Macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc. Natl. Acad. Sci. U.S.A. 95, 13141–13146 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaay9789/DC1
Fig. S1. Examination of RIBE in irradiated A549 cells.
Fig. S2. Therapeutic effect of RT-MPs.
Fig. S3. RT-MPs induce ferroptosis in tumor cells.
Fig. S4. RT-MPs promote immunogenic cell death.
Fig. S5. Macrophages show robust phagocytosis of RT-MPs.
Fig. S6. Therapeutic effect of combination treatment with RT-MPs and anti–PD-1 depends on immune system activation.
Fig. S7. Therapeutic effect of RT-MPs on A549-DDR cells.
Fig. S8. Hemanalysis and biochemical analyses.
Table S1. Sequences of primers for RT-qPCR analysis.
Table S2. Antibodies used in this article.
Movie S1. Radiated LLC-RFP released more microparticles.
Movie S2. Unradiated LLC-RFP released less microparticles.
Movie S3. Macrophages and LLC-RFP cells.
Movie S4. Macrophages and RT-MP–treated LLC-RFP cells.
Movie S5. Two hours after RT-MPs’ injection.
Movie S6. Twenty-four hours after RT-MPs’ injection.
Movie S7. Macrophages phagocytose PKH26-labeled RT-MPs in vitro.
Movie S8. RT-MP–treated macrophages and LLC-RFP cells.