

Our work uncovers a novel SUMOylation-dependent regulation of DUSP6 stability and its activity, which regulates Drp1 function.
Abstract
Imbalanced mitochondrial fission/fusion, a major cause of apoptotic cell death, often results from dysregulation of Drp1 phosphorylation of two serines, S616 and S637. Whereas kinases for Drp1-S616 phosphorylation are well-described, phosphatase(s) for its dephosphorylation remains unclear. Here, we show that dual-specificity phosphatase 6 (DUSP6) dephosphorylates Drp1-S616 independently of its known substrates ERK1/2. DUSP6 keeps Drp1-S616 phosphorylation levels low under normal conditions. The stability and catalytic function of DUSP6 are maintained through conjugation of small ubiquitin-like modifier-1 (SUMO1) and SUMO2/3 at lysine-234 (K234), which is disrupted during oxidation through transcriptional up-regulation of SUMO-deconjugating enzyme, SENP1, causing DUSP6 degradation by ubiquitin-proteasome. deSUMOylation underlies DUSP6 degradation, Drp1-S616 hyperphosphorylation, mitochondrial fragmentation, and apoptosis induced by H2O2 in cultured cells or brain ischemia/reperfusion in mice. Overexpression of DUSP6, but not the SUMOylation-deficient DUSP6K234R mutant, protected cells from apoptosis. Thus, DUSP6 exerts a cytoprotective role by directly dephosphorylating Drp1-S616, which is disrupted by deSUMOylation under oxidation.
INTRODUCTION
Oxidative cell death is characterized by mitochondrial damage, including mitochondrial arrest and time-dependent mitochondrial fragmentation. As one of the key regulators of mitochondrial fission, Drp1 is thought to play a vital role in the cellular response to oxidation that eventually results in mitochondrial fragmentation and the consequent cell demise (1, 2). The effect of Drp1 on mitochondrial fission is dependent on the phosphorylation states of two serine (S) residues at the C-terminal guanosine triphosphatase (GTPase) effector domain of Drp1. Whereas the phosphorylation of Drp1 at S616 activates mitochondrial fission, that at S637 inhibits the fission (3, 4). Several different protein kinases have been reported to phosphorylate Drp1-S616, including cyclin-dependent kinase 1 (CDK1) or CDK5 (1), protein kinase Cδ (PKCδ) (2), mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase 1/2 (ERK1/2) (5, 6), Rho-associated coiled-coil kinase (ROCK), and Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα) (7). On the other hand, Drp1-S637 is phosphorylated by protein kinase A (8, 9), as well as ROCK1, CaMKIα, and adenosine monophosphate (AMP)–activated protein kinase (AMPK) (10, 11). Thus, Drp1 is prone to phosphorylation under many different conditions, implicating the importance of dephosphorylation in keeping the optimal Drp1 activity. However, whereas the dephosphorylation of Drp1-S637 has been shown to be catalyzed by protein phosphatase 2A and/or calcineurin (8, 9), little is known about how Drp1-S616 is dephosphorylated, although mechanisms that dampen the activity of the kinases described above could help reduce Drp1-S616 phosphorylation levels.
Dual-specificity phosphatases (DUSPs) have been implicated as major modulators of signaling pathways affecting various physiological processes (12). Among them, DUSP6, also known as MAPK phosphatase-3 (MKP-3) or DUSP6/MKP-3, is a cytoplasmic enzyme reported to specifically dephosphorylate ERK1/2 at both tyrosine and serine/threonine residues, leading to inactivation of ERK1/2 MAPKs in vitro and in vivo (12, 13–16). As a key tyrosine and serine/threonine phosphatase under highly coordinated and multilayered regulation, including at the levels of transcription, posttranscription, and posttranslation (17–19), DUSP6 is thought to act as a critical regulator of the ERK signaling cascade and a cytoplasmic anchor of ERKs.
DUSP6 is a short-lived protein, with its stability affected by serum growth factors and oxidation (17, 19). In particular, it has been reported that, H2O2, a major contributor to oxidative damage, can cause DUSP6 degradation, resulting in increased phosphorylation of ERK1/2 (21, 22). This is thought to form a plausible cascade linking oxidation to cell death through DUSP6 destabilization by oxidation, which, in turn, leads to augmentation of ERK1/2 phosphorylation, Drp1-S616 phosphorylation, mitochondrial fragmentation, and then cell death. However, whether the cascade actually works in such a fashion and the molecular mechanism by which oxidation causes DUSP6 degradation remain unclear.
The degradation of DUSP6 may involve phosphorylation, which is followed by ubiquitination and proteasome-mediated degradation (17, 23). Therefore, posttranslational modifications (PTMs) may serve a vital link in oxidation-induced DUSP6 degradation. It would be of importance to know whether additional PTMs on DUSP6 also exist and how they interact with phosphorylation and ubiquitination to affect DUSP6 stability. Here, we show that DUSP6 degradation is countered by SUMOylation, a form of PTMs through covalent enzymatic conjugation of small ubiquitin-like modifier (SUMO) proteins to specific lysine residues of substrate proteins. SUMOylation is reversed by specific deSUMOylation enzymes called Sentrin-specific proteases (SENPs). By examining the SUMOylation status of DUSP6 during the treatment with H2O2, we found that DUSP6 deSUMOylation precedes its degradation by oxidation. Enhancing DUSP6 SUMOylation not only attenuated its degradation by H2O2 through disrupting its ubiquitination but also suppressed oxidative mitochondrial fragmentation and cell death. Furthermore, we demonstrate that DUSP6 is a Drp1 phosphatase that mediates dephosphorylation of Drp1-S616, which underlies the mechanism of cytoprotective effect of DUSP6 SUMOylation. Thus, DUSP6 functions as a specific negative regulator of mitochondrial damage through dephosphorylating Drp1-S616, which is disrupted by oxidation through deSUMOylation.
RESULTS
Oxidation destabilizes DUSP6 via ubiquitin-mediated degradation, and overexpression of DUSP6 protects cells against oxidative stress
As a redox-sensitive phosphatase, DUSP6 has been shown to be destabilized by the treatment of H2O2, leading to increased phosphorylation of ERK1/2 (21, 22). Known to be degraded via the ubiquitin-proteasome pathway (17), the H2O2-induced DUSP6 degradation may also involve direct ubiquitination of DUSP6 proteins. To test this, we first measured the levels of endogenous DUSP6 proteins in HeLa cells following the treatment with different concentrations of H2O2 for 1 hour (Fig. 1A) and with 0.5 mM H2O2 for different times (Fig. 1B). Consistent with previous findings (20), H2O2 treatment decreased DUSP6 levels in a concentration- and time-dependent manner (Fig. 1, A and B). Furthermore, the treatment with H2O2 (0.5 mM, 1 hour) caused caspase-3 cleavage (Fig. 1C) and increased DUSP6 ubiquitination (Fig. 1D), supporting the view that oxidation destabilizes DUSP6 by promoting its ubiquitination.
Fig. 1. H2O2 destabilizes DUSP6 via ubiquitin-mediated degradation, and overexpression of DUSP6 protects cells against oxidative stress.
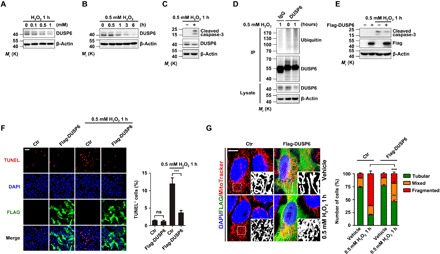
(A and B) H2O2 treatment decreased the levels of endogenous DUSP6 proteins in a dose- and time-dependent manner. Lysates from HeLa cells treated with H2O2 at different concentrations for 1 hour (A) and at 0.5 mM for different times (B) as indicated were analyzed by immunoblotting (IB) using an anti-DUSP6 antibody. β-Actin was used as the loading control. (C and D) H2O2 treatment increased caspase-3 cleavage (C) and DUSP6 ubiquitination (D). Lysates from HeLa cells untreated or treated with 0.5 mM H2O2 for 1 hour were subjected to IB with anti-cleaved caspase-3 and anti-DUSP6 antibodies (C) or they were subjected to immunoprecipitation (IP) with the DUSP6 antibody [or control immunoglobulin G (IgG)] before being used for IB with anti-Ubiquitin and anti-DUSP6 antibodies (D). β-Actin was used as the loading control. (E to G) Overexpression of DUSP6 suppressed oxidation-induced increases in caspase-3 cleavage (E), apoptosis (F), and mitochondrial fragmentation (G). HeLa cells transiently transfected with either an empty control vector (−) or a vector encoding Flag-DUSP6 (+) were untreated or treated with 0.5 mM H2O2 for 1 hour at 24 hours after transfection. In (E), cell lysates were subjected to IB with the indicated antibodies. In (A) to (E), blots are representatives of at least three independent experiments. In (F), cells were fixed for TUNEL staining (red) to identify apoptotic cells and immunofluorescence (IF) labeling of Flag (green) to assess Flag-DUSP6 expression. 4′,6-diamidino-2-phenylindole (DAPI) (blue) was used for counterstaining of nuclei. Scale bar, 100 μm. Representative confocal images are shown. Quantification of TUNEL-positive (TUNEL+) cells is shown at the right. In (G), after the H2O2 treatment, cells were incubated with MitoTracker Red (200 nM) for 50 min at 37°C before fixation, IF for Flag, and DAPI labeling. Representative confocal images are shown. Scale bar, 10 μm. Black and white insets are magnifications of the boxed areas of MitoTracker labeling. Quantification of cells with different forms of mitochondrial morphology is shown at the right. Quantification data in (F) and (G) represent means ± SEM from n = 3 independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001, control (Ctr) versus Flag-DUSP6, by one-way analysis of variance (ANOVA) with pairwise comparison using Tukey’s multiple comparisons test. ns, not significant.
To assess the possible role of DUSP6 in oxidative cell damage, we examined the effect of DUSP6 overexpression on H2O2-induced cell apoptosis and mitochondrial fragmentation. In HeLa cells transfected with Flag-DUSP6, the exposure to 0.5 mM H2O2 for 1 hour caused less increase in caspase-3 cleavage (Fig. 1E) and TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling)–positive cells (Fig. 1F) than vector-transfected control cells. H2O2-induced mitochondrial fragmentation was also attenuated by the overexpression of DUSP6, as shown by the examination of mitochondrial morphology using MitoTracker Red (Fig. 1G). Following the previous classification (24), we categorized the cells according to their mitochondrial morphology as tubular, fragmented, or mixed. The H2O2 treatment caused a marked increase in cells that contained mainly fragmented mitochondria in control (Fig. 1G). However, this change was quite moderate in Flag-DUSP6–transfected cells, with more than 46.0 ± 4.3% (n = 310 cells of three independent experiments) cells still containing mainly tubular mitochondria after the H2O2 treatment as compared to the control (20.7 ± 1.7%, n = 308 cells of three independent experiments). Together, these results indicate that DUSP6 plays a protective role against oxidative damage. However, the H2O2 treatment disrupts the protective function by degrading DUSP6 through the ubiquitination-proteasome pathway, consistent with the previous reports that DUSP6 is a substrate of ubiquitination (20–22).
DUSP6 is modified by SUMO
SUMO modification has emerged as a key PTM that regulates protein stability, protein-protein interactions, trafficking, and transcriptional activities, thereby regulating a plethora of biological processes (25). To investigate whether SUMOylation is involved in regulating DUSP6 stability, we first examined whether DUSP6 is SUMOylated using HeLa cells transiently cotransfected with Flag-DUSP6 and hemagglutinin (HA)–SUMO1 or SUMO2. As shown in Fig. 2A and fig. S1A, the anti-Flag antibody pulled down not only Flag-DUSP6, recognized by both anti-DUSP6 and anti-Flag antibodies, but also a SUMO-conjugated band that is ~20 kDa larger than the unmodified protein and detected by the anti-SUMO1 or anti-SUMO2/3 antibody. The same band was also pulled down by anti-HA antibody when immunoblotting was performed using the anti-DUSP6 antibody, indicating that DUSP6 is SUMOylated. The SUMOylated DUSP6 was abolished by the coexpression of arginine-glycine-serine (RGS)–SENP1 but not the catalytically dead SENP1 mutant (SENP1m) (Fig. 2A and fig. S2A), consistent with deSUMOylation action of SENP1. To further examine whether DUSP6 is SUMOylated in vitro, purified DUSP6 protein generated in Escherichia coli was used for in vitro SUMOylation assay. By immunoblotting with the anti-SUMO1 or anti-DUSP6 antibody, we detected the SUMOylated band of DUSP6 (Fig. 2B).
Fig. 2. DUSP6 is modified by SUMO1 at K234.
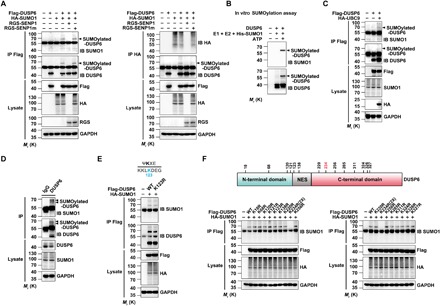
(A) DUSP6 is SUMO1-conjugated in vivo. Lysates from HeLa cells transiently transfected with empty vector (−), Flag-DUSP6, HA-SUMO1, RGS-SENP1, or RGS-SENP1m at various combinations as indicated for 24 hours were subjected to denaturing IP with anti-Flag (left) and anti-HA (right) antibodies, which was followed by IB using anti-SUMO1, anti-DUSP6, and anti-HA. The original lysates were also analyzed by IB with anti-Flag, anti-HA, anti-RGS for input, and anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for loading control. Arrowheads indicate SUMOylated DUSP6. (B) DUSP6 is SUMO1-conjugated in vitro. Purified recombinant DUSP6 was incubated with E1, E2, SUMO1, and adenosine triphosphate (ATP) in vitro at 37°C for 1 hour, and reaction was terminated with SDS loading buffer. The samples prepared above were analyzed by Western blotting with SUMO1 and DUSP6 antibodies as indicated. (C) DUSP6 conjugation by endogenous SUMO1 was enhanced by overexpression of UBC9. Lysates from HeLa cells transiently transfected with empty vector (−), Flag-DUSP6, or HA-UBC9 at various combinations as indicated for 24 hours were subjected to denaturing IP and IB as in (A) (left). (D) SUMO1 conjugation of endogenous DUSP6 in mouse brain. Lysates prepared from mouse cerebral cortices under denaturing conditions were subjected to IP with anti-DUSP6 antibody, followed by IB with anti-SUMO1 and anti-DUSP6 antibodies. The original lysates were also analyzed by IB using anti-DUSP6 and anti-SUMO1 for input and anti-GAPDH for loading control. Bands for SUMOylated-DUSP6 are indicated by arrowheads. (E) DUSP6 SUMOylation is not dependent on K123, the lysine residue that adheres consensus SUMO modification site (inset), as predicted for mouse DUSP6 using SUMOsp 2.0 software. Lysates from HeLa cells transiently cotransfected with HA-SUMO1 and Flag-DUSP6 or Flag-DUSP6K123R mutant, or vector control (−) as indicated for 24 hours, were subjected to IP with anti-Flag antibody, followed by IB with anti-SUMO1 and anti-DUSP6 antibodies. The original lysates were analyzed by IB using anti-Flag and anti-HA for input and anti-GAPDH for loading control. (F) Identification of K234 as the SUMO modification site of mouse DUSP6. Top: Diagram for domain organization of mouse DUSP6 and the locations of all lysine residues, except for K254, which is buried inside the protein and unlikely SUMOylatable based on the SUMOsp 2.0 software. NES, nuclear export signal. Bottom: Lysates from HeLa cells transiently cotransfected with HA-SUMO1, wild-type (WT) Flag-DUSP6, or one of the K➔R mutants of Flag-DUSP6, or the vector control (−) as indicated for 24 hours, were subjected to IP with anti-Flag antibody, followed by IB with anti-SUMO1. The original lysates were also analyzed by IB using anti-Flag and anti-HA for input and anti-GAPDH for loading control. Note that 2× amount of DUSP6K234R plasmid was used in the transfection to match the protein expression with the wild-type DUSP6 and other mutants. In (A) to (F), blots are representatives of at least three independent experiments.
Next, to test whether DUSP6 is conjugated by endogenous SUMO1, immunoprecipitants from HeLa cells transiently transfected with Flag-DUSP6 were subjected to immunoblotting with the anti-SUMO1 or anti-DUSP6 antibody. In both cases, a band with the size predicted for SUMO1-conjugated DUSP6 was detected with the coexpression of the SUMO E2–conjugating enzyme, UBC9 (Fig. 2C). Furthermore, to examine whether DUSP6 is SUMOylated in vivo, we performed immunoprecipitation using the anti-DUSP6 antibody against lysates prepared from mouse brain cortex. By immunoblotting with the anti-SUMO1 or anti-DUSP6 antibody, we detected two weak but nonetheless clear bands, corresponding to SUMOylated-DUSP6 forms with estimated molecular weights of 62 and 64 kDa, which represent the sum of DUSP6 [~42 and 44 kDa (26)] and SUMO1 (~20 kDa) (Fig. 2D). Together, these results suggest that DUSP6 can be SUMOylated both in vivo and in vitro.
K234 is the major SUMOylation site in DUSP6
Protein SUMOylation typically occurs at lysine (Lys) residues located within the consensus sequence Ψ-K-X-E, where Ψ is any hydrophobic amino acid and X is any amino acid (27). To map the SUMOylation sites on DUSP6, we first mutated K123 of mouse DUSP6, the only Lys residue that perfectly adheres to the consensus SUMOylation site (Fig. 2E, inset), to an arginine (R) and tested the ability of SUMO1 to conjugate it after coexpression in HeLa cells. However, K123R became SUMOylated similarly as the wild-type DUSP6, indicating that K123 is not the SUMOylation site of DUSP6. We then individually substituted the remaining lysine residues of mouse DUSP6 with arginines. Among the 13 substitutions, only K234R failed to show the SUMOylated band, indicating that K234 is the primary residue of DUSP6 subjected to SUMO1 modifications (Fig. 2F). K234 is well conserved, and it is exposed at the protein surface as a part of the C-terminal domain of DUSP6 (fig. S2D). Together, these findings indicate that DUSP6 is SUMOylated at the C-terminal residue K234.
SUMO1 modification enhances DUSP6 stability
SUMO modification has been implicated in the regulation of protein degradation (28, 29). Here, we provide several pieces of evidence to demonstrate that SUMOylation plays an important role in stabilizing DUSP6 proteins by reducing its degradation in cells. First, when expressed in HeLa cells, the protein level of the SUMOylation-deficient DUSP6K234R mutant was approximately a half of that of wild-type DUSP6 (Fig. 3A), despite the similar mRNA levels between the DUSP6K234R mutant and wild-type DUSP6 (Fig. 3B). Second, in cells cotransfected with Flag-DUSP6 and HA-SUMO1, the inhibition of de novo protein synthesis by cycloheximide (CHX; 100 μg/ml) for 24 hours resulted in a markedly higher level of DUSP6 protein than cells transfected with Flag-DUSP6 alone, but this effect was abolished by cotransfection with SENP1 or the K234R mutation (Fig. 3C). This indicates that in the presence of SUMO1 conjugation, DUSP6 degradation is slowed down. Third, the degradation of DUSP6 occurred mostly through the ubiquitin-proteasome, rather than the lysosomal, pathways as shown by the blocking effect of the specific proteasome inhibitor, MG132, but not the lysosomal inhibitor, chloroquine, on DUSP6 degradation in the presence of CHX (Fig. 3D). Noticeably, the degradation occurred faster for Flag-DUSP6K234R than Flag-DUSP6, and this was abolished by MG132 but not chloroquine (Fig. 3D). A more detailed quantitative analysis of DUSP6 levels at different times after the addition of CHX also revealed that while Flag-DUSP6K234R had a half-life of about 8 hours, Flag-DUSP6 displayed a half-life of >24 hours, and the coexpression of HA-SUMO1 markedly extended the lifetime of Flag-DUSP6 (fig. S3, A and B). Together, these results further demonstrate that SUMOylation protects DUSP6 against degradation by the ubiquitin-proteasome pathway.
Fig. 3. SUMOylation enhances DUSP6 stability by repressing its ubiquitination.
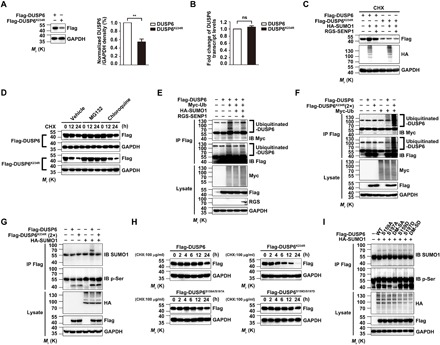
(A and B) The SUMOylation-deficient DUSP6K234R mutant exhibits reduced protein, but not RNA, expression as compared to the wild type. Lysates from HeLa cells transiently transfected with Flag-DUSP6 or Flag-DUSP6K234R were subjected to IB with the anti-Flag antibody to determine protein expression (A) and real-time quantitative reverse transcription polymerase chain reaction (RT-PCR) to measure the DUSP6 transcript levels (B). Results normalized to wild-type DUSP6 were summarized for three independent experiments and shown as means ± SEM. **P < 0.01, by two-tailed Student’s t test. (C) SUMOylation increases the stability of DUSP6 but not the DUSP6K234R mutant. Lysates from HeLa cells transiently transfected with Flag-DUSP6, Flag-DUSP6K234R, HA-SUMO1, or RGS-SENP1 at various combinations as indicated and treated with CHX (100 μg/ml) for 12 hours beginning at 24 hours after transfection to prevent de novo protein synthesis were analyzed by IB with anti-Flag, anti-HA, and anti-GAPDH. Note that, in the presence of CHX, the increase in DUSP6 level due to HA-SUMO1 overexpression indicates decreased degradation, which was reversed by RGS-SENP1; however, the level of DUSP6K234R is unaffected by HA-SUMO1 and RGS-SENP1. (D) DUSP6K234R exhibits faster degradation via the proteasome pathway than wild-type DUSP6. HeLa cells transiently transfected with Flag-DUSP6 or DUSP6K234R were treated with CHX (100 μg/ml) for 0 to 24 hours as indicated beginning at 24 hours after transfection. Dimethyl sulfoxide (vehicle control), proteasome inhibitor, MG132 (20 μM), or lysosome inhibitor, chloroquine (100 μM), was added 1 hour before CHX. Cell lysates were subjected to IB using anti-Flag and anti-GAPDH antibodies. Note the faster decrease in DUSP6K234R than DUSP6 and the blockade by MG132 but not chloroquine. (E) SUMO1 modification of DUSP6 represses its ubiquitination. Lysates from HeLa cells transiently transfected with Flag-DUSP6, HA-SUMO1, Myc-ubiquitin (Myc-Ub), or RGS-SENP1 at various combinations as indicated were subjected to IP with anti-Flag antibody under denaturing conditions, which was followed by IB with anti-Myc and anti-Flag antibodies. The original lysates were also analyzed by IB for inputs of Myc-Ub, Flag-DUSP6, and RGS-SENP1, with GAPDH as the loading control. (F) The SUMOylation-deficient DUSP6K234R mutant has enhanced ubiquitination as compared to wild-type DUSP6. Similar to (E) but the cells were transfected with Flag-DUSP6, Flag-DUSP6K234R (2×), or Myc-Ub at various combinations as indicated. Note the stronger Myc-Ub labeling samples transfected with Flag-DUSP6K234R than Flag-DUSP6. (G) DUSP6 SUMOylation has no impact on its phosphorylation. Lysates from HeLa cells transiently transfected with Flag-DUSP6, Flag-DUSP6K234R (2×), or HA-SUMO1 at various combinations as indicated were subjected to IP with the anti-Flag antibody, which was followed by IB with anti-SUMO1 and anti–p-Ser antibodies. The original lysates were also analyzed by IB for the inputs of HA-SUMO1 and Flag-DUSP6, with GAPDH as the loading control. Note the similar levels of p-Ser labels between DUSP6 and DUSP6K234R and the lack of effect of HA-SUMO1. (H) DUSP6 SUMOylation, but not phosphorylation, regulates its degradation. Lysates from HeLa cells transiently transfected with Flag-DUSP6, Flag-DUSP6K234R, or phosphorylation-defective (S159A/S197A, DM-SA) or phosphorylation-mimetic (S159D/S197D, DM-SD) DUSP6 mutant and treated with CHX (100 μg/ml) for 0 to 24 hours as indicated beginning at 24 hours after transfection were subjected to IB with anti-Flag and anti-GAPDH antibodies. Note that the phosphorylation-defective and phosphorylation-mimetic DUSP6 mutants exhibited similar stability to the wild-type protein. (I) DUSP6 phosphorylation has no impact on its SUMOylation. Lysates from HeLa cells transiently cotransfected with HA-SUMO1 and Flag-DUSP6, or one of its phosphorylation-defective or phosphorylation-mimetic mutants as indicated for 24 hours were subjected to IP and IB similarly as in (G). Note the similar levels of SUMOylation for DUSP6 and all its mutants. In (C) to (I), blots are representatives of at least three independent experiments. Note that 2× amount of DUSP6K234R plasmid was used for transfection in some experiments to ensure comparable protein expression with the wild type.
SUMO modification represses DUSP6 ubiquitination
SUMOylation and ubiquitination can act either synergistically or antagonistically (30, 31). To test how SUMOylation affects DUSP6 ubiquitination, we transfected HeLa cells with plasmids for Flag-DUSP6 and Myc-ubiquitin (Myc-Ub). Polyubiquitinated DUSP6 was readily detected by immunoblotting with either anti-Myc or anti-Flag antibody in immunoprecipitants pulled down by the anti-Flag antibody (Fig. 3E). The coexpression of HA-SUMO1 largely abolished DUSP6 ubiquitination under these conditions, and the effect of HA-SUMO1 was reversed by coexpression of the deSUMOylating enzyme RGS-SENP1 (Fig. 3E). Furthermore, when cotransfected with Myc-Ub in HeLa cells, the SUMOylation-deficient Flag-DUSP6K234R mutant exhibited more robust polyubiquitination than the wild-type Flag-DUSP6 (Fig. 3F). These results indicate that SUMO modification represses DUSP6 ubiquitination, protecting DUSP6 from degradation by proteasomes.
There is no detectable interplay between SUMOylation and phosphorylation of DUSP6
Previous studies have shown the involvement of DUSP6 phosphorylation at serine residues S159 and S197 in its degradation by proteasomes (17). Since SUMOylation suppressed DUSP6 degradation through inhibiting its ubiquitination (Fig. 3, D to F), we also investigated the possible interplay between the SUMOylation and phosphorylation of DUSP6. First, immunoblotting by the anti-phosphoserine (p-Ser) antibody against immunoprecipitants pulled down by the anti-Flag antibody revealed no change in the phosphorylation of the DUSP6K234R mutant compared to wild-type DUSP6, and the phosphorylation levels were not affected by the coexpression of HA-SUMO1 (Fig. 3G), suggesting that SUMOylation has no effect on DUSP6 phosphorylation. More unexpectedly, the phosphorylation-defective [S159A, S197A, or double mutant–serine mutated to alanine (DM-SA; S159A/S197A)] and phosphorylation-mimetic [S159D, S197D, or double mutant–serine mutated to aspartic acid (DM-SD; S159D/S197D)] mutants of DUSP6 exhibited no detectable difference from wild-type DUSP6 in the time course of degradation following the addition of CHX (Fig. 3H). Furthermore, the phosphorylation-defective and phosphorylation-mimetic mutants of DUSP6 exhibited similar levels of SUMOylation to the wild-type phosphatase when coexpressed with HA-SUMO1 (Fig. 3I). Consistent with the lack of effect of DUSP6 phosphorylation on its SUMOylation, DUSP6 contains neither a consensus phosphorylation-dependent SUMO modification motif nor negatively charged amino acid–dependent SUMO modification motif, as determined using JASSA (www.jassa.fr/). Together, our data suggest that there is no significant interplay between DUSP6 SUMOylation and its phosphorylation, at least for that at S159 and S197.
DeSUMOylation of DUSP6 reduces its phosphatase activity
DUSP6 is best known to regulate the Ras-MAPK signaling cascade due to its unusually tight substrate specificity for ERK1/2. To determine how DUSP6 SUMOylation affects its enzymatic activity, we performed the following experiments. First, serum growth factors induce phosphorylation of both ERK1/2 and DUSP6, as well as the expression of the latter, causing biphasic changes in the phosphorylation of ERK1/2 (17). Consistently, a treatment of epidermal growth factor (EGF; 50 ng/ml) led to a transient increase in ERK1/2 phosphorylation in HeLa cells, which peaked at around 15 min, and overexpression of Flag-DUSP6 suppressed the EGF-evoked ERK1/2 phosphorylation (Fig. 4A). Moreover, the inhibitory effect of DUSP6 was augmented by the coexpression of HA-SUMO1, an effect not seen with the DUSP6K234R mutant (Fig. 4B), suggesting that SUMOylation enhances the catalytic activity of DUSP6. Second, using para-nitrophenylphosphate (p-NPP) hydrolysis as a direct measure of DUSP6 phosphatase activity (32), we observed enhanced phosphatase activity when purified recombinant DUSP6 was SUMOylated in vitro and decreased phosphatase activity by addition of recombinant SENP1(419 to 644 amino acids) (Fig. 4C). Similar results were further confirmed in HeLa cells transfected with Flag-DUSP6, which was significantly increased by the coexpression of HA-SUMO1 and decreased by the coexpression of RGS-SENP1 (fig. S4A). However, although the recombinant SUMOylation-deficient DUSP6K234R still showed basal phosphatase activity, it was less than that of wild-type DUSP6 (Fig. 4D). Similar results were found in HeLa cells transfected with Flag-DUSP6K234R (fig. S4B). The phosphatase activity of Flag-DUSP6K234R approximated to 51.8 ± 8.5% (n = 3) of that of wild-type Flag-DUSP6 (fig. S4, B and F). Moreover, in vitro SUMOylation (Fig. 4D) or the coexpression of HA-SUMO1 (fig. S4B) did not further increase the catalytic activity of DUSP6K234R. Thus, SUMOylation positively affects the phosphatase activity of DUSP6. Third, to exclude the possibility that the SUMO site mutation impaired the catalytic activity of DUSP6, we generated two additional mutants around K234, DUSP6I233A and DUSP6I233N, taking into account that the hydrophobic Ile residue at the beginning of the IKYI motif in DUSP6 represents a key requirement of this crippled consensus SUMO conjugation site. As expected and similar to DUSP6K234R, the phosphatase activities of recombinant DUSP6I233A and DUSP6I233N were significantly lower than that of wild-type DUSP6 (Fig. 4E). In addition, neither Flag-DUSP6I233A nor Flag-DUSP6I233N was SUMOylated when coexpressed with HA-SUMO1, and their phosphatase activities were also similar to that of Flag-DUSP6K234R and significantly lower than that of wild-type Flag-DUSP6 (fig. S4C). These results further support the importance of DUSP6 SUMOylation in promoting its phosphatase activity. Fourth, the glycine residue, G97, of SUMO1 is critical for its conjugation of target proteins (33). The coexpression of the conjugation-deficient SUMO1 G97A mutant with Flag-DUSP6 not only failed to reveal DUSP6 SUMOylation but also resulted in a much lower phosphatase activity than the coexpression with HA-SUMO1 (fig. S4D). Last, DUSP6 phosphatase activity can be markedly increased in the presence of ERK2 (16). With increasing concentrations of recombinant ERK2, p-NPP hydrolysis increased dose dependently for wild-type DUSP6 but not for DUSP6K234R mutant and activity-deficient DUSP6C293S (Fig. 4F). Similar results were found in lysates from cells that expressed wild-type DUSP6 and DUSP6K234R mutant (fig. S4E). Together, these data strongly suggest that SUMOylation positively regulates the phosphatase activity of DUSP6.
Fig. 4. SUMOylation promotes DUSP6 phosphatase activity.
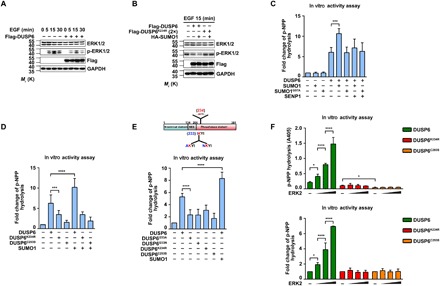
(A) Overexpression of DUSP6 suppressed growth factor–induced ERK1/2 activation. HeLa cells transiently transfected with empty vector or Flag-DUSP6 were serum-starved for 24 hours in Dulbecco’s modified Eagle’s medium (DMEM) containing 1% fetal bovine serum (FBS) beginning at 24 hours after transfection and stimulated with EGF (50 ng/ml) for the indicated times. Cell lysates were analyzed by IB with anti-ERK1/2, anti–p-ERK1/2, anti-Flag, and anti-GAPDH antibodies. Note the overall decrease in p-ERK1/2 levels in cells that overexpressed DUSP6. (B) The inhibitory effect of DUSP6 on ERK1/2 activation is regulated by SUMOylation. HeLa cells transiently transfected with HA-SUMO1, Flag-DUSP6, or Flag-DUSP6K234R in various combinations as indicated were serum-starved and treated with EGF as in (A) but for 15 min only. Cell lysates were analyzed by IB as in (A). Note that HA-SUMO1 coexpression further enhanced the inhibitory effect of DUSP6 on EGF-induced ERK1/2 activation; however, DUSP6K234R had no effect on p-ERK1/2 levels whether HA-SUMO1 was coexpressed. (C to E) SUMOylation enhances the phosphatase activity of DUSP6. Purified recombinant DUSP6, DUSP6K234R, DUSP6I233A, DUSP6I233N, DUSP6C293S, and SUMOylated DUSP6 in vitro were subjected to p-NPP hydrolysis assay for phosphatase activity. SUMOylated DUSP6 had a higher phosphatase activity compared to the wild-type DUSP6 (C), and the DUSP6K234R mutant exhibited lower catalytic activity than wild-type DUSP6 (D). The other two SUMOylation-deficient mutants, DUSP6I233A and DUSP6I233N, had similarly low phosphatase activities as DUSP6K234R (E). DUSP6C293S was used as a negative control as indicated. (F) Increasing amount of recombinant ERK2 enhanced phosphatase activity of DUSP6 but not that of DUSP6K234R. Incubation with the increasing amount of recombinant ERK2 and the phosphatase activities of DUSP6, DUSP6K234R, and DUSP6C293S were assessed by p-NPP hydrolysis assay. Data for the p-NPP assays in (C) to (F) represent means ± SEM of n = 3 independent experiments. *P < 0.05, ***P < 0.001, and ****P < 0.0001, by one-way ANOVA with pairwise comparison using Tukey’s multiple comparisons test.
Furthermore, to examine whether the interaction with ERK1/2 is required for SUMO1 to increase the phosphatase activity of DUSP6, we used the kinase interaction motif (KIM) mutant (R64, 65A) of DUSP6 (the key sites of contact for ERK1/2). The results showed that with both wild-type DUSP6 and the KIM mutant, but not the K234R mutant, in vitro SUMOylation increased the phosphatase activity, while the addition of ERK2 increased only the phosphatase activity of wild-type DUSP6 but not the KIM mutant. Therefore, the SUMO1-regulated DUSP6 phosphatase activity appears to be independent of ERK2. The ERK2- and SUMO1-induced DUSP6 phosphatase activities may be additive (fig. S5A). SUMOylation of proteins can alter their interaction properties and thereby their subcellular localization, function, and/or stability. Thus, we examined the interaction between ERK2 and DUSP6 or DUSP6K234R in an in vitro binding assay, which showed that the SUMOylation-deficient DUSP6 mutant, K234R, had decreased binding with ERK2 as compared to wild-type DUSP6 (fig. S5B), and while SUMO1 enhanced DUSP6 binding to ERK2, the SUMO1 mutant G97A did not (fig. S5C). Together, these results suggest that DUSP6 SUMOylation affects its interaction with ERK1/2. As expected, the KIM mutant (R64, 65A) did not show binding with DUSP6 (fig. S5, B and C).
Oxidation induces DUSP6 deSUMOylation
Given that DUSP6 undergoes degradation in response to H2O2 treatment (Fig. 1, A and B) but SUMOylation enhances DUSP6 stability (Fig. 3, C to F), we hypothesized that DUSP6 SUMOylation might be compromised by oxidative stress, allowing DUSP6 to be ubiquitinated and then degraded. The SUMOylation of DUSP6 was decreased over time in the presence of 1 mM H2O2, which was accompanied with a parallel decrease in the overall levels of SUMO1-conjugated proteins, as well as a decrease in DUSP6 protein levels (Fig. 5A, left). However, the same treatment did not alter the level of the SUMOylation-deficient DUSP6K234R mutant, despite the obvious decrease in the overall levels of SUMO1-conjugated proteins in the same samples (Fig. 5A, right). These indicate that deSUMOylation is the major cause of DUSP6 degradation in response to oxidation.
Fig. 5. Oxidation causes progressive deSUMOylation and degradation of DUSP6 via transcriptional up-regulation of SENP1.
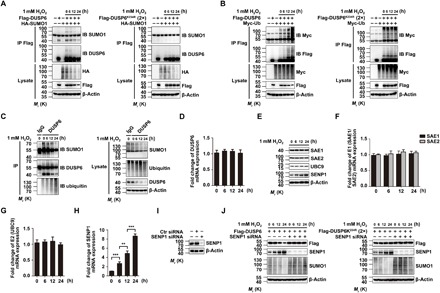
(A) H2O2 treatment induced a time-dependent loss of DUSP6 SUMOylation. HeLa cells transiently cotransfected with HA-SUMO1 and Flag-DUSP6 (left) or Flag-DUSP6K234R (2×) (right) mutant constructs were untreated or treated with 1 mM H2O2 for 0 to 24 hours as indicated beginning at 24 hours after transfection. Cell lysates were subjected to IP with the anti-Flag antibody, which was followed by IB for SUMO1 and DUSP6. The original lysates were also analyzed by IB for inputs of HA-SUMO1 and Flag-DUSP6, with β-actin as the loading control. Note that the parallel progressive decreases in the levels of SUMOylated and total DUSP6, as well as the overall protein SUMOylation (HA IB panel) after exposure to H2O2 (left); however, the levels of DUSP6K234R were unaffected by H2O2 despite the decrease in overall protein SUMOylation (right). (B) H2O2 treatment induced a time-dependent increase in the ubiquitination of DUSP6 (left) but not the SUMOylation-deficient DUSP6K234R mutant (right). Similar to (A) but the cells were cotransfected with Myc-Ub and Flag-DUSP6 or Flag-DUSP6K234R, and IB was performed using anti-Myc to assess ubiquitination and anti-Flag for the exogenously expressed Flag-DUSP6 both after IP and for total lysates. Note that the progressive increase in ubiquitination and the paralleled decrease in total protein levels after H2O2 exposure seen for wild-type DUSP6 (left) were not defected for DUSP6K234R, which exhibited a high ubiquitination level throughout (right). For IP/IB, membranes (B) after IB analysis with the anti-Myc antibody, the blots were stripped and reprobed with the anti-Flag antibody. (C) H2O2 treatment caused a time-dependent decrease in SUMOylation and paralleled increases in ubiquitination and degradation of endogenous DUSP6. Lysates from untransfected HeLa cells treated with 1 mM H2O2 for 0 to 24 hours as indicated were subjected to IP with the anti-DUSP6 antibody (or a control IgG), followed by IB analysis for SUMO1, ubiquitin, DUSP6 (left). The original lysates were also analyzed by IB for inputs of these proteins, with β-actin as the loading control (right). (D to H) H2O2 treatment caused a time-dependent up-regulation of SENP1 via a transcriptional mechanism. The same lysates as in (C) were analyzed for mRNA levels of DUSP6 (D), SAE1/SAE2 (E1, F), UBC9 (E2, G), and SENP1 (H) by real-time quantitative RT-PCR. Only SENP1 showed a progressive increase over time (H). Data were normalized to values obtained for the untreated samples for individual experiments and represent means ± SEM for n = 3 independent experiments. **P < 0.01 and ***P < 0.001, by one-way ANOVA with pairwise comparison using Tukey’s multiple comparisons test. In IB analysis for SAE1, SAE2, UBC9, and SENP1, only SENP1 showed a progressive increase over time in protein levels (E). (I) siRNA-mediated knockdown of SENP1 expression. Lysates from HeLa cells transiently transfected with either a control siRNA or SENP1-specific siRNA for 24 hours were analyzed by IB for SENP1, showing effective knockdown of SENP1. (J) Knockdown of SENP1 expression prevented H2O2-induced degradation of DUSP6. HeLa cells transiently transfected with Flag-DUSP6, Flag-DUSP6K234R (2×), or SENP1 siRNA in various combinations were untreated or treated with 1 mM H2O2 for 0 to 24 hours as indicated beginning at 24 hours after transfection. Cell lysates were analyzed by IB for Flag-DUSP6, SENP1, and SUMO1 levels, with β-actin as the loading control. The loss of SENP1 prevented the H2O2-induced decreases in Flag-DUSP6 levels (left) and the overall protein SUMOylation but had no impact on Flag-DUSP6K234R levels (right). For (A) to (C), (E), (I), and (J), blots are representatives of at least three independent experiments. Two times amount of Flag-DUSP6K234R plasmid was used for transfection to match the expression of Flag-DUSP6.
To investigate the relationship between DUSP6 SUMOylation and ubiquitination under oxidative conditions, we coexpressed Myc-Ub with Flag-DUSP6 or Flag-DUSP6K234R in HeLa cells and then treated the cells with 1 mM H2O2 for different time periods. The wild-type DUSP6 showed a progressive increase in ubiquitination over a period of 24-hour treatment with H2O2, mirroring the course of SUMOylation decrease and despite the decrease in the Flag-DUSP6 protein levels (Fig. 5B, left). By contrast, the DUSP6K234R mutant exhibited higher ubiquitination than the wild-type DUSP6 under untreated conditions, as shown before (Fig. 3F), which was not increased by the H2O2 treatment (Fig. 5B, right). Using untransfected HeLa cells, we further demonstrated that during the H2O2 treatment, the endogenous DUSP6 underwent a time-dependent deSUMOylation and a parallel progressive increase in ubiquitination, as well as a gradual loss of DUSP6 proteins (Fig. 5C). These occurred without a detectable change in DUSP6 mRNA levels (Fig. 5D). Together with the data that overexpression of SUMO1 attenuated DUSP6 ubiquitination and the effect was reversed by the coexpression of the deSUMOylating enzyme SENP1 (Fig. 3, C to F), these results demonstrate that SUMOylation protects DUSP6 from degradation by the ubiquitin-proteasome pathway, but oxidation disrupts this protective mechanism by deSUMOylating DUSP6, which then allows ubiquitination and subsequent degradation of DUSP6 proteins by the proteasomes.
Noticeably, the H2O2 treatment led to an overall decrease in SUMO1-conjugated proteins, suggesting a general deSUMOylating effect of oxidation. To define the molecular underpinning of oxidation-induced deSUMOylation, we first measured the levels of SUMO-modifying enzymes under the treatment of H2O2. Consistent with the previous study (34), H2O2 did not affect the levels of SUMO-conjugating enzymes, E1 (SAE1/SAE2), and E2 (UBC9) (Fig. 5F). However, the levels of the SUMO-deconjugating enzyme, SENP1, were progressively increased over time (Fig. 5F). Using similar cell samples used for immunoblotting, we also performed quantitative reverse transcription polymerase chain reaction (RT-PCR) and found that the mRNA levels of E1 and E2 SUMO-conjugating enzymes were not changed but that of SENP1 increased following the H2O2 treatment (Fig. 5, F to H).
To further confirm the role of SENP1 in oxidation-induced DUSP6 degradation, we knocked down SENP1 expression in HeLa cells using a small interfering RNA (siRNA)–based strategy (Fig. 5I) (35). The knockdown of SENP1 protected Flag-DUSP6 from H2O2-induced degradation (Fig. 5J, left). As a control, the knockdown of SENP1 did not affect the levels of Flag-DUSP6K234R (Fig. 5J, right), and in both cases, the overall protein SUMOylation levels in the SENP1 siRNA-transfected cells were no longer decreased by the H2O2 treatment (Fig. 5J). Examining untransfected HeLa cells, we detected the increase in SENP1 protein expression with the treatment of as low as 0.5 mM H2O2 and for as short as 1 hour, with accompanied decreases in protein SUMOylation and DUSP6 protein levels (Fig. 6, A and B). In addition, the knockdown of SENP1 protected endogenous DUSP6 from H2O2-induced degradation (fig. S7). Together, these results reveal a transcriptional mechanism that up-regulates SENP1 expression in response to oxidation, which, in turn, dampens SUMOylation of a large set of proteins, including DUSP6, allowing DUSP6 degradation by the ubiquitin-proteasome system.
Fig. 6. Increasing DUSP6 SUMOylation suppresses oxidation-induced mitochondrial fragmentation and cell apoptosis.
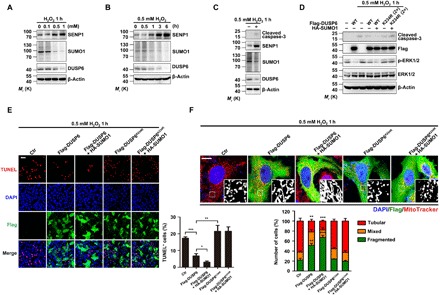
(A and B) H2O2 treatment increased SENP1 levels in a concentration- and time-dependent manner. Lysates from untransfected HeLa cells treated with H2O2 at different concentrations for 1 hour (A) and at 0.5 mM for different time periods (B) as indicated were analyzed by IB for levels of SENP1, SUMO1, and DUSP6, with β-actin used as the loading control. Note that the increases in SENP1 were accompanied with decreases in overall protein SUMOylation and endogenous DUSP6 levels. (C) H2O2-induced increase in caspase-3 cleavage is accompanied with an increase in SENP1 expression. Lysates from untransfected HeLa cells untreated or treated with 0.5 mM H2O2 for 1 hour were analyzed as in (A) and (B), with an additional analysis for cleaved caspase-3. (D) Overexpression of DUSP6 suppressed H2O2-induced caspase-3 cleavage in a SUMOylation-dependent manner. HeLa cells transiently cotransfected with an empty vector (−), a vector encoding Flag-DUSP6 (wild type) or the Flag-DUSP6K234R mutant, and HA-SUMO1 as indicated were untreated or treated with 0.5 mM H2O2 for 1 hour beginning at 24 hours after transfection. Cell lysates were analyzed by IB for levels of cleaved caspase-3, Flag-DUSP6, p-ERK1/2, and ERK1/2, with β-actin used as the loading control. The overexpression of Flag-DUSP6 decreased H2O2-induced caspase-3 cleavage and the p-ERK1/2 levels, both of which were further suppressed by HA-SUMO1; however, Flag-DUSP6K234R did not have these effects. In (A) to (D), blots are representatives of at least three independent experiments. (E and F) Overexpression of DUSP6 attenuated H2O2-induced cell apoptosis (E) and mitochondrial fragmentation (F) in a SUMOylation-dependent manner. H2O2-treated cells as described in (D) were either fixed and then stained for IF labeling of Flag for Flag-DUSP6 expression (green) and cell apoptosis (red) by TUNEL assay system (E) or incubated with MitoTracker Red (red) for 50 min at 37°C before fixation and IF staining for Flag (F). DAPI (blue) was used as the nuclear counterstaining. Representative confocal images are shown at the left (E) or at the top (F). Scale bars, 100 μm (E) and 10 μm (F). Black and white images in (F) show magnification of boxed areas for MitoTracker. Quantification data represent means ± SEM of n = 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, compared to control by one-way ANOVA with pairwise comparison using Tukey’s multiple comparisons test. The overexpression of Flag-DUSP6 decreased H2O2-induced apoptosis and mitochondrial fragmentation, both of which were further suppressed by HA-SUMO1; however, Flag-DUSP6K234R did not have these effects. To ensure comparable levels of protein expression, 2× amount of Flag-DUSP6K234R plasmid was used for transfection.
DUSP6 SUMOylation is important for its protective role against oxidation-induced apoptosis and mitochondrial fragmentation
Since the overexpression of Flag-DUSP6 in HeLa cells reduced H2O2-induced apoptosis and mitochondrial fragmentation (Fig. 1, E to G), we asked whether DUSP6 SUMOylation contributed to this cytoprotective effect against oxidative stress. As shown earlier (Fig. 1C), with cells treated by 0.5 mM H2O2 for 1 hour, cleaved caspase-3 was readily detected (Fig. 6, C and D), and this was accompanied with an increase in SENP1 and decreases in protein SUMOylation and the level of endogenous DUSP6 (Fig. 6C). While the overexpression of wild-type Flag-DUSP6 suppressed the production of the cleaved caspase-3, which was further reduced by the coexpression of HA-SUMO1, the expression of the SUMOylation-deficient Flag-DUSP6K234R mutant did not show such effects (Fig. 6D). Furthermore, the overexpression of wild-type DUSP6 reduced the number of TUNEL-positive cells induced by the H2O2 treatment (Fig. 6E). This protective effect was further enhanced by the coexpression of HA-SUMO1 but not seen with the expression of Flag-DUSP6K234R no matter if HA-SUMO1 was coexpressed (Fig. 6E). Moreover, the overexpression of wild-type DUSP6, but not the SUMOylation-deficient DUSP6K234R, also attenuated the effect of H2O2 on mitochondrial fragmentation, maintaining more cells with tubular mitochondria (51.3 ± 4.1%, n = 318 cells of three independent experiments) than those with mixed and fragmented ones after the 1-hour treatment with 0.5 mM H2O2 (Fig. 6F). Again, the protective effect was enhanced by the coexpression of HA-SUMO1 (cells with tubular mitochondria, 67.0 ± 4.6%, n = 302 cells of three independent experiments; Fig. 6F). Together, these results indicate that DUSP6 SUMOylation accounts for the cytoprotective role of this phosphatase during oxidative stress.
To determine whether the interaction with ERK1/2 is required for the cytoprotective role of DUSP6 SUMOylation, we examined the effects of the KIM mutant (R64, 65A) on H2O2-induced cell apoptosis and mitochondrial fragmentation. As shown in fig. S6, the exposure to 0.5 mM H2O2 for 1 hour caused similar increases in caspase-3 cleavage (fig. S6A) and TUNEL-positive cells (fig. S6B) in HeLa cells transfected with Flag-DUSP and Flag-DUSP6R64, 65A, and both were less than those transfected with Flag-DUSP6K234R or the vector, suggesting that the protective effects of DUSP6 are dependent on its SUMOylation and independent of its binding to ERK2.
DUSP6 physically associates with Drp1 to specifically dephosphorylate Drp1-S616
Numerous studies have established the importance of Drp1 phosphorylation in the regulation of mitochondrial fission (1–3, 5, 7), which may ultimately lead to cell apoptosis. Whereas the phosphorylation at Drp1-S616 activates the fission, that at Drp1-S637 suppresses it (3, 10). Thus, the ratio of Drp1-S616/S637 phosphorylation is indicative of the capacity of Drp1-mediated mitochondrial dynamics, and its imbalance has been implicated in pathogenesis of neurological diseases (36). While several kinases, including CDK1, ERK1/2, and PKCδ, have been shown to phosphorylate Drp1-S616 (7), the phosphatase(s) for this site is not known.
The phosphorylation-dependent Drp1 regulation on mitochondrial fission prompted us to investigate whether the cytoprotective effect of DUSP6 could be exerted through Drp1. First, we examined whether DUSP6 is directly associated with Drp1, like in the case between the phosphatase and its known substrates, ERK1/2 (16). Purified recombinant DUSP6 or DUSP6K234R and Drp1 were used for in vitro binding assay, and coimmunoprecipitation (co-IP) experiments demonstrated that anti-DUSP6 antibody was able to pull down Drp1 (Fig. 7A). Furthermore, anti-Drp1 antibody was able to reciprocally pull down DUSP6 and DUSP6K234R mutant, and there was a weaker association between DUSP6K234R and Drp1 (Fig. 7A). Similar result was found in HeLa cells cotransfected with Myc-Drp1 and Flag-DUSP6 or Flag-DUSP6K234R (fig. S8C). These indicate that DUSP6 can exist in the same complex with Drp1, and this association may depend on the SUMOylation status of DUSP6. Using tissue lysates prepared from mouse cerebral cortex, we also detected Drp1 and DUSP6 from immunoprecipitants pulled down by the anti-DUSP6 and anti-Drp1 antibodies, respectively (Fig. 7B), demonstrating that endogenous DUSP6 and Drp1 are physically associated with each other in vivo in the brain.
Fig. 7. DUSP6 dephosphorylates Drp1-S616 independently of ERK1/2.

(A) Direct association between recombinant DUSP6 and Drp1. Purified DUSP6 or DUSP6K234R and Drp1 were mixed in lysis buffer I and incubated at 4°C for 1 hour with gentle rotation. Samples were subjected to IP with either the anti-DUSP6 (left) or anti-Drp1 (right) antibody, which was followed by IB using anti-DUSP6 and anti-Drp1 antibodies as shown. (B) Physical association between endogenous DUSP6 and Drp1 in mouse brain. Lysates prepared from mouse cerebral cortices were subjected to IP with anti-DUSP6 (left) or anti-Drp1 (right) antibody, which was followed by IB analysis with anti-DUSP6 and anti-Drp1 antibodies as shown. The lysates were also directly analyzed by IB for the inputs of DUSP6 and Drp1, with β-actin used as the loading control. Native Drp1 and DUSP6 pulled down each other in reciprocal co-IP experiments. (C) Overexpression of DUSP6 reduced phosphorylation levels of Drp1-S616, but not Drp1-S637, in a dose-dependent manner. Lysates from HeLa cells transfected with increasing amounts of Flag-DUSP6 plasmid for 24 hours were analyzed by IB using anti–p–Drp1-S616, anti–phospho–Drp1-S637 (p–Drp1-S637), anti-Drp1, and anti-Flag antibodies, with anti-GAPDH used for the loading control. With increasing expression of Flag-DUSP6, p–Drp1-S616 levels were gradually reduced. (D) Drp1 increased the phosphatase activity of DUSP6 in vitro. Incubation with the recombinant Drp1 or ERK2 and the phosphatase activities of DUSP6 and DUSP6C293S were assessed by p-NPP hydrolysis assay. Data for the p-NPP assays in (D) represent means ± SEM of n = 3 independent experiments. ***P < 0.001, by one-way ANOVA with pairwise comparison using Tukey’s multiple comparisons test. (E to H) ERK1/2 phosphorylation status does not affect p–Drp1-S616 and p–Drp1-S637 levels unless cells are under oxidative stress. HeLa cells were either untransfected (E and G) or transiently transfected with increasing amounts of plasmids for T7-ERK1 and Myc-ERK2 at the 1:1 ratio (F) or Flag-DUSP6 (H) as indicated for 24 hours. For (E), the cells were serum-starved for 24 hours in DMEM containing 1% FBS and then stimulated with EGF (50 ng/ml) for the indicated time periods before the addition of with or without 0.5 mM H2O2 for 1 hour for some samples as indicated. The time of EGF addition was adjusted so that EGF and H2O2 treatments ended simultaneously. For (F) to (H), cells were untreated or treated with 0.5 mM H2O2 for 1 hour as shown. In (G) and (H), PD98059 at different concentrations as indicated was added 1 hour before the application of H2O2 and maintained throughout the H2O2 treatment. Lysates obtained from the above cells were subjected to IB with anti–p–Drp1-S616, anti–p–Drp1-S637, anti-Drp1, anti–p-ERK1/2, anti-ERK1/2, and anti-DUSP6 antibodies. β-Actin was used as the loading control. Note that the changes in p-ERK1/2 levels only affected p–Drp1-S616 levels in H2O2-treated samples. In (H), the overexpression of Flag-DUSP6 lowered p–Drp1-S616 levels even when ERK1/2 activity was inhibited by PD98059 without and with H2O2 treatment. (I to K) Overexpression of Flag-DUSP6, but not its catalytically inactive mutant, Flag-DUSP6C293S, suppressed Drp1-S616 phosphorylation regardless of the activity levels of ERK1/2. HeLa cells transiently transfected with an empty vector (−), Flag-DUSP6 (wild type), or Flag-DUSP6C293S (C293S) as indicated were treated with EGF as in (E), coexpressed with increasing amounts of T7-ERK1 and Myc-ERK2 as in (F), or treated with various concentrations of PD98059 as in (G), but without the H2O2 treatment. Cell lysates were subjected to IB as in (E). Note that p–Drp1-S616 levels were decreased by the overexpression of Flag-DUSP6, but not Flag-DUSP6C293S, under all conditions irrespective of the phosphorylation status of ERK1/2. (L) DUSP6 reduces p–Drp1-S616 levels in a SUMOylation-dependent manner. HeLa cells transiently transfected with an empty vector (−), a vector encoding Flag-DUSP6 or one of its mutants, Flag-DUSP6K234R (2×) or Flag-DUSP6C293S, or HA-SUMO1 in various combinations as indicated were untreated or treated with 0.5 mM H2O2 for 1 hour at 24 hours after transfection. Cell lysates were subjected to IB with anti–p–Drp1-S616, anti–p–Drp1-S637, anti-Drp1, anti-cleaved caspase-3, and anti-Flag antibodies. β-Actin was used as the loading control. Note that HA-SUMO1 enhanced the inhibitory effect of Flag-DUSP6 on decreasing p–Drp1-S616 levels in both the absence and presence of H2O2. Neither the SUMOylation-deficient DUSP6K234R mutant nor the catalytically inactive DUSP6C293S mutant had any detectable effect on p–Drp1-S616 levels. Also note the parallel decreases in p–Drp1-S616 and cleaved caspase-3 levels in the H2O2-treated samples. Blots are representatives of at least three independent experiments. To ensure the comparable expression levels, 2× amount of the Flag-DUSP6K234R plasmid was used for the transfection.
The physical association of DUSP6 with Drp1 impelled us to test the ability of the phosphatase to regulate Drp1 dephosphorylation. With HeLa cells transiently transfected with different amounts of the Flag-DUSP6 plasmid, the levels of phosphorylated Drp1-S616 (p–Drp1-S616) decreased as the amount of Flag-DUSP6 increased (Fig. 7C). By contrast, the phosphorylation of Drp1-S637 (p–Drp1-S637) was not affected by the expression of Flag-DUSP6 (Fig. 7C). These results suggest that DUSP6 specifically promotes the dephosphorylation of Drp1-S616, but not Drp1-S637. To further examine the functional relationship between DUSP6 and Drp1, we measured DUSP6 phosphatase activity in the absence and presence of the coexpression of Myc-Drp1 using the p-NPP hydrolysis assay. As shown above (Fig. 4F) and in a previous study (13), the inclusion of a substrate, e.g., ERK2, can increase DUSP6 catalytic activity in vitro. Unexpectedly, recombinant Drp1 also increased the phosphatase activity of DUSP6 (Fig. 7D). Similar results were also confirmed in exogenous expression system, in which Myc-Drp1 increased the phosphatase activity of DUSP6 to a similar extent to Myc-ERK2 (fig. S8D). These results suggest that Drp1 may be a novel substrate of DUSP6. Thus, DUSP6 appears to be a phosphatase that is physically associated with Drp1 and specifically involved in regulating Drp1-S616 phosphorylation.
Given the involvement of ERK1/2 in Drp1-S616 phosphorylation (5) and the high specificity of DUSP6 in dephosphorylating and inactivating ERK1/2 (17), it would be possible that in the cell, DUSP6 mainly facilitated Drp1-S616 dephosphorylation through inactivating ERK1/2 rather than directly dephosphorylating Drp1-S616. To distinguish these possibilities, we tested the following predictions. First, if DUSP6 regulated Drp1 only through ERK1/2, then the p–Drp1-S616 levels should strictly follow that of phospho-ERK1/2 (p-ERK1/2). As shown earlier (Fig. 4A), EGF treatment caused a biphasic change in p-ERK1/2 levels in untransfected HeLa cells (Fig. 7E); however, there was not a corresponding change in the p–Drp1-S616 level unless the cells were also treated with 0.5 mM H2O2 for 1 hour (Fig. 7E). These suggest that only under oxidation, ERK1/2 regulates the phosphorylation of endogenous Drp1-S616. This conclusion is further strengthened by the experiment in which HeLa cells were transfected with increasing amounts of ERK1+2 and at 24 hours later untreated or treated with 0.5 mM H2O2 for 1 hour. Despite the increasing levels of ERK1/2 phosphorylation, corresponding to the increased protein expression, under both normal and oxidative conditions, p–Drp1-S616 levels only correlated with that of p-ERK1/2 in the H2O2-treated cells (Fig. 7F). Moreover, the inhibition of ERK1/2 activation by the potent and selective inhibitor of MAPK kinases—MEK1 and MEK2, PD98059, reduced p-Drp-S616 levels only in the H2O2-treated cells, despite the decreases in p-ERK1/2 levels in both normal and oxidative conditions (Fig. 7G). Collectively, these results suggest a lack of regulation of ERK1/2 on Drp1-S616 phosphorylation under normal conditions; however, under oxidation, Drp1-S616 becomes highly sensitive to ERK1/2. As a control, p–Drp1-S637 levels were not altered by p-ERK1/2 under any of the conditions tested (Fig. 7, E to G).
Second, if DUSP6 regulated Drp1 only through ERK1/2, then inactivating ERK1/2 would abolish the ability of DUSP6 to reduce p–Drp1-S616 levels. As shown in Fig. 7C, overexpression of Flag-DUSP6 greatly reduced the phosphorylation of Drp1-S616 and ERK1/2 without affecting that of Drp1-S637 (Fig. 7H). The inhibition of ERK1/2 activation with the maximal concentration of PD98059 did not impair the ability of the overexpressed Flag-DUSP6 to decrease the p–Drp1-S616 levels, regardless of the H2O2 treatment, although with H2O2, the control p–Drp1-S616 level was already strongly reduced by the drug (Fig. 7H). These data argue that DUSP6 can dephosphorylate Drp1-S616 independently of ERK1/2. To further confirm the ERK1/2-independent regulation of Drp1-S616 phosphorylation by DUSP6, we analyzed the levels of p–Drp1-S616 and p–Drp1-S637 in HeLa cells transfected with wild-type Flag-DUSP6 or catalytically inactive Flag-DUSP6C293S mutant under conditions when ERK1/2 phosphorylation status were changed by stimulating the cells with EGF for different durations (Fig. 7I), cotransfecting with varying amounts of ERK1+2 (Fig. 7J), and the treatment with different concentrations of PD98059 (Fig. 7K). We found that only the wild-type DUSP6, but not the DUSP6C293S mutant, decreased the level of p–Drp1-S616, but not that of p–Drp1-S637, and the effect was independent of the phosphorylation status of ERK1/2. To further test the specificity of DUSP6 in dephosphorylation of Drp1-S616, we knocked down the expression of DUSP6 in HeLa cells using the siRNA. Knockdown of DUSP6 remarkably enhanced the level of Drp1-S616 phosphorylation, increased caspase-3 cleavage (fig. S7A), and TUNEL-positive cells in HeLa cells exposed to 0.5 mM H2O2 for 1 hour (fig. S7B). The H2O2-induced mitochondrial fragmentation was also increased by siRNA knockdown of DUSP6 (fig. S7C). These effects were mimicked by SENP1 overexpression, but the knockdown of SENP1 had some moderate protective effects (fig. S7, A to C). The knockdown of DUSP6 occluded the protective effects of SENP1 siRNA (fig. S7, A to C). These results not only demonstrate the importance of DUSP6 in cytoprotection against oxidative damage but also support the essential role of SUMOylation in the protective effects of DUSP6.
Third, in vitro binding assay demonstrated the interaction between recombinant Drp1 with wild-type DUSP6 and its KIM mutant, which was reduced for DUSP6K234R (fig. S8E). Furthermore, the recombinant Drp1 increased not only the phosphatase activity of wild-type DUSP6 but also that of the KIM mutant (R64, 65A), despite the lack of effect of ERK2 on such mutant (fig. S8F).
Last, the above data suggest that under normal, nonoxidative conditions, Drp1-S616 phosphorylation is kept at low levels by the phosphatase activity of DUSP6, irrespective of the phosphorylation status of ERK1/2. Thus, the loss of DUSP6 due to oxidation will impair this protective effect, rendering Drp1-S616 sensitive to phosphorylation by its kinases, such as ERK1/2. Since deSUMOylation represents a critical step of oxidation-induced degradation of DUSP6, enhancing SUMOylation should help maintain DUSP6 stability and thereby lower p–Drp1-S616 levels under oxidative conditions. The overexpression of Flag-DUSP6 in HeLa cells not only reduced p–Drp1-S616 levels under normal conditions but also suppressed the H2O2-induced increase in Drp1-S616 phosphorylation. Moreover, these effects were further strengthened by the coexpression of HA-SUMO1 (Fig. 7L). On the other hand, neither the SUMOylation-deficient DUSP6K234R nor the catalytically inactive DUSP6C293S mutant could recapitulate the protective effect of wild-type DUSP6 (Fig. 7L), demonstrating that both the phosphatase activity and SUMOylation are critical for the DUSP6 action on Drp1-S616. Consistent with the role of Drp1-S616 phosphorylation on mitochondrial fission and the subsequent cell death, only wild-type DUSP6 prevented the oxidation-induced production of cleaved caspase-3, which was further reduced by the coexpression of HA-SUMO1 (Fig. 7L). Together, the above data suggest that DUSP6 is likely a phosphatase of p–Drp1-S616, which is present in the same complex with Drp1 to keep the phosphorylation level at S616 low. However, this mechanism is disrupted under oxidation due to enhanced expression of SENP1, which leads to DUSP6 deSUMOylation and the consequent degradation via the ubiquitin-proteasome pathway.
DUSP6 protects against ischemic brain damage in a manner that depends on its SUMOylation
To provide a more direct link with the in vivo data in brain ischemia/reperfusion (I/R), we first used oxygen and glucose deprivation/reoxygenation (OGD/reox) model in primary cultured cortical neurons to mimic I/R insult, and the results support our previous conclusions that DUSP6 can protect cells from oxidative damage (fig. S9). Second, to investigate the cytoprotective function of DUSP6 and the role of its SUMOylation in vivo, we used a rodent model of ischemic brain damage, transient middle cerebral artery occlusion (tMCAO), in which oxidative stress is a known contributing factor. Mice were subjected to 40-min tMCAO, followed by reperfusion of different time periods from 0 to 24 hours. To ensure a similar degree of ischemic insult to the brain, we measured cerebral blood flow for each mouse by laser Doppler flowmetry (LDF) during tMCAO (Fig. 8A). With 40-min tMCAO and 24-hour reperfusion, significant damage was detectable in coronal brain sections stained with triphenyltetrazolium chloride (TTC) (Fig. 8A). Using lysates from cortical tissues prepared under denaturing conditions, we detected SUMO1-conjugated bands from immunoprecipitants pulled down by the anti-DUSP6 antibody, which decreased markedly within 6 hours of reperfusion (Fig. 8B). Consistent with the decrease in SUMOylation, DUSP6 protein levels also decreased gradually over the 24-hour reperfusion period (Fig. 8B). However, DUSP6 mRNA levels did not change over the same time periods (Fig. 8C). Similar to the findings in HeLa cells treated with H2O2, the I/R-induced decrease in SUMOylation was not due to changes in the expression of SUMO-conjugating enzymes (Fig. 8, D, E, and G). However, both the mRNA and protein levels of SENP1 were strongly and progressively increased over the 24-hour reperfusion period in the cerebral cortical tissues from mice subjected to tMCAO (Fig. 8, F and G), confirming a transcriptional mechanism that up-regulates SENP1 in response to I/R.
Fig. 8. DUSP6 SUMOylation at K234 is protective to brain neurons in ischemic stroke.
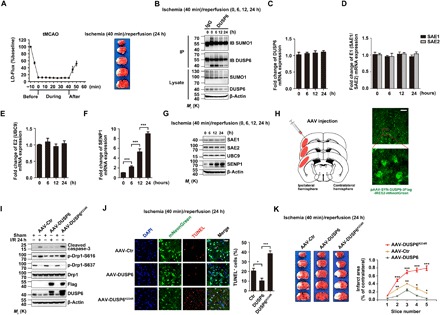
(A) Brain ischemia and brain damage in the tMCAO model. Left: Regional cerebral blood flow in the core of the middle cerebral artery territory of C57BL/6 mice before, during, and after ischemia as measured by LDF. Flow values were averaged at 5-min intervals and expressed as means ± SEM of nine experiments. Right: Representative photographs of TTC-stained brain sections (1 mm) showing brain infarction at 24 hours after the onset of ischemia. (B) Time-dependent decreases in DUSP6 SUMOylation and DUSP6 protein levels during reperfusion in mice subjected to tMCAO for 40 min. Cerebral cortices from C57BL/6 mice (wild type) that underwent 40 min tMCAO, followed by different periods of reperfusion as indicated, were lysed under denaturing conditions and subjected to IP with anti-DUSP6 (or the IgG control), which was followed by IB using anti-SUMO1 and anti-DUSP6. β-Actin was used as the loading control. Note also the time-dependent decrease in overall protein SUMOylation. (C to G) Transcriptional up-regulation of SENP1 during reperfusion of tMCAO injured mice. The original lysates as in (B) were analyzed by real-time quantitative RT-PCR for mRNA levels of DUSP6 (C), SAE1/SAE2 (D), UBC9 (E), and SENP1 (F), as well as by IB for protein levels of SAE1, SAE2, UBC9, and SENP1, with β-actin as the loading control (G). Only SENP1 showed time-dependent increases at both the mRNA (F) and protein levels (G). (H) Schematic representation of adeno-associated virus (AAV) infection into mouse primary somatosensory cortex, barrel field, and secondary somatosensory cortex (left) and representative brain images showing the expression of mNeonGreen in cerebral cortex (right). The boxed area is enhanced by high magnification to show mNeonGreen-positive neurons. Scale bar, 100 μm. (I to K) Overexpression of wild-type DUSP6 reduced, but the SUMOylation-deficient DUSP6K234R mutant exacerbated, cortical neuron apoptosis in response to brain I/R. AAV-DUSP6, AAV-DUSP6K234R, or a control AAV driven by the human synapsin promoter (hSyn) was injected into the primary somatosensory cortex, barrel field, and secondary somatosensory cortex of adult wild-type C57BL/6 mice as shown in (H). The animals were subjected to 40-min tMCAO, followed by 24-hour reperfusion at 4 weeks after stereotactic intracerebral injection. (I) The cerebral cortical tissues were lysed and subjected to IB using anti-cleaved caspase-3, anti–p–Drp1-S616, anti–p–Drp1-S637, anti-Drp1, anti-Flag, and anti-DUSP6. β-Actin was used as the loading control. Caspase-3 cleavage was reduced by AAV-DUSP6 but increased by AAV-DUSP6K234R. Note also the corresponding changes in p–Drp1-S616 levels; however, p–Drp1-S637 levels were affected by neither AAV-DUSP6 nor AAV-DUSP6K234R, despite the decrease caused by I/R. (J) The tissues were sectioned and stained by TUNEL (red) and DAPI (blue). The occurrence of TUNEL+ cells in areas with DUSP6 expression (green) was significantly less than that with DUSP6K234R expression. Representative confocal images in virus-infected cortical areas of brain sections are shown at the left. Scale bar, 100 μm. Quantification of TUNEL+ cells are shown at the right. Data represent means ± SEM of n = 3 independent experiments, with six to eight fields of each group used for statistics in each experiment. *P < 0.05 and ***P < 0.001, by one-way ANOVA with pairwise comparison using Tukey’s multiple comparisons test. (K) Brain infarction after I/R in mice injected with AAV-DUSP6, AAV-DUSP6K234R, or the control AAV. Left: Representative images of TTC-stained sequential brain sections (1 mm). Right: Quantification of infarct areas in ipsilateral hemisphere normalized to the total areas of the contralateral hemisphere in sequential coronal brain slices. Data represent means ± SEM of n = 5 mice in each group. *P < 0.05, **P < 0.01, ***P < 0.001, by one-way ANOVA with pairwise comparison using Tukey’s multiple comparisons test.
To validate the role of DUSP6 and its SUMOylation in ischemic brain damage, we ectopically expressed wild-type DUSP6 and its K234R mutant in mouse cerebral cortical neurons using adeno-associated viruses (AAVs). A control AAV that contained empty vector (AAV-control), AAV-DUSP6, or AAV-DUSP6K234R, with the expression in neurons driven by the human synapsin promoter (hSyn), was injected into the primary somatosensory cortex, barrel field, and secondary somatosensory cortex of adult mice (Fig. 8H). As shown by fluorescence microscopic imaging (Fig. 8H, right), 4 weeks after stereotactic intracerebral injection, the virus-mediated expression, based on the green IRES2-mNeonGreen fluorescence, was readily detectable in the primary and secondary somatosensory cortices. As expected, the virus-mediated DUSP6 gene transfer dampened neuronal apoptosis in response to I/R (40-min tMCAO/24-hour reperfusion), as shown by the reduced production of cleaved caspase-3 (Fig. 8I) and decreased number of TUNEL-positive cells in areas with virus-mediated mNeonGreen expression (Fig. 8J), as well as decreased number of TUNEL-positive cells, and the same result was found by immunohistochemical staining (fig. S10A). Consistent with the findings in vitro, the ectopic expression of DUSP6 also abolished the I/R-induced increase in Drp1-S616 phosphorylation, but not the decrease in p–Drp1-S637 that was seen in all I/R samples. Unexpectedly, not only did the ectopic expression of DUSP6K234R fail to mimic the protective effect of the wild-type DUSP6 but it also made the I/R-induced apoptosis and Drp1-S616 phosphorylation even more severe than the control (Fig. 8, I and J), suggesting a dominant-negative action of the SUMOylation-deficient mutant. Quantification of infarct areas from the coronal brain sections stained with TTC revealed that the expression of DUSP6K234R caused more brain damage than control viruses and that of wild-type DUSP6 protected the brain from the injury caused by I/R (Fig. 8K and fig. S10, B and C). These data further support the idea that DUSP6 SUMOylation plays an important function against I/R-induced apoptosis of cortical neurons.
DISCUSSION
Mitochondrial fragmentation resulting from deregulation of Drp1 phosphorylation is a well-known cause of oxidation-induced apoptosis (2). Although much is known about the sites and kinases involved in Drp1 phosphorylation, the phosphatase(s) involved in dephosphorylating Drp1 and its contribution in the response to oxidative stress remain mysterious. The redox-sensitive phosphatase DUSP6 has been reported to undergo degradation when treated with H2O2, resulting in increased phosphorylation of ERK1/2 (21, 22). This oxidation-induced increase in ERK1/2 activity was thought to enhance Drp1-S616 phosphorylation and the consequent mitochondrial fragmentation, linking DUSP6 instability to oxidative cell death (2). Here, we provide evidence that DUSP6 can cause Drp1-S616 dephosphorylation independently of ERK1/2. We uncover a novel regulatory mechanism by which DUSP6 SUMOylation regulates its stability, which is disrupted by oxidation, resulting in DUSP6 degradation through the ubiquitin-proteasome pathway. The degradation of DUSP6, in turn, unleashes Drp1-S616 for extravagant phosphorylation by its kinases, with ERK1/2 as the main candidates, leading to excessive mitochondrial fragmentation and cell death (fig. S12).
Our findings place DUSP6 as a central player in oxidative cell damage. This phosphatase keeps Drp1-S616 from extended phosphorylation under nonoxidative conditions to prevent excessive mitochondrial fission. Although it cannot be completely ruled out that DUSP6 may dephosphorylate Drp1-S616 through a yet-to-be-identified intermediate(s), evidence provided here suggests that Drp1-S616 is likely a direct substrate of this phosphatase. First, the DUSP6-mediated dephosphorylation of Drp1-S616 is independent of its only known substrates, ERK1/2. We show that under several different conditions, changes in ERK1/2 phosphorylation/activity did not alter p–Drp1-S616 levels unless DUSP6 levels were down-regulated by the treatment with H2O2. Expectedly, the oxidation-induced increase in Drp1-S616 phosphorylation, which is dependent on ERK1/2 (see Fig. 7H), was attenuated by the overexpression of DUSP6 and further suppressed by increasing its SUMOylation (Fig. 7L). Second, the catalytic activation of DUSP6 is achieved by binding to its substrate, e.g., ERK2 (13). We show that the catalytic activity of DUSP6 was increased in the presence of Drp1, just like in the presence of ERK2. This substrate-dependent enhancement of enzymatic activity strongly suggests that Drp1 is another substrate of DUSP6. Third, DUSP6 and Drp1 coexist in the same protein complex both in vitro and in vivo, implicating the likelihood for the phosphatase to directly act on Drp1. Therefore, we suggest that Drp1 is a novel substrate of DUSP6, which dephosphorylates Drp1 at S616.
Mitochondria are dynamic organelles that undergo fission/fusion, and the steady-state balance of fission/fusion is a key determinant of its morphology and function (37, 38). Under healthy conditions, mitochondria exist as a tubular network in equilibrium of fusion/fission. The deregulation of these processes leads to cellular dysfunction. Mitochondrial fission relies on Drp1, and Drp1 forms oligomers that wrap around the mitochondrial outer membrane and scission it. Although the involvement of Drp1 in apoptosis has been controversial and it was proposed that Drp1 is not sufficient to execute mitochondrial fission (39, 40), the mounting evidence suggested that Drp1 is essential for mitochondrial fission and mitochondrial fission–induced apoptosis (41, 42). Drp1 regulates mitochondrial dynamics by virtue of its phosphorylation at two key serine residues, S616 and S637, which alters the activity and location of the cytosolic GTPase (43, 44). It has been shown that dephosphorylation of Drp1-S637 by calcineurin facilitates its translocation to mitochondria to promote mitochondrial fission, implicating an inhibitory role of p–Drp1-S637 in the fission (9, 45). The phosphorylation of Drp1-S616, however, activates mitochondrial fission (3, 10), and this may happen easily as Drp1-S616 is phosphorylated by several different kinases, such as CDK1 or CDK5 (1), PKCδ (2), ERK1/2 (5, 6), ROCK, and CaMK IIα (7). Our finding that DUSP6 dephosphorylates Drp1-S616 provides a plausible explanation on how Drp1-S616 phosphorylation is kept in balance under normal growth conditions. Existing in the same protein complex, the phosphatase can effectively regulate the levels and duration of Drp1-S616 phosphorylation by various kinases, providing a tight restraint on the Drp1 activity and thereby the proper balance of mitochondrial fusion/fission.
Such a balance is disrupted by oxidation through degradation of DUSP6. We show a time- and concentration-dependent decrease in DUSP6 levels in response to the treatment of H2O2. The time- and concentration-dependent response to H2O2 has also been shown for mitochondrial fragmentation in a variety of cells (46, 47). We show here that both mitochondrial fragmentation and apoptotic cell death induced by H2O2 can be rescued by the overexpression of DUSP6, which also suppressed the oxidation-induced increase in Drp1-S616 phosphorylation. The virus-mediated overexpression of DUSP6 in mouse brain reduced the I/R-induced Drp1-S616 phosphorylation, caspase-3 activation, and DNA damage (revealed by TUNEL assay) in the cerebral cortex, resulting in a significant protection of the brain in the ischemic penumbra after focal cerebral ischemia. Therefore, through regulation of Drp1-S616 phosphorylation, DUSP6 plays an important role in maintaining mitochondrial dynamics for cell health, the disruption of which under stress conditions causes programmed cell death. Paradoxically, the knockout or knockdown of DUSP6 in cells or mouse models had often resulted in enhanced proliferation or differentiation (48). These have been linked to the enhanced ERK1/2 activity because of the loss of DUSP6. However, studies also show that in gastric cancer cells, the knockdown of DUSP6, as well as the treatment with a DUSP6 inhibitor, (E/Z)-BCI hydrochloride, suppressed proliferation, migration, and invasion and also caused apoptosis (49). In addition, DUSP6 exerts a protective effect against glutamate-induced cytotoxicity in mouse hippocampal neurons and procyanidin B2G2–induced cell death of prostate cancer (50, 51). The prosurvival function of DUSP6 has also been hypothesized in HeLa cells (52). These would be in line with the antiapoptotic function of DUSP6 revealed here. The consequences of DUSP6 depletion on cell survival/demise are likely dependent on the relative strengths of its substrates, ERK1/2 and Drp1, and the downstream pathways regulated by them, in different cell contexts.
The half-lives of the DUSP6 proteins as measured here at 8 hours for Flag-DUSP6K234R and >24 hours for Flag-DUSP6 do not correspond to the in vivo measurements of endogenous DUSP6 published previously (17, 18). To delineate this seemingly contradicting result, we tested the following predictions. First, the half-lives of exogenous expressed Flag-DUSP6 were examined in HeLa and Chinese hamster ovary (CHO) cells. As shown in fig. S3C, the half-lives of overexpressed Flag-DUSP6 in Hela cells were dependent on the concentration of CHX, and the higher concentrations of CHX resulted in a shorter half-life of Flag-DUSP6 (fig. S3C). Similar results were further confirmed in CHO cells using different concentrations of CHX (20 and 50 μg/ml) (fig. S3D). Second, the half-lives of endogenous DUSP6 were measured in HeLa and mouse embryonic fibroblast (MEF) cells. Consistent with a previous study (53), our observation showed that endogenous DUSP6 protein in both HeLa and MEF cells display shorter half-life (<1 hour) after being treated with CHX (100 μg/ml) (fig. S3E). Thus, this seemingly contradicting result that was not well explained requires further investigation, but many factors, including the cell type–specific expression and different concentrations of CHX application, might contribute to the observed differential half-life of DUSP6.
Our data further unveil a novel PTM mechanism that regulates DUSP6 degradation under oxidative stress. Previously, DUSP6 degradation has been shown to be regulated by ubiquitination and phosphorylation (17). However, how these respond to oxidation was unclear. We show here that under nonoxidative conditions, DUSP6 is SUMOylated at K234, which protects the phosphatase from conjugation by ubiquitin and thereby extends the half-life of the DUSP6 protein. SUMOylation has been shown to alter substrate stability (28). Many proteins are both SUMOylated and ubiquitinated, often at the same lysine residues. However, the interrelationship between these two systems is substrate specific and can act either synergistically or antagonistically (30, 31). We found that SUMOylation increases DUSP6 stability by antagonizing ubiquitination (Fig. 3). The finding that the SUMOylation-deficient DUSP6K234R mutant remains to be strongly ubiquitinated (Figs. 3F and 5B) suggests that K234 is not required for DUSP6 ubiquitination, ruling out competition for the same lysine residue as the mechanism of the antagonism. Then, it is likely that the conjugated SUMO molecule at K234 spatially blocks ubiquitination at other sites of DUSP6. Despite this, it is not known whether K234 in the wild-type DUSP6 is ubiquitinatable and whether and how such a modification, if occurs, affects ubiquitination of DUSP6 at other lysine residues. In addition, the ubiquitin complex that targets DUSP6 for degradation is unknown and requires further investigation.
Unexpectedly, we did not find DUSP6 phosphorylation at the two previously reported serine residues, S159 and S197 (17), to be critical for the half-life of this protein (Fig. 3H). The differences in the cell types used and the experimental conditions might account for the different results concerning DUSP6 phosphorylation on protein stability. Nevertheless, the phosphorylation status of DUSP6 did not affect its SUMOylation (Fig. 3I), suggesting that even if DUSP6 phosphorylation regulated its stability, it would most likely do so independently of SUMOylation. Thus, SUMOylation at K234 specifically enhances the stability of DUSP6 by interfering with its ubiquitination, implicating a cytoprotective function of SUMOylation through DUSP6, a phosphatase that helps maintain mitochondrial dynamics by restraining Drp1.
The cytoprotective role of SUMOylation is progressively disrupted in response to oxidation due to a time-dependent up-regulation of the SUMO-deconjugating enzyme, SENP1. The status of protein SUMOylation is defined by the relative activities of SUMO-conjugating and SUMO-deconjugating enzymes. Whereas we found no change in the expression of SUMO1-conjugating enzymes in HeLa cells treated with H2O2, the expression of SENP1 was increased at both the mRNA and protein levels. Mirroring the changes in SENP1, the overall protein SUMOylation levels were decreased in these cells, so were the SUMOylation and total protein levels of DUSP6. Similar changes were also detected in cerebral cortical tissues from mice subjected to I/R (Fig. 8, B to G). Thus, a time-dependent transcriptional up-regulation of SENP1 induced by oxidation underlies the mechanism of DUSP6 degradation, which ultimately leads to cell apoptosis because of the extended Drp1-S616 phosphorylation and mitochondrial fragmentation.
In summary, we demonstrate that Drp1 is a previously unidentified substrate of the redox-sensitive phosphatase, DUSP6, which restrains Drp1 phosphorylation at S616 and helps maintain the balance of mitochondrial fusion/fission. We show that under normal conditions, DUSP6 is SUMOylated at a C-terminal residue, K234, and the SUMOylation enhances DUSP6 protein stability. However, DUSP6 SUMOylation is disrupted by oxidation through up-regulation of SENP1. The deSUMOylated DUSP6 is then prone to degradation by the ubiquitin-proteasome pathway. The loss of DUSP6 unleashes Drp1-S616 for excessive phosphorylation by kinases, tipping the balance of mitochondrial dynamics toward fission. The resultant mitochondrial fragmentation then causes apoptosis. We show that enhancing DUSP6 expression and its SUMOylation protect not only cells from oxidative damage in vitro but also mouse brain from I/R insults in vivo. These actions were not recapitulated by the DUSP6 mutant that had lost its phosphatase activity or the SUMOylation site. The previously unknown function demonstrated here for DUSP6 and its regulation by SUMOylation are important because they offer novel insights into the mechanism of cytoprotection against oxidative stress. The more in-depth understanding of the DUSP6/Drp1-regulated pathway described here at the molecular level will promote the design of new strategies for treatment of diseases associated with oxidative damage, such as brain injury caused by ischemic stroke.
MATERIALS AND METHODS
Experimental animals and tMCAO
All experimental protocols with animals were carried out in accordance with the guidelines for the Care and Use of Laboratory Animals of Shanghai Jiao Tong University School of Medicine and approved by the Institutional Animal Care and Use Committee. Male C57BL/6 mice (6 to 8 weeks, 18 to 20 g) were housed under a 12-hour/12-hour light/dark cycle with free access to food and water. tMCAO was performed as previously described (54) with minor modifications. Briefly, mice were anesthetized with pentobarbital sodium. Body temperature was maintained at 37°C by a heating pad during the entire procedure. tMCAO was induced using intraluminal suture as previously described (54). The left common carotid artery and the external carotid artery were exposed by a ventral midline neck incision and clipped. The external carotid artery was ligated with 5-0 silk suture. A 2-cm length of silicone rubber–coated monofilament (7-0) was advanced from the common carotid through the internal carotid up to the level of the anterior cerebral artery. The suture was inserted 9 to 11 mm from the bifurcation of common carotid to occlude the middle cerebral artery. After 40 min of tMCAO, the suture was gently retracted to allow reperfusion for various durations (6, 12, and 24 hours). LDF (Moore Instruments Limited, Devon, UK) was used to monitor regional cerebral blood flow from 15 min before to 10 min after tMCAO. I/R were defined as a minimum of 75% decrease in cerebral blood flow at the onset of ischemia and a return to 50% of baseline blood flow measurement, respectively. Mice not achieving these criteria were excluded from the study. Among the 250 male animals that underwent the surgery for tMCAO, 70 of the ischemic animals were excluded because of insufficient decrease in cerebral blood flow following tMCAO (15), death during anesthetic induction, before completion of the surgery, during early reperfusion, or several hours before the 24-hour post-MCAO study end point (31), or lack of obvious infarct lesion at 24 hours after tMCAO (8). Owing to animal deaths before the 24-hour study end point, five animals per group were available for infarct (area) volume analysis. On the basis of a previous study (55) with minor modifications, we sectioned the mouse brain into three coronal slices beginning 1 mm from the anterior tip of the frontal lobe to 1 mm from the end of occipital lobe. The cortex of middle section (including penumbral and core regions) representing the lesion area was used for Western blot analysis and immunofluorescence staining.
Antibodies and reagents
Anti-cleaved caspase-3 (1:1000; 9661), anti-ERK1/2 (1:1000; 9102), anti–p-ERK1/2 (1:1000; 9106), anti-SAE1 (1:1000; 8688), anti–p–Drp1-S616 (1:1000; 3455), and anti–p–Drp1-S637 (1:1000; 4867) were from Cell Signaling Technology (Danvers, MA, USA). Anti-DUSP6 (1:1000; ab76310), anti-UBC9 (1:1000; ab33044), and anti-SENP1 (1:1000; ab108981) were from Abcam (Cambridge, MA, USA). Anti-Drp1 (1:500; sc-101270) was from Santa Cruz Biotechnology (Dallas, TX, USA). All other antibodies were as described previously (56). N-ethylmaleimide (NEM; 20 mM; E3876), CHX (100 μg/ml; C7698), proteasome inhibitor MG132 (20 μM; E8699), chloroquine (100 μM; C6628), EGF (50 ng/ml), and H2O2 (30%, v/v) were purchased from Sigma-Aldrich (St. Louis, MO, USA). MEK inhibitor PD98059 (52001ES08) was from Yeasen Biotechnology (Shanghai, China).
Plasmids and RNA interference
HA-SUMO1 and HA-SUMO2 were provided by J. Cheng (Shanghai Jiao Tong University School of Medicine, Shanghai, China), T7-ERK1 was purchased from Addgene (14440; Cambridge, MA, USA), and Myc-Ub and Myc-ERK2 were gifts from J. Yu (Shanghai Jiao Tong University School of Medicine, Shanghai, China). RGS-SENP1 and RGS-SENP1m plasmids were previously described (56). To obtain Flag-DUSP6, the full-length DUSP6 (mouse) was cloned into p3xFlag-Myc-CMV-24 vector. Various Flag-DUSP6 mutations (K68R, K120R, K121R, K138R, K220R, K234R, K256R, K285R, K311R, K324R, K326R, K327R, S159A, S197A, S159D, S197D, DM-SA, DM-SD, and C293S) were generated using the QuikChange Site-Directed Mutagenesis Kit (200519; Stratagene, San Diego, CA, USA). SENP1 and DUSP6 siRNA duplexes were synthesized by Sangon Biotechnology (Shanghai, China). siRNA oligos specific for SENP1 (5′-UCCUUUACACCUGUCUCGAUGUCUU-3′) and the corresponding scramble siRNA oligonucleotide (5′-CUUCCUCUCUUUCUCUCCCUUGUGA-3′) or siRNA oligos specific for DUSP6 (5′-GUGCAUUGCUUGGCAGGUA-3′) and the corresponding scramble siRNA oligonucelotide (5′-CGUACGCGGAAUACUUCGAdTdT-3′) were transfected at a final concentration of 20 nM into HeLa cells using Lipofectamine 2000 (11668019; Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol.
Cell culture and transfection
HeLa cells (TCHu187) were purchased from The Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). CHO cells (CHO-K1 line) were a gift from T. Zhou at the Department of Medicine (the University of Alabama School of Medicine, Birmingham, AL, USA). MEF cells were provided by J. Cheng (Shanghai Jiao Tong University School of Medicine, Shanghai, China). Cells were cultured at 37°C in 5% CO2 humidified incubator and grown in Dulbecco’s Modified Eagle Medium (DMEM; high glucose) supplemented with 10% fetal bovine serum (FBS). HeLa cells and CHO cells grown into ~80% confluence were transfected with the desired plasmids using Lipofectamine 2000 following the manufacturer’s instructions. In most experiments, equimolar ratios of plasmids were used for cotransfection experiments, and the total amount of plasmids was adjusted with the empty vector. To ensure that the various plasmids express comparable levels of the proteins, a twofold amount of Flag-DUSP6K234R, Flag-DUSP6I233A, or Flag-DUSP6I233N plasmid, as compared to wild-type Flag-DUSP6, was used for transfection in some experiments.
Immunoprecipitation and Western blotting
Western blotting and denaturing immunoprecipitations were performed as described previously (56). Briefly, 24 hours after transfection, HeLa cells were lysed in lysis buffer I [50 mM tris-HCl (pH 6.8), 2% SDS, 40 mM dithiothreitol (DTT), and 5% glycerol] or lysis buffer II [50 mM tris-HCl (pH 7.5), 150 mM NaCl, and 1% NP-40] after being washed with ice-cold phosphate-buffered saline (PBS). NEM (20 mM) was added to the cell lysates for detection of SUMOylated DUSP6. Then, the lysates were incubated for 20 min at 95°C before dilution with lysis buffer II. After centrifugation for 10 min at 4°C, the supernatant was collected and incubated with the desired primary antibody at 4°C overnight. Protein A/G Sepharose beads were used to purify the proteins. Then, the beads were centrifuged and washed three times with washing buffer [10 mM tris (pH 7.4), 1 mM EDTA, 1 mM EGTA (pH 8.0), 150 mM NaCl, 1% Triton X-100, and 0.2 mM sodium orthovanadate]. They were boiled in the SDS sample buffer and then subjected to SDS–polyacrylamide gel electrophoresis (PAGE) and immunoblotting. CHX chase assays were performed as described previously (56). Briefly, CHX (100 μg/ml) was added to the culture 24 hours after transfection, and samples were prepared at the indicated time points. The steady-state levels of the protein of interest were analyzed by Western blotting with appropriate antibodies as indicated.
Immunofluorescence staining
Immunofluorescence staining was performed as described previously (41) with minor modifications. Briefly, HeLa cells grown on glass coverslips in DMEM supplemented with 10% FBS were transiently transfected with desired complementary DNA (cDNA) constructs using Lipofectamine 2000. Cells were incubated at 37°C in 5% CO2 for 24 hours following transfection. Coverslips were rinsed with PBS and then incubated in 4% paraformaldehyde for 10 min at room temperature for fixation. After fixation, the cells were incubated in blocking buffer (1× PBS with 10% normal goat serum and 0.3% Triton X-100) for 1 hour at room temperature. Cells were rinsed three times with 0.01 M PBS (pH 7.4), incubated with the primary antibody (anti-Flag; 1:500; F1804, Sigma-Aldrich) overnight at 4°C, washed thrice for 10 min, and then incubated with Alexa Fluor 488–conjugated goat anti-mouse antibody (1:200; R37120, Invitrogen) for 1 hour at room temperature. Confocal images were captured on a Leica SP8 confocal microscope (Leica, Wetzlar, Germany) using a 20× objective lens. TUNEL staining was performed as described previously (54) with minor modifications. Briefly, HeLa cells and neurons grown on glass coverslips or 40-μm-thick frozen sections of mouse brain tissues were stained using In Situ Cell Death Detection Kit, TMR red (12156792910; Roche, Basel, Switzerland) according to the manufacturer’s protocols. The apoptotic cells appeared red, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). The stained cells/sections were photographed with a confocal microscope (Leica, Wetzlar, Germany). For quantification, two or three quadrants were selected from each section, and the numbers of DAPI-positive (blue) and TUNEL-positive (red) cells in each quadrant were counted. The percentage of TUNEL-positive cells was calculated using the formula red/blue × 100%.
Mitochondrial staining was performed as described previously (3). HeLa cells were plated on gelatin-coated coverslips. At 24 hours after transfection, cells were treated with or without 0.5 mM H2O2 for 1 hour and then stained with 200 nM MitoTracker Red CMXRos (M7512, Invitrogen) for 50 min at 37°C. Cells were fixed with 4% paraformaldehyde for 10 min, permeabilized, and blocked for 1 hour at room temperature with blocking buffer (1× PBS with 10% normal goat serum and 0.3% Triton X-100). Cells were rinsed three times with 0.01 M PBS (pH 7.4), incubated with the anti-Flag antibody (1:500; F1804, Sigma-Aldrich) overnight at 4°C, washed thrice for 10 min, and incubated with Alexa Fluor 488–conjugated goat anti-mouse antibody (1:200; R37120, Invitrogen) for 1 hour at room temperature. Cells were rinsed three times with PBS and then stained with DAPI. Mitochondria were imaged using a laser scanning confocal microscope (Leica SP8). For each sample, several random fields of cells (≥100 cells) were evaluated for mitochondrial morphology.
Cloning, mutagenesis, and expression of recombinant proteins
The cDNA for mouse DUSP6, mouse ERK2, human Drp1, human SUMO1, and human SENP1 were described above. Columns for protein purification were purchased from GE Healthcare. The full-length DUSP6, ERK2, and Drp1 cDNAs were cloned in E. coli expression vector pE-SUMO3. The coding sequences for amino acid residues 1 to 97 of SUMO1 and residues 419 to 644 of SENP1 were cloned in E. coli expression vector pET28a. All mutants were constructed by PCR mutagenesis using the KOD Plus Mutagenesis Kit (TOYOBO). The purification of these recombinant proteins followed similar procedures as described previously (57, 58). Briefly, the expression construct was transformed into BL21 (DE3) cells and grown in 2× YT at 37°C until optical density at 600 nm reached 0.8. Then, isopropyl-β-d-1-thiogalactopyranoside was added to a final concentration of 0.2 mM, and the culture was transferred to a 25°C shaker for a further 12 hours. The cells were collected by centrifugation, resuspended in ice-cold buffer [20 mM tris-HCl (pH 7.4), 0.5 M NaCl, and 20 mM imidazole], and disrupted by a high-pressure cell breaker. The supernatant of the cell lysate was loaded onto a 5-ml HisTrap FF column eluted by a 0.02 to 0.2 M imidazole gradient. DUSP6, ERK2, and Drp1 were further purified by a 5-ml HiTrap Q HP column and eluted by a NaCl gradient [0.05 to 1 M in 10 mM tris-HCl (pH 8.0)]. The peak fractions were collected, concentrated, and buffer exchanged into 10 mM tris-HCl (pH 7.4) and 0.1 M NaCl. The samples were snap-frozen in liquid nitrogen and stored in −80°C before use.
In vitro SUMOylation assay
In vitro SUMOylation of DUSP6 was carried out with a SUMOylation kit (BML-UW8955-0001, Enzo Life Sciences). Purified DUSP6 was suspended in SUMOylation buffer (20 μl) containing E1, E2, SUMO1, and adenosine triphosphate (ATP). Following 1-hour incubation at 37°C, reaction was terminated with SDS loading buffer. The samples prepared above were analyzed by Western blotting with SUMO1 and DUSP6 antibodies as indicated.
Phosphatase activity assay
The general substrate p-NPP (21101ES60, Yeasen Biotechnology, Shanghai, China) was used to measure the phosphatase activity of DUSP6 in vitro and in vivo. In vitro phosphatase activities and catalytic activation of DUSP6 were performed using purified DUSP6, and its mutants were generated in E. coli. The phosphatase activity of SUMOylated DUSP6 was measured after DUSP6 was SUMOylated in vitro. The purified DUSP6 or its mutants were added into the p-NPP Assay Buffer, followed by addition of the same amount of p-NPP substrate (50 μl). After incubation at 37°C for 10 min, the reaction was stopped by adding the equal volume of Stop Solution (100 μl), and the released p-NPP was measured at 405 nm by multiscan spectrum (BioTek Elx800, BioTek Instruments, Winooski, VT, USA). In vivo phosphatase activities and catalytic activation of DUSP6 were performed using cell lysates from HeLa cells expressing Flag-DUSP6 or its various mutations. HeLa cells expressing Flag-DUSP6 or one of its various mutations (Flag-DUSP6K234R, Flag-DUSP6I233A, and Flag-DUSP6I233N) were cotransfected with or without HA-SUMO1, RGS-SENP1, Myc-ERK2, or Myc-Drp1 and lysed at 24 hours after transfection. The protein content was quantified by the bicinchoninic acid assay (Pierce Chemical Co., Dallas, TX, USA), and a desired amount of the lysate was added into the p-NPP Assay Buffer, followed by addition of the same amount of p-NPP substrate (50 μl). After incubation at 37°C for 10 min, the reaction was stopped by adding the equal volume of Stop Solution (100 μl), and the released p-NPP was measured at 405 nm by multiscan spectrum (BioTek Elx800, BioTek Instruments, Winooski, VT, USA).
In vitro binding assay
Purified DUSP6 or DUSP6K234R and Drp1 were mixed in lysis buffer I [50 mM tris-HCl (pH 6.8), 2% SDS, 40 mM DTT, and 5% glycerol] and incubated at 4°C for 1 hour with gentle rotation. Samples were then incubated with the DUSP6 or Drp1 antibody at 4°C overnight. Protein complex was pulled down using protein A/G–coupled agarose beads, centrifuged, and washed three times with washing buffer [10 mM tris (pH 7.4), 1 mM EDTA, 1 mM EGTA (pH 8.0), 150 mM NaCl, 1% Triton X-100, and 0.2 mM sodium orthovanadate]. The beads were lastly boiled for 10 min in the SDS sample buffer and subjected to SDS-PAGE, followed by immunoblotting.
RNA extraction and analysis by RT-PCR
RNA was extracted from HeLa cells with or without the H2O2 treatment or uninjured or ischemic mouse brain cortex tissues using the TRIzol RNA extraction kit (DP421, Tiangen, Beijing, China). RT-PCR was performed in a final volume of 10 μl with the Power SYBR Green PCR Master Mix (CW2621M, CWBIO, Beijing, China) according to the manufacturer’s protocol. The primers used are as follows: mDUSP6, 5′-ATAGATACGCTCAGACCCGTG-3′(forward) and 5′-ATCAGCAGAAGCCGTTCGTT-3′(reverse); hDUSP6, 5′-GAAATGGCGATCAGCAAGACG-3′(forward) and 5′-CGACGACTCGTATAGCTCCTG-3′(reverse); mSAE1, 5′-CAGTATGACCGACAGATCCGC-3′ (forward) and 5′-GGCAACCTGAGCCTTTGATCT-3′(reverse); hSAE1, 5′-CTCTCGGGTGCTTCTTGTCG-3′ (forward) and 5′-CATGGTCAGTCCTTTCACTCC-3′(reverse); mSAE2, 5′-CCACATCGACCTGATTGATCTG-3′(forward): and 5′-GGCAACCTGAGCCTTTGATCT-3′(reverse); hSAE2, 5′-TTCTCCCACATCGACCTGATT-3′ (forward) and 5′-ACCTGTGCCTTTGATCTTCCA-3′(reverse); mUBC9, 5′-GGAGGAAGGACCACCCTTTTG-3′(forward) and 5′-GGATAGCGCACTCCCAGTT-3′(reverse); hUBC9, 5′-AAAAATCCCGATGGCACGATG-3′ (forward) and 5′-CTTCCCACGGAGTCCCTTTC-3′(reverse); mSENP1, 5′-CTGGGGAGGTGACCTTAGTGA-3′ (forward) and 5′-GTGATAATCTGGACGATAGGCTG-3′(reverse); hSENP1, 5′-AGTGAACCACAACTCCGTATTC-3′ (forward) and 5′-AAAAGATCGGTCCAAATGTCCTT-3′(reverse); mβ-actin, 5′-GGCTGTATTCCCCTCCATCG-3′ (forward) and 5′-CCAGTTGGTAACAATGCCATGT-3′(reverse); hβ-actin, 5′-CATGTACGTTGCTATCCAGGC-3′ (forward) and 5′-CTCCTTAATGTCACGCACGAT-3′ (reverse).
Primary neuron culture and OGD/reox
Cortical cultures were prepared from embryonic day 13.5 (E13.5) C57BL/6 mouse embryos by dissection, trypsin treatment, and mechanical dissociation. Cells were plated on poly-l-lysine–coated dishes at a density of 3 million per 6-cm dish and 0.15 million per well for 24-well plates in Neurobasal medium (Gibco) containing 2% B27 (Gibco) and 1% GlutaMAX (Gibco) and incubated at 37°C in humidified air supplemented with 5% CO2. Cortical neurons were then fed every 4 days with this serum-free medium until experimental usage at 14 days in vitro (DIV). The OGD/reox was performed as described previously (59) with minor modifications. Cortical neurons grown in 6-cm dishes or grown on glass coverslips in 24-well plates were transfected with or without Flag-DUSP6 plasmid using NeuroPORTER transfection kit (NPT01, Sigma-Aldrich), and cells were incubated at 37°C in 5% CO2 for 36 hours following transfection. Then, neurons were subjected to 2-hour OGD and 24-hour reoxygenation. Neuronal cell lysates were used for Western blotting, and neurons grown in 24-well plates were used for TUNEL staining.
Immunohistochemical staining
Immunohistochemical staining was performed as described previously (60) with minor modifications. Briefly, 20-μm-thick frozen sections of mouse brain tissues were stained using the Colorimetric TUNEL Apoptosis Assay Kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s protocols. The apoptotic cells appeared brown, and the nuclei were stained with hematoxylin (blue). The stained sections were photographed with a microscope (Leica, Wetzlar, Germany). For quantification, two or three quadrants were selected from each section, and the numbers of hematoxylin (blue) and TUNEL-positive (brown) cells in each quadrant were counted. The percentage of TUNEL-positive cells was calculated using the formula brown/blue × 100%.
TTC assay and infarct area measurement
TTC assay was performed as described previously (54) with minor modifications. At 24 hours after tMCAO, C57BL/6 mice were anesthetized, brains removed and sliced into five 1-mm-thick sections. The sections were incubated in 2% TTC for 30 min at 37°C. The photographs of TTC-stained brain sections show the brain infarction. In each slice, the area of the noninfarcted portion in the lesion side and the total area in the contralateral side were measured by an investigator blinded to the genotype using ImageJ. The areas of each side were summed over the number of sections evaluated, and the respective volumes were calculated by multiplying each sum by 1 mm (thickness of each section). The percentage of infarction volume was calculated as follows: [(volume of contralateral side − noninfarcted volume of the lesion side)/volume of contralateral side] × 100%; the percentage of cortex infarction volume was calculated as follows: (volume of infarcted cortex of lesion side/volume of total cortex of contralateral side) × 100%.
Stereotaxic AAV virion injection into mouse brain cortex
AAV virions were produced by Obio Technology Co. Ltd. (Shanghai, China). For DUSP6 expression in neurons, mouse DUSP6 or its mutant K234R cDNA was inserted into AAV vector, pAAV-hSyn-MCS-mNeonGreen-3Flag. C57BL/6 mice (male, 6 weeks) were placed in the Stereotaxic frame (RWD Life Science, San Diego, CA, USA). The AAV virions were injected into the primary somatosensory cortex, barrel field, and secondary somatosensory cortex using the coordinates according to the mouse brain atlas: anterior-posterior (AP) = ±0.00 mm; left (L) = +3.70 mm; dorsal-ventral (DV) = −2.50 mm. A total of 1 μl of DUSP6 or 2 μl of DUSP6K234R was injected by a microelectrode connected with a microinjector pump (KDS 310, KD Scientific, Holliston, MA, USA) at a rate of 0.1 μl/min, and a further 10 min after infusion was given to allow complete diffusion of AAV from the tip. After 4 weeks, brain slices from mice injected with the virion were examined directly by fluorescence microscopy, or the virus-injected mice were subjected to tMCAO.
Supplementary Material
Acknowledgments
Funding: This study was supported by grants from the National Natural Science Foundation of China (31830031, 31761163002, and 31671053 to Y.L.), the Science and Technology Commission of Shanghai Municipality (18JC1420302), and the Innovative Research Team of High-Level Local Universities in Shanghai. Author contributions: Y.L. conceived the studies and designed the experiments. R.M. carried out the biochemical and immunofluorescence experiments. L.M. and A.Z. prepared the recombinant proteins. W.W., R.G., Y.G., and J.T. performed parts of the biochemical and immunofluorescence experiments. R.M. and Y.W. prepared and performed the tMCAO model experiments. W.W. and H.L. assisted with data analysis and analyzed results. T.-L.X., J.C., A.Z., and M.X.Z. discussed the results and provided technical support. R.M., A.Z., M.X.Z., and Y.L. wrote the manuscript with inputs from the other authors. All authors discussed and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaaz0361/DC1
Fig. S1. DUSP6 plays a protective role against oxidative damage depending on its phosphatase activity but not ERK1/2.
Fig. S2. DUSP6 is modified by SUMO2/3.
Fig. S3. SUMOylation regulates stability of DUSP6.
Fig. S4. SUMOylation of DUSP6 is important for its catalytic activity.
Fig. S5. DUSP6 SUMOylation affects its interaction properties.
Fig. S6. The protective effect of DUSP6 SUMOylation is independent of its binding to ERK2.
Fig. S7. Importance of DUSP6 in cytoprotection against oxidative damage.
Fig. S8. Knockdown of SENP1 protected endogenous DUSP6 from H2O2-induced degradation, and DUSP6 can physically associated with Drp1.
Fig. S9. DUSP6 protects neurons from oxidative damage induced by OGD/reox.
Fig. S10. Overexpression of DUSP6, but not the SUMOylation-deficient DUSP6K234R mutant, protected mouse brain from damages caused by I/R.
Fig. S11. Quantification of the Western blot results (Figs. 1, 2, 6, and 7).
Fig. S12. Schematic model showing the role of DUSP6 SUMOylation in Drp1-mediated cell death under physiological and oxidative stress conditions.
REFERENCES AND NOTES
- 1.Taguchi N., Ishihara N., Jofuku A., Oka T., Mihara K., Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Qi X., Disatnik M.-H., Shen N., Sobel R. A., Mochly-Rosen D., Aberrant mitochondrial fission in neurons induced by protein kinase Cδ under oxidative stress conditions in vivo. Mol. Biol. Cell 22, 256–265 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kashatus D. F., Lim K.-H., Brady D. C., Pershing N. L., Cox A. D., Counter C. M., RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol. 13, 1108–1115 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Z., Jiang H., Chen S., Du F., Wang X., The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Kashatus J. A., Nascimento A., Myers L. J., Sher A., Byrne F. L., Hoehn K. L., Counter C. M., Kashatus D. F., Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 57, 537–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyakurel A., Savoia C., Hess D., Scorrano L., Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol. Cell 58, 244–254 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lima A. R., Santos L., Correia M., Soares P., Sobrinho-Simões M., Melo M., Máximo V., Dynamin-related protein 1 at the crossroads of cancer. Genes 9, E115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cribbs J. T., Strack S., Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cereghetti G. M., Stangherlin A., Martins de Brito O., Chang C. R., Blackstone C., Bernardi P., Scorrano L., Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang W., Wang Y., Long J., Wang J., Haudek S. B., Overbeek P., Chang B. H., Schumacker P. T., Danesh F. R., Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 15, 186–200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wikstrom J. D., Israeli T., Bachar-Wikstrom E., Swisa A., Ariav Y., Waiss M., Kaganovich D., Dor Y., Cerasi E., Leibowitz G., AMPK regulates ER morphology and function in stressed pancreatic β-cells via phosphorylation of DRP1. Mol. Endocrinol. 27, 1706–1723 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickinson R. J., Keyse S. M., Diverse physiological functions for dual-specificity MAP kinase phosphatases. J. Cell Sci. 119, 4607–4615 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Camps M., Nichols A., Gillieron C., Antonsson B., Muda M., Chabert C., Boschert U., Arkinstall S., Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 280, 1262–1265 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Muda M., Theodosiou A., Gillieron C., Smith A., Chabert C., Camps M., Boschert U., Rodrigues N., Davies K., Ashworth A., Arkinstall S., The mitogen-activated protein kinase phosphatase-3 N-terminal noncatalytic region is responsible for tight substrate binding and enzymatic specificity. J. Biol. Chem. 273, 9323–9329 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Fjeld C. C., Rice A. E., Kim Y., Gee K. R., Denu J. M., Mechanistic basis for catalytic activation of mitogen-activated protein kinase phosphatase 3 by extracellular signal-regulated kinase. J. Biol. Chem. 275, 6749–6757 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Arkell R. S., Dickinson R. J., Squires M., Hayat S., Keyse S. M., Cook S. J., DUSP6/MKP-3 inactivates ERK1/2 but fails to bind and inactivate ERK5. Cell. Signal. 20, 836–843 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Marchetti S., Gimond C., Chambard J.-C., Touboul T., Roux D., Pouysségur J., Pagès G., Extracellular signal-regulated kinases phosphorylate mitogen-activated protein kinase phosphatase 3/DUSP6 at serines 159 and 197, two sites critical for its proteasomal degradation. Mol. Cell. Biol. 25, 854–864 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurek A., Amagasaki K., Gembarska A., Heldin C.-H., Lennartsson J., Negative and positive regulation of MAPK phosphatase 3 controls platelet-derived growth factor-induced Erk activation. J. Biol. Chem. 284, 4626–4634 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Bermudez O., Pagès G., Gimond C., The dual-specificity MAP kinase phosphatases: Critical roles in development and cancer. Am. J. Physiol. Cell Physiol. 299, C189–C202 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Chan D. W., Liu V. W., Tsao G. S., Yao K.-M., Furukawa T., Chan K. K., Ngan H. Y., Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 29, 1742–1750 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Han P., Zhou X.-H., Chang N., Xiao C.-L., Yan S., Ren H., Yang X.-Z., Zhang M.-L., Wu Q., Tang B., Diao J.-P., Zhu X., Zhang C., Li C.-Y., Cheng H., Xiong J.-W., Hydrogen peroxide primes heart regeneration with a derepression mechanism. Cell Res. 24, 1091–1107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou J., Ge L., Jia C., Zheng X., Cui H., Zong R., Bao X., Yin Y., Ma J.-X., Li W., Liu Z., Zhou Y., ROS-mediated different homeostasis of murine corneal epithelial progenitor cell line under oxidative stress. Sci. Rep. 6, 36481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kucharska A., Rushworth L. K., Staples C., Morrice N. A., Keyse S. M., Regulation of the inducible nuclear dual-specificity phosphatase DUSP5 by ERK MAPK. Cell. Signal. 21, 1794–1805 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Prieto J., León M., Ponsoda X., Sendra R., Bort R., Ferrer-Lorente R., Raya A., López-García C., Torres J., Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 7, 11124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prasad S., Pal P. K., SUMOylation: One small modification for proteins, multiple giant problems for mankind. Mov. Disord. 33, 403 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Jafarnejad S. M., Chapat C., Matta-Camacho E., Gelbart I. A., Hesketh G. G., Arguello M., Garzia A., Kim S.-H., Attig J., Shapiro M., Morita M., Khoutorsky A., Alain T., Gkogkas C. G., Stern-Ginossar N., Tuschl T., Gingras A.-C., Duchaine T. F., Sonenberg N., Translational control of ERK signaling through miRNA/4EHP-directed silencing. ELife 7, e35034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez M. S., Dargemont C., Hay R. T., SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Geiss-Friedlander R., Melchior F., Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Gareau J. R., Lima C. D., The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geoffroy M.-C., Hay R. T., An additional role for SUMO in ubiquitin-mediated proteolysis. Nat. Rev. Mol. Cell Biol. 10, 564–568 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Wilkinson K. A., Henley J. M., Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 428, 133–145 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dadke S., Cotteret S., Yip S.-C., Jaffer Z. M., Haj F., Ivanov A., Rauscher F. III, Shuai K., Ng T., Neel B. G., Chernoff J., Regulation of protein tyrosine phosphatase 1B by sumoylation. Nat. Cell Biol. 9, 80–85 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Kubota Y., O'Grady P., Saito H., Takekawa M., Oncogenic Ras abrogates MEK SUMOylation that suppresses the ERK pathway and cell transformation. Nat. Cell Biol. 13, 282–291 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Bossis G., Melchior F., Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol. Cell 21, 349–357 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Li X., Luo Y., Yu L., Lin Y., Luo D., Zhang H., He Y., Kim Y.-O., Kim Y., Tang S., Min W., SENP1 mediates TNF-induced desumoylation and cytoplasmic translocation of HIPK1 to enhance ASK1-dependent apoptosis. Cell Death Differ. 15, 739–750 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Kim J.-E., Kim Y.-J., Kim J. Y., Kang T.-C., PARP1 activation/expression modulates regional-specific neuronal and glial responses to seizure in a hemodynamic-independent manner. Cell Death Dis. 5, e1362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friedman J. R., Nunnari J., Mitochondrial form and function. Nature 505, 335–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mishra P., Chan D. C., Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 15, 634–646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sheridan C., Martin S. J., Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 10, 640–648 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Lee J. E., Westrate L. M., Wu H., Page C., Voeltz G. K., Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540, 139–143 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prudent J., Zunino R., Sugiura A., Mattie S., Shore G. C., McBride H. M., MAPL SUMOylation of Drp1 Stabilizes an ER/mitochondrial platform required for cell death. Mol. Cell 59, 941–955 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Fonseca T. B., Sánchez-Guerrero Á., Milosevic I., Raimundo N., Mitochondrial fission requires DRP1, but not dynamins. Nature 570, E34–E42 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Chang C.-R., Blackstone C., Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 1201, 34–39 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otera H., Ishihara N., Mihara K., New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 1833, 1256–1268 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Campello S., Scorrano L., Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 11, 678–684 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jendrach M., Mai S., Pohl S., Vöth M., Bereiter-Hahn J., Short- and long-term alterations of mitochondrial morphology, dynamics and mtDNA after transient oxidative stress. Mitochondrion 8, 293–304 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Fan X., Hussien R., Brooks G. A., H2O2-induced mitochondrial fragmentation in C2C12 myocytes. Free Radic. Biol. Med. 49, 1646–1654 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beaudry K., Langlois M.-J., Montagne A., Cagnol S., Carrier J. C., Rivard N., Dual-specificity phosphatase 6 deletion protects the colonic epithelium against inflammation and promotes both proliferation and tumorigenesis. J. Cell. Physiol. 234, 6731–6745 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu Q.-N., Liao Y.-F., Lu Y.-X., Wang Y., Lu J.-H., Zeng Z.-L., Huang Q.-T., Sheng H., Yun J.-P., Xie D., Ju H.-Q., Xu R.-H., Pharmacological inhibition of DUSP6 suppresses gastric cancer growth and metastasis and overcomes cisplatin resistance. Cancer Lett. 412, 243–255 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Huang X., Liao W., Huang Y., Jiang M., Chen J., Wang M., Lin H., Guan S., Liu J., Neuroprotective effect of dual specificity phosphatase 6 against glutamate-induced cytotoxicity in mouse hippocampal neurons. Biomed. Pharmacother. 91, 385–392 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Kumar R., Deep G., Wempe M. F., Surek J., Kumar A., Agarwal R., Agarwal C., Procyanidin B2 3,3″-di-O-gallate induces oxidative stress-mediated cell death in prostate cancer cells via inhibiting MAP kinase phosphatase activity and activating ERK1/2 and AMPK. Mol. Carcinog. 57, 57–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacKeigan J. P., Murphy L. O., Blenis J., Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat. Cell Biol. 7, 591–600 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Xie X.-L., Nie X., Wu J., Zhang F., Zhao L.-L., Lin Y.-L., Yin Y.-J., Liu H., Shu Y.-N., Miao S.-B., Li H., Chen P., Han M., Smooth muscle 22α facilitates angiotensin II-induced signaling and vascular contraction. J. Mol. Med. (Berl) 93, 547–558 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Zhang H., Wang Y., Zhu A., Huang D., Deng S., Cheng J., Zhu M. X., Li Y., SUMO-specific protease 1 protects neurons from apoptotic death during transient brain ischemia/reperfusion. Cell Death Dis. 7, e2484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ashwal S., Tone B., Tian H. R., Cole D. J., Pearce W. J., Core and penumbral nitric oxide synthase activity during cerebral ischemia and reperfusion. Stroke 29, 1037–1046 (1998). [PubMed] [Google Scholar]
- 56.Wang Y., Wang Y., Zhang H., Gao Y., Huang C., Zhou A., Zhou Y., Li Y., Sequential posttranslational modifcations regulate PKC degradation. Mol. Biol. Cell 27, 410–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Robbins D. J., Zhen E., Cheng M., Xu S., Ebert D., Cobb M. H., MAP kinases ERK1 and ERK2: Pleiotropic enzymes in a ubiquitous signaling network. Adv. Cancer Res. 63, 93–116 (1994). [DOI] [PubMed] [Google Scholar]
- 58.Wiland A. M., Denu J. M., Mourey R. J., Dixon J. E., Purification and kinetic characterization of the mitogen-activated protein kinase phosphatase rVH6. J. Biol. Chem. 271, 33486–33492 (1996). [DOI] [PubMed] [Google Scholar]
- 59.Guo C., Hildick K. J., Luo J., Dearden L., Wilkinson K. A., Henley J. M., SENP3- mediated deSUMOylation of dynamin-related protein 1 promotes cell death following ischaemia. EMBO J. 32, 1514–1528 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Owen R. P., White M. J., Severson D. T., Braden B., Bailey A., Goldin R., Wang L. M., Ruiz-Puig C., Maynard N. D., Green A., Piazza P., Buck D., Middleton M. R., Ponting C. P., Schuster-Böckler B., Lu X., Single cell RNA-Seq reveals profound transcriptional similarity between Barrett’s oesophagus and oesophageal submucosal glands. Nat. Commun. 9, 4261 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/13/eaaz0361/DC1
Fig. S1. DUSP6 plays a protective role against oxidative damage depending on its phosphatase activity but not ERK1/2.
Fig. S2. DUSP6 is modified by SUMO2/3.
Fig. S3. SUMOylation regulates stability of DUSP6.
Fig. S4. SUMOylation of DUSP6 is important for its catalytic activity.
Fig. S5. DUSP6 SUMOylation affects its interaction properties.
Fig. S6. The protective effect of DUSP6 SUMOylation is independent of its binding to ERK2.
Fig. S7. Importance of DUSP6 in cytoprotection against oxidative damage.
Fig. S8. Knockdown of SENP1 protected endogenous DUSP6 from H2O2-induced degradation, and DUSP6 can physically associated with Drp1.
Fig. S9. DUSP6 protects neurons from oxidative damage induced by OGD/reox.
Fig. S10. Overexpression of DUSP6, but not the SUMOylation-deficient DUSP6K234R mutant, protected mouse brain from damages caused by I/R.
Fig. S11. Quantification of the Western blot results (Figs. 1, 2, 6, and 7).
Fig. S12. Schematic model showing the role of DUSP6 SUMOylation in Drp1-mediated cell death under physiological and oxidative stress conditions.