

Abstract
Voltage-gated sodium channels (VGSCs) are responsible for the initiation and propagation of action potentials in neurons. The human genome includes ten human VGSC α-subunit genes, SCN(X)A, encoding Nav1.1–1.9 plus Nax. To understand the unique role that each VGSC plays in normal and pathophysiological function in neural networks, compounds with high affinity and selectivity for specific VGSC subtypes are required. Toward that goal, a structural analog of the VGSC pore blocker tetrodotoxin, 4,9-anhydrotetrodotoxin (4,9-ah-TTX), has been reported to be more selective in blocking Na+ current mediated by Nav1.6 than other TTX-sensitive VGSCs, including Nav1.2, Nav1.3, Nav1.4, and Nav1.7. While SCN1A, encoding Nav1.1, has been implicated in several neurological diseases, the effects of 4,9-ah-TTX on Nav1.1-mediated Na+ current have not been tested. Here, we compared the binding of 4,9-ah-TTX for human and mouse brain preparations, and the effects of 4,9-ah-TTX on human Nav1.1-, Nav1.3- and Nav1.6-mediated Na+ currents using the whole-cell patch clamp technique in heterologous cells. We show that, while 4,9-ah-TTX administration results in significant blockade of Nav1.6-mediated Na+ current in the nanomolar range, it also has significant effects on Nav1.1-mediated Na+ current. Thus, 4,9-ah-TTX is not a useful tool in identifying Nav1.6-specific effects in human brain networks.
Keywords: Voltage-gated sodium channels; 4,9-anhydrotetrodotoxin; Nav1.1
GRAPHICAL ABSTRACT
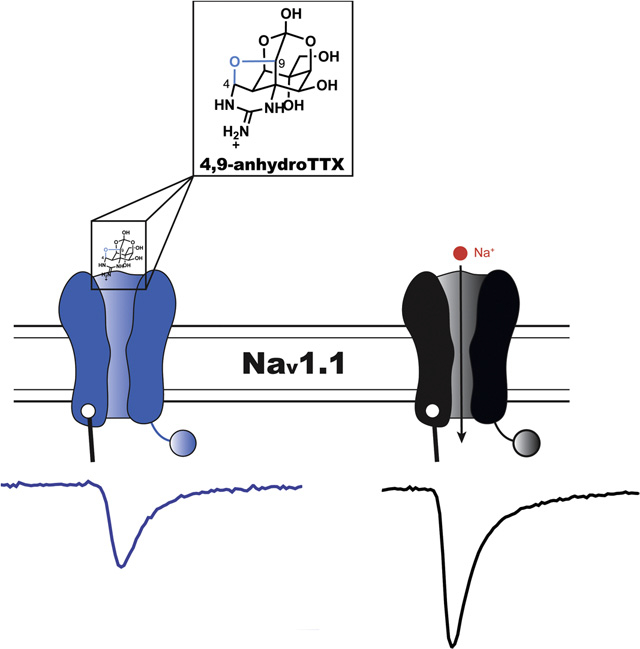
1. Introduction
Voltage-gated sodium channels (VGSCs) are transmembrane protein complexes that drive the initiation and propagation of action potentials in excitable cells. VGSCs consist of a single, pore-forming α-subunit (220–260 kDa) and two smaller β-subunits (30–40 kDa) that have many roles, including the modulation of channel expression, location, and function. To date, ten major human VGSC α-subunit subtypes (Nav1.1–1.9 plus Nax), encoded by the SCN(X)A genes have been identified [1]. Toxins have long served as pharmacological probes, playing an instrumental role in elucidating the structure and function of VGSCs [2]. Among the most widely used is tetrodotoxin (TTX), a potent guanidinium neurotoxin found in many species of aquatic and terrestrial organisms [3]. TTX binds with high affinity to Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.6, and Nav1.7. These channels are deemed “TTX sensitive” due to their susceptibility to half-maximal conduction blockade at nanomolar concentrations of TTX. To further understand the unique role each VGSC plays in normal and pathophysiological function in neural networks, compounds with high affinity for a specific VGSC subtype are required [4]. A structural analog of TTX, 4,9-an-hydro-TTX (4,9-ah-TTX) has been reported to be more selective in blocking Na+ current (INa) mediated by Nav1.6 (IC50 = 7.8 nM) than the other TTX sensitive VGSCs, including Nav1.2 (IC50 = 1260 nM), Nav1.3 (IC50 = 341 nM), Nav1.4 (IC50 = 988 nM), and Nav1.7 (IC50 = 1270 nM) [5]. While SCN1A, encoding Nav1.1, has been implicated in neurological diseases [6], the effect of 4,9-ah-TTX on Nav1.1-mediated INa has not been tested. Despite this, numerous studies have utilized 4,9-ah-TTX as a Nav1.6 selective inhibitor following the initial report of its selective properties [7–11]. In this study, we determine the affinity of 4,9-ah-TTX for human and mouse brain VGSCs using radioligand competition binding. We investigate the effects of 4,9-ah-TTX on human Nav1.1-, Nav1.3- and Nav1.6-mediated INa using the whole-cell patch clamp technique in HEK cells. Finally, we use mass spectrometry to evaluate the pH and time-dependence of 4,9-ah-TTX conversion to 4-epiTTX and TTX, as previous reports have indicated these factors can influence the pharmacological analysis of 4,9-ah-TTX [12,13]. Our work shows that, while 4,9-ah-TTX administration results in significant blockade of Nav1.6-mediated INa at nanomolar concentrations, it also has significant effects on Nav1.1-mediated INa at similar concentrations. Thus, 4,9-ah-TTX is not useful to resolve Nav1.6-specific effects in brain networks.
2. MATERIALS & METHODS
2.1. Cell lines
Human Embryonic Kidney (HEK) 293 T cells stably expressing human VGSC α subunits Nav1.1 (GenBank accession number NP_001159435.), Nav1.3 (GenBank accession number NP_008853.3) or Nav1.6 (GenBank accession number NP_055006.1) cDNAs were cultured in Dulbecco’s Modified Eagle Medium containing (4.5 g/L D-glucose, L-glutamine, 110 mg/L sodium pyruvate, 400 μg/mL G418, 100 U/mL penicillin/streptomycin). All cells were maintained in an incubator at 37 °C with 5% CO2 until the time of recording.
2.2. Mouse models
Mouse studies were performed in compliance with protocols approved by the University of Michigan IACUC and were in accordance with the policies of the NIH Guide for the Care and Use of Laboratory Animals. Scn1atm1Kea mice were a generous gift from the laboratory of Dr. Jennifer Kearney at Northwestern University [14]. The Scn1atm1Kea colony was maintained by breeding heterozygous (129S6.Scn1a+/−) animals to 129S6/SvEvTac mice (Taconic #129SVE). For all experiments, 129S6.Scn1a+/− mice were crossed to generate −/−, +/−, and +/+ offspring. Genotyping of F1 pups was performed by PCR amplification of mouse genomic DNA. Primers used in genotyping were: 5′-AGTCTGTACCAGGCAGAACTTG-3′; 5′-CTGTTTGCTCCATCTTGTC ATC-3′ and 5′-GCTTTTGAAGCGTGCAGAATGC-3′. The PCR products are 1 kb for the Scn1a WT allele and 650 bp for the transgenic allele. All mice were maintained on a 12:12 h light:dark cycle and had ad libitum access to food and water throughout the experiments. Male and female mice were used in all experiments.
2.3. Human brain samples
De-identified, frozen, human cortical white matter samples from neurologically normal patients were obtained from the Human Brain and Spinal Fluid Resource Center in Los Angeles, CA under IRB approval (HUM00012142).
2.4. [3H]-Saxitoxin competition binding
To measure the competitive binding of 4,9-ah-TTX to VGSCs, whole brain membranes were prepared from postnatal day (P) 10–17 mice or from frozen human brain samples as described previously [15]. Equilibrium [3H]-saxitoxin (STX) binding in the presence of 4,9-ah-TTX was measured at 4 °C following an incubation period of one hour using a vacuum filtration assay with a saturating concentration (5 nM) of C-11 labelled [3H]-STX (20 Ci/mmol, American Radiolabeled Chemicals Inc.). In a subset of samples, 10 μM unlabeled TTX (Alomone Labs) was added to assess nonspecific binding. To quantify [3H]-STX binding, counts per minute (CPM) values obtained from liquid scintillation counting (Packard Tri-carb 1900TR) were corrected for specific binding by subtraction of nonspecific (10 μM TTX) values and then converted to decays per minute (DPM) before normalization to total protein concentration using the bicinchoninic acid (BCA) protein assay (Thermo-Fisher). A stock of 4,9-ah-TTX was diluted in 1XP binding buffer (50 nM HEPES pH 7.4, 130 mM choline chloride, 5.4 mM KCl, 80 μM MgSO4, 0.1 % dextrose) to give a final concentration of 10000, 1000, 100, 10, 1, 0.1 or 0.01 nM. A 7-point concentration-response curve was generated for 4,9-ah-TTX in competition with 5 nM [3H]-STX. Four independent experiments were conducted, and in each experiment, each sample was tested in duplicate. The average DPM reading of duplicate values was used for analysis. Competitive binding data were analyzed using Microsoft Excel and Graph Pad Prism v8.2 (San Diego, CA). The specific binding curve in [3H]-STX (fmol)/protein (mg) was initially generated using the following one-site IC50 equation:
where Top and Bottom are plateaus in the units of the Y-axis. IC50 represents the concentration in nanomolar of unlabeled competitor that results in binding half-way between Top and Bottom values. Data were then normalized for each concentration point to the Top value obtained in the assay for each unlabeled toxin to obtain fraction of total binding. Each Y value thus represents the fraction of maximal [3H]-STX displacement at the corresponding concentration (X value). To obtain Ki values from the IC50, the following equation was used:
where [3H-STX] = 5 nM in each assay at equilibrium. Kd of [3H]-STX = 2.5 nM. Ki = equilibrium dissociation constant of the unlabeled competitor in nM.
2.5. Whole-cell patchclamp electrophysiology
INa was measured at room temperature using the whole-cell patch clamp technique using previously described electrophysiological methods [17]. Cells were plated on 35 × 10 mm treated polystyrene culture dishes (Thermo-Fisher) and used for electrophysiological recordings within 1–3 days after plating. Cells were identified using an Eclipse TE300 upright microscope (Nikon). Micropipettes were obtained from 1.5 mm outer diameter capillary glass tubing (Warner Instruments) using a P-97 horizontal puller (Sutter Instrument Co.). Micropipettes were then polished using a MF-830 micro forge (Narishige) to obtain a resistance between 2.0–6.0 MΩ. The intracellular solution contained the following (in mM): 115 CsCl, 5 EGTA, 0.4 GTP, 2 MgATP, 0.5 CaCl2, 10 HEPES, 5 Na+ phosphocreatine, 20 tetraethylammonium chloride, pH 7.2 with CsOH. Extracellular solution contained the following (in mM): 120 NaCl, 1 BaCl2, 2 MgCl2, 0.2 CdCl2, 1 CaCl2, 20 sucrose, 10 glucose, 10 HEPES, 20 tetraethylammonium chloride, pH 7.35 with NaOH. Signals were amplified using a Multiclamp 700B amplifier (Molecular Devices). Data were acquired with a Digidata 1440A interface (Molecular Devices) and analyzed using pClamp10 offline. Pipette and whole-cell capacitance were compensated at 70 %. Signals were low pass-filtered at 5 kHz, and data were sampled at 40 kHz. For the data generated in Figs. 2–4 and Tables 1 and 2 4,9-ah-TTX (Cayman Chemical) was evenly dissolved into the static extracellular bath solution at a concentration of 100 nM prior to recording. Cells were bathed in 4,9-ah-TTX for at least 10 min prior to recording. Peak INa was normalized to cell capacitance to obtain current density, used to plot I–V curves and calculate conductance with the following equation:
where g is conductance, I is current, V is the test potential, and Vrev is the measured reversal potential. Peak currents were normalized to the maximum peak INa amplitude. The V1/2 of activation represents the voltage of the membrane at which half-maximal conductance occurred. To determine the INa amplitude and the voltage dependence of activation, Na+ currents were evoked by 250 ms depolarizing test pulses (from −100 to +30 mV at 5 and 10 mV intervals) from a holding potential of −120 mV. Voltage-dependence of inactivation was determined by applying a 50 ms test pulse to 0 mV after 250 ms prepulses to the same voltages as described for the voltage dependence of activation. Normalized activation and inactivation curves were fit with a Boltzmann equation:
where V1⁄2 is the membrane potential in the midpoint of the curve, and k is the slope factor. Persistent sodium current was analyzed as the average current in a 2 ms time period exactly 50 ms after the peak current was observed at a depolarizing step from −120 to 0 mV. Persistent current values were normalized to cell capacitance to obtain current density. The mean of all values obtained in the absence of 4,9-ah-TTX (control) were compared to the mean of all values obtained in the presence of 100 nM 4,9-ah-TTX. For data generated in Figs. 5 and 6 and Table 3, cells were plated on coverslips (Thermo-Fisher) and extracellular solution containing 4,9-ah-TTX was perfused into the RC-26 recording chamber (Warner Instruments) by a gravity-driven perfusion system with a flow rate of 2–3 mL/min. Peak INa was evoked via a 250 ms depolarizing pulse to 0 mV from a holding potential of −120 mV. Baseline recordings were established, followed by a 3 min perfusion of 4,9-anhydro-TTX at the indicated concentrations. Extracellular recording solution was then perfused on for ≥ 3 min and a washout recording obtained. The effect of 4,9-ah-TTX was evaluated as a decrease in peak INa compared to baseline control of that cell. Series resistance was monitored throughout perfusion recordings. If series resistance changed ≥ 20 % at any time, the recording was not included in the analysis. Signals were low pass-filtered at 10 kHz, and data were sampled at 20 kHz. Capacitive transients and leak conductance were subtracted using a P/4 protocol.
Fig. 2.
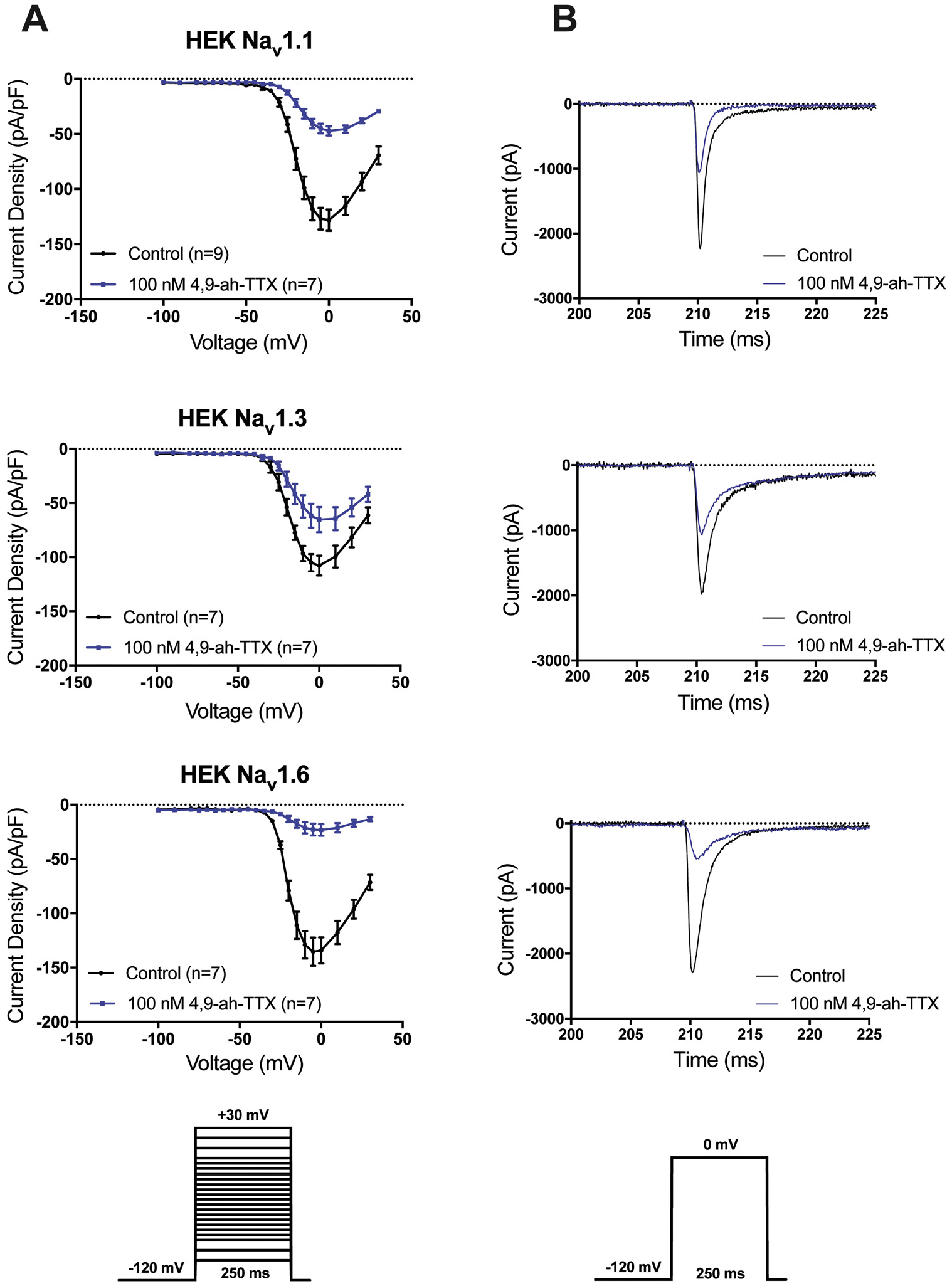
Nav1.1, Nav1.3 and Nav1.6 I-V curves and INa traces in the presence and absence of 100 nM 4,9-ah-TTX. (A) Current-voltage (I-V) relationship obtained from recordings in the presence and absence of 100 nM 4,9-ah-TTX via whole-cell patch clamp of HEK cells stably expressing human Nav1.1, Nav1.3 or Nav1.6. INa was evoked by stepping to different 250 ms test pulses in 5–10 mV intervals. Peak INa was normalized to cell capacitance to obtain current density. Values represent the mean ± S.E. (B) Representative peak INa traces recorded in the presence of 100 nM 4,9-ah-TTX and absence (control) via whole-cell patch clamp from HEK cells stably expressing Nav1.1, Nav1.3 or Nav1.6. Cells were voltage-clamped at −120 mV and peak INa was elicited via stepping to a test pulse of 0 mV for 250 ms.
Fig. 4.

Normalized conductance to voltage and voltage dependence of availability relationships. Curves represent the voltage-dependence of steady-state activation (A) and voltage dependence of availability (B) in the presence (blue) and absence (black) of 100 nM 4,9-ah-TTX via whole-cell patch clamp from HEK cells stably expressing human Nav1.1, Nav1.3 or Nav1.6. (B) Data points represent the mean ± S.E. Data obtained from recordings depicted in Fig. 2.
Table 1.
Average peak and persistent INa density and recorded in the presence of 100 nM 4,9-ah-TTX and absence (control) via whole-cell patch clamp from HEK cells stably expressing human (h) Nav1.1, Nav1.3 or Nav1.6. Values represent the mean ± S.E.
| Peak INa Density (pA/pF) | Persistent INa Density (pA/pF) | ||||
|---|---|---|---|---|---|
| Control | 4,9-anhydro-TTX (100 nM) | Control | 4,9-anhydro-TTX (100 nM) | ||
| hNavl.l | −131.6 ± 9.47 | −47.24 ± 4.04 | −5.25 ± 0.39 | −2.58 ± 0.94 | |
| hNav1.3 | −108.7 ± 8.54 | −66.78 ± 11.61 | −2.77 ± 0.82 | −3.71 ± 0.26 | |
| hNav1.6 | −136.9 ± 12.72 | −23.63 ± 4.45 | −1.52 ± 0.13 | −1.84 ± 0.54 | |
Table 2.
Voltage-dependence of activation and inactivation parameters recorded in the presence of 100 nM 4,9-ah-TTX and absence (control) via whole-cell patch clamp from HEK cells stably expressing human (h) Nav1.1, Nav1.3 or Nav1.6. Values represent the mean ± S.E.
| V1/2 of Activation (mV) | k (mV−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| control | N | 4,9-anhydro-TTX (100nM) | N | control | N | 4,9-anhydro-TTX (100nM) | N | ||
| hNav1.1 | − 17.36 ± 1.05 | 9 | −15.31 ± 0.83 | 7 | 5.501 ± 0.27 | 9 | 5.705 ± 0.14 | 7 | |
| hNav1.3 | − 16.88 ± 1.58 | 7 | −14.47 ± 1.25 | 7 | 5.261 ± 0.24 | 7 | 5.332 ± 0.28 | 7 | |
| hNav1.6 | − 17.97 ± 0.80 | 7 | −17.28 ± 0.99 | 7 | 4.857 ± 0.28 | 7 | 6.265 ± 0.41* | 7 | |
| V1/2 of Activation (mV) | k (mV−1) | ||||||||
| control | N | 4,9-anhydro-TTX (100nM) | N | control | N | 4,9-anhydro-TTX (100nM) | N | ||
| hNav1.1 | − 50.41 ± 2.37 | 9 | −45.14 ± 1.55 | 7 | − 5.04 ± 0.22 | 9 | − 4.83 ± 0.55 | 7 | |
| hNav1.3 | − 53.41 ± 2.22 | 7 | −55.97 ± 1.95 | 6 | − 5.85 ± 0.59 | 7 | − 9.16 ± 1.51 | 6 | |
| hNav1.6 | − 50.73 ± 1.54 | 7 | −55.83 ± 2.79 | 5 | − 4.63 ± 0.41 | 7 | − 8.61 ± 2.17 | 5 | |
p = 0.0158.
Fig. 5.

Percent reduction in Nav1.1 and Nav1.6 peak INa by 4,9-ah-TTX. Percent reduction from control in peak INa mediated by Nav1.1 and Nav1.6 following perfusion of 10 nM 4,9-ah-TTX (light blue), 100 nM 4,9-ah-TTX (cyan), and 500 nM (dark blue). Data points represent the mean ± S.E.
Fig. 6.

Nav1.1 and Nav1.6 peak INa traces before, during and after perfusion of 10, 100 and 500 nM 4,9-ah-TTX. Representative INa traces for the control (black), perfusion of 10 nM, 100 nM, and 500 nM 4,9-ah-TTX (blue) and after washout (red) via whole-cell patch clamp from HEK cells stably expressing human Nav1.1 (A-C) or human Nav1.6 (D-F). Cell were voltage-clamped at −120 mV and peak INa was elicited via stepping to a test pulse of 0 mV for 250 ms.
Table 3.
Percent reduction in Nav1.1 and Nav1.6 peak INa from control following the perfusion of 10, 100 or 500 nM 4,9-ah-TTX. Values represent the mean ± S.E.
| Reduction in Peak INa Following Perfusion of 4,9-ah-TTX | ||||
|---|---|---|---|---|
| [4,9-ah-TTX] | (% of Control) | |||
| Nav 1.1 | N | Nav 1.6 | N | |
| 10nM | 11 % (± 4) | 4 | 18 % ( ±2) | 4 |
| 100nM | 46 % ( ± 5) | 5 | 39 % ( ±2) | 4 |
| 500 nM | 64 % ( ± 4) | 5 | 76 % ( ±2) | 3 |
2.6. Mass spectrometry
A 500 μM stock solution of 4,9-ah-TTX (Cayman Chemical) was prepared in 1XP binding buffer. Duplicate 10 μL samples containing 100 μM 4,9-ah-TTX were prepared in plastic 1.7 mL centrifuge tubes according to the conditions outlined in Fig. 9 varying buffer solution (1XP or external recording solution), pH (range 6.4–8.4), incubation time, and storage temperature. Samples were diluted with 30 μL acetonitrile, vortexed to mix, and centrifuged at 12,000 x g for 20 min. 10 μL each sample was further diluted with 80 μL acetonitrile and 10 μL 0.1 % formic acid in MilliQ water with 2.5 μg/mL [15N]-arginine as an internal standard (Cambridge Isotopes). Samples 2 and 6, indicated as being incubated for 60 min, were incubated at room temperature for 1 h prior to the first acetonitrile dilution. For assessment of time dependence, triplicate samples of 10 μL volume were prepared with 1XP buffer at pH 7.4 and incubated at ambient temperature (22 °C) for 0, 1, and 5 weeks. Samples were quenched by the addition of 30 μL acetonitrile and stored at −20 °C until prep as described previously and analysis of all samples after the 5-week timepoint.
Fig. 9.
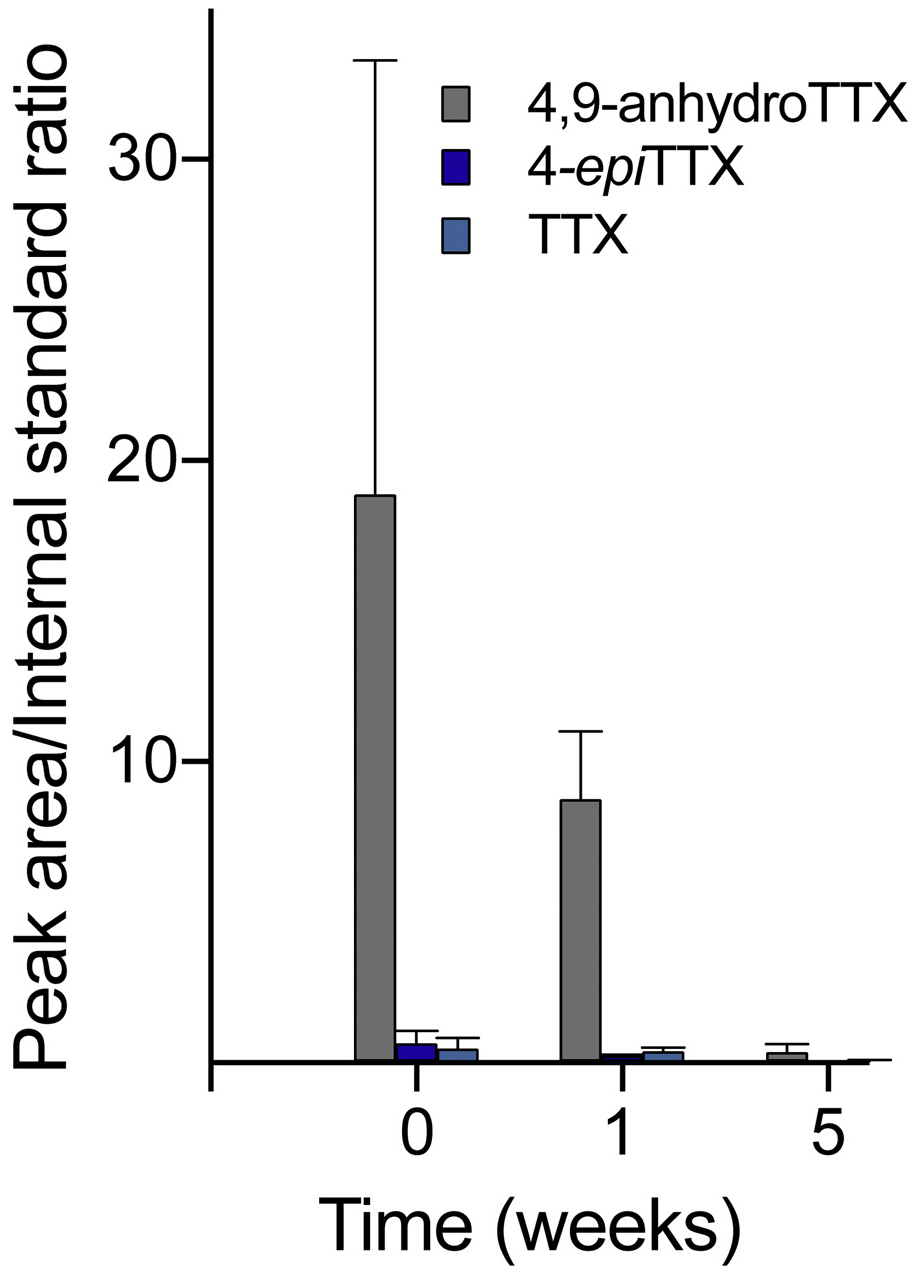
4,9-ah-TTX, 4-epiTTX and TTX in solution over time. Plot of mass spectrometry data assessing the relative abundances of 4,9-ah-TTX, 4-epi-TTX, and TTX over time comparing ratios of the analyte peak area to the peak area of the internal standard.
Samples were injected in 0.5 μL volumes on a hydrophobic liquid interaction chromatography (HILIC) BEH Amide 1.7 μM, 2.1 × 100 mm UPLC column (Waters) and analyzed using an Agilent G6545A quadrupole-time of flight (Q-TOF) mass spectrometer equipped with a dual AJS ESI source and an Agilent 1290 Infinity series diode array detector, autosampler, and binary pump. Solvent A was water with 0.1 % formic acid and solvent B was 95 % acetonitrile, 5% water, and 0.1 % formic acid. Separations were performed using a 20 % solvent A isocratic method for 5 min followed by a 1 min gradient from 20 % to 40 % A and an 8 min re-equilibration at 20 % A. Data was analyzed using MassHunter software (Agilent). The peak areas of 4,9-ah-TTX, [15N]-arginine, epi-TTX, and TTX were obtained by integration.
2.7. Statistical analysis
All data analysis of the recorded INa was performed using the software packages Clampfit v10.4 (Molecular Devices), Microsoft Excel, or Graph Pad Prism v8.2 (San Diego, CA). The statistical significance of differences between mean values was evaluated using Student’s unpaired t-test, or one-way ANOVA with p < 0.05 considered significant. Results are presented as mean ± S.E unless indicated otherwise.
3. Results
3.1. 4,9-ah-TTX binding affinity for human and mouse VGSCs
The binding curve of unlabeled 4,9-ah-TTX in competition with 5 nM [3H]-STX for the VGSCs present in Scn1a+/+ mouse whole brain membranes is shown in Fig. 1 A. 4,9-ah-TTX showed normal steepness (Hill slope of −1.04) and was determined to have an inhibitory dissociation constant (Ki) of 63.02 nM with 95 % CI (47.42–84.59 nM). At 10 μM, 4,9-ah-TTX displaced > 95 % of the [3H]-STX present. Fig. 1B depicts the binding curve of 4,9-ah-TTX to Scn1a−/− mouse whole brain membranes. The Ki was determined to be 87.67 nM 95 % CI (68.8–113.5), IC50 of 248.6 nM with 95 % CI (186.6–333.4), and a Hill slope of −1.08. Fig. 1C shows the results of 4,9-ah-TTX binding to human cortical white matter membrane preparations. The human brain binding data resulted in a Ki of 62.27 nM with 95 % CI (29.2–136.3), IC50 of 172.4 nM 95 % CI (85.8–463.4) and a Hill slope of −1.21. No significant differences in the Ki, IC50 or Hill slope of 4,9-ah-TTX were detected between the Scn1a+/+, Scn1a−/− mouse, or human brain membranes (one-way ANOVA p = 0.05).
Fig. 1.
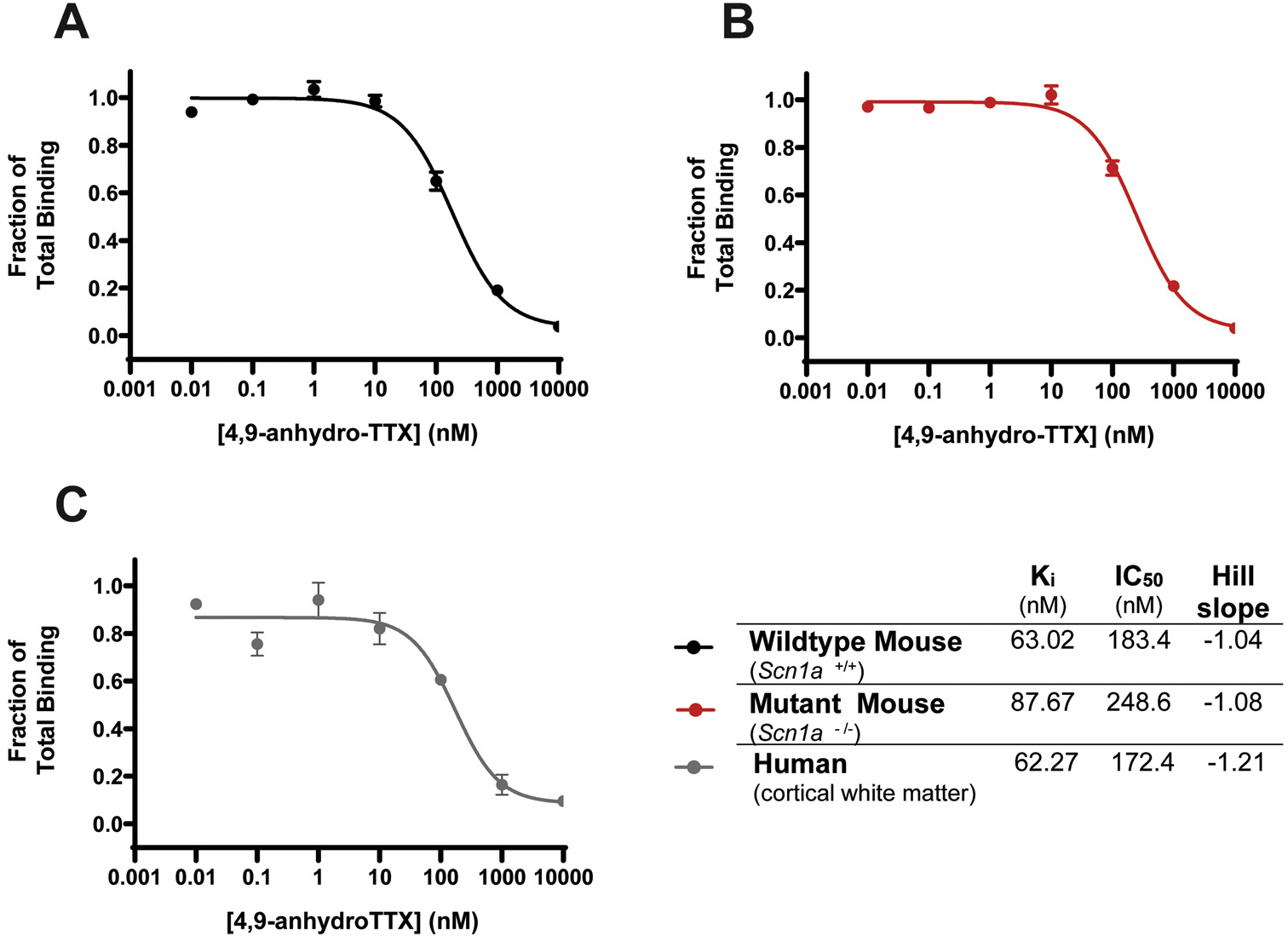
[3H]-Saxitoxin competition binding. Concentration response curves showing binding of 4,9-ah-TTX to human and mouse brain membrane samples in competition with 5 nM [3H]-saxitoxin. Unlabeled 4,9-ah-TTX was tested at 10 μM, 1000 nM, 100 nM, 10 nM, 1 nM, 0.1 nM and 0.01 nM. (A) Binding of 4,9-ah-TTX to Scn1a+/+ mouse whole brain membranes (N = 4). (B) Binding of 4,9-ah-TTX to Scn1a−/− mouse whole brain membranes (N = 4). (C) Binding of 4,9-ah-TTX to human cortical white matter membranes (N = 2). Each data point represents the mean fraction of total specific binding ± S.E.
3.2. Effects of 100 nM 4,9-ah-TTX on INa density in human HEK-Nav1.1, -Nav1.3 or -Nav1.6 cells
Fig. 2. shows the current-voltage (I–V) relationships for HEK-Nav1.1, -Nav1.3 or -Nav1.6 cells in the presence and absence of 100 nM 4,9-ah-TTX. Differences in mean INa density evoked at 0 mV following the bathing of cells in 100 nM 4,9-ah-TTX compared to control for each cell line are shown in Table 1. Mean INa density in the absence of 4,9-ah-TTX (control) for HEK-Nav1.1 was −131.6 ± 9.47, HEK-Nav1.3–108.7 ± 8.54 and HEK-Nav1.6–136.9 ± 12.72. Exposure of cells to 100 nM extracellular 4,9-ah-TTX reduced the mean peak INa density in HEK-Nav1.1 cells by 64 ± 3% (84.31 pA/pF ± 11.39 S.E.), in HEK-Nav1.3 cells by 39 ± 11 % (41.87 pA/pF ± 14.41 S.E.), and in HEK-Nav1.6 cells by 83 ± 3% (113.3 ± 13.79 S.E.). The conductance-voltage (G–V) relationships for HEK-Nav1.1, HEK-Nav1.3 or HEK Nav1.6 cells in the presence and absence of 4,9-ah-TTX are shown in Fig. 4. The V1/2 of activation was not significantly different in any of the cell lines tested in the presence of 100 nM extracellular 4,9-ah-TTX compared to control (Table 2). The slope factor (k) was not significantly different when comparing control to 100 nM 4,9-ah-TTX conditions for HEK-Nav1.1 or HEK-Nav1.3. In contrast, the slope factor for HEK Nav1.6 was significantly increased when comparing control (k = 4.857 ± 0.280) to the 100 nM 4,9-ah-TTX condition (k = 6.265 ± 0.415) (p = 0.015). The voltage dependence of availability relationship for HEK-Nav1.1, HEK-Nav1.3 or HEK-Nav1.6 cells in the presence and absence of 100 nM 4,9-ah-TTX is also shown in Fig. 4. The V1/2 of inactivation and slope factor were not significantly different between the control and 4,9-ah-TTX conditions for HEK-Nav1.1, HEK-Nav1.3 or HEK-Nav1.6 (Table 2). An analysis of persistent current at a test pulse to 0 mV in the presence and absence of 100 nM 4,9-ah-TTX revealed a significant reduction (49.2 %) in HEK-Nav1.1 persistent INa density (Fig. 3). There was no significant difference in the persistent INa density observed in the control versus 100 nM 4,9-ah-TTX condition for HEK-Nav1.3 or HEK-Nav1.6.
Fig. 3.
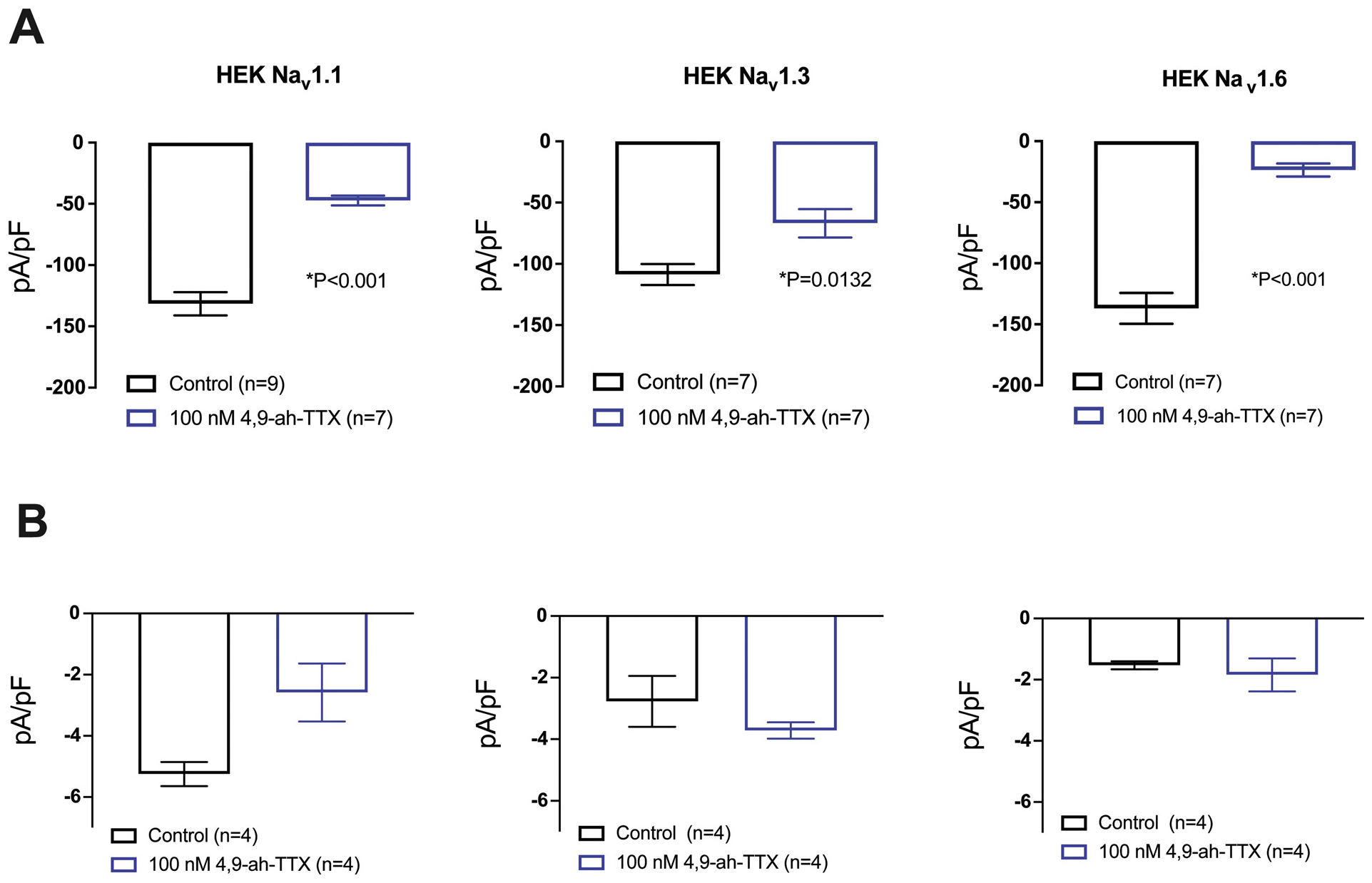
Average peak and persistent INa density recorded in the presence and absence of 100 nM 4,9-ah-TTX in HEK cells stably expressing human Nav1.1, Nav1.3 or Nav1.6. (A) Cells were voltage-clamped at −120 mV and peak INa was elicited via stepping to a test pulse of 0 mV for 250 ms. (B) Persistent INa density present in a 2 ms time period 50 ms after peak INa was observed at a test pulse to 0 mV. Bar graphs represent mean ± S.E. Significant differences between control and toxin treated conditions were tested using a Student’s unpaired t-test. Data obtained from recordings depicted in Fig. 2.
3.3. Reduction in Nav1.1 and Nav1.6 mediated peak INa following perfusion of 4,9-ah-TTX
Perfusion of 4,9-ah-TTX onto HEK-Nav1.1 and HEK-Nav1.6 cells resulted in a concentration dependent reduction in peak INa (Figs. 5 and 6). Nav1.1 peak INa was reduced by (11 % ± 4) following the perfusion of 10 nM 4,9-ah-TTX. At 100 nM, the mean reduction in Nav1.1 peak INa was (46 % ± 5). The perfusion of 500 nM 4,9-ah-TTX reduced Nav1.1 peak INa by (64 % ± 4) (Table 3). Washout of 4,9-ah-TTX restored peak INa on average to 94.5 % (range 84–106 %) of baseline control in all recordings. Nav1.6 peak INa was reduced by (18 % ± 2) following the perfusion of 10 nM 4,9-ah-TTX. At 100 nM, the mean reduction in Nav1.6 peak INa was (39 % ± 2). The perfusion of 500 nM 4,9-ah-TTX reduced Nav1.6 peak INa by (76 % ± 2) (Table 3).
3.4. Time and pH dependent effects of 4,9-ah-TTX conversion to 4-epiTTX and TTX
Relative abundances of 4,9-ah-TTX, 4-epiTTX, and TTX were monitored at different pH conditions and incubation times at ambient temperature. The results shown in Fig. 8 indicate that differences in pH over the range 6.4–8.4 do not significantly impact the distribution of 4,9-ah-TTX, 4-epiTTX, and TTX, suggesting 4,9-ah-TTX was not significantly converted to 4-epiTTX or TTX under our analysis conditions. Assessing the relative abundances of these molecules over time similarly did not result in increased amounts of 4-epiTTX or TTX relative to 4,9-ah-TTX; however, a significant decrease in all three molecules was observed over time, with a minute amount remaining after 5 weeks of incubation at ambient temperature, as shown in Fig. 9.
Fig. 8.

Impact of pH on 4,9-ah-TTX, 4-epiTTX and TTX. (Top) Plot of mass spectrometry results comparing peak area/internal standard ratio based on peak integration corresponding to 4,9-ah-TTX, 4-epiTTX, and TTX. (Inset) Zoomed-in graph of represented data. (Below) Reaction conditions used in analysis. ER: external recording solution used in electrophysiology experiments. 1XP: binding buffer used in competition binding experiments.
4. Discussion
TTX binds with high specificity to the VGSC pore in a 1:1 stoichiometry to occlude the conduction of Na+ ions by forming a web of electrostatic interactions with the channel. A large body of evidence from numerous studies strongly supports that TTX interacts with a narrow region of residues within the channel involved in permeation and the selectivity of Na+ ions [19]. Major coordinating residues are located on the membrane re-entrant P-loops (pore loops) of segments S5-S6 on all 4 domains. This region of the channel heavily influences channel conductance and ion selectivity, forming an inner and outer ring of charged amino acid residues (six carboxylates and one lysine) known as the selectivity filter ([20]; Favre et al. [21]). The asymmetric charge distribution of each ring coordinates solvated Na+ ions as they pass through the pore [16]. Mutagenesis and electrophysiological studies have revealed four key residues (D: aspartate, E: glutamate, K: lysine, and A: alanine), each present in one of the domains, that form the inner ring of the mammalian VGSC selectivity filter. The DEKA motif found in human VGSCs is conserved across all subtypes. Mutating K and A of the DEKA motif to glutamates, generating DEEE, abolishes Na+ selectivity and permits Ca2+ to permeate the channel [22]. Three or four residues downstream of the DEKA selectivity filter in each domain lie the outer ring of the human VGSC selectivity filter composed of an EE(M/D)D motif (see Fig. 10). The outer EE(M/D)D ring is comprised of four negatively charged carboxylates shown to attract Na+ ions into the outer vestibule of the pore (Ding et al. [23]). Neutralizing mutations in the inner or outer ring of the selectivity filter, and any residues linking the two regions, can dramatically reduce TTX binding [19,24–26].
Fig. 10.

VGSC schematic with TTX binding site and selectivity filter residue alignment. (Top) Schematic of the mammalian voltage-gated sodium channel α and β subunit complex. Segments are labelled numerically (1–6) and domains are labelled in roman numerals (I-IV). Voltage-sensing S4 segments in each domain are labelled in light red. The pore loop regions formed by segments S5 and S6 are labelled in green. The amino acid residues representing the Na+ ion selectivity filter and canonical TTX binding site are depicted in dark red circles. (Below) Amino acid residues of the selectivity filter in human Nav1.1, Nav1.3, Nav1.6 or mouse Nav1.6. The selectivity filter is −2 through +4 residues from the DEKA locus. The inner DEKA ring residues of each domain are shown in red. The outer EE(M/D)D ring residues of each domain are shown in blue. The following accession numbers were used to generate the basic local alignment (hNav1.1: NP_001159435.1; hNav1.3: NP_008853.3; hNav1.6: NP_055006.1; mNav1.6 NP_001070967.1).
Another key residue in TTX binding is Y401 (Nav1.4 numbering). TTX-sensitive channels have an aromatic Y/F (tyrosine or phenylala-nine) at this position that is crucial for coordinating TTX in the pore [27]. Nav1.5, Nav1.8, and Nav1.9 achieve “TTX-resistance” through replacement of the Y/F residue with a nonaromatic cystine (C) or serine (S) [28]. Intriguingly, all the TTX-resistant VGSC subtypes are located on human chromosome 3. Adaptive resistance strategies in TTX-sensitive channels have been observed in many TTX-producing predators and their prey (Soong and Venkatesh [29]). Various species of puffer-fish also possess nonaromatic substitutions for the Y/F residue present in domain I, dramatically reducing their sensitivity to TTX. Many species of newts and snakes have evolved TTX resistant Nav1.4 channels via substitutions in the pore loops of domain III and IV [30]. Amino acid substitutions of the inner and outer selectivity filter ring of domain III and IV that alter TTX sensitivity have been identified [31,32]. A threonine (T) substitution of the methionine (M) in the domain III outer selectivity filter ring on Nav1.4 is observed in all TTX-harboring newts and the Thamnophis couchii garter snake [18].
Recent high-resolution structural evidence has confirmed that TTX restricts Na+ conduction via binding to the vestibule of the VGSC selectivity filter [33,34]. Cryogenic electron microscopy studies showing TTX bound to the American cockroach sodium channel NavPaS reveal hydrogen bonds and salt bridges formed between the polar groups on the TTX molecule and acidic residues in the pore loop P2 helix segment of domains I, II and IV [33]. The DE of the inner DEKA selectivity filter also directly binds to TTX in the NavPaS channel [33]. In a subsequent study, the authors obtained high-resolution structural data of TTX bound to hNav1.7 [34]. Inner and outer selectivity filter residues identified by previous studies were implicated in the coordination of TTX within the pore. The coordination of TTX in hNav1.7 was nearly identical to that observed in NavPaS, differing only in the P2 segment of domain III [34]. Nav1.7 contains an isoleucine in place of an aspartate that is present in the other 8 human VGSC subtypes. This outer ring aspartate likely contributes to TTX binding in hNav1.1. Aside from that difference, similar TTX coordination strategies observed by Shen et al. [34] would be anticipated in hNav1.1, hNav1.3 and hNav1.6.
It is not known which amino acid residues of VGSCs form the binding site for 4,9-ah-TTX, although it is postulated that 4,9-ah-TTX interacts with the same residues as TTX [12]. Xu et al. explored the differences in binding of TTX and 4,9-ah-TTX to human Nav1.2 using computational modeling and molecular dynamics. The authors proposed that the H-bond interactions made between the C4 and C9 hydroxyl of TTX and the outer ring carboxylates of the human Nav1.2 selectivity filter are key factors in the greater inhibitory activity of TTX relative to 4,9-ah-TTX (Xu and Li [35]). It is important to distinguish the species used in an experimental sodium channel preparation when comparing results across studies. The original study reporting 4,9-ah-TTX to be selective for Nav1.6, utilized the expression of mouse cDNA in Xenopus oocytes [5]. Amino acid sequence alignments shown in Fig. 10 reveal no differences when comparing the selectivity filter regions of human Nav1.1, Nav1.3, and Nav1.6 to mouse Nav1.6. Despite the high homology seen in human and rodent VGSCs, it is possible that alternative 4,9-ah-TTX binding conformations are achieved in mouse, rat, and human VGSCs, leading to different results observed across studies. To detect possible species-selective differences in 4,9-ah-TTX for mouse and human brain VGSCs, we conducted [3H]-STX binding with cortical white matter membranes from neurologically normal humans (Fig. 1C). The lack of significant differences found in affinity and potency suggest there is no differential sensitivity of 4,9-ah-TTX for human or mouse VGSCs. Further structural studies analyzing the precise amino acid residues of each human TTX-sensitive VGSCs responsible for the coordination of 4,9-ah-TTX are required to understand potential subtype specific binding contributions.
Previous studies conducted on 4,9-ah-TTX, performed in markedly different experimental systems, have yielded variable results. Recording INa using voltage-clamp in the squid giant axon, Kao and Yasumoto reported 4,9-ah-TTX to have an IC50 of 298 nM [36]. These experiments were performed at 7 °C with an external solution pH of 7.8. The authors noted the importance of this external pH, as the C-10 hydroxyl of 4,9-ah-TTX has a reported pKa of 7.95 (Goto et al. 1965). Using [3H]-STX binding in rat brain synaptosomes, Yotsu-Yamashita et al. reported an equilibrium dissociation constant for 4,9-ah-TTX of 180 ± 11 nM [37]. Using the two-electrode voltage clamp technique in Xenopus oocytes, Rosker et al. reported that 4,9-ah-TTX selectively inhibits mouseNav1.6 with an IC50 of 7.8 ± 2.3 nM [5]. Teramoto et al. reported that resurgent-like INa present in isolated mouse vas deferens myocytes is abolished in the presence of 3 μM 4,9-ah-TTX [11]. Further investigation showed that native INa in mouse vas deferens myocytes was sensitive to 4,9-ah-TTX, with a Ki of 512 nM [10]. The authors then compared this to the effects of 4,9-ah-TTX in HEK cells transiently transfected with mouse Nav1.6 cDNA. In this system, the authors reported 4,9-ah-TTX to have a Ki of 112 nM for Nav1.6-mediated INa and a Ki of 92 nM when Nav1.6 was co-expressed with β1 subunits. Measuring INa using an automated patch clamp system in CHO and HEK cells, Rogers et al. reported an IC50 for 4,9-ah-TTX of 32.9 nM for Nav1.6- and 1.6 μM for Nav1.7-mediated INa. [9].
4,9-ah-TTX was determined to have an inhibitory dissociation constant (Ki) of 62.03 nM for Scn1a+/+ mouse brain membrane preparations in our [3H]-STX binding assay, consistent with its reduced affinity for brain VGSCs compared to TTX. In Scn1a−/− mice, global expression of Nav1.1 is deleted [14]. While not statistically significant, 4,9-ah-TTX shows a reduced affinity for Scn1a−/− mouse brain compared to Scn1a+/+ (Ki of 87.67 nM vs. 63.02 nM), suggesting Nav1.1 contributes to the binding activity of 4,9-ah-TTX. A heterogenous mixture of Nav1.1, Nav1.2, Nav1.5, Nav1.6, and Nav1.7 are known to be present in the adult mouse whole-brain membrane preparations utilized to test 4,9-ah-TTX (Wu et al. [38], [39–41], Kanellopoulos et al. [42]).
We showed that bathing cells in 100 nM 4,9-ah-TTX significantly reduced INa density in HEK-Nav1.1 (64 ± 3%), HEK-Nav1.3 (39 ± 11 %), and HEK-Nav1.6 cells (83 ± 3%) compared to control. No significant changes in the V1/2 of activation were observed when comparing control to the presence of 100 nM 4,9-ah-TTX, consistent with 4,9-ah-TTX exerting its inhibitory effects through pore-blocking action, like TTX. To confirm the observed effect was both acute and reversible, as is the case with TTX, we recorded peak INa mediated by Nav1.1 and Nav1.6 before, during perfusion, and after washout of 4,9-ah-TTX (Fig. 5). Consistent with the results we observed from the bath exposure to 4,9-ah-TTX, the perfusion of 500 nM 4,9-ah-TTX reduced Nav1.1-mediated peak INa by 64 % from baseline, 100 nM 4,9-ah-TTX induced a 46 % reduction from baseline, and 10 nM reduced Nav1.1-mediated peak INa by 11 % from baseline (Table 3). 4,9-ah-TTX reduced Nav1.6 peak INa by (76 %), (39 %), and (18 %) at 500 nM, 100 nM and 10 nM concentrations respectively (Table 3). Together, these data suggest 4,9-ah-TTX has comparable inhibitory activity for human Nav1.1 and Nav1.6 at nanomolar concentrations.
Tetrodotoxin has long been considered a voltage and state-independent blocker, inhibiting sodium current without altering the voltage-dependence of activation or inactivation, regardless of the original state of the channel [43]. Reports of TTX causing a modest hyperpolarizing shift in the voltage-dependence of inactivation do exist [44]. 4,9-ah-TTX was reported to induce a hyperpolarizing shift in the voltage-dependence of inactivation of Nav1.6 [5]. We did not detect any shifts from baseline in the voltage-dependence of inactivation for Nav1.1, Nav1.3 or Nav1.6 in the presence of 100 nM 4,9-ah-TTX (Table 2). Possible state-dependent effects of 4,9-ah-TTX on Nav1.1 were not directly examined. Neurons expressing Nav1.1 in vivo would rest at a more depolarized resting membrane potential (~ −65 mV) than the holding potential used in our experiments (−120 mV). Further studies examining the inhibitory activity of 4,9-ah-TTX on hNav1.1 at holding potentials from −55 to −70 mV would provide a more physiologically relevant interpretation of our findings.
An important consideration when testing the activity of 4,9-ah-TTX is the spontaneous equilibration that can occur between 4,9-ah-TTX, 4-epiTTX and TTX (Fig. 7: [45]), which can dramatically influence the results of pharmacological testing. Tsukamoto et al. tested 4,9-ah-TTX on human Nav1.6 mediated INa in HEK cells and reported an IC50 value of 294 ± 25 nM from a freshly prepared working solution. However, using a solution prepared from a frozen stock solution that had been placed in storage for 6 months, the authors found a much greater reduction in INa following the application of 100 nM 4,9-ah-TTX, presumably due to the conversion of 4,9-ah-TTX to TTX [13]. We performed analytical authentication of 4,9-ah-TTX prior to using it in our experiments to determine the level of contamination present and to validate the presence of 4,9-ah-TTX over a pH range from 6.4–8.4, including the pH of our electrophysiological recording solution (pH = 7.35). The relative amounts of 4,9-ah-TTX compared to 4-epiTTX and TTX were assessed in the electrophysiologicalrecording and binding buffer solutions using mass spectrometry. In contrast to previous findings, in our hands we found little to no evidence of 4,9-ah-TTX conversion to 4-epiTTX or TTX under the different conditions. We also assessed the relative abundances of these molecules over time at pH 7.4 and did not observe interconversion. We did find that incubating 4,9-ah-TTX at ambient temperature over the course of 5 weeks resulted in a significant decrease in the total amount of 4,9-ah-TTX without corresponding increases in 4-epiTTX or TTX, suggesting that 4,9-ah-TTX is not stable under these conditions for extended periods of time. However, the 4,9-ah-TTX used in our experiments had negligible levels of contamination from 4-epiTTX and TTX under multiple conditions.
Fig. 7.
Molecular structures and equilibrium of tetrodotoxin, 4,9-anhydrotetrodotoxin, and 4-epi-tetrodotoxin.
5. Conclusion
Identifying a Nav1.6 selective inhibitor is of great interest to VGSC biology. Nav1.6 is widely expressed in the mammalian peripheral and central nervous systems as well as in the heart, although at lower levels [46,47]. In the adult brain, Nav1.6 is the most abundantly expressed VGSC subtype [1]. Within neurons, Nav1.6 is highly concentrated at the distal axon initial segment of excitatory and inhibitory neurons and nodes of Ranvier in myelinated axons [48,49]. Variants in SCN8A, which encodes human Nav1.6, have been implicated in epilepsy, movement disorders, and cognitive impairment [50]. A compound with the selective properties previously reported for 4,9-ah-TTX holds great value. However, caution should be exercised when using this reagent. Our findings demonstrate that, in addition to blocking Nav1.6-mediated INa, 4,9-ah-TTX has significant inhibitory effects on Nav1.1-mediated INa in the nanomolar range and thus cannot be used as a selective Nav1.6 inhibitor in human cells or tissues where Nav1.1 is also expressed.
Acknowledgements
Supported by R37 NS076752 to L.L.I., R35 GM124880 to A.R.H.N., F31 NS111906 to A.L.L., and F31HL144047 to A.A.B..
Abbreviations:
- 4,9-ah-TTX
4,9-anhydro-tetrodotoxin
- HEK
human embryonic kidney
- IC50
half-maximal inhibitory concentration
- INa
Na+ current
- STX
Saxitoxin
- TTX
tetrodotoxin
- VGSC
voltage-gated sodium channel
References
- [1].Goldin AL, Resurgence of sodium channel research, Annu. Rev. Physiol 63 (2001) 871–894. [DOI] [PubMed] [Google Scholar]
- [2].Cestele S, Catterall WA, Molecular mechanisms of neurotoxin action on voltage-gated sodium channels, Biochimie 82 (2000) 883–892. [DOI] [PubMed] [Google Scholar]
- [3].Noguchi T, Arakawa O, Tetrodotoxin - Distribution and accumulation in aquatic organisms, and cases of human intoxication, Mar. Drugs 6 (2008) 220–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kwong K, Carr MJ, Voltage-gated sodium channels, Curr. Opin. Pharmacol 22 (2015) 131–139. [DOI] [PubMed] [Google Scholar]
- [5].Rosker C, Lohberger B, Hofer D, Steinecker B, Quasthoff S, Schreibmayer W, The TTX metabolite 4,9-anhydro-TTX is a highly specific blocker of the Na-v1.6 voltage-dependent sodium channel, Am. J. Physiol.-Cell Physiol 293 (2007) C783–C789. [DOI] [PubMed] [Google Scholar]
- [6].Nolan D, Fink J, Genetics of epilepsy, Handb. Clin. Neurol 148 (2018) 467–491. [DOI] [PubMed] [Google Scholar]
- [7].Hargus NJ, Nigam A, Bertram EH, Patel MK, Evidence for a role of Na(v)1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis, J. Neurophysiol 110 (2013) 1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Meredith FL, Rennie KJ, Regional and developmental differences in Na+ currents in vestibular primary afferent neurons, Front. Cell. Neurosci 12 (2018) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rogers M, Zidar N, Kikelj D, Kirby RW, Characterization of Endogenous Sodium Channels in the ND7–23 Neuroblastoma Cell Line: Implications for Use as a Heterologous Ion Channel Expression System Suitable for Automated Patch Clamp Screening, Assay Drug Dev. Technol 14 (2016) 109–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Takahara K, Yamamoto T, Uchida K, Zhu HL, Shibata A, Inai T, Noguchi M, Yotsu-Yamashita M, Teramoto N, Effects of 4,9-anhydrotetrodotoxin on voltage-gated Na+ channels of mouse vas deferens myocytes and recombinant Na(V)1.6 channels, Naunyn Schmiedebergs Arch. Pharmacol 391 (2018) 489–499. [DOI] [PubMed] [Google Scholar]
- [11].Teramoto N, Zhu HL, Yotsu-Yamashita M, Inai T, Cunnane TC, Resurgent-like currents in mouse vas deferens myocytes are mediated by Na(V)1.6 voltage-gated sodium channels, Pflugers Archiv-Eur. J. Physiol 464 (2012) 493–502. [DOI] [PubMed] [Google Scholar]
- [12].Teramoto N, Yotsu-Yamashita M, Selective Blocking Effects of 4,9-Anhydrotetrodotoxin, Purified from a Crude Mixture of Tetrodotoxin Analogues, on Na(V)1.6 Channels and Its Chemical Aspects, Mar. Drugs 13 (2015) 984–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tsukamoto T, Chiba Y, Wakamori M, Yamada T, Tsunogae S, Cho Y, Sakakibara R, Imazu T, Tokoro S, Satake Y, Adachi M, Nishikawa T, Yotsu-Yamashita M, Konoki K, Differential binding of tetrodotoxin and its derivatives to voltage-sensitive sodium channel subtypes (Na(v)1.1 to Na(v)1.7), Br. J. Pharmacol 174 (2017) 3881–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL, Kearney JA, Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice, Neurobiol. Dis 65 (2014) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA, Functional coexpression of the beta-1 and type iia alpha-subunits of sodium-channels in a mammalian-cell line, J. Biol. Chem 270 (1995) 3306–3312. [DOI] [PubMed] [Google Scholar]
- [16].Catterall WA, Voltage-gated sodium channels at 60: structure, function and pathophysiology, J. Physiol.-Lond 590 (2012) 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen CL, Calhoun JD, Zhang YQ, Lopez-Santiago L, Zhou N, Davis TH, Salzer JL, Isom LL, Identification of the cysteine residue responsible for disulfide linkage of Na+ channel alpha and beta 2 subunits, J. Biol. Chem 287 (2012) 39061–39069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hanifin CT, Gilly WF, Evolutionary history of a complex adaptation: Tetrodotoxin resistance in salamanders, Evolution 69 (2015) 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fozzard HA, Lipkind GM, The Tetrodotoxin Binding Site Is within the Outer Vestibule of the Sodium Channel, Mar. Drugs 8 (2010) 219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chiamvimonvat N, PerezGarcia MT, Tomaselli GF, Marban E, Control of ion flux and selectivity by negatively charged residues in the outer mouth of rat sodium channels, J, Physiol.-Lond 491 (1996) 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Favre I, Moczydlowski E, Schild L, On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel, Biophys. J 71 (1996) 3110–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Heinemann SH, Teriau H, Stuhmer W, Imoto K, Numa S, Calcium-channel characteristics conferred on the sodium-channel by single mutations, Nature 356 (1992) 441–443. [DOI] [PubMed] [Google Scholar]
- [23].Ding JJ, Pan L, Ding X, Insight into tetrodotoxin blockade and protein in the mouse brain, Neuroreport 13 (2002) 2547–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Noda M, Suzuki H, Numa S, Stuhmer W, A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel-ii, FEBS Lett. 259 (1989) 213–216. [DOI] [PubMed] [Google Scholar]
- [25].Penzotti JL, Fozzard HA, Lipkind GM, Dudley SC, Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule, Biophys. J 75 (1998) 2647–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Terlau H, Heinemann SH, Stuhmer W, Pusch M, Conti F, Imoto K, Numa S, Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel-ii, FEBS Lett. 293 (1991) 93–96. [DOI] [PubMed] [Google Scholar]
- [27].Heinemann SH, Terlau H, Imoto K, Molecular-basis for pharmacological differences between brain and cardiac sodium-channels, Pflugers Archiv-Eur. J. Physiol 422 (1992) 90–92. [DOI] [PubMed] [Google Scholar]
- [28].Satin J, Kyle JW, Chen M, Bell P, Cribbs LL, Fozzard HA, Rogart RB, A mutant of ttx-resistant cardiac sodium-channels with ttx-sensitive properties, Science 256 (1992) 1202–1205. [DOI] [PubMed] [Google Scholar]
- [29].Soong TW, Venkatesh B, Adaptive evolution of tetrodotoxin resistance in animals, Trends Genet. 22 (2006) 621–626. [DOI] [PubMed] [Google Scholar]
- [30].Toledo G, Hanifin C, Geffeney S, Brodie ED, Convergent evolution of tetrodotoxin-resistant sodium channels in predators and prey, in: French RJ, Noskov SY (Eds.), Na Channels from Phyla to Function, Vol. 78 Elsevier Academic Press; Inc, San Diego, 2016, pp. 87–113. [DOI] [PubMed] [Google Scholar]
- [31].Geffeney S, Brodie ED, Ruben PC, Mechanisms of adaptation in a predator-prey arms race: TTX-resistant sodium channels, Science 297 (2002) 1336–1339. [DOI] [PubMed] [Google Scholar]
- [32].Geffeney SL, Fujimoto E, Brodie ED, Ruben PC, Evolutionary diversification of TTX-resistant sodium channels in a predator-prey interaction, Nature 434 (2005) 759–763. [DOI] [PubMed] [Google Scholar]
- [33].Shen HZ, Li ZQ, Jiang Y, Pan XJ, Wu JP, Cristofori-Armstrong B, Smith JJ, Chin YKY, Lei JL, Zhou Q, King GF, Yan N, Structural basis for the modulation of voltage-gated sodium channels by animal toxins, Science 362 (2018) 8. [DOI] [PubMed] [Google Scholar]
- [34].Shen HZ, Liu DL, Wu K, Lei JL, Yan N, Structures of human Na(v)1.7 channel in complex with auxiliary subunits and animal toxins, Science 363 (2019) 1303-+. [DOI] [PubMed] [Google Scholar]
- [35].Xu L, Li DY, Resistance mechanisms of Na(v)1.2 sodium channel by theoretical approaches, Chem. Biol. Drug Des 92 (2018) 1445–1457. [DOI] [PubMed] [Google Scholar]
- [36].Kao CY, Yasumoto T, Actions of 4-epitetrodotoxin and anhydrotetrodotoxin on the squid axon, Toxicon 23 (1985) 725–729. [DOI] [PubMed] [Google Scholar]
- [37].Yotsu-Yamashita M, Sugimoto A, Takai A, Yasumoto T, Effects of specific modifications of several hydroxyls of tetrodotoxin on its affinity to rat brain membrane, J. Pharmacol. Exp. Ther 289 (1999) 1688–1696. [PubMed] [Google Scholar]
- [38].Wu L, Nishiyama K, Hollyfield JG, Wang Q, Localization of Na(v)1.5 sodium channel. [DOI] [PMC free article] [PubMed]
- [39].Cheah CS, Westenbroek RE, Roden WH, Kalume F, Oakley JC, Jansen LA, Catterall WA, Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome, Channels 7 (2013) 468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ogiwara I, Miyamoto H, Tatsukawa T, Yamagata T, Nakayama T, Atapour N, Miura E, Mazaki E, Ernst SJ, Cao DZ, Ohtani H, Itohara S, Yanagawa Y, Montal M, Yuzaki M, Inoue Y, Hensch TK, Noebels JL, Yamakawa K, Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice, Commun. Biol 1 (2018) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Katz E, Stoler O, Scheller A, Khrapunsky Y, Goebbels S, Kirchhoff F, Gutnick MJ, Wolff F, Fleidervish IA, Role of sodium channel subtype in action potential generation by neocortical pyramidal neurons, Proc. Natl. Acad. Sci. U.S.A 115 (2018) E7184–E7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kanellopoulos AH, Koenig J, Huang HL, Pyrski M, Millet Q, Lolignier S, Morohashi T, Gossage SJ, Jay M, Linley JE, Baskozos G, Kessler BM, Cox JJ, Dolphin AC, Zufall F, Wood JN, Zhao J, Mapping protein interactions of sodium channel Na(V)1.7 using epitope-tagged gene-targeted mice, EMBO J. 37 (2018) 427–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Catterall WA, Neurotoxins that act on voltage-sensitive sodium-channels in excitable-membranes, Annu. Rev. Pharmacol. Toxicol 20 (1980) 15–43. [DOI] [PubMed] [Google Scholar]
- [44].Heggeness ST, Starkus JG, Saxitoxin and Tetrodotoxin - electrostatic effects on sodium-channel gating current in crayfish axons, Biophys. J 49 (1986) 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mosher HS, The chemistry of tetrodotoxin, Ann. N. Y. Acad. Sci 479 (1986) 32–43. [DOI] [PubMed] [Google Scholar]
- [46].Schaller KL, Krzemien DM, Yarowsky PJ, Krueger BK, Caldwell JH, NOVEL A, Abundant sodium-channel expressed in neurons and glia, J. Neurosci 15 (1995) 3231–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Frasier CR, Wagnon JL, Bao YO, McVeigh LG, Lopez-Santiago LF, Meisler MH, Isom LL, Cardiac arrhythmia in a mouse model of sodium channel SCN8A epileptic encephalopathy, Proc. Natl. Acad. Sci. U.S.A 113 (2016) 12838–12843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hu WQ, Tian CP, Li T, Yang MP, Hou H, Shu YS, Distinct contributions of Na (v)1.6 and Na(v)1.2 in action potential initiation and backpropagation, Nat. Neurosci 12 (2009) 996–U961. [DOI] [PubMed] [Google Scholar]
- [49].Lorincz A, Nusser Z, Molecular Identity of Dendritic Voltage-Gated Sodium Channels, Science 328 (2010) 906–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wagnon JL, Bunton-Stasyshyn RK, Meisler MH, Mutations of sodium channel SCN8A (Na(v)1.6) in neurological disease, in: Pitt GS (Ed.), Ion Channels in Health and Disease, Academic Press Ltd-Elsevier Science Ltd, London, 2016, pp. 239–264. [Google Scholar]