

Abstract
Background
This study aimed at evaluating the imaging findings of phosphaturic mesenchymal tumors and tumor-induced osteomalacia and assess the clinical and biochemical profiles of patients with tumor-induced osteomalacia.
Materials and methods
Imaging findings in six patients with tumor-induced osteomalacia and histopathologically proven phosphaturic mesenchymal tumors were evaluated. Clinical and biochemical profiles of these patients were also assessed.
Results
Along with having a characteristic biochemical profile, patients with phosphaturic mesenchymal tumors also have certain imaging findings which can aid in the diagnosis such as increased uptake on DOTA PET-CT and homogeneous post-contrast enhancement on CT and MRI.
Conclusion
Patients with tumor-induced osteomalacia have characteristic symptoms, imaging and biochemical profiles. For radiologists, raising the suspicion of a phosphaturic mesenchymal tumor in patients with refractory hypophosphatemic osteomalacia as well as localizing the tumor on imaging is crucial, as complete excision of the tumor leads to resolution of the osteomalacia and the patient’s clinical symptoms.
Keywords: Phosphaturic mesenchymal tumor, Tumor-induced osteomalacia, Radiology, Nuclear medicine, DOTA PET, Bone tumors
Introduction
Phosphaturic mesenchymal tumor (PMT) is a rare neoplasm which is frequently associated with tumor-induced osteomalacia (TIO), also known as oncogenic osteomalacia. TIO is a paraneoplastic syndrome which occurs as a result of expression of a biologically active substance by the tumor, called FGF23 or fibroblast growth factor 23, which induces renal phosphate excretion [1]. FGF23 acts on kidneys in two ways: first by decreasing proximal tubular re-absorption of phosphate and second by inhibiting 1-alpha-hydroxylase, which decreases the levels of 1-alpha, 25 dihydroxyvitamin D3 [2]. Thus increased phosphate excretion as a result of FGF23 causes mobilization of calcium and phosphate from bones, decrease in osteoblastic activity and ultimately generalized osteomalacia. Patients usually present with gradual muscle weakness and multiple bone pains due to pathological fractures. The diagnosis is delayed many a times as it is a rare condition which is often not included in the list of differentials by the treating physician and also due to the non-specific nature of the patient’s symptoms [3]. While biochemical tests such as serum calcium, phosphorus and FGF23 testing can help in diagnosing tumor-induced osteomalacia, the causative tumor itself (PMT) is difficult to localize within the body. Even if localized and biopsied, the relative rarity of PMT results in pathologists being unfamiliar with its characteristic histologic appearance. Accordingly, diagnosis is difficult if not impossible for the pathologist in many cases if the clinical presentation of the lesion is not known or if the pathologist is not aware of the entity at all. This difficulty is further enhanced by the fact that several variants of PMT exist and recently a variant of the PMT which is hormonally inactive has been described in literature [4]. While patients can suffer for years on end if the diagnosis is missed, correctly diagnosing a phosphaturic mesenchymal tumor and the resulting oncogenic osteomalacia and excising the tumor leads to near-complete clinical improvement and a dramatic reversal of symptoms and biochemical abnormalities. Even though a rare diagnosis, tumor-induced osteomalacia should be considered in the list of differential diagnoses in every patient with hypophosphatemic rickets or hypophosphatemic osteomalacia. Lesions are usually seen involving bone and soft tissue in the extremities and head and neck, and occurrence of primary abdominal, pelvic or thoracic visceral organ parenchymal lesions is not reported.
Here we describe six cases of phosphaturic mesenchymal tumor presenting to our institute, five of which were associated with tumor-induced osteomalacia. The final diagnosis was delayed by 2–5 years in four cases and by almost 20 years in one case. 68Ga-DOTA PET-CT is the most sensitive imaging modality in localizing phosphaturic mesenchymal tumors [5]. It is necessary for radiologists to be aware of this condition and keep a diagnosis of phosphaturic mesenchymal tumors in mind when reviewing imaging of a patient with a clinical and biochemical picture of hypophosphatemic osteomalacia.
Imaging characteristics of PMT are diverse and remain incompletely defined, as the majority of previous publications are not in radiologic literature.
The purpose of our study was to evaluate the imaging findings of phosphaturic mesenchymal tumors and tumor-induced osteomalacia and to assess the clinical and biochemical profiles of patients with this condition.
Materials and Methods
A retrospective evaluation of the cases of phosphaturic mesenchymal tumors with proven histopathology was conducted. All consecutive such cases presenting to our institute from January 2010 to March 2018 were evaluated and obtained by searching the hospital’s electronic medical records and pathology database. All patients with PMTs proven on histopathology and imaging of the tumor available on the hospital’s picture archiving and communications system (PACS) were included in the study. Cases of PMTs without available imaging of the primary tumor were excluded. Six such cases of histopathologically proven PMTs which also had imaging of the primary tumor available were found. Imaging findings and clinical and biochemical data of these six patients were studied. Frequency of various imaging findings were tabulated, using the Statistical Package for Social Sciences (SPSS) version 20.
Results and Analysis
Mean age of the patients diagnosed with PMT was 43 years, with a range of 19 years to 53 years. The duration for diagnosis of the tumor from onset of symptoms ranged from 2 years (minimum) to 38 years (maximum). Locations for the PMT included the chest wall (n = 2) (Fig. 1a–d), foot (n = 1) (Fig. 2a–d), paranasal sinus (n = 1; this patient had recurrence in the paranasal sinus after a primary PMT occurring in the hand) (Fig. 3), acetabulum (n = 1) (Fig. 4) and hand (n = 2) (Fig. 5). One patient with primary PMT in the hand had a recurrence of the disease after 2 years in the ethmoid sinus. Serum calcium levels varied from low (n = 3), normal (n = 2) to high (n = 1). Serum phosphorus levels were low in five patients and normal in one patient. Serum parathormone levels were high in three patients, normal in two patients and not obtained in one patient (as patient was biochemically asymptomatic). Serum FGF23 levels were raised in all patients (except the patient who was biochemically asymptomatic in whom the test was not performed).Clinical and biochemical data of the six patients are summarized in Table 1.
Fig. 1.

a 19-year-old male, with radiograph of both hands showing subperiosteal resorption along radial margin of middle phalanges of multiple fingers (yellow arrows), a feature consistent with hyperparathyroidism. Radiograph of both knee joints showing bowing of bilateral femori, tibia and fibulae; a feature seen in osteomalacia. b (a) Non-contrast axial CT shows a mass arising from the left posterior chest wall with erosion of the left 10th rib. It shows dense stippled calcification. (b) The mass shows intense and heterogeneous enhancement. (c) Incidental note is made of nephrocalcinosis involving both kidneys (yellow arrows), keeping with the patient having hyperparathyroidism. (d) Multiple rib fractures are seen (orange arrows) along with erosion of the left 10th rib adjacent to the mass (blue arrow). c Multiple lytic lesions are seen involving the right humeral head, left scapula, right iliac bone, sacrum and left acetabulum. These were reported as Brown tumors, in a patient with hyperparathyroidism. Axial post-contrast CT showing enlargement of bilateral parathyroid glands (orange arrows). d (a) Post-contrast axial CT done 2 years later shows increase in size of the mass, and now associated with a left-sided pleural effusion. (b) The rib fractures (orange arrows) are unchanged. Blue arrow shows rib erosion associated with the mass. (c) CT guided biopsy of the left posterior chest wall mass was performed
Fig. 2.

a 50-year-old female patient (a) Chest radiograph showing insufficiency rib fractures (blue arrows). (b) Radiograph of right hip joint with proximal femur shows Looser’s zone (pseudo-fracture) along medial margin of right femur, a characteristic feature of osteomalacia. (c) Radiograph of both legs shows pseudo-fractures of distal ends of both tibia and fibulae. (d) Radiograph of foot shows pseudo-fractures involving the 4th and 5th metatarsal bones. b (a) MLO view of left breast showing a lobulated mass. (b) On USG it corresponds to a hypoechoic mass with multiple cystic clefts within (yellow arrows), a finding commonly seen in Phyllodes tumors (c) Fused PET-CT image showing low-grade uptake in the left breast mass (d) Post-contrast CT image showing homogeneous enhancement of the mass. c (a) Axial non-contrast CT image shows no significant abnormality in the foot. (b) DOTA PET image shows a focus of increased tracer uptake involving the base of right 4th toe. d (a) Ultrasound guided biopsy of the foot lesion was performed. The lesion appears hypoechoic and lobulated on ultrasound. (b) Intra-operative ultrasound of the foot lesion being performed to assess adequacy of resection. (c) Gross surgically resected specimen of the phosphaturic mesenchymal tumor resected from right foot. Despite having a size of hardly 1 cm, the tumor wrecked biochemical havoc within the patient’s body
Fig. 3.

49-year-old male with chest radiograph showing multiple rib fractures. a Coronal T1W image shows a lobulated mass (yellow arrowhead) in the left nasal cavity extending into the left ethmoid air cells. The mass is isointense on T1WI. The left maxillary content is isointense. b Coronal STIR image shows the mass with a heterogeneous hyperintense appearance on T2 (yellow arrowhead). The left maxillary sinus shows mucosal thickening, with the cavity contents being T1 and T2 hypointense. c Coronal post contrast T1W image shows the mass demonstrating mild homogeneous enhancement (yellow arrowhead). The left maxillary sinus contents are non-enhancing. d Axial diffusion weighted image shows the mass not demonstrating diffusion restriction. Fused DOTA PET-CT images showing uptake within the left nasal cavity and ethmoid sinus mass. The left nasal cavity and ethmoid sinus mass was suspected to be a recurrence of the phosphaturic mesenchymal tumor which the patient previously had. The left maxillary sinus contents were reported as fungal sinusitis (the tumor and the fungal infection confirmed intra-operatively and on histopathology)
Fig. 4.

Radiographs showing multiple rib fractures and scoliosis, multiple fractures and deformities of bilateral femori and pelvic bones. Rugger jersey spine with end-plate sclerosis suggestive of renal osteodystrophy is also noted
Fig. 5.

Radiograph of right hand showing a soft tissue swelling near the second metacarpophalangeal joint, with no bone involvement. Coronal post-contrast image showing homogeneous enhancement within the lobulated soft tissue mass. Coronal T1W image showing the mass with isointense signal to skeletal muscle. Axial T2W image showing the mass as mildly hyperintense to skeletal muscle. It shows few hypointense foci within. FFE image shows the hypointense foci blooming, suggesting the presence of calcification or hemorrhage. Diffusion weighted image does not show diffusion restriction within the mass. Histopathologically proven hormonally inactive PMT
Table 1.
Clinical and biochemical details—laboratory values given in parentheses
| Age (years) | Gender | Duration between start of symptoms and final diagnosis | Location of phosphaturic mesenchymal tumor | Serum calcium (normal 8.6–10 mg/dl) | Serum phosphorus (normal 2.7–4.5 mg/dl) | Serum parathormone (normal 8–24 pg/ml) | Serum FGF23 (normal 5–195 RU/ml) | |
|---|---|---|---|---|---|---|---|---|
| Case 1 | 19 | Male | 2 years | Chest wall | Low (8.4) | Low (0.8) | High (36.7) | High (865.4) |
| Case 2 | 50 | Female | 6 years | Web space between fourth and fifth toes of right foot | Low (8.4) | Low (1.8) | High (154.4) | High (580.6) |
| Case 3 | 49 | Male | 15 years for primary; 2 years for recurrence | Right hand index finger (primary); left ethmoid sinus (recurrence) | Normal (9.2) | Low (2.4) | Normal (15.5) | High (3087) |
| Case 4 | 53 | Male | 20 years | Right acetabulum | High (10.3) | Low (2.3) | Normal (20.2) | High (1021) |
| Case 5 | 38 | Male | 38 years | Right hand index finger | Normal (9) | Normal (3.2) | Not done | Not done |
| Case 6 | 53 | Male | 2 years | Pleura/chest wall | Low (8.2) | Low (2.0) | High (152.6) | High (930) |
DOTA PET-CT showed uptake within the PMT in all four patients in whom it was performed. The MRI, CT and Ultrasound features were variable and are described in Table 2.
Table 2.
Imaging findings
| Findings of osteomalacia on radiographs | Uptake on DOTA PET-CT | MR imaging characteristics | CT imaging characteristics | Ultrasound | Image guided intervention performed | |
|---|---|---|---|---|---|---|
| Case 1 | Present | Not done | Not done | Calcified enhancing chest wall mass arising from rib | Not done | CT guided biopsy |
| Case 2 | Present | Present | T2 intermediate signal intensity lesion with enhancement | Not visualized on CT | Hypoechoic lobulated lesion | USG guided biopsy and intra-operative USG |
| Case 3 | Present | Present | Hypointense on T1 and hyperintense on T2 with mild enhancement | Hypodense lesion with mild enhancement | Not done | None (endoscopic biopsy) |
| Case 4 | Present | Present | Not done | Hypodense lesion | Not done | CT guided biopsy |
| Case 5 | No | Not done | Isointense with skeletal muscle on T1, mildly hyperintense on T2 with homogeneous enhancement | Not done | Not done | None (direct core biopsy) |
| Case 6 | Present | Present | Not done | Pleural-based nodule | Not done | CT guided biopsy |
Discussion
Phosphate plays a vital role in cellular metabolism and DNA synthesis. Acute hypophosphatemia can cause weakness, hypotension, rhabdomyolysis, respiratory failure, congestive cardiac failure, seizures and altered mental status. Because these symptoms are non-specific, the diagnosis of hypophosphatemia may be delayed [6]. Chronic hypophosphatemia results in defective bone mineralization leading to osteomalacia.
Phosphaturic mesenchymal tumors (PMTs) cause renal phosphate excretion and decrease vitamin D synthesis, by secreting a peptide hormone like substance fibroblast growth factor-23 (FGF23). FGF23 acts on kidneys by decreasing proximal tubular re-absorption of phosphate and by inhibiting 1-alpha-hydroxylase, which converts vitamin D to its active form. This ultimately leads to the paraneoplastic syndrome of tumor-induced hypophosphatemic hyperphosphaturic osteomalacia. PMTs are rare tumors that occur most frequently in middle aged adults and can involve essentially any bone or soft tissue location including the paranasal sinuses, skull base, chest wall and extremities. Till date, around 300 cases of PMT have been reported in literature. A recent 2017 study has shown that PMTs are a single disease entity unified by immunohistochemical expression of CD56, ERG, SATB2 and somatostatin receptor 2A [7]. PMTs may appear histologically similar to other mesenchymal tumors such as solitary fibrous tumor, hemangiopericytoma or chondromyxoid fibroma among others; however, they are a specific neoplasm with varying morphotypes and a common molecular profile, rather than different neoplasms causing tumor induced osteomalacia [7].
Histologically, PMTs are characterized by a proliferation of bland spindle cells associated with a variable amount of ‘grungy’ calcified matrix [8] (Fig. 6). A small subset of PMTs exhibit malignant histologic features and may behave in a clinically malignant fashion [9].We also report a case of histopathologically proven PMT which was hormonally inactive, matching another case recently described in the literature [4]. PMTs are usually small in size and not clinically palpable. Because PMTs are rare, the diagnosis is often not considered by physicians, surgeons, radiologists or pathologists, even in patients presenting with osteomalacia [3]. Thus, most patients ultimately diagnosed with PMT have experienced osteomalacia for years before the tumor is identified. Measurement of serum FGF23 can assist in diagnosis and management of disorders of phosphate and bone metabolism. A high FGF23 level in a patient with normal renal function, hypophosphatemia and with or without osteomalacia, is suggestive of PMT. FGF23 is located on chromosome 12 and is composed of three exons. Mutations in FGF23 that render the protein resistant to proteolytic cleavage lead to increased activity of FGF23 and renal phosphate loss [2].
Fig. 6.

Phosphaturic mesenchymal tumor: histology reveals a hypocellular tumor composed of bland spindle cells with small nuclei, indistinct nucleoli and hemangiopericytoma-like vasculature. These cells are intermixed with the distinctive “grungy” calcification (marked by asterisk) of the stromal matrix (hematoxylin and eosin, 200 × original magnification)
Clinical features of tumor-induced osteomalacia include bone pains, generalized weakness and pathological fractures. The biochemical abnormalities that characterize tumor-induced osteomalacia consist of depressed levels of serum phosphate and 1,25-dihydroxyvitamin D3 (calcitriol—the active form of vitamin D), together with elevated levels of urine phosphate [10]. Since active vitamin D promotes intestinal phosphate absorption, patients with oncogenic osteomalacia have difficulty absorbing phosphate from their diet. The definitive treatment involved early and total resection of the causative phosphaturic mesenchymal tumor. Subtotal resection may be associated with local recurrence or distant metastases [3, 11].
Tertiary hyperparathyroidism is a known association of long-standing tumor-induced osteomalacia, reported in 5% cases of TIO [12]. The cause is direct action by FGF23 on the parathyroid glands, which initially decreases parathormone levels. However, the glands eventually develop resistance and undergo hyperplasia, leading to the state of tertiary hyperparathyroidism. A rare cause of tertiary hyperparathyroidism in TIO cases would be chronic over-correction of phosphate levels which would lead to the parathyroid glands secreting excess of parathormone [12, 13].
The differential diagnosis of TIO includes other renal phosphate wasting diseases such as X-linked hypophosphatemic rickets, autosomal dominant hypophosphatemic rickets, hereditary hypophosphatemic rickets with hypercalciuria, and Fanconi syndrome [10]. Fanconi syndrome causes general dysfunction of the proximal renal tubule and may manifest during childhood or adulthood. The clinical expression of hypophosphatemic rickets with hypercalciuria usually occurs between 6 months and 7 years of age. Hypophosphatemic rickets with hypercalciuria and Fanconi syndrome may be dismissed, as these diseases also cause hypercalciuria and aminoaciduria, respectively. Although X-linked hypophosphatemic rickets and autosomal dominant hypophosphatemic rickets are biochemically indistinguishable from oncogenic osteomalacia, these genetic diseases usually manifest during childhood, while TIO is typically diagnosed in middle age. A lack of family history of bone disease and identification of a tumor on PET-CT are indicators of TIO. The clinical work-up for TIO is as summarized in Fig. 7.
Fig. 7.
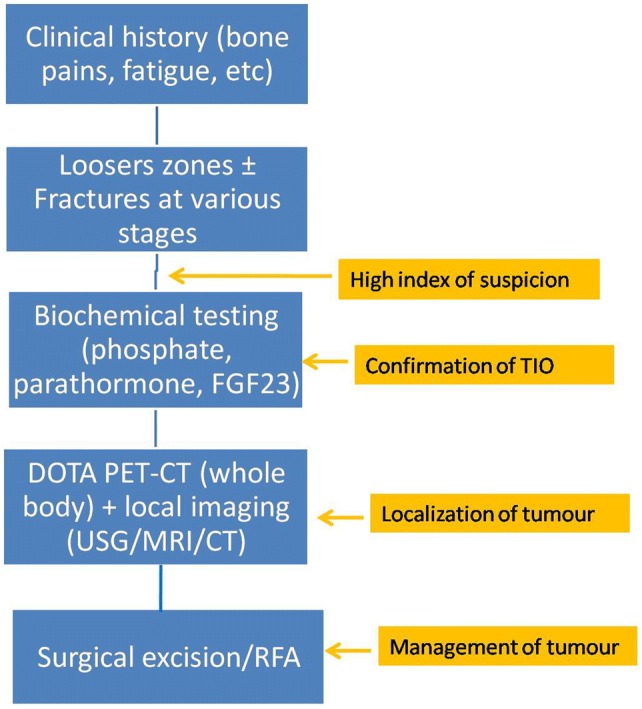
Flow-chart for clinical work-up of TIO
Since phosphaturic mesenchymal tumors show diverse appearances on histopathology, conventional imaging modalities such as radiography, ultrasound, CT and MRI also show variable characteristics on imaging [14].
The most sensitive imaging modality to localize PMTs is PET CT and 68 Ga-DOTA PET-CT scores over FDG PET CT, which will show increased tracer uptake within the tumor [5].
The skeletal manifestations of oncogenic osteomalacia are the same as osteomalacia due to other causes, with radiographs showing osteopenia, coarse trabeculae, Looser’s zones, insufficiency fractures and bowing of long bones [15].
In patients who have developed associated hyperparathyroidism, the imaging findings may match those of renal osteodystrophy (which itself is a combination of secondary hyperparathyroidism and osteomalacia). Imaging findings in renal osteodystrophy include those of osteomalacia plus other findings of secondary hyperparathyroidism including ‘salt and pepper’ appearance of skull, diffuse bone sclerosis, rugger jersey spine (sclerosis of vertebral body end plates) and soft tissue calcification [15].
Conclusion
Phosphaturic mesenchymal tumor (PMT) is a rare distinct mesenchymal neoplasm with variable but recognizable histopathology and a common immunological profile. It frequently elicits a clinical paraneoplastic syndrome consisting of tumor-induced hypophosphatemic hyperphosphaturic osteomalacia due to increased secretion of FGF23. The patients present with gradual muscular weakness, bone pain and pathologic fractures. Many a times the diagnosis is delayed for years due to the non-specific nature of these symptoms, lack of clinical suspicion, failure to include serum phosphorus levels in routine blood chemistry testing, and difficulty in identifying the responsible tumor. Additionally, these tumors are often missed histopathologically because of their rarity and morphologic overlap with other mesenchymal neoplasms. For radiologists, raising the suspicion of a PMT in patients with refractory hypophosphatemic osteomalacia as well as localizing the tumor on imaging is crucial, as complete excision of the tumor leads to resolution of the osteomalacia and the patient’s clinical symptoms.
Author Contributions
Concepts: ASK, AKJ, MSD, KBG. Design: ASK, AKJ, MSD, KBG. Definition of intellectual content: ASK, AKJ, MSD. Literature search: ASK, AKJ, MSD. Clinical studies: ASK, AKJ, KBG. Experimental studies. Data acquisition: ASK. Data analysis: ASK, AKJ, MSD, KBG. Statistical analysis: ASK. Manuscript preparation: ASK. Manuscript editing: ASK, AKJ, MSD, KBG. Manuscript review: ASK, AKJ, MSD, KBG. Guarantor: ASK, AKJ.
Funding
None.
Compliance with Ethical Standards
Conflict of Interest
All authors declare that they have no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ameya S. Kawthalkar, Email: ameya005@gmail.com
Amit K. Janu, Email: amiteasy10@gmail.com
Mrunmayee S. Deshpande, Email: mrunmayee2703@gmail.com
Kunal B. Gala, Email: drgala@gmail.com
Ashish Gulia, Email: ashishgulia@gmail.com.
Ajay Puri, Email: docpuri@gmail.com.
References
- 1.Ghorbani-Aghbolaghi A, Darrow MA, Wang T. Phosphaturic mesenchymal tumor (PMT): exceptionally rare disease, yet crucial not to miss. Autopsy & Case Reports. 2017;7(3):32–37. doi: 10.4322/acr.2017.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quarles LD. Role of FGF23 in vitamin D and phosphate metabolism: implications in chronic kidney disease. Experimental Cell Research. 2012;318(9):1040–1048. doi: 10.1016/j.yexcr.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ledford CK, Zelenski NA, Cardona DM, Brigman BE, Eward WC. The phosphaturic mesenchymal tumor: why is definitive diagnosis and curative surgery often delayed? Clinical Orthopaedics and Related Research. 2013;471(11):3618–3625. doi: 10.1007/s11999-013-3178-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiu L, Heim-Hall J. Nonphosphaturic variant of phosphaturic mesenchymal tumor, mixed connective tissue type: a case report. American Journal of Clinical Pathology. 2018;149(1):S118. doi: 10.1093/ajcp/aqx123.277. [DOI] [Google Scholar]
- 5.Ho CL. Ga68-DOTA peptide PET/CT to detect occult mesenchymal tumor-inducing osteomalacia: a case series of three patients. Nuclear Medicine and Molecular Imaging. 2015;49(3):231–236. doi: 10.1007/s13139-015-0328-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. American Journal of Medicine. 2005;118(10):1094–1101. doi: 10.1016/j.amjmed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 7.Agaimy A, Michal M, Chiosea S, Petersson F, Hadravsky L, Kristiansen G, et al. Phosphaturic mesenchymal tumors: clinicopathologic, immunohistochemical and molecular analysis of 22 cases expanding their morphologic and immunophenotypic spectrum. The American Journal of Surgical Pathology. 2017;41(10):1371–1380. doi: 10.1097/PAS.0000000000000890. [DOI] [PubMed] [Google Scholar]
- 8.Shustik DA, Ng DC, Sittampalam K. Phosphaturic mesenchymal tumour mixed connective tissue variant: report of three cases with unusual histological findings. International Journal of Clinical and Experimental Pathology. 2015;8(6):7506–7517. [PMC free article] [PubMed] [Google Scholar]
- 9.Qiu S, Cao L-L, Qiu Y, Yan P, Li Z, Du J, et al. Malignant phosphaturic mesenchymal tumor with pulmonary metastasis: a case report. Medicine. 2017;96(17):6750. doi: 10.1097/MD.0000000000006750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jagtap VS, Sarathi V, Lila AR, Bandgar T, Menon P, Shah NS. Hypophosphatemic rickets. Indian Journal of Endocrinology and Metabolism. 2012;16(2):177–182. doi: 10.4103/2230-8210.93733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnaoutakis D, Naseri I. Sinonasal phosphaturic mesenchymal tumor: a rare and misinterpreted entity. Journal of Neurological Surgery Reports. 2015;76(2):233–238. doi: 10.1055/s-0035-1562852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tartaglia F, Minisola S, Sgueglia M, Blasi S, Brunelli D, DegliEffetti E, et al. Tumor-induced hypophosphatemicosteomalacia associated with tertiary hyperparathyroidism: a case report. Giornale di Chirurgia. 2006;27(1–2):9–13. [PubMed] [Google Scholar]
- 13.Silver J, Naveh-Many T. FGF23 and the parathyroid. Endocrine FGFs and Klothos: Springer; 2010. pp. 92–99. [Google Scholar]
- 14.Shi Z, Deng Y, Li X, Li Y, Cao D, Coossa VS. CT and MR imaging features in phosphaturic mesenchymal tumor-mixed connective tissue: a case report. Oncology Letters. 2018;15:4970–4978. doi: 10.3892/ol.2018.7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang C, Rosenthal D, Mitchell D, Handa A, Kattapuram S, Huang A. Imaging findings of metabolic bone disease. RadioGraphics. 2016;36(6):1871–1887. doi: 10.1148/rg.2016160004. [DOI] [PubMed] [Google Scholar]