

Abstract
In recent years, new protein engineering methods have produced more than a dozen symmetric, self‐assembling protein cages whose structures have been validated to match their design models with near‐atomic accuracy. However, many protein cage designs that are tested in the lab do not form the desired assembly, and improving the success rate of design has been a point of recent emphasis. Here we present two protein structures solved by X‐ray crystallography of designed protein oligomers that form two‐component cages with tetrahedral symmetry. To improve on the past tendency toward poorly soluble protein, we used a computational protocol that favors the formation of hydrogen‐bonding networks over exclusively hydrophobic interactions to stabilize the designed protein–protein interfaces. Preliminary characterization showed highly soluble expression, and solution studies indicated successful cage formation by both designed proteins. For one of the designs, a crystal structure confirmed at high resolution that the intended tetrahedral cage was formed, though several flipped amino acid side chain rotamers resulted in an interface that deviates from the precise hydrogen‐bonding pattern that was intended. A structure of the other designed cage showed that, under the conditions where crystals were obtained, a noncage structure was formed wherein a porous 3D protein network in space group I213 is generated by an off‐target twofold homomeric interface. These results illustrate some of the ongoing challenges of developing computational methods for polar interface design, and add two potentially valuable new entries to the growing list of engineered protein materials for downstream applications.
Keywords: protein design, protein cages, nanotechnology, protein assembly, computational biology, protein crystallography
1. INTRODUCTION
A long‐standing goal in the field of protein engineering has been to develop reliable methods to create designed protein nanocages with high symmetry, which could find applications in wide‐ranging fields such as drug delivery, imaging, energy, and nanotechnology.1, 2, 3, 4 Since 2012, more than a dozen examples of successful cage designs have been validated in atomic detail by X‐ray crystallography,5, 6, 7, 8 and several others have been verified at lower resolution by electron microscopy.8, 9 Recent studies are beginning to generate novel cages with specific applications in mind, including the ability to encapsulate other molecules such as nucleic acids,10, 11, 12, 13 present viral antigens,14 or rigidly bind other proteins for cryo‐EM imaging.15, 16
Notwithstanding the impressive successes that have been reported for designed protein cages, considerable experimental challenges remain. Reviewing available literature suggests that success rates for designed cages are around 10%.7, 8, 17 Different strategies for designing cages present distinct challenges. Methods based on genetic fusion between different oligomeric components face difficulties in flexibility.5, 18, 19, 20, 21, 22, 23 Methods based on the computational design of novel interfaces between oligomeric units have led to numerous successes, but they have revealed a tendency for proteins with computationally designed interfaces to fail during protein expression trials or to aggregate (e.g., by misfolding or assembling in indefinite fashion) upon expression. This has motivated efforts to improve solubility. In one study, increasing the net charge on the solvent‐exposed surfaces of the designed protein subunits had a positive effect.24
Further efforts to improve the success rates for designed assemblies are needed, including to improve their solubility. One approach that would provide a general solution would be to design more polar surfaces at the protein–protein interfaces that drive assembly of the material. Hydrogen‐bonding interactions across the interface could offset the cost of desolvating the polar groups upon interface formation. This approach could reduce solubility problems, but at the expense of more demanding design, as hydrogen bonding is geometrically more exacting than hydrophobic burial. Here we report crystal structures of two protein cages that were designed with a greater emphasis on interfacial hydrogen bonding, with some surprising findings.
2. RESULTS AND DISCUSSION
2.1. Protein components and design principles
We designed two novel two‐component assemblies with tetrahedral symmetry, T33‐51 and T33‐53 (Figure 1a; see Section 4) using a preliminary version of HBNet, a Rosetta protocol that designs extensive hydrogen‐bonding networks at protein–protein interfaces.25 In each case, the cage is intended to assemble from two different trimeric protein building blocks. Four trimers of each type occupy alternating vertex positions of a cube. A computationally designed interface holds the two different trimeric components together at the edges, resulting in a cage with tetrahedral symmetry and A12B12 stoichiometry. The diameters of the designed cages are 13 nm, with molecular masses of about 480 kDa.
Figure 1.
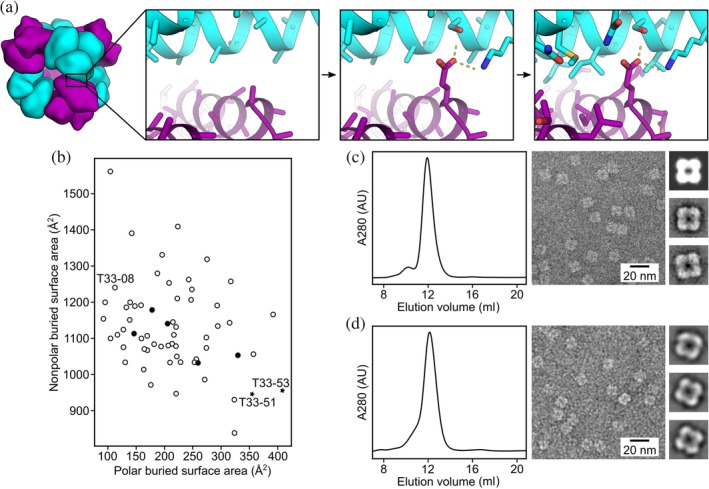
Design and characterization of T33‐51 and T33‐53. (a) Graphical depiction of the hydrogen bond network‐focused design protocol. (Left) Docked configurations for T33‐51 and T33‐53, based on the previously unsuccessful design T33‐08, featured (second from left) contacts between the trimeric building blocks based on well‐anchored elements of secondary structure. (Second from right) In an initial sequence design step, hydrogen bond networks compatible with the docked backbone were identified and placed. (Right) Subsequently, the full protein–protein interface was designed, keeping the identities of the hydrogen bond network residues fixed. (b) A comparison of designed interface hydrophobicities for T33‐51 and T33‐53 versus previous designs. Unsuccessful designs from King et al.7 are shown as open circles, and the previously designed insoluble assembly, T33‐08, is labeled. Successful, structurally validated designs from King et al.7 are shown as filled circles. The two new designs, T33‐51 and T33‐53, are shown as stars and labeled. (c) SEC and EM of T33‐51. (d) SEC and EM of T33‐53. Both SEC profiles show strong single peaks at the expected elution volume for the ~480 kDa tetrahedral assemblies. Representative negative stain electron micrographs for each assembly are shown. Uniform particles of ~13 nm were observed for both T33‐51 and T33‐53. Insets: four‐lobed, square‐shaped particle averages closely resemble a projection of T33‐51 calculated from the computational design model along its twofold symmetry axis (top inset in B)
The two designs studied here were based on the same pair of trimeric building blocks (PDB 1NOG and PDB 1WY1). These two naturally trimeric protein components are homologs of each other, sharing 38% sequence identity and similar folds. The design of a tetrahedral cage (called T33‐08) constructed from these components had been attempted before, prior to development of the HBNet protocol, but the resulting construct proved insoluble.7 The new protocol led to different sequences compared to the earlier effort (see Section 4 for sequences) featuring more polar character at the designed interfaces (Figure 1b). Being based on the same underlying components and closely related docked configurations, the two constructs examined in the present study were similar to each other, differing only in the amino acid sequences at the designed interfaces intended to drive cage assembly.
2.2. Expression and preliminary structural characterization
For production of the designed cages, both subunit types were expressed together in the same Escherichia coli cells so that assembly of the A12B12 cages could occur in vivo. Both pairs of proteins exhibited high levels of soluble expression and were easily purified by Ni2+‐affinity chromatography. For both designs, the two coexpressed proteins could be concentrated after Ni2+‐affinity purification up to about 60 mg/ml in aqueous buffer without noticeable aggregation. Thus, the new sequence designs based on the preliminary HBNet protocol were confirmed to have better solubility behavior than the initial design (T33‐08). We also tested the solubility of the individual protein components (i.e., subunits A or B) when expressed in the absence of their designed binding partners. For both designs, one component (T33‐51A and T33‐53A) was found to be soluble and well‐behaved, while the other (T33‐51B and T33‐53B) exhibited only insoluble expression (data not shown). All the protein subunits are apparently soluble long enough to allow proper assembly when their cognate partners are present in cells, though some of the components are not soluble indefinitely by themselves. SEC analysis showed a strong peak for an assembly with an apparent molecular weight of about 500 kDa (elution volume of ~12 ml on a Superdex 200 10/300 column) for both constructs, consistent with the expected tetrahedral assembly state (Figure 1c,d). After purification, the two designed assemblies were examined by negative stain electron microscopy to see whether the formation of geometrically regular structures could be confirmed visually. Protein assemblies resembling the ~13 nm designed structures were readily observed for both constructs, and 2D averages of each particle matched low‐resolution projections calculated from the computational design model of T33‐51 (Figure 1c,d). Together, these data indicate that the computationally designed interfaces of T33‐51 and T33‐53 successfully drive assembly to the intended tetrahedral complexes.
2.3. Crystal structure of the T33‐51 tetrahedral cage
In an effort to assess the designed materials in atomic detail, both cage constructs were crystallized by hanging drop vapor diffusion and examined by X‐ray diffraction.
The diffraction data for T33‐51 were processed in space group P23 with unit cell dimensions a = b = c = 106.7 Å, with data extending to 3.40 Å resolution. Data collection and processing statistics are shown in Table 1. Because the two oligomeric protein components of the cage are designed based on proteins that are homologous to each other, there exists a pseudo‐four‐fold axis along the tetrahedral twofold axis, which caused the data to also process reasonably well in space group P432. If this were the true space group, then only one of the components could be present in the crystal in order for there to be fourfold symmetry. Fortunately, this space group was ruled out because the value for R merge was 12.8% in P432, compared to 9.2% in P23, indicating that the fourfold axis was imperfect or pseudosymmetric.
Table 1.
T33‐51 crystallographic data
| T33‐51 | |
|---|---|
| Data collection | |
| PDB accession code | http://firstglance.jmol.org/fg.htm?mol=5CY5 |
| Beamline | APS‐NECAT‐24‐ID‐C |
| Space group | P23 |
| Unit cell dimensions | |
| a = b = c (Å) | 106.7 |
| α = β = γ (°) | 90.0 |
| Reflections observed | 154,895 |
| Unique reflections | 5,827 |
| Wavelength (Å) | 0.9795 |
| Resolution (Å) | 75.45–3.40 |
| Highest resolution shell (Å)a | 3.48–3.40 |
| R sym (%) | 9.2 (135.9) |
| CC(1/2) | 100.0 (70.4) |
| I/σ | 29.0 (2.8) |
| Completeness (%) | 99.9 (99.5) |
| Wilson B value (Å2) | 107.1 |
| Refinement | |
| Resolution (Å) | 75.45–3.40 |
| Resolution (Å) (last shell) | 3.74–3.40 |
| Reflections used | 5,825 |
| R work(%)/R free(%) | 15.6 (17.8)/19.7 (23.0) |
| Protein molecules in asymmetric unit | 2 (1 of each subunit type) |
| Number of non‐H atoms | 2,255 |
| RMS deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.00 |
| Average B‐factor (Å2) | 81.1 |
| Ramachandran plot regions | |
| Favored (%) | 95.4 |
| Allowed (%) | 4.6 |
| Outliers (%) | 0.00 |
A hypothetical atomic model for the tetrahedral cage was available at the outset, since the design protocol outputs a full‐atom description of the intended assembly in PDB format. The unit cell of the crystal we obtained was only large enough to contain a single copy of the designed cage, giving a solvent content of 51%. Therefore, according to the P23 crystal symmetry, the cage had to sit at the origin of the unit cell (i.e., at the point of tetrahedral symmetry), with the cage oriented so that its symmetry axes were aligned with the symmetry axes of the crystal space group. The molecular replacement problem was therefore greatly simplified. There were, however, two distinct orientations of the cage that had to be considered, related to each other by 90° rotation about a principal axis; this is because the rotational point group symmetry of the cubic lattice is octahedral or 432 while the crystal point group is only tetrahedral or 23. Of the two possible orientations for the cage in the unit cell, one gave a lower starting R‐value between calculated and observed structure factor amplitudes (33% compared to 38%) and was used as the starting point for restrained refinement. The asymmetric unit, which comprised the atomic components to be refined, consisted of one copy of each subunit type. In the later stages it was discovered that the crystal specimen was partially hemihedrally twinned, which was consistent with an earlier observation that the diffraction data could be reduced in P432 with a value for R merge that was only moderately worse than for P23 (12.8% vs. 9.2%). The presence of twinning was also supported by a statistical analysis of the intensity data according to the L‐test, which gave a value of 0.41 for data between 8 and 4 Å, whereas the theoretically expected values for untwinned and perfectly twinned data are 0.5 and 0.375 respectively.26 The value for the twin fraction refined to 33% under an appropriate twin law (l, −k, h).
The final model for this first cage construct, T33‐51, could be refined to R work/R free of 0.156/0.197 at 3.40 Å resolution. The arrangement of subunits in the cage closely matched the design: the rmsd between the refined model and the design was only 1.4 Å on C‐alpha atoms over the entire 24‐subunit assembly (Figure 2a). Agreement at the amino acid side chain level at the designed interface was mixed (Figure 2b,c). Overall, many of the designed atomic contacts at the interface were observed in the intended configuration: for 27 out of 36 amino acids within 6 Å of the other subunit, the deviation at chi1 between the design model and refined structure was ≤25°. However, the agreement was lower for the residues intended to form the hydrogen‐bonding network. For example, residue S90 of component A and E81 of component B form an unexpected hydrogen bond network with residue K94 of component A, whereas by design residue N151 of component A was intended to form a hydrogen bond network with S90 and E81. In the crystal structure, N151 is flipped such that it no longer comes close to the other residues with which it was designed to form hydrogen bonds (Figure 2c). Although completely unambiguous determination of sidechain rotamers was not possible with 3.40 Å resolution and B‐values in the range of 70–80, the orientations of residues involved in this hydrogen‐bonding network interaction were reasonably clear in the calculated electron density map, and without indication of alternative conformations. This suggested that the designed hydrogen bonds were not making critical contributions to the stability of the tetrahedral material, though the polarity of the designed interface residues may have helped mitigate aggregation problems during expression.
Figure 2.
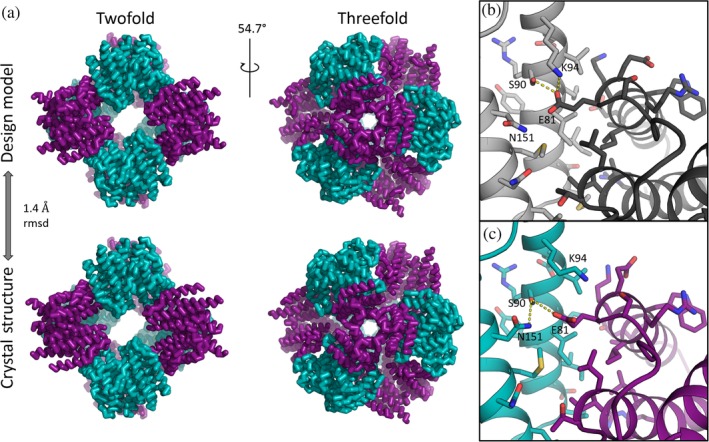
Crystal structure and comparison with design for cage T33‐51. (a) Views of the fully assembled 24‐subunit T33‐51 cage along the twofold/pseudo‐four‐fold axis (left) and along one of the threefold symmetry axes (right). The crystal structure of the 24‐subunit T33‐51 cage aligns to the design model with 1.4 Å rmsd on C‐alphas. The asymmetric unit consisted of one monomer of each subunit type: T33‐51A (purple) and T33‐51B (cyan). (b and c) Both similarities and differences between the designed (b) and experimental (c) interfaces are seen at the sidechain level. Flipped rotamers in the crystal structure compared to the design led to unanticipated hydrogen bond networks, yet overall agreement between designed and experimental residues at the designed interface is high
2.4. Crystal structure of the T33‐53 accidental minimum contact lattice
The X‐ray diffraction data for the second design, T33‐53, initially appeared to process well in space group I23. Similar to the situation for the structure of the first cage (in P23), space group I23 also supports a position at the origin with 23 (T) point symmetry where a cage with symmetry T could sit. However, the body‐centered unit cell dimensions of a = b = c = 138.4 Å in this case appeared to be slightly too small to accommodate the designed cage situated at the origin. We considered that the trimeric components had perhaps rotated relative to the design, which then would have allowed a slightly deformed cage to pack in the observed unit cell. However, molecular replacement with the trimeric components as search models failed to identify any alternate solutions with rotated trimers. We considered that our observed diffraction was in fact consistent with either I23 or I213; this is a rare case where systematic absences do not distinguish between space groups. Our reasons for favoring I23 at the outset were noted above; I213 does not possess any point of 23 (T) symmetry and therefore could not support a tetrahedral cage (given that the unit cell was not large enough to fit multiple copies of a subunit with noncrystallographic symmetry). When we repeated the molecular replacement calculations in space group I213, a noncage solution was immediately apparent.
The correct structure of the T33‐53 crystal is shown in Figure 3, and the data collection and processing statistics are listed in Table 2. Structure determination in the correct space group resulted in a porous, interconnected 3D crystal lattice. Remarkably, the structure is made up of only one component from the original design; the asymmetric unit consists of just one copy of subunit B (see Section 4). The natural trimeric unit for component B is correctly formed, and sits on a crystallographic threefold axis, but there is an accidental crystallographic twofold interaction that arises where two trimers make contact using regions of their surfaces that were intended to form the designed heterotypic interface with subunit A, which is absent from the crystal (Figure 3a). Nearly all of the 11 residues mutated from the native protein sequence of 1NOG lie at or near the unintended dimeric interface, where they create a large and relatively hydrophobic patch on the protein surface which sticks to the same region on an adjacent protein subunit and gives rise to the unintended twofold axis (Figure 3c). Additionally, a loop region of the native protein sequence (residues 76–81), which should have been solvent‐exposed by design, appears to form new electrostatic interactions that help hold the twofold interface together as well.
Figure 3.
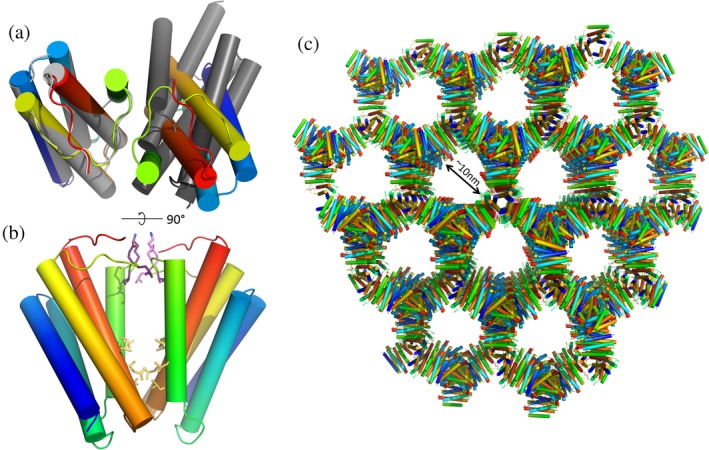
Structure of the T33‐53 crystal lattice. (a) Ribbon models of the design (gray) versus crystal structure (rainbow), viewed down the twofold axis of the unanticipated homotypic interface. The orientation of the adjacent protein subunit in the observed crystal structure is significantly rotated from that in the design model. (b) View perpendicular to twofold axis. Hydrophobic sidechains of residues that were mutated from the native sequence of 1NOG are shown in yellow. A hydrophobic patch on T33‐53B at the designed interface forms an unintended twofold homotypic interaction. Another interaction surface, held together by electrostatic interactions between sidechains, occurs at the C2 symmetry axis between residues 76–81 of each chain (sidechains colored in purple). This segment is a loop region, which contains no mutations from the native protein sequence and was predicted to remain solvent‐exposed in the design model. (c) Lattice structure of the I213 crystal of T33‐53B oriented along the C3 symmetry axis
Table 2.
T33‐53 crystallographic data
| T33‐53 | |
|---|---|
| Data collection | |
| PDB accession code | http://firstglance.jmol.org/fg.htm?mol=5VL4 |
| Beamline | APS‐NECAT‐24‐ID‐C |
| Space group | I2(1)3 |
| Unit cell dimensions | |
| a = b = c (Å) | 138.4 |
| α = β = γ (°) | 90.0 |
| Reflections observed | 21,295 |
| Unique reflections | 3,554 |
| Wavelength (Å) | 0.9795 |
| Resolution (Å) | 97.85–4.10 |
| Highest resolution shell (Å) | 4.20–4.10 |
| R sym (%) | 14.7 (77.6) |
| CC(1/2) | 100.0 (74.2) |
| I/σ | 6.57 (2.31) |
| Completeness (%) | 98.9 (94.9) |
| Wilson B value (Å2) | 135.5 |
| Refinement | |
| Resolution (Å) | 97.85–4.10 |
| Resolution (Å) (last shell) | 4.58–4.10 |
| Reflections used | 3,551 |
| R work(%)/R free (%) | 21.0 (27.9)/26.4 (32.4) |
| Protein molecules in asymmetric unit | 1 |
| Number of non‐H atoms | 1,179 |
| RMS deviations | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.02 |
| Average B‐factor (Å2) | 267.2 |
| Ramachandran plot regions | |
| Favored (%) | 97.9 |
| Allowed (%) | 1.4 |
| Outliers (%) | 0.7 |
In principle, trimers of subunit B might conceivably have come together to make a cage structure, but the orientation of the trimers with respect to one another in the 3D lattice places their threefold symmetry axes in incompatible positions. Instead, the arrangement is a rather remarkable network structure (Figure 3c). The natural threefold symmetry axis of the original protein trimer and the fortuitous twofold axis between trimers are nonintersecting, with an angle of 54° between them. Those two interactions—the (natural) threefold interface and the fortuitous twofold—are the only molecular contacts present in the crystal. The solvent content for the crystal is 76.6%. Interestingly, this situation of a connected 3D lattice in space group I213 formed by nonintersecting threefold and twofold axes was discussed in 2001 by Padilla et al. in the context of the minimum contact requirements for designing three‐dimensional crystalline materials from simpler oligomeric units.18
The observed crystal structure stands in contrast to the solution (SEC) data and electron microscopy results for the T33‐53 construct, which supported a cage structure. A likely explanation is that the computationally designed interface was not stable to the crystallization conditions employed, and that an alternate homotypic interface involving drastically rotated trimers (of component B) was favored and selected by crystal growth. It is perhaps notable that the pH of the crystallization buffer was 5.6, and unexpected protonation (e.g., of histidines) could have impacted the energetics. Although the unusual network structure seen in the crystal structure was not the intended outcome for this redesigned protein oligomer, engineered porous 3D protein crystals such as this could find diverse applications, such as in creating catalytic reaction vessels for preparing inorganic materials27 or in immobilizing enzymes to create biosensors or microbioreactors.28
3. CONCLUSION
This study was motivated by a common problem of low solubility that often afflicts designed proteins bearing novel protein–protein interfaces; such interfaces tend to be somewhat hydrophobic. More hydrophilic interfaces based on geometrically specific hydrogen‐bonding networks offer a potential advantage, but those interactions require highly accurate design. A designed tetrahedral cage (called T33‐08) from an earlier study,7 which expressed insolubly, was taken a starting point for improving the design using a new Rosetta protocol (a preliminary version of HBNet) that emphasizes favorable hydrogen bonding. Two computationally designed sequences were produced experimentally. Both formed the intended cages in solution, which led to crystallographic work to investigate their atomic details.
The crystal structure data on the two designed variants revealed two new interesting results for designed protein assemblies, one matching closely to its intended computational design (a tetrahedrally symmetric cage) and one forming an unexpected kind of extended 3D lattice. The first case, which crystallized in the form of a cage, was surprising at the atomic level. Owing to side chain differences compared to the design, some of the intended hydrogen‐bonding interactions do not occur in the interface, yet the proteins were clearly more soluble than an earlier design based on the same building blocks. One interpretation of this finding is that the designed sequences from the preliminary HBNet protocol were indeed more hydrophilic in the interfaces, thus mitigating the previous solubility problem and enabling the intended cage structures to form with the majority of the specifically intended atomic interactions made. The second designed construct presented other surprises. In this case, the cage structure evident in solution was apparently not stable to crystallization conditions (i.e., pH 5.6). Nonetheless, the features introduced into the trimeric protein surface introduced a general tendency toward association. As a result, under specific crystallization conditions, one of the two trimeric components that was intended to assemble into the cage instead self‐associated to make a porous and highly unusual 3D crystalline network held together by the natural threefold trimeric contacts and a single fortuitous twofold contact between trimeric units.
In broad terms, the computational design efforts here were successful. Using a new protocol, two new protein cages were designed, and solution experiments indicated assembly to the intended configurations; a previous design attempt using the same proteins as building blocks had yielded only insoluble protein. Yet finer considerations show that current design methods still leave room for improvements, including in designing hydrogen‐bonding interactions. We can identify two factors that likely contributed to the failure to design a hydrogen‐bonding network in T33‐51 that was recapitulated in the crystal structure (it is unknown whether the hydrogen‐bonding network in T33‐53 is present as intended in the tetrahedral complex in solution). First, we used a preliminary version of the HBNet protocol that was incomplete. Subsequent refinement of the protocol has led to the design of numerous structures with extended hydrogen‐bonding networks that have been faithfully recapitulated in high‐resolution crystal structures, including pH‐responsive hydrogen‐bonding networks rooted by histidine residues.25, 29, 30 Second, we applied the preliminary design protocol to an extremely limited design space. We searched only the docked configurations that we experimentally characterized in our previous report on two‐component tetrahedral complexes,7 and we did not allow backbone movement at any point during the HBNet or RosettaDesign portions of our design protocol. It is therefore somewhat unsurprising that we were unable to identify hydrogen‐bonding networks of exceptional quality or stability. Future efforts to design more polar protein–protein interfaces that drive assembly of protein nanomaterials should search as broad a design space as possible, and should incorporate the lessons learned from successful instances of HBNet‐based design mentioned above. Among others, these include requiring the absence of buried unsatisfied hydrogen‐bonding groups and constraining the possible conformations of the hydrogen‐bonding residues through burial. Achieving stability across experimental conditions—or controlled responsiveness to changes in conditions—is another challenge for future efforts. In practical terms, the present work adds two new two‐component protein nanomaterials to the growing repertoire available for potential applications in medicine and nanotechnology.
4. MATERIALS AND METHODS
4.1. Protein scaffold design
Computational design of two‐component tetrahedral cages was performed using Rosetta as described earlier7 with some modifications. Briefly, two different trimeric components of known structure were chosen as building blocks for a given candidate cage. For each pair of components, four copies of each trimer were placed at alternating vertices of a cube of sufficient size to assure that the components did not collide. Then the single rotational degree of freedom and the single translational degree of freedom for each trimer type (i.e., four degrees of freedom in total) were sampled to identify docked configurations featuring high numbers of Cβ‐Cβ contacts in proximity between the two components, without interpenetration. These docked configurations were then subjected to computational amino acid sequence design at the novel trimer–trimer interface. The sequence design procedure employed an early prototype of the recently developed HBNet protocol that favors the formation of extended hydrogen‐bonding networks.25 Two candidate designs with low calculated Rosetta energies were chosen to test experimentally. These two test cases were based on the same component trimers, but with slightly different rigid body degrees of freedom and different amino acid sequences at the designed interfaces. Polar and nonpolar surface area buried at the designed intercomponent interface (presented in Figure 1b) was calculated using built‐in SASA functions in Rosetta.
4.2. Protein expression and purification
Genes encoding the designed protein components of each cage design were cloned into the pETDuet™‐1 expression vector (Novagen) as reported previously.7 The two components for each cage were coexpressed in E. coli from the same plasmid. The B component in each design contains a C‐terminal His6‐tag, which was used for Ni‐affinity purification. Pooled and concentrated nickel elution fractions were further purified by size exclusion chromatography (SEC) using a Superdex 200 10/300 gel filtration column (GE Life Sciences) with 25 mM TRIS pH 8.0, 150 mM NaCl, 1 mM DTT as running buffer. SEC fractions containing pure protein in the desired assembly state were pooled, concentrated, and stored at 4°C for subsequent use in analytical SEC, electron microscopy, and X‐ray crystallography.
4.3. Analytical SEC
Analytical SEC was performed on a Superdex 200 10/300 gel filtration column using the running buffer described above. The designed materials were each loaded onto the column at a concentration of 50 μM (of each subunit). The apparent molecular weights of the designed assemblies were estimated by comparison to previously determined nanocage standards.7
4.4. Negative stain electron microscopy
Six microliters of purified T33‐51 and T33‐53 samples at concentrations ranging from 0.006 to 0.02 mg/ml were applied to glow discharged, carbon coated 400‐mesh copper grids (Ted Pella, Inc.), washed with Milli‐Q water and stained with 0.075% uranyl formate based on methods described previously.31 Grids were visualized for assembly validation and optimized for data collection. Screening and sample optimization were performed on a 100 kV Morgagni M268 transmission electron microscope (FEI, Hillsboro, OR) equipped with a Gatan Orius CCD camera (Gatan, Pleasanton, CA). Data collection was performed on a 120 kV Tecnai G2 Spirit transmission electron microscope (FEI, Hillsboro, OR). All images were recorded using a Gatan Ultrascan 4000 4k × 4k CCD camera (Gatan, Pleasanton, CA) at 52,000× magnification at the specimen level. The contrast of all micrographs was enhanced in ImageJ32 for clarity.
Coordinates for 20,388 (T33‐51) and 37,425 (T33‐53) unique particles were obtained for averaging using EMAN2.33 Boxed particles were used to obtain 2D class averages by refinement in EMAN2. Back‐projection images at 20 Å were computed in EMAN2 based on coordinates of the design models.
4.5. Crystallization, data collection, and processing
Crystals of the T33‐51 protein assembly were obtained using the hanging drop vapor diffusion crystallization method with a reservoir of 0.1 mM HEPES, pH 8.8 and 15% PEG 3350. Initial crystals were obtained at the UCLA Crystallization Facility using a Mosquito liquid handling device (TTP LabTech). Optimized crystals were obtained using a microbatch crystallization method in which a 600 nl drop containing 12 mg/ml protein and the previous reservoir solution in a 2:1 ratio was overlaid with 20 μl of a 6:5 ratio of paraffin oil to silicone oil on top of the drop to slow down evaporation. Cubic crystals of about 150 μm in length were observed after 7 days at room temperature. T33‐51 crystals were cryo‐protected with 33% glycerol and flash frozen in liquid nitrogen prior to data collection.
T33‐53 crystals were obtained using the hanging drop vapor diffusion crystallization method with a protein concentration of 12 mg/ml and a reservoir of 0.1 M Na citrate, pH 5.6, 2.5 M 1,6‐hexanediol, and 0.01 M manganese chloride. Cubic crystals ranging from 100 to 200 μm in length were obtained from initial crystallization screens. Data were collected from these crystals without further optimization.
All datasets were collected at the Advanced Photon Source (APS), beamline 24‐ID‐C (NE‐CAT). Data were indexed, integrated, and scaled using XDS/XSCALE.34 Structures were determined by molecular replacement using the known trimeric structures as the search models. A nonstandard molecular replacement analysis was required in one case as described in Section 2.
4.6. Structure solution and refinement
The T33‐51 model was refined to 3.40 Å with iterative rounds of model building and refinement carried out using Coot35 and PHENIX.36 After each cycle of model rebuilding, reciprocal space refinement, including refinement of coordinates and atomic displacement parameters, was carried out using phenix.refine. Subsequent cycles of refinement were performed using Buster37 and included TLS refinement,38 with the final cycle being done using phenix.refine, leading to a model with R work/R free of 0.156/0.197 at a resolution of 3.40 Å. These crystals were partially hemihedrally twinned, and the statistical averaging effects of twinning presumably contribute to the favorable refinement R‐value obtained. Note that residues 98–103 of T33‐51A could not be reliably modeled and were therefore omitted from the final model. Coordinates and structure factors for the T33‐51 protein cage have been deposited with the PDB code http://firstglance.jmol.org/fg.htm?mol=5CY5.
The T33‐53 model was refined to 4.10 Å in a similar fashion to T33‐51, except that Buster was used for all refinement cycles. Symmetry considerations (discussed in Section 2) made it clear that the crystal contained only one of two coexpressed protein components (and did not comprise the two‐component cage structure), so two initial models were generated by molecular replacement based on each of the distinct components and refined separately. Note that the two proteins A and B are homologous and structurally similar to each other, so either could have represented the crystallized component. Refinement against the B component of the design model (PDB: 1NOG) resulted in a significantly lower R‐factor (23% vs. 35%), so further refinement was pursued based on this. The R work/R free of the final model was 0.210/0.264. Coordinates and structure factors for the T33‐53 noncage assembly have been deposited with the PDB code http://firstglance.jmol.org/fg.htm?mol=5VL4.
4.7. Protein sequences
Residues mutated from native sequence are underlined.
Residues added to native sequence are in bold.
A components are based on PDB 1WY1 and B components are based on PDB 1NOG.
T33‐51A (13 mutations):
MRITTKVGDKGSTRLFGGEEVWKDDPIIEANGTLDELTSFIGEAKHYVDEEMKGILEEIQNDIYKIMGEIGSKGKIEGISEERIKWLAGLIERYSEMVNKLSFVLPGGTLESAKLDVCRTIARRAERKVATVLREFGIGTLAAIYLALLSRLLFLLARVIEIEKNKLKEVRS
T33‐51B (13 mutations + C‐terminal His6‐tag):
MFTRRGDQGETDLANRARVGKDSPVVEVQGTIDELNSFIGYALVLSRWDDIRNDLFRIQNDLFVLGEDVSTGGKGRTVTMDMIIYLIKRSVEMKAEIGKIELFVVPGGSVESASLHMARAVSRRLERRIKAASELTEINANVLLYANMLSNILFMHALISNKRLNIPEKIWSIHRVSLEHHHHHH
T33‐53A (11 mutations):
MRITTKVGDKGSTRLFGGEEVWKDDPIIEANGTLDELTSFIGEAKHYVDEEMKGILEEIQNDIYKIMGEIGSKGKIEGISSERIKWLAGLISRYEEMVNKLSFVLPGGTLESAKLDVCRTIARRAERKVATVLREFGIGTNAAIYLAALSDLLFLLARVIEIEKNKLKEVRS
T33‐53B (11 mutations + C‐terminal His6‐tag):
MFTRRGDQGETDLANRARVGKDSPVVEVQGTIDELNSFIGYALVLSRWDDIRNDLFRIQNDLFVLGEDVSTGGKGRTVTLEMILYLVERVTEMKAEIGKIELFVVPGGSVESASLHMARAVSRRLERRIKAASRLTEINDNVLLYAAMLSSILFMHALISNKRLNIPEKIWSIHRVSLEHHHHHH
4.8. Protein design models
Atomic coordinates of the two computational design models described here are provided in Supporting Information.
AUTHOR DECLARATIONS
N.P.K. and D.B. are cofounders and equity holders of Icosavax, Inc., a biotech company commercializing vaccines based on computationally designed protein nanomaterials.
Supporting information
Data S1 Supporting Information
Data S2 Supporting Information
ACKNOWLEDGMENTS
This work was supported by NSF grant CHE 1629214 (T.O.Y, N.P.K., D.B.), a grant from Takeda Pharmaceuticals (N.P.K.), and the Howard Hughes Medical Institute (D.B.). Initial screening and X‐ray diffraction analysis was performed at UCLA core facilities supported by grant DE‐FC02‐02ER63421 from the BER program of the US Department of Energy Office of Science. We thank Mike Collazo, Mike Sawaya and Duilio Cascio at the UCLA‐DOE X‐ray Crystallization and Crystallography Core Facilities for assistance with crystallization screening, model building and refinement. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on 24‐ID‐C beam line is funded by a NIH‐ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357.
Cannon KA, Park RU, Boyken SE, et al. Design and structure of two new protein cages illustrate successes and ongoing challenges in protein engineering. Protein Science. 2020;29:919–929. 10.1002/pro.3802
Kevin A. Cannon and Rachel U. Park contributed equally to this study.
Funding information Howard Hughes Medical Institute; National Science Foundation, Grant/Award Number: CHE 1629214; Takeda Pharmaceuticals North America; U.S. Department of Energy, Grant/Award Number: DE‐FC02‐02ER63421
Contributor Information
Neil P. King, Email: neilking@uw.edu.
Todd O. Yeates, Email: yeates@mbi.ucla.edu.
REFERENCES
- 1. Cannon KA, Ochoa JM, Yeates TO. High‐symmetry protein assemblies: Patterns and emerging applications. Curr Opin Struct Biol. 2019;55:77–84. [DOI] [PubMed] [Google Scholar]
- 2. Howorka S. Rationally engineering natural protein assemblies in nanobiotechnology. Curr Opin Biotechnol. 2011;22:485–491. [DOI] [PubMed] [Google Scholar]
- 3. Aumiller WM, Uchida M, Douglas T. Protein cage assembly across multiple length scales. Chem Soc Rev. 2018;47:3433–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Y, Ardejani MS, Orner BP. Design and applications of protein‐cage‐based nanomaterials. Chemistry. 2016;11:2814–2828. [DOI] [PubMed] [Google Scholar]
- 5. Lai YT, Cascio D, Yeates TO. Structure of a 16‐nm cage designed by using protein oligomers. Science. 2012;336:1129. [DOI] [PubMed] [Google Scholar]
- 6. King NP, Sheffler W, Sawaya MR, et al. Computational design of self‐assembling protein nanomaterials with atomic level accuracy. Science. 2012;336:1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. King NP, Bale JB, Sheffler W, et al. Accurate design of co‐assembling multi‐component protein nanomaterials. Nature. 2014;510:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bale JB, Gonen S, Liu Y, et al. Accurate design of megadalton‐scale two‐component icosahedral protein complexes. Science. 2016;353:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hsia Y, Bale JB, Gonen S, et al. Design of a hyperstable 60‐subunit protein icosahedron. Nature. 2016;535:136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butterfield GL, Lajoie MJ, Gustafson HH, et al. Evolution of a designed protein assembly encapsulating its own RNA genome. Nature. 2017;552:415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Azuma Y, Edwardson TGW, Terasaka N, Hilvert D. Modular protein cages for size‐selective RNA packaging in vivo. J Am Chem Soc. 2018;140:566–569. [DOI] [PubMed] [Google Scholar]
- 12. Edwardson TGW, Mori T, Hilvert D. Rational engineering of a designed protein cage for siRNA delivery. J Am Chem Soc. 2018;140:10439–10442. [DOI] [PubMed] [Google Scholar]
- 13. Terasaka N, Azuma Y, Hilvert D. Laboratory evolution of virus‐like nucleocapsids from nonviral protein cages. Proc Natl Acad Sci U S A. 2018;115:5432–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marcandalli J, Fiala B, Ols S, et al. Induction of potent neutralizing antibody responses by a designed protein nanoparticle vaccine for respiratory syncytial virus. Cell. 2019;176:1420–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu Y, Gonen S, Gonen T, Yeates TO. Near‐atomic cryo‐EM imaging of a small protein displayed on a designed scaffolding system. Proc Natl Acad Sci U S A. 2018;115:3362–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y, Huynh DT, Yeates TO. A 3.8 Å resolution cryo‐EM structure of a small protein bound to an imaging scaffold. Nat Commun. 2019;10:1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yeates TO. Geometric principles for designing highly symmetric self‐assembling protein nanomaterials. Annu Rev Biophys. 2017;46:23–42. [DOI] [PubMed] [Google Scholar]
- 18. Padilla JE, Colovos C, Yeates TO. Nanohedra: Using symmetry to design self assembling protein cages, layers, crystals, and filaments. Proc Natl Acad Sci U S A. 2001;98:2217–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lai YT, Tsai KL, Sawaya MR, Asturias FJ, Yeates TO. Structure and flexibility of nanoscale protein cages designed by symmetric self‐assembly. J Am Chem Soc. 2013;135:7738–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lai Y‐T, Reading E, Hura GL, et al. Structure of a designed protein cage that self‐assembles into a highly porous cube. Nat Chem. 2014;6:1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sciore A, Su M, Koldewey P, et al. Flexible, symmetry‐directed approach to assembling protein cages. Proc Natl Acad Sci U S A. 2016;113:8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Badieyan S, Sciore A, Eschweiler JD, et al. Symmetry‐directed self‐assembly of a tetrahedral protein cage mediated by de novo‐designed coiled coils. Chembiochem. 2017;18:1888–1892. [DOI] [PubMed] [Google Scholar]
- 23. Cristie‐David AS, Chen J, Nowak DB, et al. Coiled‐coil‐mediated assembly of an icosahedral protein cage with extremely high thermal and chemical stability. J Am Chem Soc. 2019;141:9207–9216. [DOI] [PubMed] [Google Scholar]
- 24. Bale JB, Park RU, Liu Y, et al. Structure of a designed tetrahedral protein assembly variant engineered to have improved soluble expression. Protein Sci. 2015;24:1695–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boyken SE, Chen Z, Groves B, et al. De novo design of protein homo‐oligomers with modular hydrogen‐bond network–mediated specificity. Science. 2016;352:680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Padilla JE, Yeates TO. A statistic for local intensity differences: Robustness to anisotropy and pseudo‐centering and utility for detecting twinning. Acta Cryst. 2003;D59:1124–1130. [DOI] [PubMed] [Google Scholar]
- 27. Ueno T. Porous protein crystals as reaction vessels. Chemistry. 2013;19:9096–9102. [DOI] [PubMed] [Google Scholar]
- 28. Kowalski AE, Johnson LB, Dierl HK, Park S, Huber TR, Snow CD. Porous protein crystals as scaffolds for enzyme immobilization. Biomater Sci. 2019;7:1898–1904. [DOI] [PubMed] [Google Scholar]
- 29. Chen Z, Boyken SE, Jia M, et al. Programmable design of orthogonal protein heterodimers. Nature. 2019;565:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boyken SE, Benhaim MA, Busch F, et al. De novo design of tunable, pH‐driven conformational changes. Science. 2019;364:658–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nannenga BL, Iadanza MG, Vollmar BS, Gonen T. Overview of electron crystallography of membrane proteins: Crystallization and screening strategies using negative stain electron microscopy. Curr Protoc Prot Sci. 2013;72:17.15.1–17.15.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang G, Peng L, Baldwin PR, et al. EMAN2: An extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. [DOI] [PubMed] [Google Scholar]
- 34. Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 36. Adams PD, Grosse‐Kunstleve RW, Hung L‐W, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Cryst. 2002;D58:1948–1954. [DOI] [PubMed] [Google Scholar]
- 37. Bricogne G, Blanc E, Brandl M, et al. Buster. Cambridge, United Kingdom: Global Phasing Ltd, 2011. [Google Scholar]
- 38. Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Cryst. 2006;D62:439–450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting Information
Data S2 Supporting Information