

Abstract
Much of our understanding of protein structure and mechanistic function has been derived from static high‐resolution structures. As structural biology has continued to evolve it has become clear that high‐resolution structures alone are unable to fully capture the mechanistic basis for protein structure and function in solution. Recently Hydrogen/Deuterium‐exchange Mass Spectrometry (HDX‐MS) has developed into a powerful and versatile tool for structural biologists that provides novel insights into protein structure and function. HDX‐MS enables direct monitoring of a protein's structural fluctuations and conformational changes under native conditions in solution even as it is carrying out its functions. In this review, we focus on the use of HDX‐MS to monitor these dynamic changes in proteins. We examine how HDX‐MS has been applied to study protein structure and function in systems ranging from large, complex assemblies to intrinsically disordered proteins, and we discuss its use in probing conformational changes during protein folding and catalytic function.
Statement for a Broad Audience
The biophysical and structural characterization of proteins provides novel insight into their functionalities. Protein motions, ranging from small scale local fluctuations to larger concerted structural rearrangements, often determine protein function. Hydrogen/Deuterium‐exchange Mass Spectrometry (HDX‐MS) has proven a powerful biophysical tool capable of probing changes in protein structure and dynamic protein motions that are often invisible to most other techniques.
Keywords: conformational switching, conformational transitions, hydrogen/deuterium‐exchange mass spectrometry (HDX‐MS), intrinsic disorder, structural dynamics, structural rearrangements
1. INTRODUCTION
While high‐resolution three‐dimensional structures of macromolecular complexes have deepened our understanding of their assembly and construction, proteins are dynamic molecules that are constantly in motion. These motions range from small fluctuations in local structure to large‐scale conformational rearrangements between distinct structural states. Despite the fact that these dynamic motions and structural rearrangements are critical for protein function, we do not yet fully understand how these motions and conformational changes contribute to protein structure and function.1 Furthermore, high‐resolution models are usually only available for the endpoints of dynamic processes and one can only infer what takes place during protein motion and conformational changes. In order to fully understand how a protein functions, we need to understand the dynamic structural changes that lead to functionally distinct conformations. In recent years Hydrogen/Deuterium‐exchange Mass Spectrometry (HDX‐MS) has developed into a powerful solution‐state structural technique that enables the study of macromolecular protein complexes under native conditions. HDX‐MS enables one to interrogate protein structure and function by directly monitoring backbone amide solvent accessibility in solution, which is sensitive to protein structural conformation and dynamics.2
Due to advances in mass spectrometry and liquid chromatography, the complexity and size of the systems amenable to analysis by HDX‐MS has greatly expanded. Compared to other high‐resolution structural techniques, information about protein conformation can be obtained using HDX‐MS for samples spanning a range of sizes and complexities, provided one's analyte of interest is homogeneous in composition and well‐behaved in solution.3 While HDX can also be probed by NMR with residue level resolution, this approach is generally restricted to small soluble proteins that are amenable to solution state NMR analysis.2, 4 By contrast, mass spectrometers have no inherent size limit and have been used to study systems as large and complex as intact ribosomal complexes.5, 6 HDX‐MS experiments require submicromolar concentrations of sample and can, under certain circumstances, provide residue level information.2
In this review, we will discuss how HDX‐MS has been used to interrogate challenging biological problems and extract information that was otherwise not available. We will show how the HDX‐MS data enabled the researchers to reach their conclusions and highlight the power of HDX‐MS as a tool to study diverse biological systems and protein structures. In addition, pulse labeling HDX‐MS has recently been used to study protein folding mechanisms and the structures of intrinsically disordered proteins; areas of structural biology where conventional high‐resolution structural techniques have shown limited success. We will also review how HDX‐MS can be used to resolve and monitor conformational changes in integral membrane proteins and large protein complexes. These recent works demonstrate the power and versatility of HDX‐MS as a solution state structural technique and highlight how the technique has rapidly evolved over the recent years.
2. THE BIOPHYSICS OF HDX‐MS
Protein motion is constant and ranges from large‐scale, slower structural rearrangements to rapid, transient fluctuations in local structure.7 These innate, often subtle, dynamic motions across the protein backbone are fundamental to protein function but cannot be probed readily by most structural techniques.8 The intrinsic chemical rate of deuterium exchange of a residue's amide group is primarily dependent on temperature, pH, and the particular type of amino acid. Under physiological conditions the exchange rates for fully exposed amide hydrogens range from 101 to 103 s−1.9 On top of this, HDX‐MS applied to proteins is sensitive to the accessibility, or exposure, of backbone amide hydrogens resulting from a protein's conformation and can change in response to protein motion and structural dynamics.9, 10 In native proteins, the accessibility of a backbone amide hydrogen is largely dependent on hydrogen bonding and local secondary structure, as well as solvent accessibility.10, 11 Backbone amide hydrogens more exposed to deuterated solvent will exchange faster than those occluded in the protein core. Similarly, those amide hydrogens engaged in stable hydrogen bonds are protected from deuterium and will exchange more slowly. Local fluctuations in protein structure transiently expose backbone amide hydrogens to deuterium. The propensity for exposed amide hydrogens in a peptide segment to exchange with deuterium is related to how frequently and for how long they exist in an exposed, exchange‐competent state with respect to the chemical rate of exchange.10 These transitions are monitored under equilibrium conditions during continuous labeling HDX‐MS experiments.12 Briefly, in such an experiment, a protein is diluted into a deuterated solution for various amounts of time, ranging from seconds to hours or days. The exchange reaction is then quenched by dropping the solution pH to 2.5 and 0°C, where the labeling rate is at its minimum.10, 13, 14, 15 The labeled protein is digested into peptide fragments that are separated and analyzed by liquid chromatography coupled to mass spectrometry (LC‐MS). The reverse phase LC separation must be performed quickly and under quench conditions to limit back exchange of the deuterium label with water in solution. The extent of labeling is straightforward to analyze by mass spectrometry as uptake of a single deuteron produces a Dalton increase in mass.
Under physiological conditions, the majority of local protein motion is faster than the HDX labeling rate and thus leads to gradual incorporation of deuterium onto the peptide backbone (Figure 1a). So‐called EX2 kinetics is observed when the interconversion rate between a peptide's protected, exchange‐incompetent and exposed, exchange‐competent state is faster than the labeling rate.10, 16, 17 In this case, rapid opening and closing events allow only a subset of the collectively exposed amide hydrogens to exchange during any single transition to the exchange competent state. These rapid fluctuations are characterized by unimodal mass spectra envelopes that gradually shift to higher masses with time. Over time the entire peptide segment will become deuterated after repeatedly sampling the exposed, exchange‐competent state. The rate and extent of a peptide's deuterium uptake under EX2 kinetics is interpretable in terms of local structural dynamics, motion, and flexibility (Figure 1a). Furthermore, by characterizing how this exchange profile changes with environment, upon ligand binding, or between homologous proteins, one can begin to appreciate and dissect the structural basis for protein function and behavior.
Figure 1.
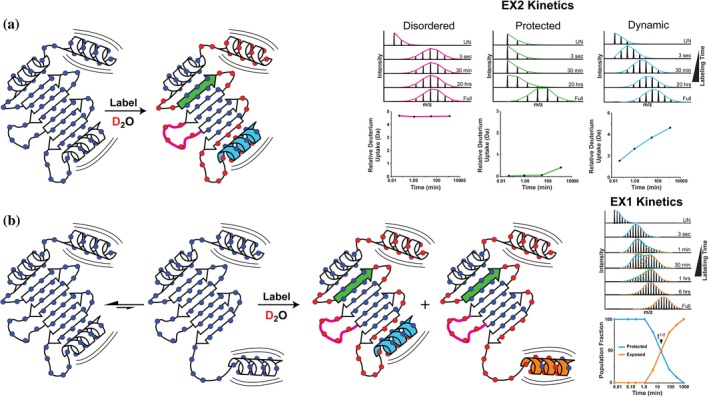
Protein structural dynamics and motion monitored by HDX‐MS. Continuous labeling HDX‐MS probes the accessibility of backbone amide hydrogens (blue circles) by their exchange with deuterium in solution. Under equilibrium conditions the majority of protein structural dynamics and motion manifests as “EX2 kinetics” (a) where deuterium is gradually incorporated across the protein backbone in a manner directly related to the local structural dynamics and changes in amide accessibility. Examples of peptide specific HDX‐MS data with representative mass spectra show the gradual incorporation of deuterium over time. Peptide segments in highly structured regions with strong hydrogen bonding networks (green beta sheet) take up deuterium much slower than regions with exposed and accessible amides (pink loop) and those undergoing dynamic motions (blue helix) that occur faster than the labeling rate. (b) When these dynamic structural changes are slower than the labeling rate (e.g., a reversible interconversion or conformational change) they produce unique and resolvable HDX states that manifest as “EX1 kinetics.” Here the protein reversibly interconverts between conformations with a protected (blue) or exposed (orange) helical domain. Analysis of the mass spectra reveals the equilibrium distribution of these two states and their respective half‐lives
Large‐scale structural rearrangements, including reversible interconversion between conformations, can also be monitored by HDX‐MS (Figure 1b). As a protein changes conformation, the rate and extent of HDX will shift in accordance with the local structure of each state. These types of protein motion are typically slow (on the order of milliseconds to seconds) and highly correlated across segments of the amide backbone. In an HDX labeling experiment, this behavior manifests as multimodal isotopic distributions with unique HDX populations for each structural state, and is termed EX1 kinetics.17, 18, 19, 20 For example, if a region in a protein reversibly interconverts between a “closed” protected (exchange‐incompetent) and an “open” exposed (exchange‐competent) state, with the residues in this segment switching conformations in a concerted manner, this would be readily apparent by HDX‐MS (Figure 1b). The amide hydrogens in the “closed” state would be protected from exchange and visible as a lower mass population in the spectra, whereas those that had transitioned to the exposed state would be evidenced as a deuterated higher mass population in the spectra. As the protein reversibly transitions between these states, the individual HDX populations will ultimately converge on the fast exchanging exposed state, enabling one to determine the rate of conversion between states and their relative half‐lives (Figure 1b). More generally, EX1 kinetics is indicative of correlated local structural changes or motion occurring slower than the labeling rate; meaning multiple amide hydrogens in a peptide segment adopt a relatively long‐lived exchange competent state and become deuterated together.17, 21 In reality, EX1 exchange kinetics are rarely observed under physiological conditions because one's ability to resolve EX1 kinetics is dependent on the rate of interconversion between the protected‐exchange incompetent and exposed‐exchange competent state being slower than the labeling rate.17, 21
EX1 kinetics are also often misidentified during analysis.18, 22, 23, 24 Determining the presence and root cause of EX1 behavior requires careful interrogation and analysis of the data using the proper analytical tools. Most commonly, HDX‐MS data is analyzed by centroid fitting, where the centroid of a mass envelope is identified and tracked over time.25 While centroid fitting may be sufficient for analysis of clean EX2 kinetics data, it is insufficient for the analysis and extraction of accurate structural dynamic information when EX1 kinetics are present. More importantly, centroid fitting fails to identify all but the most blatantly obvious EX1 exchange data where the two HDX states are dramatically different. EX1 exchange kinetics commonly manifests as abnormally broad mass envelopes, where the two HDX states are not dissimilar enough from one another to produce visually distinct populations. Analysis by binomial fitting is sensitive to changes in mass envelope width and can therefore detect the presence of two similar, coexisting HDX states and characterize their individual exchange profiles through bimodal deconvolution. (For a more expansive explanation of bimodal deconvolution and binomial fitting the reader is referred here.18, 24) The presence of bimodal spectra in HDX‐MS data does not always correspond to EX1 kinetics arising from large scale conformational changes, reversible interconversion between states, or local coordinated motion. It can also be indicative of conformational heterogeneity resulting from sample impurity or degradation, mixed populations from unsaturated ligand binding, and even back exchange from chromatographic carryover and improper sample handling.14, 26, 27 Similarly where EX2 kinetics are observed, it does not necessarily prove that the local protein motion across those residues is uncorrelated, only that it is occurring faster than the labeling rate.17, 28 Critical analysis of HDX‐MS data using the proper and most robust analytical tools is essential for making meaningful conclusions about protein structural dynamics and motion.
Below, we highlight some notable examples where HDX‐MS has been used to investigate protein motions and conformational changes, revealing novel insights into their behavior and functions.
2.1. Conformational sampling and protein folding
The mechanism by which proteins fold in solution remains the subject of intense debate.29, 30, 31 An unfolded protein must rearrange in solution to find its final, correct, conformation amongst a vast number of incorrect alternatives. Exactly how proteins find this proverbial “needle in a haystack” is still unclear; are there multiple discrete folding pathways for a given protein, or does folding occur through a single distinct pathway?31, 32 Biology has evolved a solution to the protein folding problem and encoded it in each protein's sequence. However, we have yet to elucidate these solutions for ourselves. Classical high‐resolution structural techniques, such as cryo‐EM and X‐ray crystallography, are not amenable to studying protein folding. While folding can often be directly monitored by spectroscopic methods, these approaches lack the structural and temporal resolution needed to resolve and appreciate the mechanism of protein folding.16, 29
Pulse labeling HDX‐MS enables direct monitoring of active events such as protein folding with high structural and temporal resolution. During folding the protein is rapidly pulsed with deuterium, effectively capturing a snapshot of the protein's structure as it transitions to the native folded state. Using a short, fixed, often millisecond, labeling time abrogates the contributions of structural dynamics to exchange as the labeling rate is faster than folding can occur; thus the acquisition of structure during folding is the primary factor contributing to HDX.12, 33 Walters et al. sought to determine the mechanism of protein folding for maltose binding protein (MBP), a large multidomain protein, using pulse labeling HDX‐MS.32 An obligate folding intermediate was observed soon after folding was initiated (Figure 2A). This intermediate was comprised of two structurally adjacent helices that form long‐range contacts in the native folded state, as seen in the crystal structure (Figure 2c,d). Together with these helices, the adjacent sheets form the structural core of MBP. As these helices formed, they became protected from exchange resulting in clear bimodal isotopic distributions representing both a protected helical state and an exposed unfolded state (Figure 2e,f). After formation of this intermediate, multiple slow but distinct folding events were observed. These sequential folding events began nearest to the obligate intermediate's structural core and concluded in the most distant parts of the protein (Figure 2b). While Walters et al. were unable to resolve individual discrete folding intermediates from within these events, it was clear that folding radiated outward from the structural core that was present in the fast folding intermediate. The authors of this protein folding pulse deuteration study concluded that for MBP, folding occurs through a single distinct pathway where the obligate fast folding intermediate acts as a seed for the subsequent folding events. Bimodal deconvolution allowed the researchers to resolve how, and on what time scale, each peptide segment folds. If MBP folded by multiple discrete pathways, they would have manifested in the data as uncorrelated and temporally blurred folding events. Each discrete folding event would not be resolvable from one another, therefore the observable folding kinetics for each peptide would be the combination of all folding pathways. Their conclusions are further supported by a subsequent study where Ye et al. examined the effect of mutations on the folding mechanism for MBP by HDX‐MS.34 Mutations made in the hydrophobic cluster of the fast folding intermediate and structural core resulted in dramatically slower folding for the intermediate and all subsequent folding events. The folding rate for the intermediate was roughly 20× slower, whereas the subsequent folding events were only twofold slower thus supporting their earlier conclusions that formation of the intermediate is required for folding to occur.34 They also demonstrated using HDX‐MS, that folding aided by a chaperone abrogated the mutation's effect on folding. HDX‐MS is a sensitive tool to probe the conformational space proteins navigate through. It has been successfully used to identify partially folded intermediate conformations during the protein folding process. The time‐resolved monitoring of structure formation throughout the protein can be used to understand the mechanism of protein folding and unfolding in solution and deepen our understanding of how large, multidomain proteins fold and how this process is modulated by chaperones. HDX‐MS combines high structural resolution with the temporal resolution of quench‐flow spectroscopic techniques enabling for the precise elucidation of how and when folding occurs in proteins.
Figure 2.
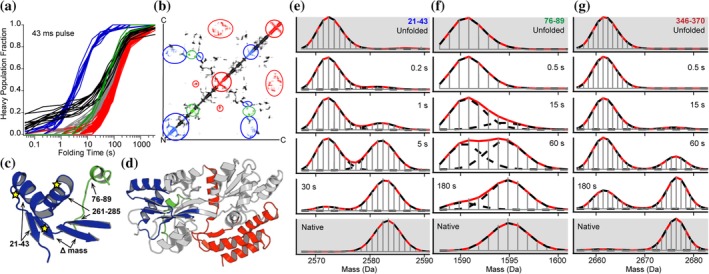
Elucidating the mechanism of protein folding by pulse labeling HDX‐MS. The folding of maltose binding protein (MBP) was monitored by pulse labeling HDX‐MS where unfolded and fully deuterated MBP was diluted to initiate folding. Using a quench flow system the samples were rapidly labeled during folding with a ~millisecond pulse of H2O (D–H exchange) resulting in bimodal isotopic distributions showing the formation of secondary structure, and resistance to exchange, as each peptide folds (e–g). Analysis of all peptides folding kinetics revealed the formation of an obligate “fast folding” intermediate state (blue traces and highlights a–e). Subsequent folding events radiated outward from the structural core formed by the fast folding intermediate and only a single distinct folding pathway was observed (a and b). The contact map in (b) highlights the long‐range contacts formed by the fast folding intermediate and how each subsequent folding event was seeded by the formation of this structural core. Peptide specific folding kinetics mapped onto the crystal structure for the native folded state (c and d) further highlight how peptides nearest to the fast folding intermediate and structural core (c, d, and e blue highlights) fold sequentially radiating outward from the core (c, d, f, and g). Source: Adapted from Reference 32 with permission
3. MONITORING TRANSIENTLY PREFERRED CONFORMATIONAL STATES IN INTRINSICALLY DISORDERED PROTEINS
While it had long been believed that proteins require a well‐folded, ordered native structure to carry out their functions, recent research revealed the ubiquitous nature of intrinsically disordered proteins (IDPs) that play essential roles in cellular signaling, regulation, and have been implicated in disease pathogenesis.35 IDPs are critical for a number of diverse cellular processes and individual IDPs are often functionally promiscuous. The large range of IDP functionalities are likely derived from their inherent conformational plasticity.36 IDPs are challenging to study using HDX‐MS for a number of reasons; significant flexibility and a lack of secondary structure means IDPs require very short, rapid deuteration time points under physiological conditions. IDPs have been shown to rapidly interconvert between ensembles of conformational states and this structural heterogeneity poses additional hurdles during data analysis.37 For example, if an ensemble of conformations are present in solution, multimodal spectra may make ascribing which population in the spectra belongs to which conformer a challenging task.38
The combination of rapid conformational switching along with a lack of secondary structure often means most IDPs are maximally deuterated in mere seconds. In order to get any dynamic information from proteins with such rapid deuteration times researchers have made use of quench‐flow devices to detect transient conformational states of IDPs at very short timeframes.39, 40 Even without access to quench‐flow instrumentation, adjusting the pH can dramatically increase the time window covered by HDX.41, 42 Decreasing the pH by one unit slows the exchange rate 10‐fold, enabling one to probe differences in HDX for even the most exposed amide hydrogens.2, 10, 11 By reducing the pH during the exchange reaction, Goswami et al. were able to compare the dynamics of an IDP responsible for regulating nuclear receptors, PGC‐1α, in the presence and absence of the nuclear receptor PPARγ.41 Initially, they were unable to detect differences in HDX at physiological conditions, however, at pH 6.0 where the labeling rate is dramatically slower, they were able to see how regions of PGC‐1α rapidly fold upon receptor binding. This study is an excellent example of how labeling times may be adjusted to capture protein dynamics that occur on very fast time‐scales; however, care should be taken to ensure the protein's structural dynamics and conformational bias are not altered by lowering the pH.
Pulse labeling HDX has enabled researchers to better understand the mechanism of aggregation for the clinically important IDPs Aβ (amyloid beta) and AS (alpha‐synuclein).43, 44 Aggregation of Aβ and AS is closely linked with progression of their neurological diseases, Alzheimer and Parkinson's disease respectively. By monitoring the structure of these IDPs during aggregation, researchers were able to understand how the appearance of early structural features contribute to the initiation and kinetics of the aggregation cascade. By HDX‐MS these structural seeds for aggregation are visualized as the formation of a long‐lived protected population. Identification of these preferential structural features is critical for understanding the progression of disease and developing effective therapeutics.43, 44 The presence of multiple conformers/oligomeric states frequently generates complex multimodal spectra. While discerning which population belongs to a certain conformer may be very challenging, typically a shift in mass to more protected populations is observed as a result of aggregation.43, 44 Aggregation, especially fibril formation, can cause poor protein sequence coverage due to decreased proteolysis efficiency in bottom‐up HDX‐MS approaches where the protein is proteolyzed before analysis.37, 43, 45, 46 In addition, typically, HDX‐MS data for a given peptide is an average of all states present in the population, meaning if multiple unique states exist but are not resolvable by bottom‐up HDX‐MS workflows, they remain undetected. Furthermore, if all protein states are not proteolyzed with similar efficiencies the resulting distributions seen during analysis can be artificially skewed and misrepresentative of what existed in solution. More recent technological innovations have allowed for combining top‐down HDX‐MS workflows with the gas phase fragmentation technique, electron capture dissociation (ECD), to isolate and analyze individual conformers from within a population. In a top‐down HDX‐MS workflow the whole intact protein is ionized into the mass spectrometer for analysis. Typically, this workflow would be applied to small soluble proteins with a high degree of conformational homogeneity in solution. However, when combined with ECD the capabilities are greatly expanded. ECD functions by fragmenting a protein or peptide inside the mass spectrometer that has been selected for based on mass and charge. ECD enables analysis of the peptide fragments without proteolytic digestion having occluded which unique solution state conformer they originated from. When ionized in a mass spectrometer different protein conformations take on charge differently due to their size and shape, thus allowing one to isolate and analyze target conformers and their peptide fragments.47, 48, 49 ECD also enables fragmentation of a peptide without scrambling of the deuterium label, as is the case with other fragmentation methods, meaning deuterium uptake can be analyzed with single amino acid resolution.1, 47, 48 Pan et al. were able to take advantage of different conformers’ preferences for different charge states and select for specific isotopically labeled Aβ42 conformers using precursor ion selection enabling analysis of the Aβ42 oligomer's HDX profile with single amino acid resolution using ECD.48
The innate conformational plasticity of IDPs enables them to engage multiple partners and modulates a diverse range of functionalities. However, it has proven difficult to understand or identify specific structural features or conformations present in IDPs that give rise to these functions. Interactions with IDPs are often accompanied by a disorder‐to‐order transition, which can be directly monitored by HDX‐MS.50, 51 Like many proteins, CyaA, an adenylate cyclase toxin from bacteria, contains a large intrinsically disordered region (IDR).52 Binding of calmodulin (CaM) to CyaA activates its enzymatic activity, generating toxic levels of cAMP. HDX‐MS was used by O'Brien et al. to elucidate the mechanism of CyaA activation through ligand binding. While no crystal structure of unbound CyaA was available, HDX‐MS revealed an IDR encompassing a 75 amino acid stretch of fast exchanging peptides previously thought to primarily be helical based on the ligand bound crystal structure. Upon binding CaM, large regions of the C‐terminus displayed significant increases in backbone amide protection, suggesting formation of secondary structure confirmed to be helical via CD. This ligand‐induced gain of structure is likely important for adopting a catalytically active conformational state. Regions distal to the binding location, such as around the catalytic site became rigidified upon CaM binding, while the catalytic loop remained highly disordered and solvent accessible (Figure 3a,b). The authors posit that balancing a more ordered catalytic site with a flexible loop may optimize substrate binding and product release, and that structural disorder in CyaA is a mechanism used to inhibit enzymatic activity. To illustrate the benefit of pairing high‐resolution atomic models with a solution state technique, the authors noted a rigid G helix in the ligand bound crystal structure was shown to be highly flexible in solution by HDX‐MS (Figure 3c,d). They surmise the rigidity in the crystal structure may be an artifact of symmetric crystal packing and not representative of the native environment.52 HDX‐MS can be used to characterize structural transitions that often define protein function and control protein–protein interactions. Though HDX data do not provide a three‐dimensional structure of a protein, it provides useful information on how proteins dynamically move in solution and is a more powerful tool when combined with high resolution structures. This ligand induced structural rearrangement is an excellent example of how HDX‐MS can be used to characterize structural transitions that often define protein function and control protein–protein interactions.
Figure 3.
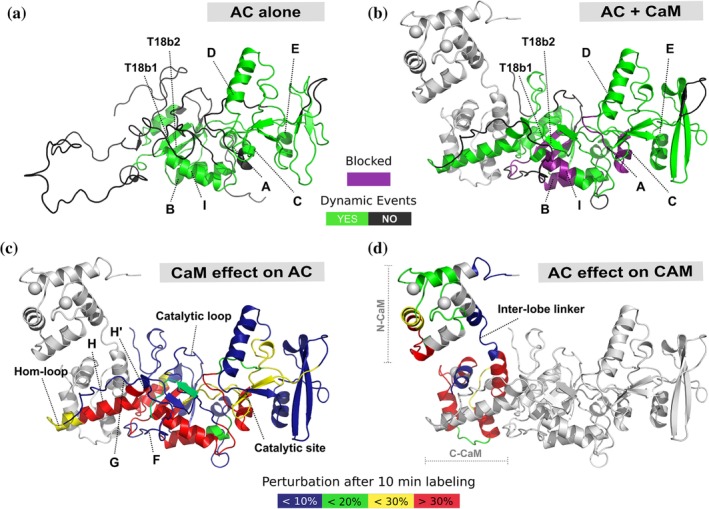
Ligand induced disorder to order transition is critical for catalytic function in CyaA. The structure of free CyaA (AC) eluded previous structural characterization as large sections of the protein are intrinsically disordered. By HDX‐MS it was observed that a 75 amino acid long region, previously seen as helical in the ligand bound structure, was intrinsically disordered in the apo state and becomes ordered upon binding with CaM (a and b). Purple indicates helices with dramatic increases in protection from deuteration, and green indicates regions where a dynamic event occurred upon ligand binding. Differences in percent deuteration on AC and CaM upon binding CaM and AC, respectively, mapped onto the crystal structure (c and d). These data highlight how CaM induces formation of structure in AC upon binding, however that structure is highly dynamic across the H helicies. In contrast the catalytic site remains largely unperturbed as it is primed for high catalytic turnover. Source: Adapted from Reference 52 with permission
4. CONFORMATIONAL SWITCHING THROUGH CORRELATED MOTIONS
It is well appreciated that a protein's three‐dimensional structure dictates its function and induced or spontaneous changes in that structure are critical to that function. Typically, static high‐resolution models are used to visualize protein structure and its related function. However, functionally relevant dynamic changes in protein structure are not always apparent by these methods. One of the powers of HDX‐MS is the ability to identify and resolve induced conformational changes distal to the ligand binding site. A better understanding of allosteric effects can provide important details on protein function. Conversely it can complicate epitope mapping as it is not always apparent what changes in HDX result from protection at the binding site or long‐range compensatory structural changes. Deng et al. demonstrated that HDX data generated from a typical HDX‐MS experiment, where an antibody was preincubated with myoglobin (Mb) and allowed to reach equilibrium, provided data that was unable to differentiate changes in peptide amide protection caused by antibody binding at the immediate binding site from distal allosteric changes.53 In order to better differentiate between the antigen binding site and allosteric effects, the authors utilized a novel kinetic labeling approach using microfluidic chips to perform mixing of small solution volumes. In this configuration, the antibody, antigen, and deuterium were mixed simultaneously prior to quenching and enzymatic digestion. The authors demonstrate the kinetic labeling approach can distinguish the epitope of an antibody against Mb from distal allosteric changes because local allosteric changes in HDX were generally not detectable until ~200 ms, where peptides in regions involved directly in binding Ab displayed an immediate decrease in HDX. It thus appears that the allosteric communication leading to detectable changes in structure may occur more slowly than the immediate increase in protection resulting from a ligand binding event.53 The authors note that this approach proved successful here; however, it has limited scope as the allosteric change they observed was markedly slower than the initial binding event. It will be interesting to determine if this kinetic labeling approach is generalizable to other protein systems as well, or if rates of allosteric propagation in other proteins takes place on timescales too rapid to parse out allosteric changes from ligand binding using HDX‐MS.
Protein–protein interactions can alter a protein's conformational landscape giving rise to new conformational states, and the subsequent changes are often critical for a diverse range of protein functions, including those involved in cellular signaling.7 Tulsian et al. used HDX‐MS along with supporting techniques to probe such binding induced conformational changes in the ternary complex of protein kinase A (PKA), phosphodiesterase (PDE), and cAMP to better understand how cells terminate cellular signaling.54 The two cAMP binding sites on PKA have been well characterized; however, the mechanism of cAMP hydrolysis by PDE was poorly understood. It had puzzled researchers how cAMP tightly bound to PKA could be hydrolyzed by PDE. Monitoring peptides in the cAMP binding sites confirmed PKA alone could only bind cAMP but not hydrolyze it. Following the addition of PDE, they observed hydrolysis and release of cAMP resulting in bimodal mass spectra representing a protected, cAMP bound state, and an exposed apo state. However, over time they observed increased broadening of the mass envelope indicating the presence of a unique conformational state distinct from the apo and cAMP bound states of this ternary complex. The observed HDX profile could not be attributable to linear combinations of spectra corresponding to any individual assembly (Figure 4 top right spectrum). The authors suggest this unique HDX state may arise from an additional structural rearrangement in the RIα‐PDE‐cAMP complex that forms a substrate channel linking the cAMP binding site to the PDE catalytic site as a method to shuttle bound cAMP to the catalytic site for enzymatic hydrolysis.
Figure 4.
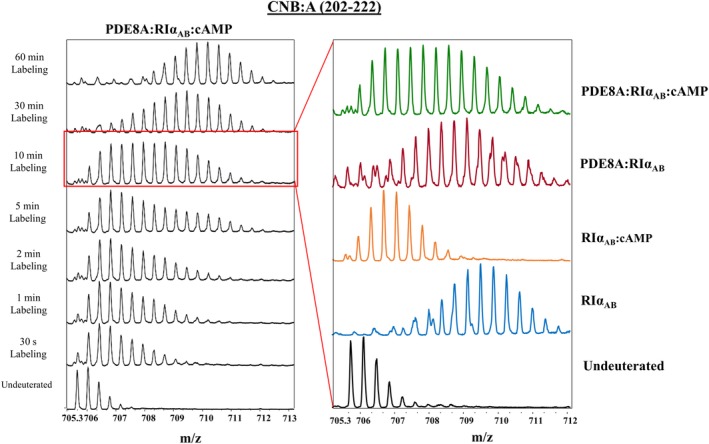
HDX‐MS reveals a novel functional state in protein kinase A ternary complex. Hydrolysis of cAMP by the protein kinase A (PKA) and phosphodiesterase (PDE) ternary complex was monitored in real time by HDX‐MS. A highly broad mass envelope appeared over time in the ternary complex (top right spectra). Analysis of each component and their possible assemblies (spectra shown on the right) could not explain the distribution seen in the ternary complex indicating this is a unique and catalytically active structural state. Source: Adapted from Reference 54 with permission
It has been shown that many proteins require a lipid environment to properly fold into their native functional conformation and many essential protein–protein interactions occur on the surface of membranes with integral and peripheral membrane proteins.50, 55 It is known that this amphipathic environment can have profound effects on the native structure and dynamics of proteins.56 Researchers have tried to recapitulate the native environments of integral and peripheral membrane proteins in vitro using detergent solubilization, lipid micelles and bicelles, liposomes, and lipid nanodiscs.55, 57, 58 While it is not well understood how each of these synthetic lipid systems influence or perturb a protein's structure, dynamics, or function these synthetic systems are invaluable experimental tools for the study of membrane protein structure and function. Recently, the atomic structures of an increasing number of integral and peripheral membrane proteins, often solubilized in detergent, have been solved; and while these structures have proved invaluable in furthering our understanding of membrane protein structure they do not fully convey how these dynamic proteins function. One such integral membrane protein, the P‐glycoprotein ABCB1 transporter (Pgp) facilitates the active transport of a myriad of small molecules across the cell membrane, including drugs, peptides, and lipid‐like molecules through a dynamic conversion between inward and outward facing conformations (Figure 5c).59 Using HDX‐MS to study the conformational dynamics of Pgp, Li et al. identified multiple peptides across Pgp displaying bimodal spectra likely resulting from the inward and outward facing conformations (Figure 5a).59 HDX‐MS was performed in both detergent and lipid nanodiscs to mimic the native environment of this integral membrane protein and to attempt to understand how different synthetic lipid systems influence protein structure, dynamics, and function. A larger number of peptides exhibiting bimodal spectra suggestive of mixed EX1/EX2 kinetics were identified in the nanodiscs compared to detergent (Figure 5b). This observation may suggest the more native membrane‐like nanodisc environment slowed the conformational interconversion rate in local regions enough to resolve multiple conformational states. It is also possible that the lipid packing in nanodiscs restricts protein motion in a way that is not present in detergent.60 Meanwhile, the conformational switching in detergent likely takes place on a faster time scale effectively masking certain peptides indicative of dynamic conformational transitions. Peptides from ligand‐free Pgp displayed bimodals on different timescales; fast conformationally interconverting peptides only displayed bimodals at the earliest time points (10 s and 1 min), moderate peptides displayed bimodals up to 1 hr, and peptides suggesting the slowest conformational transitions displayed bimodal spectra up to 4 hr (Figure 5a). The presence of mixed EX1/EX2 kinetics enabled the authors to extract approximate half‐lives for these intermediate conformations. The authors deduced that the presence of peptides exhibiting structural transitions on different timescales likely implies that Pgp exists not only as the inward or outward facing conformation but instead is more dynamic and fluidly undergoes multiple conformational transitions.
Figure 5.
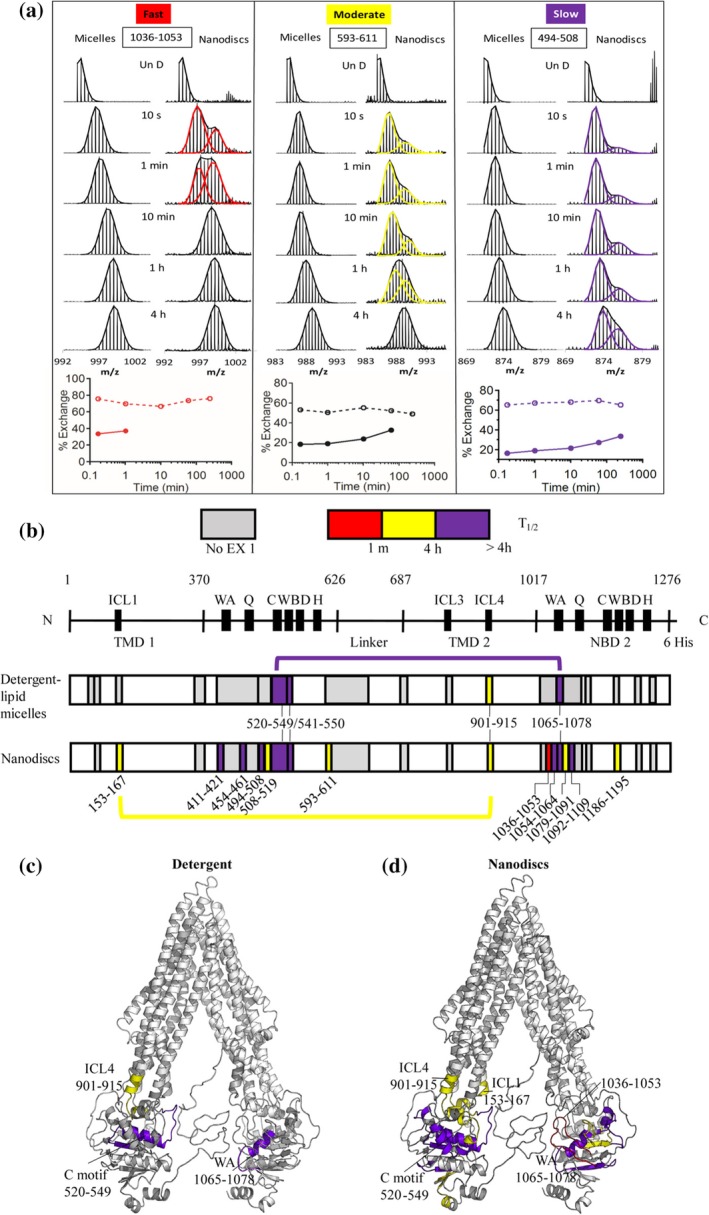
Dynamic conformational changes in the integral membrane protein Pgp. The ABCB1 transporter P‐glycoprotein P (Pgp) reversibly interconverts between inward and outward facing conformational states to facilitate transport of ligand across the cell membrane. HDX‐MS of Pgp in lipid nanodics and detergent micelles revealed conformational transitions occurring on multiple timescales across Pgp indicating the presence of more than two discrete conformational states (a–c). Binomial fitting of bimodal isotopic distributions enabled the deconvolution of multiple coexisting HDX populations and analysis of their conformational kinetics (a). In nanodiscs peptides throughout Pgp exhibited EX1 kinetics on three distinct timescales; fast (red), moderate (yellow), and slow (purple). In contrast Pgp reconstituted in detergent micelles contained fewer peptides exhibiting EX1 kinetics and did not contain those peptides where fast transitions were observed in nanodiscs (a and b). Peptides exhibiting EX1 kinetics mapped onto Pgp structure by their relative timescales (c). Source: Adapted from Reference 59 with permission
While often a protein's functional structure is visualized as a single discrete state, this static image does not represent the ensemble of states present in solution that contribute to that protein's functionality. This is especially relevant for promiscuous proteins, such as the Pgp transporter, which has an incredibly diverse substrate pool or may interact with multiple partners and ligands. By using HDX‐MS to study changes in protein structure, it becomes possible to start to identify and link specific structural changes to specific functional events, to characterize the timescales of protein motion during a conformational change, and to shift the perspective on protein dynamics and conformational change away from a static image to one that captures a fluid, dynamic entity.
5. CONCLUSIONS
HDX‐MS has evolved into a powerful and versatile solution state biophysical and structural technique capable of probing protein structure and motion under native conditions and in complex assemblies. Recent advancements have enabled the investigation of protein folding intermediates, the characterization of transient structure in IDPs, and the real‐time observation of large integral membrane proteins during catalysis. These studies demonstrate how HDX‐MS can be readily applied to study protein structural dynamics in systems with a scale and complexity where other techniques have struggled. As the field continues to evolve, the technique is becoming increasingly powerful and accessible, further expanding the scale and complexity of systems capable of being studied. For example, HDX‐MS has recently been used to study the structural dynamics of proteins on intact enveloped viruses such as dengue virus and influenza virus.61, 62, 63 Similarly, the structural characterization of integral membrane proteins in native membrane environments by HDX‐MS has become commonplace. What were once considered pipedreams of structural biology are now within reach. Furthermore, as HDX‐MS becomes used by a larger population of researchers, significant efforts have been made across the field to standardize experimental procedures and data processing and reporting practices.3, 26
Hodge EA, Benhaim MA, Lee KK. Bridging protein structure, dynamics, and function using hydrogen/deuterium‐exchange mass spectrometry. Protein Science. 2020;29:843–855. 10.1002/pro.3790
Funding information National Institute of General Medical Sciences, Grant/Award Number: R01‐GM099989; National Institutes of Health, Grant/Award Number: T32GM008268‐27; National Institutes of Allergies and Infectious Diseases, Grant/Award Number: R01‐AI140868
REFERENCES
- 1. Benhaim M, Lee KK, Guttman M. Tracking higher order protein structure by hydrogen‐deuterium exchange mass spectrometry. Protein Pept Lett. 2019;26:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Englander WS. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom. 2006;17:1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Masson GR, Burke JE, Ahn NG, et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (hdx‐ms) experiments. Nat Methods. 2019;16:595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sugiki T, Kobayashi N, Fujiwara T. Modern technologies of solution nuclear magnetic resonance spectroscopy for three‐dimensional structure determination of proteins open avenues for life scientists. Comput Struct Biotech J. 2017;15:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fowler ML, McPhail JA, Jenkins ML, et al. Using hydrogen deuterium exchange mass spectrometry to engineer optimized constructs for crystallization of protein complexes: Case study of pi4kiiiβ with rab11. Protein Sci. 2016;25:826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Inglis AJ, Masson GR, Shao S, et al. Activation of gcn2 by the ribosomal p‐stalk. Proc Natl Acad Sci U S A. 2019;116:4946–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bu Z, Callaway D. Proteins move! Protein dynamics and long‐range allostery in cell signaling. Advan Prot Chem Struct Biol. 2011;83:163–221. [DOI] [PubMed] [Google Scholar]
- 8. Percy AJ, Rey M, Burns KM, Schriemer DC. Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry—A review. Analyt Chim Acta. 2012;721:7–21. [DOI] [PubMed] [Google Scholar]
- 9. Bai Y, Milne JS, Mayne L, Englander WS. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weis DD. Hydrogen exchange mass spectrometry of proteins: Fundamentals, methods, and applications. Hoboken, NJ, USA: John Wiley & Sons, Ltd, 2016;p. 295–321. [Google Scholar]
- 11. Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: A new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deng Y, Zhang Z, Smith DL. Comparison of continuous and pulsed labeling amide hydrogen exchange/mass spectrometry for studies of protein dynamics. J Am Soc Mass Spectrom. 1999;10:675–684. [DOI] [PubMed] [Google Scholar]
- 13. Marcsisin SR, Engen JR. Hydrogen exchange mass spectrometry: What is it and what can it tell us? Analyt Bioanalyt Chem. 2010;397:967–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hamuro Y, Coales SJ. Optimization of feasibility stage for hydrogen/deuterium exchange mass spectrometry. J Am Soc Mass Spectrom. 2018;29:623–629. [DOI] [PubMed] [Google Scholar]
- 15. Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein–ligand interactions. Expert Rev Proteom. 2014;8:43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chamberlain AK, Handel TM, Marqusee S. Detection of rare partially folded molecules in equilibrium with the native conformation of rnaseh. Nat Struct Biol. 1996;3:782–787. [DOI] [PubMed] [Google Scholar]
- 17. Ferraro DM, Lazo ND, Robertson AD. Ex1 hydrogen exchange and protein folding. Biochemistry. 2004;43:587–594. [DOI] [PubMed] [Google Scholar]
- 18. Weis DD, Wales TE, Engen JR, Hotchko M, Eyck LF. Identification and characterization of ex1 kinetics in h/d exchange mass spectrometry by peak width analysis. J Am Soc Mass Spectrom. 2006;17:1498–1509. [DOI] [PubMed] [Google Scholar]
- 19. Smith DL, Deng Y, Zhang Z. Probing the non‐covalent structure of proteins by amide hydrogen exchange and mass spectrometry. J Mass Spectrom. 1997;32:135–146. [DOI] [PubMed] [Google Scholar]
- 20. Sivaraman T, Robertson AD. Kinetics of conformational fluctuations by ex1 hydrogen exchange in native proteins. Methods Mol Biol. 2001;168:193–214. [DOI] [PubMed] [Google Scholar]
- 21. Malhotra P, Udgaonkar JB. Tuning cooperativity on the free energy landscape of protein folding. Biochemistry. 2015;54:3431–3441. [DOI] [PubMed] [Google Scholar]
- 22. Engen JR, Wales TE. Analytical aspects of hydrogen exchange mass spectrometry. Ann Rev Analyt Chem. 2015;8:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wales TE, Eggertson MJ, Engen JR. Considerations in the analysis of hydrogen exchange mass spectrometry data. Methods Mol Biol. 2013;1007:263–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guttman M, Weis DD, Engen JR, Lee KK. Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J Am Soc Mass Spectrom. 2013;24:1906–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J, Ramachandran P, Kumar R, Gross ML. H/d exchange centroid monitoring is insufficient to show differences in the behavior of protein states. J Am Soc Mass Spectrom. 2013;24:450–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hudgens JW, Huang R, D'Ambro E. Method validation and standards in hydrogen/deuterium exchange mass spectrometry. Hoboken, NJ, USA: John Wiley & Sons Ltd, 2016;p. 40–50. [Google Scholar]
- 27. Wu Y, Kaveti S, Engen JR. Extensive deuterium back‐exchange in certain immobilized pepsin columns used for h/d exchange mass spectrometry. Analyt Chem. 2006;78:1719–1723. [DOI] [PubMed] [Google Scholar]
- 28. Li K, Chen G, Mo J, et al. Orthogonal mass spectrometry‐based footprinting for epitope mapping and structural characterization: The il‐6 receptor upon binding of protein therapeutics. Analyt Chem. 2017;89:7742–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Englander WS. Protein folding intermediates and pathways studied by hydrogen exchange. Ann Rev Biophys Biomol Struct. 2000;29:213–238. [DOI] [PubMed] [Google Scholar]
- 30. Englander WS, Mayne L, Kan Z‐Y, Hu W. Protein folding—how and why: By hydrogen exchange, fragment separation, and mass spectrometry. Annu Rev Biophys. 2015;45:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Finkelstein AV. 50+ years of protein folding. Biochemistry. 2018;83:S3–S18. [DOI] [PubMed] [Google Scholar]
- 32. Walters BT, Mayne L, Hinshaw JR, Sosnick TR, Englander WS. Folding of a large protein at high structural resolution. Proc Natl Acad Sci. 2013;110:18898–18903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gardner NW, McGinness SM, Panchal J, Topp EM, Park C. A cooperative folding unit as the structural link for energetic coupling within a protein. Biochemistry. 2017;56:6555–6564. [DOI] [PubMed] [Google Scholar]
- 34. Ye X, Mayne L, Z‐y K, Englander WS. Folding of maltose binding protein outside of and in groel. Proc Natl Acad Sci U S A. 2018;115:519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dunker KA, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. FEBS J. 2005;272:5129–5148. [DOI] [PubMed] [Google Scholar]
- 36. Fonin AV, Darling AL, Kuznetsova IM, Turoverov KK, Uversky VN. Intrinsically disordered proteins in crowded milieu: When chaos prevails within the cellular gumbo. Cell Mol Life Sci. 2018;75:3907–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mandell JG, Baerga‐Ortiz A, Falick AM, Komives EA. Measurement of solvent accessibility at protein‐protein interfaces. Methods Mol Biol. 2005;305:65–80.15939994 [Google Scholar]
- 38. Wang G, Abzalimov RR, Bobst CE, Kaltashov IA. Conformer‐specific characterization of nonnative protein states using hydrogen exchange and top‐down mass spectrometry. Proc Natl Acad Sci U S A. 2013;110:20087–20092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Al‐Naqshabandi MA, Weis DD. Quantifying protection in disordered proteins using millisecond hydrogen exchange‐mass spectrometry and peptic reference peptides. Biochemistry. 2017;56:4064–4072. [DOI] [PubMed] [Google Scholar]
- 40. Keppel TR, Weis DD. Analysis of disordered proteins using a simple apparatus for millisecond quench‐flow h/d exchange. Analyt Chem. 2013;85:5161–5168. [DOI] [PubMed] [Google Scholar]
- 41. Goswami D, Devarakonda S, Chalmers MJ, Pascal BD, Spiegelman BM, Griffin PR. Time window expansion for hdx analysis of an intrinsically disordered protein. J Am Soc Mass Spectrom. 2013;24:1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coales SJ, E SY, Lee JE, Ma A, Morrow JA, Hamuro Y. Expansion of time window for mass spectrometric measurement of amide hydrogen/deuterium exchange reactions. Rapid Commun Mass Spectrom. 2010;24:3585–3592. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Y, Rempel DL, Zhang J, Sharma AK, Mirica LM, Gross ML. Pulsed hydrogen–deuterium exchange mass spectrometry probes conformational changes in amyloid beta (aβ) peptide aggregation. Proc Natl Acad Sci U S A. 2013;110:14604–14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Illes‐Toth E, Rempel DL, Gross ML. Pulsed hydrogen‐deuterium exchange illuminates the aggregation kinetics of α‐synuclein, the causative agent for Parkinson's disease. ACS Chem Nerosci. 2018;9:1469–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kheterpal I, Zhou S, Cook KD, Wetzel R. Abeta amyloid fibrils possess a core structure highly resistant to hydrogen exchange. Proc Natl Acad Sci U S A. 2000;97:13597–13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kheterpal I, Lashuel HA, Hartley DM, Walz T, Lansbury PT, Wetzel R. Abeta protofibrils possess a stable core structure resistant to hydrogen exchange. Biochemistry. 2003;42:14092–14098. [DOI] [PubMed] [Google Scholar]
- 47. Pan J, Han J, Borchers CH, Konermann L. Characterizing short‐lived protein folding intermediates by top‐down hydrogen exchange mass spectrometry. Analyt Chem. 2010;82:8591–8597. [DOI] [PubMed] [Google Scholar]
- 48. Pan J, Han J, Borchers CH, Konermann L. Conformer‐specific hydrogen exchange analysis of aβ(1–42) oligomers by top‐down electron capture dissociation mass spectrometry. Analyt Chem. 2011;83:5386–5393. [DOI] [PubMed] [Google Scholar]
- 49. Pan J, Zhang S, Chou A, Borchers CH. Higher‐order structural interrogation of antibodies using middle‐down hydrogen/deuterium exchange mass spectrometry. Chem Sci. 2015;7:1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. [DOI] [PubMed] [Google Scholar]
- 51. Keppel TR, Howard BA, Weis DD. Mapping unstructured regions and synergistic folding in intrinsically disordered proteins with amide h/d exchange mass spectrometry. Biochemistry. 2011;50:8722–8732. [DOI] [PubMed] [Google Scholar]
- 52. O'Brien DP, Durand D, Voegele A, et al. Calmodulin fishing with a structurally disordered bait triggers cyaa catalysis. PLoS Biol. 2017;15:e2004486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deng B, Zhu S, Macklin AM, et al. Suppressing allostery in epitope mapping experiments using millisecond hydrogen/deuterium exchange mass spectrometry. MAbs. 2017;9:1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tulsian N, Krishnamurthy S, Anand G. Channeling of camp in pde‐pka complexes promotes signal adaptation. Biophys J. 2017;112:2552–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vadas O, Jenkins ML, Dornan GL, Burke JE. Chapter seven using hydrogen–deuterium exchange mass spectrometry to examine protein–membrane interactions. Methods Enzymol. 2017;583:143–172. [DOI] [PubMed] [Google Scholar]
- 56. Müller DJ, Wu N, Palczewski K. Vertebrate membrane proteins: Structure, function, and insights from biophysical approaches. Pharmacol Rev. 2008;60:43–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martens C, Shekhar M, Borysik AJ, et al. Direct protein‐lipid interactions shape the conformational landscape of secondary transporters. Nat Commun. 2018;9:4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Redhair M, Clouser AF, Atkins WM. Hydrogen‐deuterium exchange mass spectrometry of membrane proteins in lipid nanodiscs. Chem Phys Lipids. 2019;220:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Li M, Guttman M, Atkins WM. Conformational dynamics of p‐glycoprotein in lipid nanodiscs and detergent micelles reveal complex motions on a wide time scale. J Biol Chem. 2018;293:6297–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harrison RA, Engen JR. Conformational insight into multi‐protein signaling assemblies by hydrogen‐deuterium exchange mass spectrometry. Curr Opin Struct Biol. 2016;41:187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Garcia NK, Guttman M, Ebner JL, Lee KK. Dynamic changes during acid‐induced activation of influenza hemagglutinin. Structure. 2015;23:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lim X‐XX, Chandramohan A, Lim X‐YE, Crowe JE, Lok S‐MM, Anand GS. Epitope and paratope mapping reveals temperature‐dependent alterations in the dengue‐antibody interface. Structure. 2017;25:1391–1402. [DOI] [PubMed] [Google Scholar]
- 63. Wijesinghe KJ, Urata S, Bhattarai N, et al. Detection of lipid‐induced structural changes of the Marburg virus matrix protein vp40 using hydrogen/deuterium exchange‐mass spectrometry. J Biol Chem. 2017;292:6108–6122. [DOI] [PMC free article] [PubMed] [Google Scholar]