

Abstract
Every method used to quantify biomolecular interactions has its own strengths and limitations. To quantify protein‐DNA binding affinities, nitrocellulose filter binding assays with 32P‐labeled DNA quantify K d values from 10−12 to 10−8 M but have several technical limitations. Here, we considered the suitability of biolayer interferometry (BLI), which monitors association and dissociation of a soluble macromolecule to an immobilized species; the ratio k off/k on determines K d. However, for lactose repressor protein (LacI) and an engineered repressor protein (“LLhF”) binding immobilized DNA, complicated kinetic curves precluded this analysis. Thus, we determined whether the amplitude of the BLI signal at equilibrium related linearly to the fraction of protein bound to DNA. A key question was the effective concentration of immobilized DNA. Equilibrium titration experiments with DNA concentrations below K d (equilibrium binding regime) must be analyzed differently than those with DNA near or above K d (stoichiometric binding regime). For ForteBio streptavidin tips, the most frequent effective DNA concentration was ~2 × 10−9 M. Although variation occurred among different lots of sensor tips, binding events with K d ≥ 10−8 M should reliably be in the equilibrium binding regime. We also observed effects from multi‐valent interactions: Tetrameric LacI bound two immobilized DNAs whereas dimeric LLhF did not. We next used BLI to quantify the amount of inducer sugars required to allosterically diminish protein‐DNA binding and to assess the affinity of fructose‐1‐kinase for the DNA‐LLhF complex. Overall, when experimental design corresponded with appropriate data interpretation, BLI was convenient and reliable for monitoring equilibrium titrations and thereby quantifying a variety of binding interactions.
Keywords: allosteric regulation, binding affinity, biolayer interferometry, fructose repressor protein, fructose‐1‐kinase, lactose repressor protein, protein‐DNA binding
1. INTRODUCTION
Biomolecular interactions—such as transcription factors binding to DNA, protein–protein complex formation, and protein small‐molecule interactions—are ubiquitous in living organisms. Therefore, the quantification of biomolecular interactions is central to basic research and drug discovery. The various methods for measuring these interactions, such as nitrocellulose filter binding assays, isothermal titration calorimetry, surface plasmon resonance, enzyme‐linked immunosorbent assay (ELISA), and myriad spectroscopic assays have their own strengths and limitations.1, 2, 3 Some require multiple washes and long incubations, others require high sample consumption, still others have very low through‐put. Some binding reactions do not produce the signal monitored by a given technique. Furthermore, a signal change must be linearly related to the fraction of the bound species,3, 4, 5 which can limit the range of binding affinities that each technique can accurately measure.
We have a long‐standing interest in measuring K d values for various homologs of the LacI/GalR transcription repressor family binding to their cognate DNA operators. Historically, these reactions have been quantified with a nitrocellulose filter binding assay that relies upon 32P‐DNA.6 In that pseudo‐equilibrium assay, various concentrations of protein are mixed with a constant amount of radio‐labeled DNA, allowed to reach equilibrium, and then quickly filtered through negatively‐charged nitrocellulose paper. The protein and protein‐DNA complexes bind to the paper whereas unbound DNA is washed away, and the fraction of 32P‐DNA bound to the protein is used to quantify binding affinity (Figure 1).7 For variants of the lactose repressor protein (LacI) and its homologs, filter binding assays can reliably measure protein‐DNA affinities that range from ~10−13 to 10−8 M.6, 8, 9, 10, 11, 12, 13, 14
Figure 1.
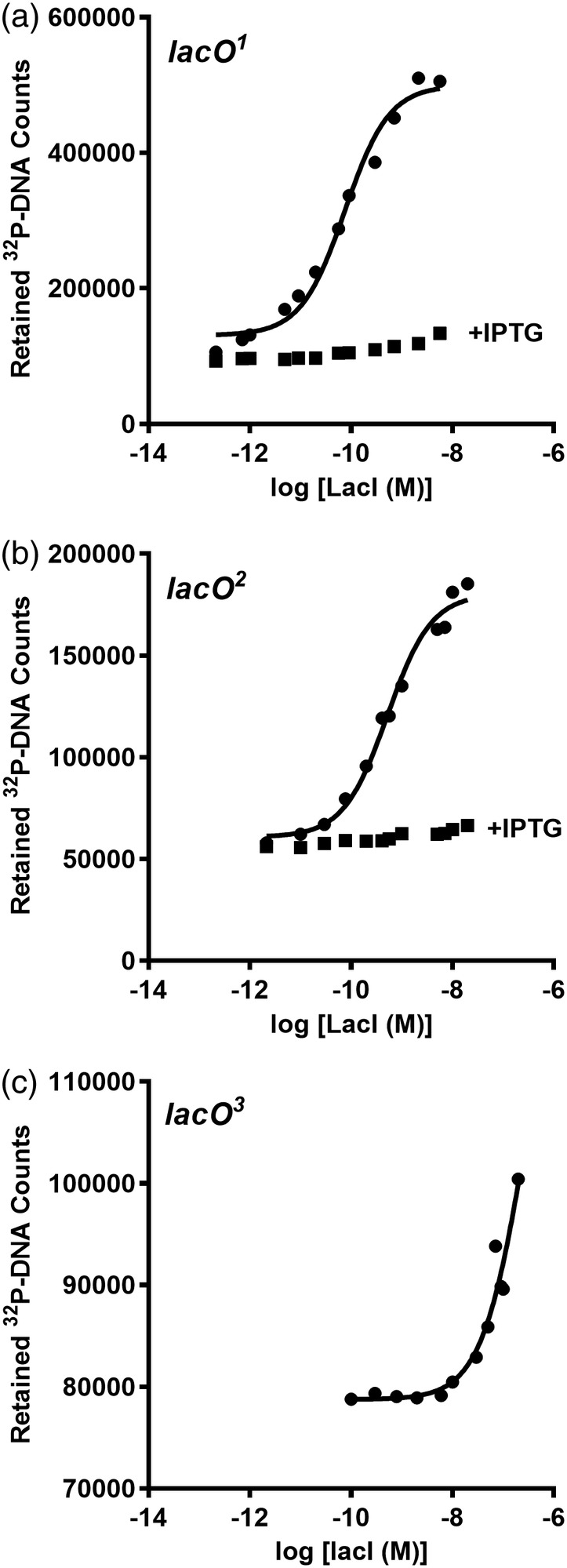
LacI‐DNA binding assayed with filter binding. The filter binding assay was used to monitor LacI binding to (a) lacO 1, (b) lacO 2, and (c) lacO 3 in the absence (●) and presence (■) of 1 mM IPTG. In these experiments, DNA concentration was fixed at 3 × 10−12 M. The solid line represents the best fit of Equation (1) and the values reported in Table 2 are the average and standard deviations of at least three independent experiments with at least two different protein preparations. Filter binding could not be used to determine K d values for the natural lacO 3 operator (c) because the filter was saturated with protein at concentrations above those shown. A lower limit for K d was estimated from the partial binding curve
The filter binding assay has several drawbacks. High protein concentrations can oversaturate the nitrocellulose filter, which limits the ability to measure low‐affinity interactions. The act of filtering perturbs the equilibrium, making it a challenge to quantify interactions with fast dissociation kinetics. The 32P half‐life is short and radiation damages the DNA, which leads to the need to frequently replenish this reagent. Since protein binds to the paper, the method cannot be used to quantify protein–protein interactions. Conversely, some proteins do not bind well to the nitrocellulose paper, so the technique does not work for all protein‐DNA interactions.15 Last, this method is time‐consuming, which is challenging when multiple variants of the repressor proteins need to be evaluated.
Given the limitations of the nitrocellulose filter binding assay, we explored whether we could quantify protein‐DNA interactions using biolayer interferometry (BLI). This technique uses a biosensor tip comprising an optical fiber to which biomolecules can be immobilized. When light is passed through the tip, the wavelength of the reflected light is proportional to the thickness of the bound layer.16 The physical principles underlying BLI are similar to those of surface plasmon resonance (SPR). A key difference is that SPR continuously flows solution over the binding surface, whereas the BLI sensor tip is dipped into successive, constant volumes of solution for each of the binding steps (Figures S1 and S2).17
The first experimental step in a BLI assay is to immobilize one species of the binding reaction, leading to a change in the reflected wavelength (Δλ; Figure S2). Subsequent steps contain additional biomolecules. If they bind to the immobilized species, further changes in the reflected wavelength are observed. Sensor transfer to buffer (or to buffer plus soluble competitor ligand) facilitates dissociation, leading to the opposite effect on Δλ. Thus, the original design of a BLI experiment was to monitor association and dissociation reactions in real‐time to quantify K d from the ratio k off/k on 17 (Figure S1).
To use BLI to monitor protein‐DNA interactions, we immobilized biotinylated‐DNA on streptavidin‐coated biosensor tips and then transferred it to a protein solution. Benchmark experiments used the well‐characterized lactose repressor protein (LacI) and an engineered LacI homolog named “LLhF”18 (Figure 2). Initial binding experiments showed a strong BLI signal change but also revealed a limitation: The association and dissociation curves could not be fit with simple, single‐exponential equations (Figure S3), which precluded the determination of K d from the rate constants.
Figure 2.
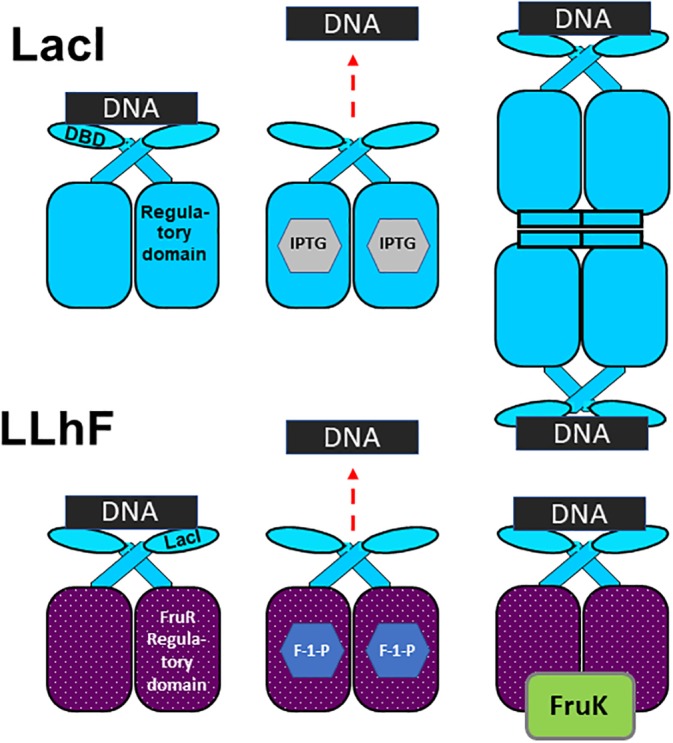
Cartoon depictions of LacI and LLhF binding to DNA operator, small molecules, and heteroprotein. The top row depicts LacI binding to DNA. (Top left) One LacI homodimer binds one DNA operator. The DNA binding domain (“DBD”) and regulatory binding domain are shown on one monomeric subunit. (Top middle) When allosteric inducer IPTG (gray hexagons in the center of each regulatory domain) binds to LacI, DNA binding affinity is diminished. (Top right) The full‐length wild‐type LacI used in this work contains a C‐terminal tetramerization domain that facilitates tetramer formation; each dimer of the tetramer binds one DNA operator. The bottom row depicts the engineered LLhF repressor protein. (Bottom left) One monomer of LLhF comprises the DNA binding domain of LacI and the regulatory domain of the fructose repressor protein (FruR); one homodimer of LLhF binds one operator DNA. (Bottom middle) The small molecule F‐1‐P is shown as hexagons in the regulatory domains of the LLhF dimer. F‐1‐P interactions with LLhF diminish DNA binding affinity. (Bottom right) The LLhF‐DNA complex binds the heteroprotein fructose‐1‐kinase (FruK)
Nevertheless, once repressor‐DNA associations reached equilibrium, we observed that the amplitude of the signal's plateau correlated with the expected fraction of bound protein (Figure 3). Thus, we were motivated to determine whether the amplitude of the BLI plateau at equilibrium—which is reported to be a function of changes to the average optical thickness16 —could be used to monitor equilibrium titration experiments. Previously, Thieker et al. used BLI amplitudes to monitor carbohydrate‐protein titrations and thereby determine binding affinities.19 However, they did not have a well‐established technique available by which to benchmark their measurements. In particular, one must establish whether the concentration of the immobilized substrate is above or below the K d of the binding reaction, because the two scenarios require different data fitting and interpretation.20
Figure 3.
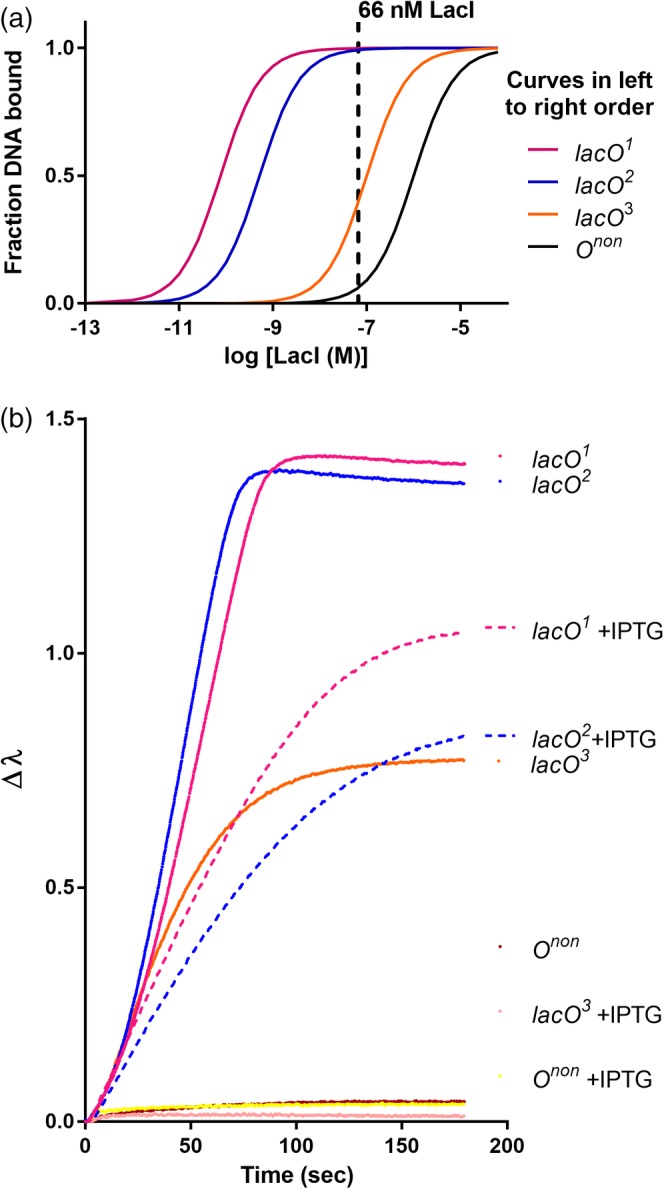
The BLI signal at equilibrium correlates with the fraction of LacI bound to DNA operators. (a) Idealized equilibrium binding curves for LacI binding to different DNA sequences, using the K d values reported in Table 2 for lacO 1 and lacO 2 and estimated K d values of 10−7 M for lacO 3 and 10−6 M for nonspecific DNA (“Onon”; Table SII). The dashed vertical line illustrates how, at a given LacI concentration, each operator has a different percent of LacI bound. (b) BLI was used to monitor 66 nM LacI binding to the same operators, in the presence and absence of 1 mM IPTG inducer. The change in BLI signal (Δλ) was monitored as a function of time until equilibrium was reached. All biosensors were loaded to a similar density of immobilized DNA; for simplicity, the DNA immobilization step is not shown. As expected, signals for lacO 3 + IPTG and Onon ± IPTG and were extremely low in these conditions (bottom of the plot)
Here, we empirically estimated the effective concentration of DNA immobilized on ForteBio High Precision streptavidin (SAX) tips and validated results for protein‐DNA binding reactions against results from filter binding assays. We next validated the BLI approach for quantifying the effects of binding allosteric small molecules to LacI and LLhF, which diminishes DNA binding by several orders of magnitude.21, 22, 23 Finally, we assessed the affinity of DNA‐bound LLhF for the heteroprotein fructose‐1‐kinase (FruK), which could not be assessed using the filter binding assay. Our results demonstrate that when experimental design and data interpretation are appropriately considered, BLI can be used to monitor equilibrium titration experiments and thereby quantify binding affinities for a variety of DNA, protein, and small molecule interactions.
2. RESULTS AND DISCUSSION
To assess whether BLI assays are useful for quantifying repressor‐operator titrations, we first chose to use the well‐characterized, wild‐type lactose repressor protein (LacI8, 9, 10, 11, 12, 13, 14). LacI binds a variety of lac operator DNA sequences (Table 1) with a range of affinities (10−12–10−7 M). Among these are the three natural operators of the lac operon, which bind to LacI with the rank order of lacO 1 > lacO 2 > lacO 3 (Figure 1, Table 2). LacI is a homotetramer composed of two homodimers that are each capable of binding DNA24, 25, 26 (Figure 2). Filter binding assays with dilute, short DNA oligomers isolate the dimer binding event because the flanking DNA is too short for the other dimer of a tetramer to bind. LacI‐DNA binding affinity is allosterically diminished when inducer isopropyl‐1‐thio‐β‐D‐galactopyranoside (“IPTG”) binds to the LacI regulatory domains (Table 3).8, 9, 10, 11, 12, 13, 14, 21, 27
Table 1.
lac operator sequencesa
| Name | Sequence |
|---|---|
| lacO 1 | 5′‐ t g t t g t g t g g A A T T G T G A G C G G A T A A C A A T T t c a c a c a g g‐3' |
| lacO 2 | 5′‐ t g t t g t g t g g A A A T G T G A G C G A G T A A C A A C C t c a c a c a g g‐3' |
| lacO 3 | 5′‐ t g t t g t g t g g A A C A G T G A G C G C A A C G C A A T T t c a c a c a g g‐3′ |
lacO 1, lacO 2, and lacO 3 are naturally‐occurring operators in the lac operon.48, 49, 50, 52 Bases shown in capital letters were protected from DNase foot printing by LacI binding53, 54; sequences shown in lower case comprise the flanking sequences of the double stranded 40‐mer oligos used in filter binding assays. The bases shown in red differ from the analogous positions in lacO 1. 45‐mer oligos were used in the BLI assay and all three operators were biotinylated at the 5′ end of one strand (Table SII).
Table 2.
K d values for repressor‐DNA bindinga
| Assay | Filter binding | BLI | Fold‐change | ||||
|---|---|---|---|---|---|---|---|
| Repressor | DNA | Avg (M) | Error | Avg (M) | Error | Avg (M) | Error |
| LacI | lacO 1 | 4.5 × 10−11 | 2.8 × 10−11 | n.d | — | — | — |
| lacO 2 | 5.2 × 10−10 | 0.3 × 10−10 | 2.7 × 10‐11 b | 1.6 × 10−11 | 0.05 | 0.14 | |
| lacO 3 | ≥10−7 | — | 0.5 × 10−7 | 0.2 × 10−7 | — | — | |
| LLhF | lacO 1 | 0.4 × 10−8 | 0.1x10−8 | 1.8x10−8 | 0.3 × 10−8 | 4.80 | 0.54 |
| lacO 2 | 4.1 × 10−8 | 3.2 × 10−8 | 4.8 × 10−8 | 1.9 × 10−8 | 1.15 | 0.94 | |
| lacO 3 | ≥10−6 | — | ≥10−5 | — | — | — | |
Abbreviations: BLI, biolayer interferometry; n.d., not determined.
All values are reported in M dimeric repressor. Average (Avg) values were determined using at least three independent determinations comprising at least two independent protein purifications. Reported errors represent one standard deviation of the mean. Fold‐change values were determined by dividing the BLI value by the filter binding value, and errors were propagated from the experimental data. All filter binding assays used a fixed DNA concentration of ~3 × 10−12 M DNA; all BLI assays used 80 nM of the indicated DNA for the immobilization step.
Estimated from simultaneous fits of replicate assays in the intermediate binding regime (Figure S7); the error shown is the standard error of the fit; as described in the text, the 95% lower confidence limit was unbounded, the upper limit was 7.2 × 10−11 M.
Table 3.
Midpoint values for allosteric ligands and fructose‐1‐kinase (FruK) binding to repressor‐DNA complexesa
| Assay | [Repressor] (M) | Filter binding | BLI | Fold‐change | |||
|---|---|---|---|---|---|---|---|
| Avg (M) | Error | Avg (M) | Error | Avg (M | Error | ||
| lacO 1 /LacI + IPTG | 3.0 × 10−9 | 2.8 × 10−6 | 0.9 × 10−6 | 23 × 10−6 | 4 × 10−6 | 8.34 | 1.08 |
| lacO 1 /LLhF + F‐1‐P | 10.0 × 10−9 | 15 × 10−6 | 9 × 10−6 | 16 × 10−6 | 5 × 10−6 | 1.04 | 0.68 |
| lacO 1 /LLhF + FruK | 10.0 × 10−9 | n.d. | — | 1.1x10−8 | 0.3 × 10−8 | — | — |
Abbreviations: BLI, biolayer interferometry; n.d., not determined.
Average (Avg) values were determined using at least three independent determinations using at least two independent protein purifications. Reported error values represent one standard deviation of the mean/error propagation. All filter binding assays used a fixed DNA concentration of 3 × 10−12 M DNA; all BLI assays used 80 nM of the indicated DNA for the immobilization step.
As a second repressor‐operator example, we chose the engineered “LLhF” chimera comprising the DNA binding domain of LacI fused to the regulatory domain of the fructose repressor protein (“FruR,”18 Figure 2). Similar to LacI, LLhF affinity for various lac operator sequences is allosterically diminished when the inducer fructose‐1‐phosphate (“F‐1‐P") binds to its FruR regulatory domains.18, 23 Extrapolating the relationship between in vivo repression assays and in vitro binding affinities,12, 13, 18, 28 LLhF was expected to have weaker DNA binding affinity than LacI. Since the homodimer is the minimal DNA binding unit for all characterized LacI/GalR proteins (reviewed in Reference 27), we presumed that LLhF behaved accordingly. LLhF also binds the hetero‐protein FruK (Supplementary Methods, Table SI 18, 29), and this interaction was used as a model system for quantifying the interactions of a heteroprotein binding to a protein‐DNA complex with BLI.
As noted in the introduction, our attempts to use association and dissociation kinetics to determine k on, k off, and K d showed that both association and dissociation kinetics were multi‐phasic (Figure S3) and that the dissociation reaction did not return to the baseline. Despite extensive efforts (briefly discussed in the legend to Figure S3), we were unable to confidently assign the kinetic phases to specific molecular events, match previously‐published rate constants,30, 31 or achieve complete dissociation. Nevertheless, we noted that when LacI association reactions reached equilibrium, the amplitudes of the BLI plateaus (Figure 3b) correlated with the expected fraction bound DNA determined from filter binding assays (Figure 3a); similar results were obtained for LLhF (Figure S4). Thus, we explored whether the amplitudes of the BLI equilibrium plateaus could be used in titration experiments to assess binding affinities.
To that end, we explored five pairs of binding partners with affinities expected to be in a range measurable by both filter binding and BLI techniques: LacI‐lacO 2, LacI‐lacO 3; LLhF‐lacO 1, LLhF‐lacO 2, and LLhF‐lacO 3. Results are shown in Figures 1, 4, 5, 6 and Table 2. A variety of technical limitations and assessments of reproducibility are discussed in Methods. Of particular note, in some experiments, the BLI plateaus appeared to decrease over time (Figure S5). We attributed this to protein degradation over the course of the experiment, which may also occur undetectably in the filter binding assays. However, when the BLI data were sampled for equilibrium fitting at different time points, the decrease had very little effect on K d values (Figure S5).
Figure 4.
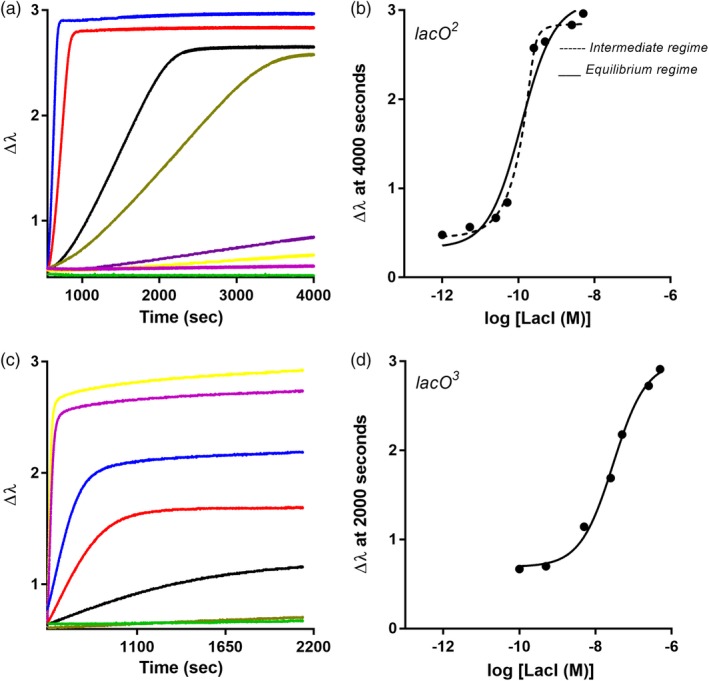
LacI‐DNA binding assayed with BLI. Biolayer interferometry was used to monitor LacI binding to (a,b) lacO 2, and (c,d) lacO 3 using an eight‐channel BLI instrument. Similar to filter binding assays, the concentration of LacI was varied (different color curves in a and c) and the DNA concentration was constant. In each experiment, DNA was loaded onto the BLI sensor tips from a solution of 80 nM; for simplicity, this step is not shown on the plot. When the repressor‐operator binding reactions reached equilibrium, plateau values at (a) 4,000 s and (c) 2000 s were plotted against log[LacI] to generate the titration curves shown in (b) and (d), respectively. Solid lines are the best fit of Equation (1) to the data, and the dashed line represents the best fit of Equation (2). The experiments shown are representative of at least three replicates with at least two different protein preparations
Figure 5.
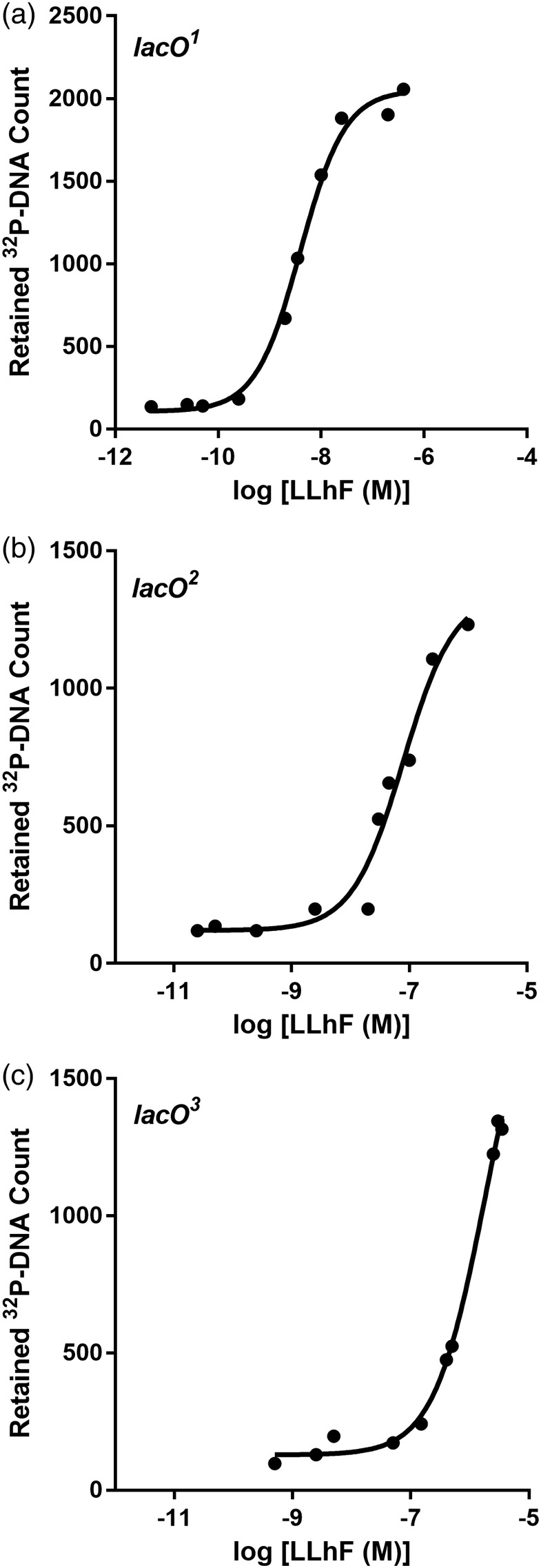
LLhF‐DNA binding assayed with filter binding. The filter binding assay was used to monitor LLhF binding to (a) lacO 1, (b) lacO 2, and (c) lacO 3. In these experiments, DNA concentration was fixed at 3 × 10 −12 M. Only partial binding curves could be obtained for lacO 3 due to limitations on the LLhF concentration that could be obtained from a single protein preparation; this partial curve was used to determine a lower limit for K d. The curves shown are representative of at least three replicates with at least two different protein preparations. The solid lines are the best fit of Equation (1) to the data
Figure 6.
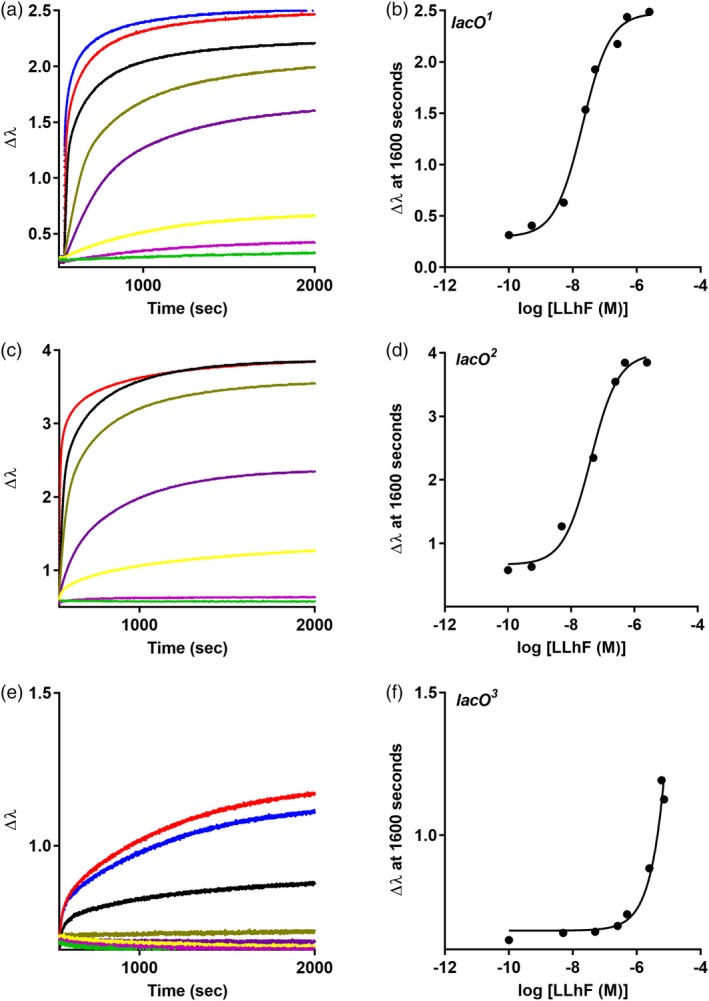
LLhF‐DNA binding assayed with BLI. Biolayer interferometry was used to monitor LLhF binding to (a‐b) lacO 1, (c‐d) lacO 2, and (e‐f) lacO 3 using an eight‐channel BLI instrument. Similar to filter binding assays, the concentration of LLhF was varied (colored curves in a, d, and e) and the DNA concentration was kept constant. In each experiment, DNA was loaded onto the BLI sensor tips from a solution of 80 nM; for simplicity, the DNA immobilization step is not shown. When the repressor‐operator binding reactions reached equilibrium, plateau values at 1600 s were plotted against log[LLhF] to generate the titration curves shown in (b), (d), and (f). Solid lines are the best fit of Equation (1) to the data. The experiments shown are representative of at least three replicates with at least two different protein preparations
Qualitatively, all repressor‐DNA binding isotherms showed the expected rank order of operator binding for each protein. Likewise, for each operator, LacI had tighter binding than LLhF. Most of the binding curves showed the expected sigmoidal character (see Reference20 and discussed further below) and fits of replicate experiments yielded highly similar values. The exception to these trends was LacI‐binding to lacO 2. Thus, in the sections below that discuss the individual reactions, LacI‐lacO2 is presented first to provide the context necessary to confidently interpret all other binding reactions. Note that for all repressor‐DNA binding reactions, K d values are reported in M dimer.
2.1. LacI binding to lacO2
By the filter binding assay, the affinity of LacI‐lacO 2 is the tightest of the five pairs described above, with a K d of 5 × 10−10 M (Figure 1a, 1b). However, titrations monitored with BLI deviated from the expected sigmoidal binding curve (Figure 4a,b), which indicated that the experimental conditions precluded direct determination of K d from the midpoint. For any binding reaction, the relationship between the concentration of the fixed component (here immobilized DNA) and the K d value affects the curve shape and the subsequent data fitting and interpretation. Depending upon the experimental design, an equilibration titration reaction can be carried out in the “equilibrium binding regime” or in the “stoichiometric binding regime” or in a regime intermediate to the two conditions.
The equilibrium binding regime is achieved when the “constant” component in the binding reaction (in this case, immobilized DNA) is ≤10‐fold than the K d.20 This allows several simplifying assumptions to be made in the derivation of Equation (1) (see legend to Figure S6). When the binding partner (repressor protein) is varied and the binding data are plotted as a semi‐log graph, the result is a sigmoidal curve (Figure S6) that can be fit with:
| (1) |
where “Y” is the observed signal, “c” is the baseline value when the concentration of protein is zero, “Y max” is the signal observed at saturation, “X” is the log of the protein concentration (transformed to improve data point weighting in nonlinear regression) and “K d” is the equilibrium dissociation constant. In filter binding assays, 32P‐DNA allows extremely low concentrations (10−12–10−13 M) of DNA to be used, which ensures that assays are in the equilibrium binding regime.
The stoichiometric binding regime occurs when the concentration of the constant component is >10‐fold greater than K d.20 In these conditions, each addition of the varied component (repressor) results in its complete binding until the constant component is saturated, and the signal no longer changes; the concentration at which saturation occurs can be used to determine binding stoichiometry. The intermediate binding regime occurs when the fixed concentration is near the K d value. Because the simplifying assumptions used to derive Equation (1) do not apply to the stoichiometric or intermediate binding regimes, these binding curves must be fit with an expanded equation32:
| (2) |
where “Y,” “c,” “Y max,” “X,” and “K d” are as defined above, “n” is the stoichiometry of the complex (here, n = 1), and “DT” is the total concentration of DNA.
In BLI assays, the LacI‐lacO 2 binding curve was not well fit with Equation (1) (Figure 4b), which indicated that the effective DNA concentration was near or above the K d for this binding event. ForteBio reports that each fiber optic tip has ~109 streptavidin molecules dispersed across its surface.33 Assuming that all streptavidin molecules bind biotinylated DNA and considering the total reaction volume of 250 μL, the DNA concentration was calculated to be 1.6 × 10−14 M, which is well below the tightest known K d values of LacI‐DNA interactions. However, when bound to the biosensor tip, DNA is immobilized into a very small local volume, which increased its effective concentration by an unknown amount. Thus, we used Equation (2) to estimate the effective DNA concentration and K d.
When Equation (2) was used to independently fit individual LacI‐lacO 2 binding curves, the resulting curve shape was a better fit to the data than that obtained with Equation (1) (Figure 4b). However, the number of parameters in Equation (2), combined with the limited number of data points per experiment, precluded reliable parameter estimation; the 95% confidence limits of the fits were unbounded for both the DNA concentration and K d. Furthermore, the curves from replicate experiments varied on different days (Figure S7), which would occur if different batches of sensor tips had varied amounts of immobilized DNA available for binding repressor. Thus, we globally fit 11 replicate experiments (carried out over multiple months with multiple lots of sensor tips) using Equation (2) with n = 1, a globally shared parameter for K d, and independent parameters for the DNA concentration (Figure S7).
From this analysis, the effective DNA concentration of replicate experiments ranged from 2 × 10−10 to 6 × 10−9 M (Figure S7F). The most frequent value was ~2 × 10−9 M. As expected, all DNA concentrations were near or greater than the K d for LacI‐lacO 2 determined with filter binding (5 × 10−10 M, Table 2).
The variation in effective DNA concentration may arise from the uncertainties of using Equation (2), and its four floating parameters, to fit the data. However, the fit errors were reasonable for this parameter (Figure S7F) and the 95% confidence limits were bounded. Thus, we find it more likely that the variation arose from lot‐to‐lot variation or aging sensor tips. The variation could not be detected from the BLI amplitude of the DNA loading step; these values were generally within 10% of each other. Assuming that similar amplitudes indicate that similar amounts of DNA were immobilized, the variation in effective concentration may arise from altered orientations or spacings of the DNA‐streptavidin complexes or from interactions of DNA with the tip surface. Reassuringly, for a given lot of sensor tips, the effective DNA concentration could be purposefully diminished by diluting the biotinylated DNA 1:1 with free biotin prior to immobilization (Figure S7D, E) to block half of the streptavidin sites from binding DNA. The decrease in the immobilized DNA could be observed in both the amplitude of the binding curve (“Y max”) and in the fitted value for DNA concentration (Figure S7F).
The lot‐to‐lot variation in the DNA concentration should not be a problem for any binding event in the equilibrium binding regime. For these types of events, the variation should be detected in the maximum signal (“Y max”) but would not alter K d. Inspection of other repressor‐operator binding events showed this to be the case (Figure S8). Indeed, the variation in maximal BLI signal parallels the variation in maximum radioactivity in a filter binding assay that arises from 32P decay.
Next, we considered the K d value determined with BLI for LacI‐lacO 2 binding, which was 2.7 ± 1.6 × 10−11 M. This value was ~20‐fold tighter than the value obtained with filter binding (5 × 10−10 M, Table 2). We first considered whether the “aberrant” BLI value was an artifact of fitting with Equation (2), as considered by Reinhart et al.32 However, equivalent K d values were consistently obtained for LacI‐lacO 2, regardless of which subsets of experiments were used for fitting. Alternatively, the LacI BLI K d value could indeed be tighter if the immobilized DNA molecules were positioned sufficiently close to allow simultaneous binding from two dimers of a LacI tetramer. The high local concentration for binding the second site would enhance overall binding. As described more extensively in the section below (for LacI binding to lacO 3), we concluded that this was indeed occurring.
In conclusion, both the shape of the semi‐log plots and any of the K d values (obtained with filter binding or BLI) support the conclusion that the LacI‐lacO 2 reaction was not in the equilibrium binding regime in BLI assays. Notably, non‐sigmoidal binding curves would be extremely hard to detect if the data were on a linear rather than a semi‐log plot. Since the DNA density is dictated by the density of streptavidin binding sites on the SAX BLI tips, we expect that the range of effective DNA concentrations will extrapolate to any other immobilized species. Thus, these tips should be suitable to assess binding affinities using Equation (1) for events with K d values >10−8 M. For binding events with K d < 10−8 M, estimates could be obtained from global fitting of multiple replicates with Equation (2).
2.2. LacI binding to lacO3
As shown by its incomplete binding curve, LacI binding to lacO 3 was too weak to quantify with the filter binding assay (Figure 1c); based on the highest concentration of protein used, the binding affinity appeared to be ≥10−7 M (Table 2). In contrast, BLI assays for LacI binding to lacO 3 were well fit with Equation (1) and yielded a K d value of 0.5 ± 0.2 × 10−7 M (Figure 4c, d, Table 2). This value would place the binding event in the equilibrium binding regime for BLI, but the binding affinity was again tighter than expected from filter binding results. Thus, we further considered the possibility that the K d of LacI for lacO 3 was affected by two dimers of a LacI tetramer simultaneously binding two immobilized DNA in the BLI assay.
According to ForteBio,33 each fiber optic sensor tip has ~109 streptavidin molecules distributed across the 0.6 mM tip, which has a surface area of 2.8 × 1013 Å2. From this, we estimated that the distance between equally distributed streptavidin molecules is ~170 Å. Each dimer of a LacI tetramer is ~75 Å long34; the two dimers of a tetramer are tethered by a helical bundle34 and form a flexible V‐shape with an angle that can vary from ~23° to >120°35; and the length of each 45‐mer DNA is >140 Å. Furthermore, LacI also has reasonable affinity for nonspecific DNA,36, 37, 38 and the second binding event is not limited to the 16 base pair operator centered within the 45‐mer DNA (Table SII). Thus, we concluded that a LacI tetramer could easily bind two immobilized DNA molecules and that this increased the apparent affinity in the BLI assay for LacI binding to either lacO 2 or to lacO 3 (Table 2).
The LacI‐lacO 3 binding event also illustrated a technical limitation of BLI that is likely to be protein specific. We attempted several experiments with an uppermost LacI concentration of 3 × 10−5 M; this concentration always yielded a “bad” point (Figure S9). We concluded that these concentrations of LacI were beyond the reliable range of the technique. Since the BLI signal is related to the average optical density, and since tetrameric LacI is a large protein (154 kDa), we expect that the upper concentration limit would be different for smaller proteins.
2.3. LLhF binding to lacO1, and lacO2, and lacO3
Unlike LacI, the chimeric repressor LLhF lacks a C‐terminal tetramerization domain and has weaker DNA binding for the dimeric unit. Thus, DNA binding reactions for this chimera should be in the equilibrium binding regime for BLI assays, and results for both filter binding and BLI assays should reflect dimer (not tetramer) binding to DNA. Like LacI, the association and dissociation curves for LLhF binding to DNA were multiphasic and could not be used to determine K d. Thus, we again examined K d values determined from the equilibrium amplitudes of the BLI signals.
Indeed, the K d values were in good agreement for the two techniques (Table 2). Using filter binding, complete LLhF binding curves were obtained for lacO 1 and lacO 2, but lacO 3 binding was too weak to reach saturation (Figure 5, Table 2). Using BLI, full titration curves were obtained for LLhF binding to lacO 1 and lacO 2 (Figure 6, Table 2). LLhF‐lacO 2 binding was in excellent agreement between the two methods. The filter binding value for LLhF‐lacO 1 (a) was slightly tighter than the BLI value and (b) did not quite meet the criteria required for the equilibrium binding regime. However, the shape of the curve was well fit with Equation (1), and < 5‐fold change between the two values from different methods was not striking. Again, LLhF binding to lacO 3 did not reach saturation in the BLI assay; in this case, it was because the maximum concentration was limited by the amount of LLhF protein that could be purified from a single preparation. This limited the range of K d values that any technique (not only BLI) could determine for this protein.
2.4. LacI and LLhF DNA binding in the presence of allosteric ligands
DNA binding by both LacI and LLhF is allosterically diminished by several orders of magnitude in the presence of small‐molecule inducers (Figures 7 and 8).8, 9, 10, 11, 12, 13, 14, 18, 21, 23, 27 The loss of DNA binding can be monitored as a function of inducer concentration, and the midpoint of this “operator release” curve is a composite of both the repressor's affinity for the inducer molecule and the allosteric linkage to DNA binding.10, 14, 39 Prior studies used the filter binding assay to determine the midpoint of operator release.10, 14, 39 This assay has been used to determine whether an amino acid mutation alters allosteric regulation in LacI.9, 10, 14 Experimental design for operator release studies must consider multiple binding events: The repressor‐operator complex—which is the fixed component of this reaction—should have a concentration that (a) in the absence of inducer, most of the protein binds DNA; (b) in the presence of inducer, most of the protein dissociates from DNA; and (c) is at least 10‐fold below the K d for inducer binding to the complex. To determine whether BLI assays could recapitulate these measurements, we again immobilized the DNA but fixed the repressor protein concentration and varied the inducer concentration over the range known to bind repressor protein.9, 10, 23
Figure 7.
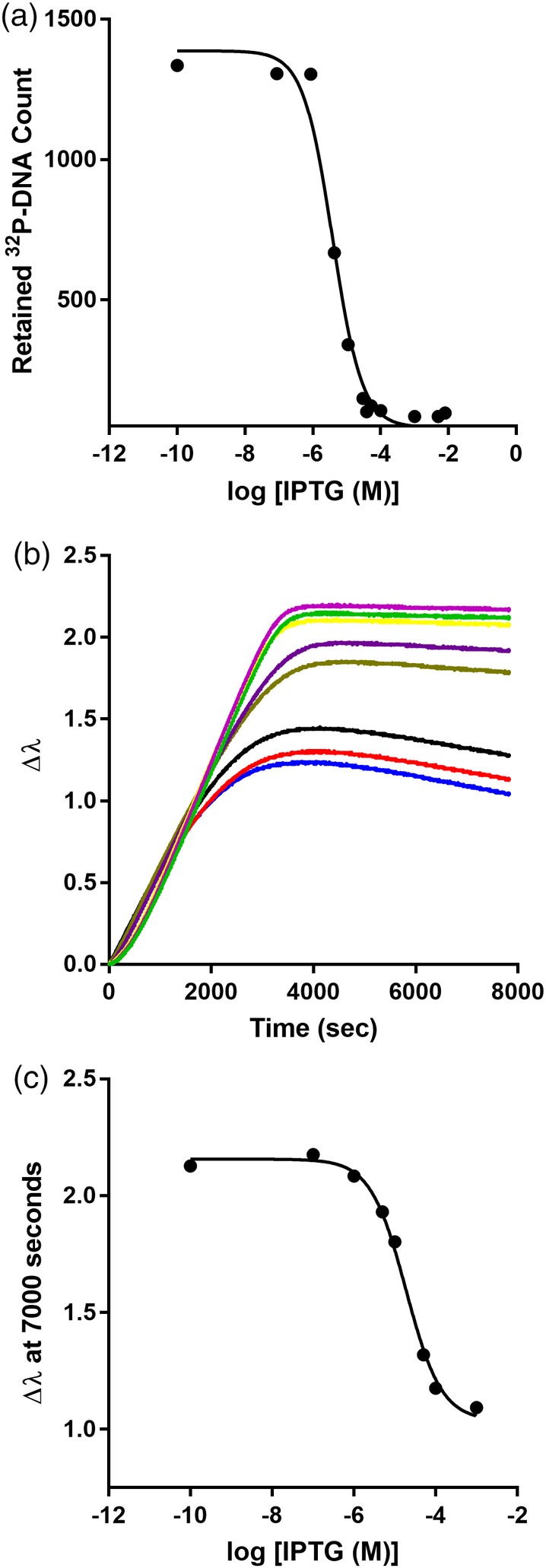
LacI‐DNA binding is diminished upon binding inducer IPTG. (a) IPTG titrations monitored with filter binding were carried out using 3 × 10−12 M lacO 1 DNA and 3 nM LacI. (b) IPTG titration monitored with an eight‐channel BLI instrument. DNA was loaded onto the BLI sensor tips from a solution of 80 nM and the concentration of LacI was fixed at 3 nM. When the reactions reached equilibrium, plateau values at 7000 s were used to obtain the titration curve in (c). The solid lines in (a) and (c) represent the best fit of Equation (1). Data shown are representative of at least three independent measurements with at least two different protein preparations
Figure 8.
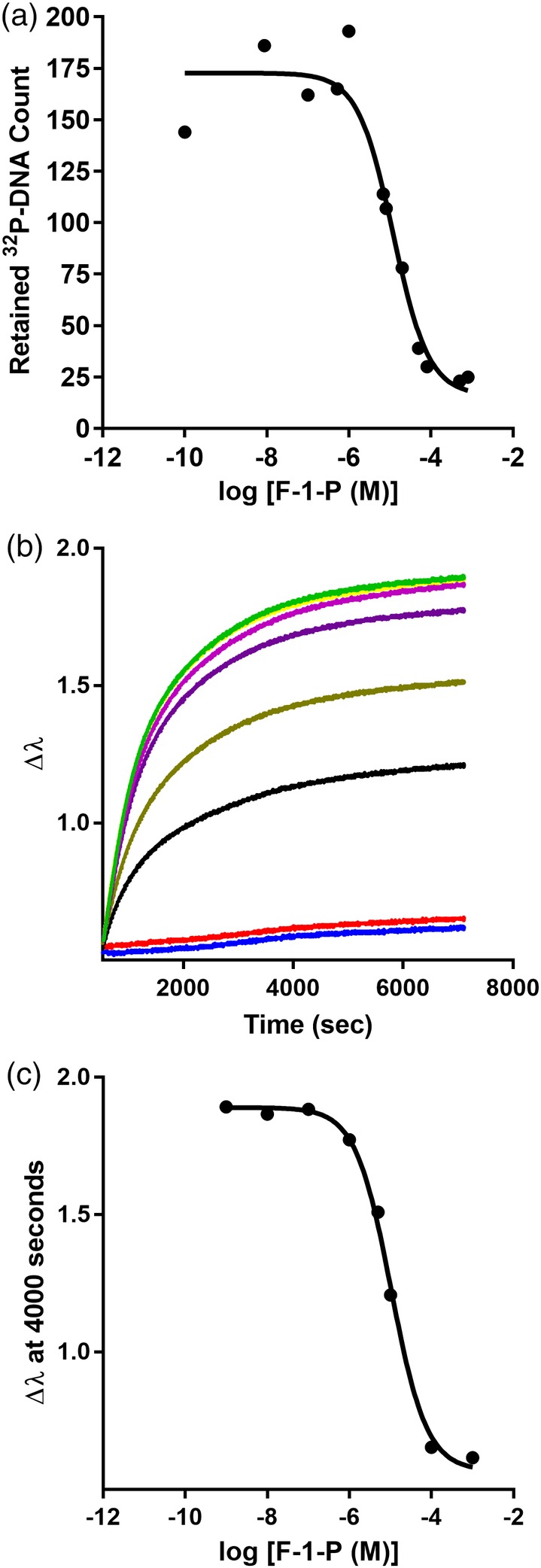
LLhF‐DNA binding is diminished upon binding inducer F‐1‐P. (a) F‐1‐P titrations monitored with filter binding were carried out using 1.5 × 10−12 M lacO 1 DNA and 10 nM LLhF. (b) IPTG titration monitored with an eight‐channel BLI instrument. DNA was loaded onto the BLI sensor tips from a solution of 80 nM and the concentration of LLhF was fixed at 10 nM. When the reactions reached equilibrium, plateau values at 4000 s were plotted to obtain the titration curve in (c). The solid lines in (a) and (c) represent the best fit of Equation (1). Data shown are representative of at least three independent measurements with at least two different protein preparations
Historically, LacI filter binding evaluations of midpoint of operator release have been performed with 10−10–10−11 M repressor protein and < 10−11 M lacO 1 operator.9, 10, 14 However, these repressor concentrations were too low to generate signal in BLI. Thus, we used 3 nM LacI—the lowest value that could be monitored with BLI—and compared results from the filter binding and BLI amplitude techniques (Table 3). The filter binding midpoint was in good agreement with previously published values.8, 9, 10, 11, 12, 13, 14 However, the filter binding and BLI midpoints differed by nearly 10‐fold (Table 3). We expect that this complication arises from the BLI LacI‐lacO 1 assay being in the intermediate or stoichiometric binding regime in both the absence and presence of inducer: LacI‐lacO 1 has even tighter binding than LacI‐lacO 2 (Table 2), and LacI‐lacO 1 + IPTG has tighter binding than LacI‐lacO 3 (Figure 3).
Fortunately, we were able to explore operator release in the equilibrium binding regime using LLhF and lacO 1 (Table 3). In this regime, the values for the two types of assays were statistically equivalent and comparable to values previously estimated for F‐1‐P releasing wild‐type FruR from its DNA operator.23 Thus, when the repressor‐DNA concentrations are in the equilibrium binding regime, BLI appears to be well‐suited for operator release experiments.
2.5. FruK binding to LLhF‐DNA complex
In prior experiments with LLhF, DNA pull‐down experiments from crude cell extracts18 and mass spectrometry (see Methods) showed that this protein formed a complex with FruK. We wished to verify the interaction by a second technique using purified proteins. Since hetero‐protein interactions cannot be quantified with filter binding assays (both free and bound protein would stick to the filter), we used BLI to monitor equilibrium titrations of the LLhF‐DNA complex and FruK.
For the LLHF‐FruK titration, the concentration of LLhF was fixed at 10 nM and pre‐incubated with various concentrations of FruK prior to binding immobilized lacO 1. From these data, the [FruK]midpoint was 1.1 ± 0.3 × 10−8 M (Figure 9). This midpoint is ~10‐fold greater than the maximum possible concentration of immobilized protein‐DNA, so binding was likely in the equilibrium binding regime. Note that, as with the operator release experiments, the FruK binding assays did not discriminate whether the midpoint (a) corresponded to K d for FruK binding to the LLhF‐DNA complex or (b) was a composite of FruK binding and altered LLhF‐DNA binding in the presence of this heteroprotein. Interestingly, since FruK also binds to wild‐type FruR (the LLhF parent protein also known as “Cra”29), this reasonably strong affinity indicates that FruK might play a physiological role in regulating central E. coli metabolism.
Figure 9.
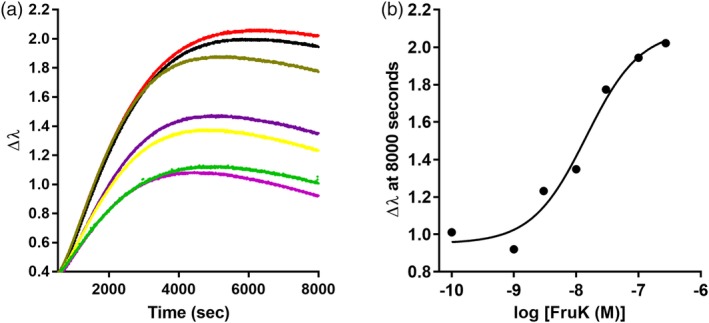
Fructose‐1‐kinase (FruK) binding to the LLhF‐DNA complex assayed with BLI. (a) Biolayer interferometry was used to monitor FruK binding to the LLhF‐lacO 1 complex using an eight‐channel instrument. Varied concentrations of FruK were pre‐mixed with a constant concentration of LLhF (10 nM); DNA concentration was also constant. DNA was loaded onto the sensor tip from a solution of 80 nM and then incubated with LLhF‐FruK mixtures. When the reactions came to equilibrium, plateau values at 8000 s were plotted against the log[FruK] to obtain the titration shown in (b). The solid line in (b) is the best fit of Equation (1) to the data. The experiments shown are representative of at least three replicates with at least two different protein preparations
3. CONCLUSION
Biolayer interferometry is becoming an important biophysical tool for quantifying biomolecular interactions. The availability of multi‐channel instruments—such as the eight‐channel BLI instrument—make sophisticated experiments reasonably easy. However, the traditional use of this technique requires that the kinetics of a given biomolecular binding event follow simple, single‐exponential kinetics.
Here, we demonstrated that thermodynamic parameters for repressor‐DNA titrations could be reliably quantified using the BLI signal after the reaction reached equilibrium. Although the two methods have very different limitations (the act of filtering perturbs the equilibrium in filter binding assays, whereas one species is immobilized in BLI assays), agreement was quite remarkable when the experimental design of the two techniques was matched, as in the LLhF experiments. On a practical note, each BLI assay required approximately one‐third of the pipetting and reagents needed to generate a comparable curve in the filter binding assay (see Methods), and the BLI assay was not subject to the time constraints of 32P decay.
As noted above, knowledge of the effective concentration of immobilized DNA was critical to data interpretation. For ForteBio High Precision Streptavidin (SAX) tips, we determined this value was most frequently ~2 × 10−9 M. Thus, for a reaction to be in the equilibrium binding regime, its K d value must be >10−8 M. The binding affinities measured by Theiker et al19 for a sulfotransferase binding to hexasaccharides ranged from 10−8 to 10−7 M. These experiments used slightly different Forte‐Bio streptavidin tips than the current work (SA rather than SAX), but if the tips have comparable numbers of binding sites, these reactions should have been in the equilibrium binding regime. In addition, we obtained a reasonable estimate of K d for an experiment in the intermediate binding regime using Equation (2) for global fitting of multiple replicates. When planning future experiments in the intermediate binding regime, we recommend systematically altering the concentration of immobilized DNA by diluting it in fixed ratios with free biotin (Figure S7E); variation in the DNA concentration should facilitate better estimation of K d.
In future experiments, the reliability of fitting with Equation (2) will define the lower limit of BLI K d values that can be obtained. Such fits would likely be impossible if the stoichiometry of the complex (here, one repressor dimer per one operator DNA) is not independently known; this limitation is shared by the filter binding assay. The upper limits we observed for BLI K d values were likely protein dependent: High concentrations of tetrameric LacI appeared to be out of the reliable range of the BLI assay, and we could not generate enough dimeric LLhF to quantify weaker binding affinities. We expect that binding events for other proteins with weaker K d values (above 10−7 M) will be amenable to BLI assays and data fitting with Equation (1), which would be an advantage over filter binding assays. A final consideration is the valency of an interaction: when one binding partner was multi‐valent (e.g., tetrameric LacI) and large enough to span the distance between two immobilized species, K d values determined with BLI equilibrium titrations were affected.
For the allosteric experiments (operator release), results from filter binding and BLI assays were in good agreement when protein‐DNA binding conditions were in the equilibrium binding regime. Finally, the LLhF‐FruK experiments showed BLI amplitudes could also be used to monitor hetero‐protein binding events that are inaccessible to the filter binding assay. Overall, the use of BLI amplitudes to monitor equilibrium titrations promise to be a convenient and reproducible assay for calculating K d and midpoint values for many types of biomolecular interactions.
4. MATERIALS AND METHODS
4.1. Protein purification and validation
For these benchmark repressor‐DNA binding studies, we used the well‐characterized E. coli lactose repressor protein (“LacI”) and a chimeric homolog (“LLhF”) that comprised the DNA binding domain of LacI and the regulatory domain of the E. coli fructose repressor protein (“FruR,” which is also known as “Cra”).18 The protein–protein interaction between LLhF‐DNA and FruK was also evaluated. Each of the three proteins was purified as described in the Supplementary Methods, and FruK identity was confirmed with mass spectrometry (Table SI). The references associated with these methods include references.10, 18, 29, 40, 41, 42
The purity of all three proteins was confirmed with SDS‐PAGE. LacI and FruK were generally >90% pure. LLhF showed trace amounts of co‐purifying FruK. However, since a strong signal could be detected for LLhF binding to FruK that was independently purified (Figure 9), the trace amounts of co‐purifying FruK must be well‐below saturation levels. For wild‐type LacI, the protein concentration was determined from the A 280 using an extinction coefficient of 0.6 ([mg/ml] • cm)−1.43, 44 For LLhF and FruK, protein concentration was determined by Bradford assays (Bio‐Rad Laboratories, Inc. Hercules, California).
For both LacI and LLhF, since one homodimer is known to bind one DNA operator, the concentration of active repressor protein could be confirmed using filter binding assays in the stoichiometric binding regime (with the DNA concentration at a concentration at least 10 times greater than K d).20 The fraction of active repressor was found to be >90% for LacI and > 70% for LLhF. FruK activity was confirmed by assessing its known enzymatic activity45:
| (3) |
Experimental details of the enzymatic assay are described in the Supplementary Methods; the references associated with these Methods include references.42, 46 K m values were in good agreement with previously published values. We assessed activity in two buffers (Supplementary Methods; Figure S10) and chose to carry out experiments in “PYK buffer” (50 mM HEPES, pH 7.0, 5 mM MgCl2, and 50 mM KCl, with 3 mM ATP and 0.335 mM NADH).
4.2. Operator DNA
Oligomeric DNA was purchased from Integrated DNA Technologies (Skokie, Illinois). Binding experiments used the natural operator sequences lacO 1, lacO 2, and lacO 3 (Table 1)47, 48, 49, 50 and a non‐specific DNA sequence.11 All operator sequences were embedded in the center of a 40 base pair oligomer for filter binding assays or a 45 base pair oligomer for BLI assays (Table SII). For binding to the BLI streptavidin biosensor tips, DNA was 5′ biotinylated on one strand. Some early BLI assays used longer DNA oligos (69mer), but no DNA length dependence was observed. For both assays, the single‐stranded DNA was annealed using the protocol of Swint‐Kruse et al.10 For filter binding assays, annealed DNA was labeled with 32P‐ATP using the protocol of Swint‐Kruse et al.10 As a control for FruK binding experiments, we also used the Cra operator fruB‐proximal (Figure S11).
4.3. Small molecule allosteric effectors
The LacI inducer, IPTG, was purchased from GoldBio (St. Louis, Missouri). At the onset of this study, the LLhF inducer, F‐1‐P, was purchased as the sodium salt from Sigma‐Aldrich (St. Louis, Missouri); in later studies, it was only available as the poorly‐soluble barium salt (Sigma‐Aldrich). Thus, to obtain the sodium counterion form of F‐1‐P, we performed ion‐exchange chromatography of the barium salt using a Dowex 50WX4 (Sigma) column pre‐charged with 2 N HCl and then equilibrated in ddH2O. The barium F‐1‐P was passed through this column several times and the pH of the final eluant was adjusted to 7 with NaOH. Eluted sodium•F‐1‐P was lyophilized and used to make stock solutions in ddH2O. Concentrations of the stock solutions were determined by stoichiometric implementations of the FruK enzyme assay (Supplementary Methods). This assay used a high concentration of FruK so that catalysis of the F‐1‐P substrate was quickly completed. The change in 340 nm before and after FruK addition was used to determine the total amount of NADH consumed during the reaction using an extinction coefficient of 6,317 M−1 • cm−1 51; the amount of NADH consumed corresponds 1:1 with the F‐1‐P concentration.
4.4. Nitrocellulose filter binding assays
The nitrocellulose filter binding assay was used to monitor LacI‐DNA binding, LacI‐IPTG “operator release,” LLhF‐DNA binding, and LLhF‐F‐1‐P “operator release” reactions. LacI experiments used filter binding buffer (“FBB”: 10 mM Tris, 150 mM KCl, 10 mM EDTA, 0.3 mM DTT, pH 7.4, 0.1 mg/mL bovine serum albumin) and LLhF experiments used the LLhF binding buffer (50 mM HEPES, 0.1 M KCl, 0.1 mM EDTA, 10 mM MgCl2, pH 7.0, 0.3 mM DTT, 0.1 mg/ml bovine serum albumin). Note that FBB for the LacI experiments excluded the 5% DMSO that was used in many historical studies of LacI,8, 9, 10, 11 which we have shown accounts for the slight difference in DNA binding affinities compared to those reported in prior publications. For equilibrium assays, the DNA concentration was fixed at least 10‐fold less than the value of K d 20 and repressor protein concentration was varied.
Within individual assays, each repressor concentration was pipetted in triplicate, which was required to compensate for scattering in the signals (e.g., see figures in Swint‐Kruse et al.10 and Falcon and Matthews11). Each type of assay was repeated at least three times, using at least two different preparations of protein. To quantify the radioactively‐labeled repressor‐DNA complexes retained on the nitrocellulose filter, we used a Fuji Typhoon phosphorimager or a Beckman LS 6500 scintillation counter. For scintillation counting, the triplicate technical replicates were counted as a single sample. For consistency among the current figures, any phosphorimager data are shown as the sums of triplicate replicates; data fitting results were identical when triplicates were treated separately (as in References10 or11) or when the triplicates were summed.
4.5. Biolayer interferometry
Initial BLI experiments used the single‐channel BLItz instrument (ForteBio, Menlo Park, California) to establish loading times and wash conditions. Titration experiments were carried out on an Octet RED96 instrument (ForteBio) that can simultaneously monitor eight samples in parallel. All BLItz measurements were carried out at room temperature with shaking at 2200 rpm; Octet measurements were carried out at 22°C and shaken at 1,000 rpm. Sample volumes were 250 in 500 μL black microcentrifuge tubes (Celltreat Scientific Products, China) for the BLItz or in black 96‐well plates (Greiner Bio‐one, Germany) for the eight‐channel BLI instrument.
For DNA binding assays, we used sensor tips derivatized with streptavidin (BLItz; ForteBio, Pall Life Sciences, Menlo Park, California) to which various biotinylated DNA sequences were attached. Early experiments used Super Streptavidin tips (“SSA,” ForteBio, Pall Life Sciences). Control experiments with these tips in the absence of DNA showed low nonspecific binding to repressor protein; however, tip‐to‐tip results were less reproducible over repeated experiments. For later experiments, a “High‐Precision Streptavidin” (“SAX”) tip became available from ForteBio that had better tip‐to‐tip reproducibility; nonspecific binding in the absence of DNA was again low (Figure S2) and was blocked by the 0.1 mg/ml bovine serum albumin present in FBB hydration buffer or by adding 1.3 μM LacI to the FBB hydration buffer. No difference in the repressor‐DNA binding curves was observed among the various blocking agents, so we chose not to use LacI for most experiments because it might interfere with small molecule experiments (see below). Finally, we demonstrated here and in a previous study29 that, in the absence of repressor protein, FruK did not bind immobilized DNA (Figure S11).
In parallel with filter binding assays, LacI titrations were carried out in FBB and LLhF assays were carried out in LLhF binding buffer. BLI tips were first hydrated at least 10 minutes in the appropriate binding buffer, loaded with biotinylated DNA (80 nM in binding buffer for 120 s), moved to a tube of binding buffer (30 s), and then moved to a solution of repressor protein that was pre‐equilibrated with any small molecules or hetero‐proteins. Protein solutions were kept on ice until added to the final reaction mixture, which was then allowed to warm to room temperature (~15 min) prior to introducing the DNA‐bound BLI tips. (BLI signals are highly temperature dependent.) The protein concentrations used in each assay are indicated in the figures and/or legends. Resulting changes in the BLI signal were monitored as a function of time until equilibrium was reached.
Each set of experiments was repeated in triplicate using at least two separate preparations of LacI, LLhF, and FruK. The within‐day reproducibility of the equilibrium plateau was usually <20% for equivalent samples, although as discussed above, day‐to‐day (and lot‐to‐lot) variation was often greater (Figure S8). The filter binding assay has a similar limitation since 32P counts differ among assays. Titrations carried out simultaneously in the Octet had much less noise than those performed with the single‐channel BLItz. Within a single 8‐tip assay, the average differences in signal values observed after the DNA load (before protein incubation) were ~10%, but as discussed above, this value is not a reliable indicator of the effective number of accessible DNA molecules. All trends observed within 1 day's work were reproducible on separate days. These technical limitations were very similar to those encountered in the day‐to‐day variation in 32P‐DNA dilutions used in filter binding assays.
During experiments, we noted that ~1 out of every 20 tips performed differently from the others. This was detected by unequal DNA loading, unusually high or low plateaus after repressor association, and/or oddly‐shaped association curves. Data values measured with “bad” tips were excluded from downstream calculations and analyses. Octet experiments have the potential to comprise eight concentrations per titration; the minimum number of data points used to define a titration curve was seven (if two tips had to be excluded, the titration was repeated). For most titration experiments, the first sample was used to determine the zero point and did not contain any of the varied component. This was always in good agreement with average signal after immobilizing the DNA, and later repressor‐DNA binding experiments used this average value as the “zero” so that the eighth channel could be used to generate an extra data point. Notably, BLI curves generated from seven to nine, singlet concentrations (each measured only once) were of similar quality to curves from filter binding assays that were generated using 12 concentrations, each pipetted in triplicate (and as described above, counted as a single sample in the current work).
4.6. Analyses of binding curves
For filter binding and BLI assays in the equilibrium binding regime, titration data were analyzed by first converting LacI, LLhF, and FruK concentrations to log scale to facilitate better weighting during nonlinear regression and fitting the data with Equation (1). To include data for the “zero” concentration in the fitting process, the value of X was set ≥10‐fold below the lowest concentration used. Following the log transformation, nonlinear regression was carried out with the program GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, California). Operator release fits were analyzed similarly, by first converting IPTG and F‐1‐P concentrations to log scale and then fitting to Equation (1); in this case, the “K d” parameter determined from the midpoint of the curve is not a true equilibrium constant (described in Results). Unless otherwise indicated, the values reported in Table 2 are the averages and standard deviations of the fits from at least three independent experiments. Essentially identical values were obtained by carrying out global fits of replicate experiments using a shared K d value and independent c and Y max values for each curve (Figure S8); global fit errors were equivalent to the standard deviations reported in Table 2.
When repressor‐operator binding appeared to be either in the stoichiometric binding regime or intermediate between the equilibrium and stoichiometric binding regimes, data were fit with the expanded Equation (2), which contains five parameters. The BLI titrations were limited to 7–9 data points and fitting with a five‐parameter equation certainly qualifies as “over‐fitting.” However, since the stoichiometry of the dimeric repressor‐DNA complex is known to be 1, we fixed this parameter and carried out nonlinear regression to estimate the remaining parameters, including K d and the effective total DNA concentration (Figure 4b and Figure S7). As described in Results and Figure S7, global fitting was also carried out with this equation.
In some BLI experiments, the equilibrium “plateaus” of all samples decreased instead of remaining constant (Figure 7b). We presume this arises from protein degradation occurring over the two‐hour experiment or adsorption to the wells. Fortunately, this had little impact on the final data analysis: When different time slices were used to generate the Y values for Equation (1), the K d values determined were essentially identical (Figure S5). Indeed, the protein decay could also be occurring in the filter binding assay but would not be detectable. This did illuminate one additional interesting observation: In the filter binding assay, the protein‐DNA mixture was generally equilibrated for 30 min prior to filtration. In BLI, some of the ternary interactions (DNA‐repressor binding small molecule or hetero‐protein) required up to 2 hr for the plateau to reach equilibrium. Thus, being able to monitor a reaction's approach to equilibrium is an advantage of BLI.
Supporting information
Appendix S1: Supporting information
ACKNOWLEDGEMENTS
We thank Drs. Mark Fisher and Joe Lutkenhaus (KUMC) for the loan of their single‐channel BLI instruments and Mr. Pierce O'Neil and Dr. Sishen Du for their assistance learning the technique. Ms. Brae Bigge (KUMC) assisted with repetitions of the single‐channel BLI data. The KUMC Octet RED96 instrument is housed in the PreClinical Models Core of the Kansas Intellectual and Developmental Disabilities Research Center under the supervision of Ms. Michelle Winters. Drs. Antonio Artigues and Maite Villar in the KUMC Mass Spectrometry facility identified the components of the LLhF‐FruK complex. We thank Drs. Maria Veiga‐da‐Cunha and Emile Van Schaftingen (Université Catholique de Louvain, Belgium) for the gift of the FruK expression plasmid and Mr. Benjamin Rau for assistance with ion exchange of F‐1‐P. Dr. Abby Hodges (MidAmerica Nazarene University) provided valuable assistance editing the manuscript. We dedicate this work to the memory of Dr. Mark Fisher, who owned the first BLItz sold by ForteBio and enthusiastically introduced us to this technology.
Weeramange CJ, Fairlamb MS, Singh D, Fenton AW, Swint‐Kruse L. The strengths and limitations of using biolayer interferometry to monitor equilibrium titrations of biomolecules. Protein Science. 2020;29:1018–1034. 10.1002/pro.3827
Present address:
Dipika Singh, The Sydney Kimmel Comprehensive Cancer Center, Johns Hopkins University, 401 N. Broadway, Baltimore, MD 21287
Funding information National Institute of General Medical Sciences, Grant/Award Numbers: GM079423, GM110761, GM118589
REFERENCES
- 1. Pollard TD. A guide to simple and informative binding assays. Mol Biol Cell. 2010;21:4061–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hulme EC, Trevethick MA. Ligand binding assays at equilibrium: Validation and interpretation. BritJ Pharmacol. 2010;161:1219–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oberfelder RW, Lee JC. Measurement of ligand‐protein interaction by electrophoretic and spectroscopic techniques. Methods Enzymol. 1985;117:381–399. [DOI] [PubMed] [Google Scholar]
- 4. Ladokhin AS, Jayasinghe S, White SH. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal Biochem. 2000;285:235–245. [DOI] [PubMed] [Google Scholar]
- 5. Lohman TM, Bujalowski W. Thermodynamic methods for model‐independent determination of equilibrium binding isotherms for protein‐DNA interactions: Spectroscopic approaches to monitor binding. Methods Enzymol. 1991;208:258–290. [DOI] [PubMed] [Google Scholar]
- 6. O'Gorman RB, Dunaway M, Matthews KS. DNA binding characteristics of lactose repressor and the trypsin‐resistant core repressor. J Biol Chem. 1980;255:10100–10106. [PubMed] [Google Scholar]
- 7. Wong I, Lohman TM. A double‐filter method for nitrocellulose‐filter binding: Application to protein‐nucleic acid interactions. Proc Natl Acad Sci U S A. 1993;90:5428–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhan H, Camargo M, Matthews KS. Positions 94−98 of the lactose repressor N‐subdomain monomer−monomer interface are critical for allosteric communication. Biochemistry. 2010;49:8636–8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhan H, Swint‐Kruse L, Matthews KS. Extrinsic interactions dominate helical propensity in coupled binding and folding of the lactose repressor protein hinge helix. Biochemistry. 2006;45:5896–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Swint‐Kruse L, Zhan H, Matthews KS. Integrated insights from simulation, experiment, and mutational analysis yield new details of LacI function. Biochemistry. 2005;44:11201–11213. [DOI] [PubMed] [Google Scholar]
- 11. Falcon CM, Matthews KS. Operator DNA sequence variation enhances high affinity binding by hinge helix mutants of lactose repressor protein. Biochemistry. 2000;39:11074–11083. [DOI] [PubMed] [Google Scholar]
- 12. Tungtur S, Schwingen KM, Riepe JJ, Weeramange CJ, Swint‐Kruse L. Homolog comparisons further reconcile in vitro and in vivo correlations of protein activities by revealing over‐looked physiological factors. Protein Sci. 2019;28:1806–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhan H, Taraban M, Trewhella J, Swint‐Kruse L. Subdividing repressor function: DNA binding affinity, selectivity, and allostery can be altered by amino acid substitution of nonconserved residues in a LacI/GalR homologue. Biochemistry. 2008;47:8058–8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swint‐Kruse L, Zhan H, Fairbanks BM, Maheshwari A, Matthews KS. Perturbation from a distance: Mutations that alter LacI function through long‐range effects. Biochemistry. 2003;42:14004–14016. [DOI] [PubMed] [Google Scholar]
- 15. Oehler S, Alex R, Barker A. Is nitrocellulose filter binding really a universal assay for protein‐DNA interactions? Anal Biochem. 1999;268:330–336. [DOI] [PubMed] [Google Scholar]
- 16.Octet® and BLItz® Systems: Thickness display in the Y‐axis, https://mdc.custhelp.com/app/answers/detail/a_id/20559/kw/optical%20thickness: Molecular Devices, ForteBio; 2019.
- 17. Abdiche Y, Malashock D, Pinkerton A, Pons J. Determining kinetics and affinities of protein interactions using a parallel real‐time label‐free biosensor, the octet. Anal Biochem. 2008;377:209–217. [DOI] [PubMed] [Google Scholar]
- 18. Meinhardt S, Manley MW, Becker NA, Hessman JA, Maher LJ, Swint‐Kruse L. Novel insights from hybrid LacI/GalR proteins: Family‐wide functional attributes and biologically significant variation in transcription repression. Nucleic Acids Res. 2012;40:11139–11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thieker DF, Xu Y, Chapla D, et al. Downstream products are potent inhibitors of the heparan sulfate 2‐O‐sulfotransferase. Sci Rep. 2018;8:11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Swint‐Kruse L, Matthews KS. Thermodynamics, protein modification, and molecular dynamics in characterizing lactose repressor protein: Strategies for complex analyses of protein structure‐function. Methods Enzymol. 2004;379:188–209. [DOI] [PubMed] [Google Scholar]
- 21. Barkley MD, Riggs AD, Jobe A, Burgeois S. Interaction of effecting ligands with lac repressor and repressor‐operator complex. Biochemistry. 1975;14:1700–1712. [DOI] [PubMed] [Google Scholar]
- 22. Mikulskis A, Aristarkhov A, Lin EC. Regulation of expression of the ethanol dehydrogenase gene (adhE) in Escherichia coli by catabolite repressor activator protein Cra. J Bacteriol. 1997;179:7129–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramseier TM, Negre D, Cortay JC, Scarabel M, Cozzone AJ, Saier MH Jr. In vitro binding of the pleiotropic transcriptional regulatory protein, FruR, to the fru, pps, ace, pts and icd operons of Escherichia coli and salmonella typhimurium. J Mol Biol. 1993;234:28–44. [DOI] [PubMed] [Google Scholar]
- 24. Bell CE, Lewis M. A closer view of the conformation of the lac repressor bound to operator. Nat Struct Biol. 2000;7:209–214. [DOI] [PubMed] [Google Scholar]
- 25. Chen J, Matthews KS. Subunit dissociation affects DNA binding in a dimeric lac repressor produced by C‐terminal deletion. Biochemistry. 1994;33:8728–8735. [DOI] [PubMed] [Google Scholar]
- 26. Chen J, Matthews KS. Deletion of lactose repressor carboxyl‐terminal domain affects tetramer formation. J Biol Chem. 1992;267:13843–13850. [PubMed] [Google Scholar]
- 27. Swint‐Kruse L, Matthews KS. Allostery in the LacI/GalR family: Variations on a theme. Curr Opin Microbiol. 2009;12:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tungtur S, Skinner H, Zhan H, Swint‐Kruse L, Beckett D. In vivo tests of thermodynamic models of transcription repressor function. Biophys Chem. 2011;159:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singh D, Fairlamb MS, Harrison KS, et al. Protein‐protein interactions with fructose‐1‐kinase alter function of the central Escherichia coli transcription regulator. Cra bioRxiv. 2017. 10.1101/201277. [DOI] [Google Scholar]
- 30. Whitson PA, Matthews KS. Dissociation of the lactose repressor‐operator DNA complex: Effects of size and sequence context of operator‐containing DNA. Biochemistry. 1986;25:3845–3852. [DOI] [PubMed] [Google Scholar]
- 31. Riggs AD, Bourgeois S, Cohn M. The lac repressor‐operator interaction. 3. Kinetic studies. J Mol Biol. 1970;53:401–417. [DOI] [PubMed] [Google Scholar]
- 32. Riley‐Lovingshimer MR, Reinhart GD. Examination of MgATP binding in a tryptophan‐shift mutant of phosphofructokinase from Bacillus stearothermophilus . Arch Biochem Biophys. 2005;436:178–186. [DOI] [PubMed] [Google Scholar]
- 33.Octet® and BLItz® Systems: How many binding sites are on the biosensor?, https://mdc.custhelp.com/app/answers/detail/a_id/20558/kw/how%20many%20binding%20sites%20are%20on%20the%20the%20biosensor: Molecular Devices, ForteBio; 2019.
- 34. Lewis M, Chang G, Horton NC, et al. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science. 1996;271:1247–1254. [DOI] [PubMed] [Google Scholar]
- 35. Taraban M, Zhan H, Whitten AE, et al. Ligand‐induced conformational changes and conformational dynamics in the solution structure of the lactose repressor protein. J Mol Biol. 2008;376:466–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. von Hippel PH, Revzin A, Gross CA, Wang AC. Non‐specific DNA binding of genome regulating proteins as a biological control mechanism: 1. The lac operon: Equilibrium aspects. Proc Natl Acad Sci U S A. 1974;71:4808–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin S, Riggs AD. The general affinity of lac repressor for E. coli DNA: Implications for gene regulation in procaryotes and eucaryotes. Cell. 1975;4:107–111. [DOI] [PubMed] [Google Scholar]
- 38. Elf J, Li GW, Xie XS. Probing transcription factor dynamics at the single‐molecule level in a living cell. Science. 2007;316:1191–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riggs AD, Newby RF, Bourgeois S. Lac repressor—operator interaction. II effect of galactosides and other ligands. J Mol Biol. 1970;51:303–314. [DOI] [PubMed] [Google Scholar]
- 40. Swint‐Kruse L, Elam CR, Lin JW, Wycuff DR, Shive Matthews K. Plasticity of quaternary structure: Twenty‐two ways to form a LacI dimer. Protein Sci. 2001;10:262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wycuff DR, Matthews KS. Generation of an Ara‐C‐araBAD promoter‐regulated T7 expression system. Anal Biochem. 2000;277:67–73. [DOI] [PubMed] [Google Scholar]
- 42. Veiga‐da‐Cunha M, Hoyoux A, Van Schaftingen E. Overexpression and purification of fructose‐1‐phosphate kinase from Escherichia coli: Application to the assay of fructose 1‐phosphate. Protein Expr Purif. 2000;19:48–52. [DOI] [PubMed] [Google Scholar]
- 43. Gardner JA, Matthews KS. Characterization of two mutant lactose repressor proteins containing single tryptophans. J Biol Chem. 1990;265:21061–21067. [PubMed] [Google Scholar]
- 44. Sommer H, Lu P. Lac repressor. Fluorescence of the two tryptophans. J Biol Chem. 1976;251:3774–3779. [PubMed] [Google Scholar]
- 45. Ferenci T, Kornberg HL. The utilization of fructose by Escherichia coli. Properties of a mutant defective in fructose 1‐phosphate kinase activity. Biochem J. 1973;132:341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fenton AW, Alontaga AY. The impact of ions on allosteric functions in human liver pyruvate kinase. Methods Enzymol. 2009;466:83–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gilbert W, Maxam A. The nucleotide sequence of the lac operator. Proc Natl Acad Sci U S A. 1973;70:3581–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pfahl M, Gulde V, Bourgeois S. "second" and "third operator" of the lac operon: An investigation of their role in the regulatory mechanism. J Mol Biol. 1979;127:339–344. [DOI] [PubMed] [Google Scholar]
- 49. Reznikoff WS, Winter RB, Hurley CK. The location of the repressor binding sites in the lac operon. Proc Natl Acad Sci U S A. 1974;71:2314–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Winter RB, Von Hippel PH. Diffusion‐driven mechanisms of protein translocation on nucleic acids. 2. The Escherichia coli lac repressor‐operator interaction: Equilibrium measurements. Biochemistry. 1981;20:6948–6960. [DOI] [PubMed] [Google Scholar]
- 51. McComb RB, Bond LW, Burnett RW, Keech RC, Bowers GN Jr. Determination of the molar absorptivity of NADH. Clinic Chem. 1976;22:141–150. [PubMed] [Google Scholar]
- 52. Hsieh WT, Whitson PA, Matthews KS, Wells RD. Influence of sequence and distance between two operators on interaction with the lac repressor. J Biol Chem. 1987;262:14583–14591. [PubMed] [Google Scholar]
- 53. Brenowitz M, Mandal N, Pickar A, Jamison E, Adhya S. DNA‐binding properties of a lac repressor mutant incapable of forming tetramers. J Biol Chem. 1991;266:1281–1288. [PubMed] [Google Scholar]
- 54. Pfahl M, Hendricks M. Interaction of tight binding repressors with lac operators. An analysis by DNA‐footprinting J Mol Biol. 1984;172:405–416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting information