

Abstract
A distinguishing feature of camel (Camelus dromedarius) VHH domains are noncanonical disulfide bonds between CDR1 and CDR3. The disulfide bond may provide an evolutionary advantage, as one of the cysteines in the bond is germline encoded. It has been hypothesized that this additional disulfide bond may play a role in binding affinity by reducing the entropic penalty associated with immobilization of a long CDR3 loop upon antigen binding. To examine the role of a noncanonical disulfide bond on antigen binding and the biophysical properties of a VHH domain, we have used the VHH R303, which binds the Listeria virulence factor InlB as a model. Using site directed mutagenesis, we produced a double mutant of R303 (C33A/C102A) to remove the extra disulfide bond of the VHH R303. Antigen binding was not affected by loss of the disulfide bond, however the mutant VHH displayed reduced thermal stability (T m = 12°C lower than wild‐type), and a loss of the ability to fold reversibly due to heat induced aggregation. X‐ray structures of the mutant alone and in complex with InlB showed no major changes in the structure. B‐factor analysis of the structures suggested that the loss of the disulfide bond elicited no major change on the flexibility of the CDR loops, and revealed no evidence of loop immobilization upon antigen binding. These results suggest that the noncanonical disulfide bond found in camel VHH may have evolved to stabilize the biophysical properties of the domain, rather than playing a significant role in antigen binding.
Keywords: antigen, disulfide bond, dynamics, flexibility, Nanobody, protein aggregation, VHH
1. INTRODUCTION
VHH (also known as nanobodies) are a class of single domain antibodies derived from the antigen‐binding domains of Camelid heavy‐chain antibodies (HCAbs).1 A variety of biophysical properties distinguishes VHHs from the conventional IgGs found in jawed vertebrates, including their small size (∼15 kDa), convex paratope structure, long CDR3 length, thermostability, and reversible refoldability following denaturation.1 These unique biophysical properties, combined with the ease of selecting high affinity binders, has generated a great deal of interest in using VHHs as research reagents,2 environmental sensors,3 as well as for therapeutic and biomedical applications.4, 5, 6 The recent clinical success and FDA approval of caplacizumab, a VHH for the treatment of acquired thrombic thrombocytopenic purpura,7 highlights the future potential therapeutic value of VHH reagents in medicine.
Compared to the VH domain found in conventional IgG antibodies, a series of sequence mutations have evolved in the camelid VHH to compensate for the loss of the light chain. In a typical IgG antibody, the pairing of the heavy chain and light chain is mediated through a series of hydrophobic interactions. The hydrophobic patch on the heavy chain which mediates the interaction with the light chain (residues V37, G44, L45, W47, Y95, and W117) is prone to aggregation in the absence of a paired light chain partner.8 In the camelid VHH, to prevent heavy chain aggregation, several of the residues within the hydrophobic patch have mutated to more hydrophilic residues (G44E and L45R), while other mutations (V37F and W47F/G) are shielded from solvent by a long CDR3 loop.9, 10 The solubility of the VHH domain is also enhanced by an increased number of charged residues found in FR3 compared to VH domains.11
Typical antibodies bind antigen at the interface between the light and heavy chain, forming a interaction site with up to six CDR loops.12 As VHHs consist solely of a heavy chain, the antigen binding site is formed by only three CDR loops. To compensate for the loss in sequence diversity by having fewer CDR loops, the VHH domain has evolved a longer CDR3 loop, as well as greater structural variability in the CDR1 and CDR2 loops compared to murine and human antibodies.11, 13 The long CDR3 loop found in camelid VHH appears to serve dual functions: to shield an aggregation prone hydrophobic patch and to increase the paratope‐epitope interaction surface. A potential liability to having a long CDR3 loop is the entropic penalty associated with fixing a longer CDR loop upon antigen binding.
Early‐on, sequence analysis of camelid VHHs noted the presence of additional disulfide bonds typically not found in antibody VH domains.13 In addition to the conserved, canonical intra‐domain disulfide bond common to antibody variable domains (C23–C104), several noncanonical disulfide bonds have been described in VHH. The most common is a disulfide bond between CDR1 and CDR3 (C33–C102).13, 14, 15 The cysteine residue on CDR1 is germline encoded, suggesting that the noncanonical disulfide bond between CDR1 and CDR3 may serve a conserved function in VHH domains.16 Noncanonical disulfide bonds may be an important factor contributing to VHH affinity by stabilizing the structure CDR3, and reducing the entropic penalty associated with fixing a long loop upon antigen binding.17, 18 In support of this hypothesis, mutational analysis of several VHHs suggested a role for the CDR1–CDR3 disulfide bond in antigen binding affinity.19
We have previously isolated and characterized a VHH called R303 which is specific for InlB, a protein virulence factor involved in host‐cell invasion by the pathogen Listeria monocyotgenes.20, 21, 22 R303 is a camel (Camelus dromedarius) VHH isolated from a naïve phage display library. The CDR3 loop in R303 is 16 residues long (as classified by North et al.23) and contains a noncanonical disulfide bond between CDR1 and CDR3 (C33–C102). As a model to better understand the role of the camelid noncanonical disulfide bond between CDR1 and CDR3 we have produced a double mutant (C33A/C102A) and examined the effect of the mutation on the affinity, biophysical properties, structure, and dynamics of the VHH.
2. RESULTS
2.1. Affinity and inhibitory properties of VHH R303 and R303C33A/C102A
The camel VHH R303 binds the leucine rich repeat domain of InlB with high affinity.20, 22 To investigate if the noncanonical disulfide bond between CDR1 and CDR3 influences the affinity of the VHH R303 toward its antigen, a double cysteine to alanine mutation (C33A/C102A) was introduced by site directed mutagenesis. The resultant double mutant (R303C33A/C102A) was purified and used in a solid phase (ELISA) binding assay to compare the relative affinities of the wild type and mutant R303 VHH to InlB. In the assay, a fixed concentration of the LRR domain of InlB (Residues 31–249; referred to as InlB249 throughout the text) was immobilized on a 96 well plate. A variable concentration of biotinylated R303 and R303C33A/C102A was applied and detected with streptavidin‐HRP (Figure 1a). The KD of R303 for InlB249 as determined by SPR was previously reported to be 0.15 nM.20 The ELISA based assay used here, found a similar affinity range, with R303 and R303C33A/C102A exhibiting an identical affinity for InlB249 (EC50 = 0.17 μg/m lor 12.6 nM) indicating that the noncanonical C33‐C102 disulfide bond found in R303 was not crucial for high affinity binding to antigen (Figure 1a).
Figure 1.
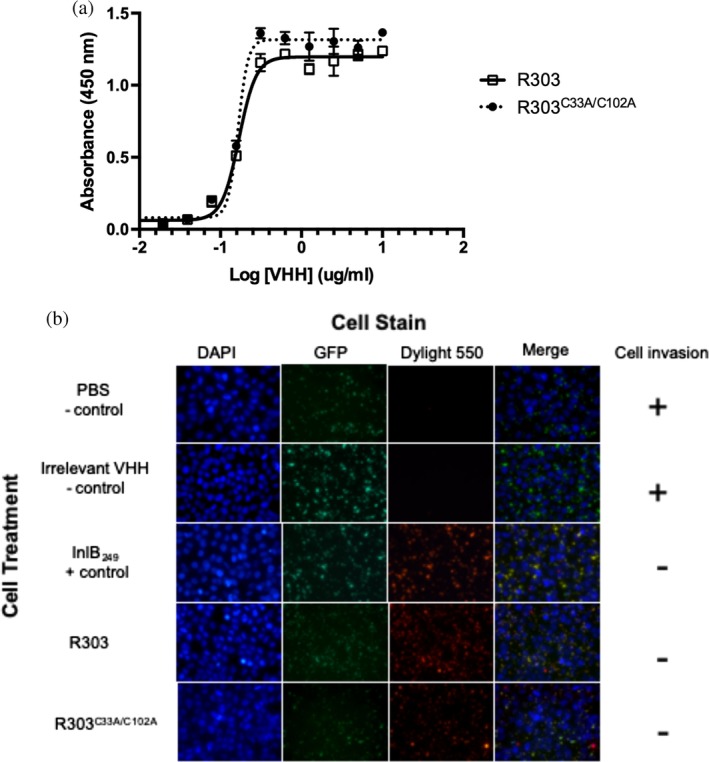
Impact of C33–C102 noncanonical disulfide bond on VHH R303 affinity and inhibitory activity. (a) Solid phase binding assay of VHH R303 and R303C33A/C102A toward immobilized inlb249. (b) Fluorescent microscopy invasion assay measuring VHH mediated inhibition of Listeria invasion.22 HeLa cells were infected with biotinylated L. monocytogenes expressing GFP in the presence of PBS buffer control, InlB2 49 (positive control to inhibit Listeria invasion), VHH negative control,25 and R303, and R303C33A/C102A Only extracellular bacteria are stained red, indicating that both R303, and R303C33A/C102A inhibit Listeria invasion
R303 has previously been shown to inhibit the in vitro invasion of HeLa cells by Listeria monocytogenes by inhibiting the interaction of Listeria InlB with the host cell receptor c‐MET.22 A qualitative microscopy‐based infection assay was previously used to determine if R303 and R303C33A/C102A were capable of inhibiting HeLa cell invasion by L. monocytogenes 22, 24 (Figure 1b). Constitutively GFP expressing and biotinylated L. monocytogenes were used to infect HeLa cells in the presence of PBS, an irrelevant VHH (negative controls), InlB249, wild type R303 (positive controls) and R303C33A/C102A. Streptavidin conjugated to DyLight550 was then used to detect extracellular bacteria (i.e., inhibition of Listeria invasion). Both wild type R303 and R303C33A/C102A inhibited Listeria invasion of HeLa cells (Figure 1b). Although the assay was qualitative, the results suggested that the C33–C102 disulfide bond not does dramatically alter the inhibitory activity of R303 toward Listeria invasion of cells in vitro.
2.2. Biophysical properties of R303 and R303C33A/C102A
Disulfide bonds are well known to increase the thermal stability of proteins.26 The role of the C33–C102 noncanonical disulfide bond in the stability of VHH R303 was examined by monitoring the temperature induced denaturation of R303 and R303C33A/C102A using CD spectroscopy (Figure 2a). The unfolding transition temperature (T m) was used to compare the relative thermal stabilities of VHHs R303 and R303C33A/C102A. The T m of R303 was calculated to be 75.0°C (±0.2°C), which is at the higher end of the typically observed VHH thermostability range (50–80°C).27 Removal of the disulfide bond between CDR1 and CDR3 in R303C33A/C102A resulted in a greater than 10°C reduction in T m (T m = 63.0 ± 0.2°C; Figure 2a).
Figure 2.
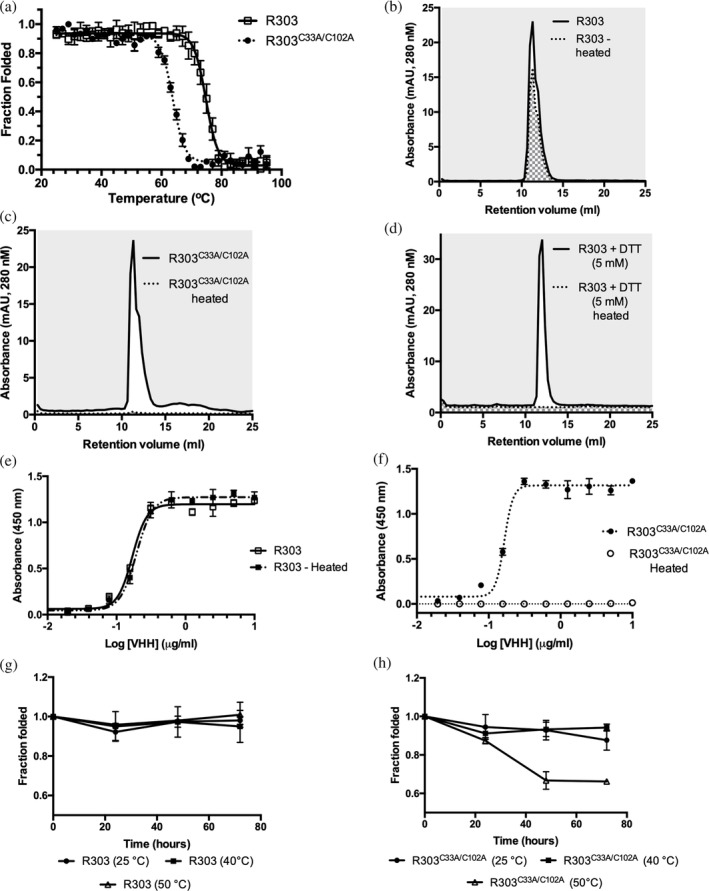
Biophysical properties of VHH R303 and R303C33A/C102A (a) Thermal denaturation of R303 and R303C33A/C102A monitored by circular dichroism spectroscopy. (b) Aggregation resistance and reversible folding of R303C33A/C102A determined using size exclusion chromatography (SEC). The area under the peak of a sample of VHH was measured before heating and following heating then cooling to allow refolding. (c) Aggregation resistance and reversible folding of R303 determined using SEC. (d) Aggregation resistance and reversible folding of R303 in the presence of 5 mM DTT determined using SEC (e) Solid phase binding assay of VHH R303 before heating and following heating then cooling to allow refolding. R303 retains full binding to InlB indicating reversible folding. (f) Solid phase binding assay of VHH R303C33A/C102A before heating and following heating then cooling to allow refolding. R303C33A/C102A loses complete binding to InlB indicating no reversible folding. (g) Time dependence of heat induced aggregation and reversible folding of R303. (h) Time dependence of heat induced aggregation and reversible folding of R303C33A/C102A
As VHHs have been known to display reversible denaturation,28, 29 the refolding and heat induced aggregation propensities of R303 and R303C33A/C102A were monitored using a size exclusion chromatography (SEC) based assay.30, 31 Both proteins were heated and the recovery of the sample following cooling was determined by calculating the area underneath the peaks following injection onto an analytical SEC column (Figure 2b,c). While a significant fraction of R303 refolded (71 ± 2% refolded, Figure 2b), no protein was recovered following heating of R303C33A/C102A (0% refolded, Figure 2c). A similar result was observed when the disulfide bonds in wild type R303 were reduced by treatment agent DTT (0% refolded, Figure 2d). These results suggest that removal of the CDR1–CDR3 disulfide bond in R303 results in irreversible heat induced aggregation of the protein and that disulfide bonds are required for reversible refolding of the R303 VHH.
As a final measure of the influence of the CDR1–CDR3 disulfide bond on the reversible denaturation of R303, the binding activity of the wild type VHH and the R303C33A/C102A mutant to InlB249 were determined following heating and refolding by cooling (Figure 2e,f). As expected, wild type R303 exhibited reversible denaturation, with full binding activity being restored after a cycle of heating and cooling (Figure 2e), while the binding activity of R303C33A/C102A was completely and irreversible abolished by heating (Figure 2f).
We also examined the aggregation propensity of R303 and R303C33A/C102A as a function of time at temperatures (25, 40, and 50°C) closer to the ranges the VHH may experience in vivo. While R303 did not aggregate at any of the temperatures tested over the course of a 72‐hr incubation (Figure 2g), on the other hand, R303C33A/C102A displayed significant aggregation after 48 hr at 50°C (Figure 2h). This indicated that R303C33A/C102A is more prone to aggregation at moderate temperatures than R303.
2.3. Structure of R303C33A/C102A and R303C33A/C102A in complex with InlB249
The structure of R303C33A/C102A was solved to a resolution of 1.20 Å (Table 1). Well diffracting crystals of R303C33A/C102A could not be obtained in the same conditions as the previously reported structure of the R303 wild type protein.22 Crystals of R303C33A/C102A grew in the orthorhombic space group P212121, rather than the monoclinic space group P21 for crystals of R303 (Table 1). The structure of R303C33A/C102A contained a dimer in the asymmetric unit, with one monomer rotated approximately 90° relative to the other (Figure 3a). The C33A and C102A mutations were clearly present in the structure, with no disulfide bond linking CDR1–CDR3 (Figure 3a). Each monomer in the dimer was nearly identical in structure (r.m.s.d of 0.3 Å), however there were slight differences in the region of CDR1 (Residues 23–35). In particular, Chain A contained a disordered region with poor electron density surrounding Residues 27 and 28, located within CDR1, while the corresponding residues in Chain B were well ordered.
Table 1.
Data collection and refinement statistics
| R303C33A/C102A | R303C33A/C102A‐ InlB249 | |
|---|---|---|
| Data collection | ||
| Diffraction source | CLS 08ID‐1 | CLS 08ID‐1 |
| Wavelength (Å) | 0.9795 | 0.9795 |
| Resolution range (Å) | 34.28–1.30 (1.35–1.30) | 51.19–1.56 (1.62–1.56) |
| Space group | P212121 | P43 |
|
Unit cell (a,b,c) (α,β,γ) |
47.28 47.39 99.34 90 90 90 |
83.21 83.21 64.93 90 90 90 |
| Total reflections | 253,689 (26010) | 384,394 (22476) |
| Unique reflections | 53,995 (5298) | 62,665 (5837) |
| Multiplicity | 4.7 (4.9) | 6.1 (3.9) |
| Completeness (%) | 96.86 (95.87) | 99.24 (93.12) |
| Mean I/σI | 15.28 (4.51) | 13.48 (1.60) |
| R merge | 0.050 (0.337) | 0.068 (0.589) |
| Refinement and model quality | ||
| R work/R Free | 0.16/0.18 | 0.17/0.20 |
| Number of non‐H atoms | ||
| Protein | 1867 | 2,519 |
| Ions | 15 | 0 |
| Solvent | 301 | 404 |
| R.m.s. bonds (Å) | 0.02 | 0.011 |
| R.m.s angles (°) | 1.62 | 1.00 |
| Ramachandran plot | ||
| Favored (%) | 98.31 | 95.95 |
| Allowed (%) | 1.69 | 4.05 |
| Outliers (%) | 0 | 0 |
| Average B‐factor | ||
| Protein | 17.52 | 28.13 |
| Ions | 42.79 | N/A |
| Solvent | 30.26 | 41.27 |
| PDB code | http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6U14 | http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6U12 |
Figure 3.
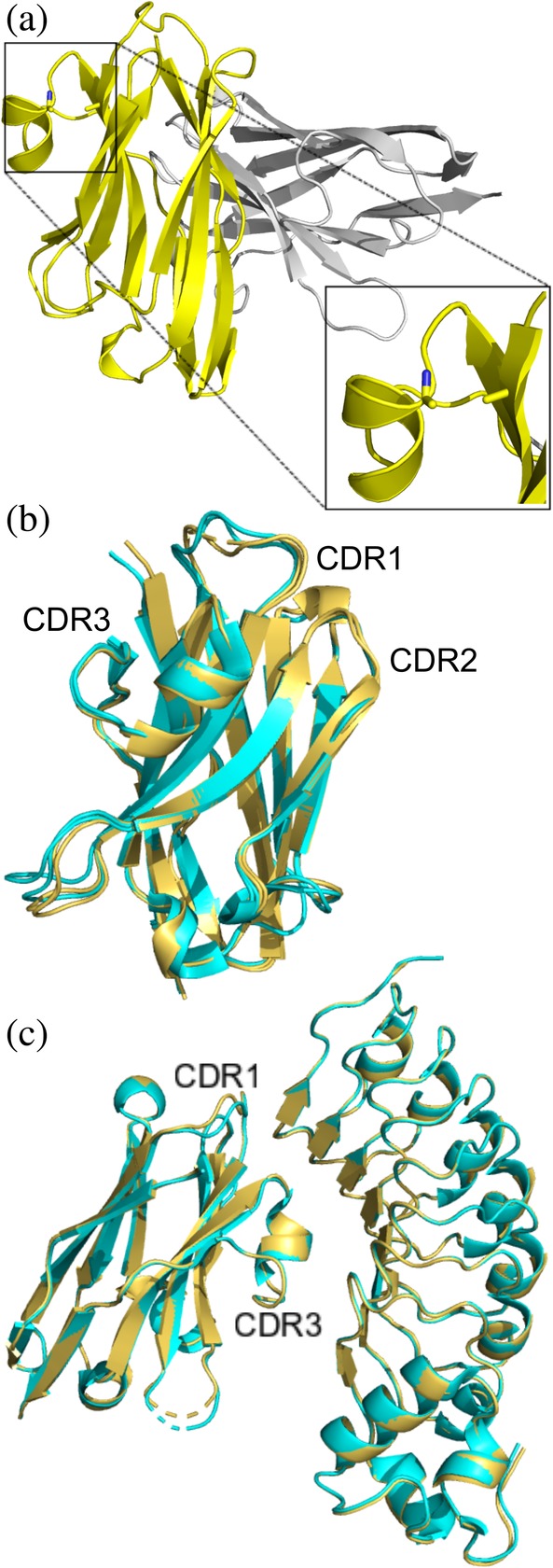
X‐ray structure of unliganded R303C33A/C102A and in complex with InlB249 (a) Structure of unliganded R303C33A/C102A (PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6U14) contains a dimer in the asymmetric unit. The C33A/C102A double mutation is clearly visible (highlighted in the zoomed in box). (b) Overlay of R303C33A/C102A (yellow ribbon structure) with wild type R303 (cyan, PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6DBA) showing only subtle changes in the structure of CDR1. (c) Structure of R303C33A/C102A in complex with InlB249 (yellow, PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6U12) aligned with the structure of R303 in complex with InlB249 (cyan, PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6DBF). The CDR loops and VHH‐antigen interactions are nearly identical in the two structures
Comparison of the structure of R303C33A/C102A with that of wild type R303 (PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6DBA 22) revealed only subtle differences, possibly due to the loss of the disulfide bond between CDR1 and CDR3 (r.m.s.d = 0.4 Å). The conformations of CDR2 and CDR3 remained virtually unaffected by the mutation. On the other hand, the conformation of CDR1 was slightly different in the mutant structure (Figure 3b). However, despite this slight difference in CDR1 conformation, in both structures the loop fell into the same canonical cluster class (cluster 13‐6 or 13‐7 as classified by North et al.23, 32).
The structure of R303C33A/C102A in complex with Inlb249 was solved to 1.56 Å (Table 1). Crystals of the complex were obtained in different conditions than the previously reported structure of wild type R303 in complex with InlB249 and crystallized in a different space group22 (Table 1). The complex structure contained a single molecule of R303C33A/C102A and InlB249, with the VHH binding to the concave face of the LRR domain of InlB, exactly as found previously with wild type R303.22 Comparison of the structures of R303C33A/C102A‐InlB249 with that of R303‐InlB249 (PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6DBF 22) revealed nearly identical structures (r.m.s.d, 0.17 Å; Figure 3c), and no change in the interactions between the VHH and InlB (Table S1). While in the unliganded structures, differences in the structure of CDR1 were observed between the mutant and wildtype R303, no such difference was observed in the complex structures, with CDR1 being in an identical conformation in both structures (Figure 3c).
2.4. B‐factor analysis of R303 and R303C33A/C102A
One potential role of the disulfide bond between CDR1 and CDR3 is to reduce the conformational flexibility of the CDR loops participating in the bond. One approach to compare the relative rigidity and flexibility of proteins in X‐ray structural models is to compare the distribution of normalized B‐factors in the structures.33 The normalized B‐factor Z‐score of the wild type (R303, PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=3DBA) and mutant (R303C33A/C102A) structures were calculated.34
As each X‐ray structure contained two molecules in the asymmetric unit, the normalized B‐factors for each chain (Chain A and Chain B) in the noncrystallographic symmetry (NCS) dimer were plotted independently to compare global differences in normalized B factors across molecules in the structures, as well as to look specifically at the flexibility of CDR1, CDR2, and CDR3 (Figure 4a). The B‐factors within each chain of the dimers were also replaced by the normalized B‐factor values, and the structures were color coded by B‐factor to visualize the relatively mobile regions within the structures (Figure 4b).
Figure 4.
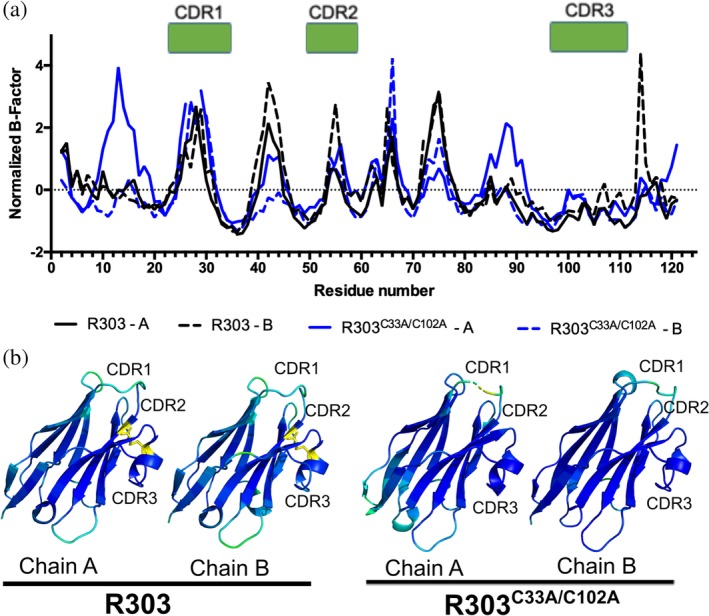
B‐factor analysis of R303C33A/C102A and R303. (a) Plot of normalized B‐factor Z‐scores for R303C33A/C102A (blue) and R303 (black). There is no major difference in the normalized B‐factors within the 3 CDR loops between the wild type and mutant structures. (b) Structures of R303C33A/C102A and R303 colored by normalized B‐factor. Darker blue indicates lower relative B‐factor, lighter blue to green indicates increasing B‐factors values. The location of the C33A‐C102 disulfide bond is shown in yellow. The C33A/C102A mutation does not dramatically alter the mobility of the CDR loops
Within each dimer, one monomer displayed lower normalized B‐factors compared to the other (Chain A in R303 and Chain B in R303C33A/C102A). The effect was more pronounced in the structure of R303C33A/C102A with Chain A having a trend of generally higher B factors than Chain B within the dimer (Figure 4a). To examine if removal of the C33‐C102 disulfide bond resulted in localized changes in structural flexibility of the CDR loops, the distribution of normalized B factors within the each of the CDR loops was compared (Figure 4a,b). The most noticeable difference in the B‐factors occurred in CDR1, where in chain A of R303C33A/C102A the residues at the apex of loop (Residues 27 and 28) were disordered raising the B‐factors of adjacent residues (Figure 4a). Chain B of R303C33A/C102A also had higher B‐factors than either chain of R303, especially near Residues 26–30 (Figure 4a). However, despite this observation, there was no statistically significant difference in the mean normalized B‐factor of CDR1 when comparing the two structures (Table S2).
There was no overall discernable difference in the normalized B‐factors of CDR2 and CDR3 when comparing R303C33A/C102A and R303 (Figure 4), and again no statistically significant difference in the mean normalized B‐factor of CDR2 and CDR3 between the two structures (Table S2) The results of the B‐factor analysis suggest that removal of the C33‐C102 disulfide bond may only have a small effect on the localized flexibility of CDR1. No major influence on the flexibility of CDR2 and CDR3 was observed and overall there was no significant difference in the flexibility of any of the three CDR loops.
2.5. B‐factor analysis of R303 and R303C33A/C102A in complex with InlB249
B‐factor analysis was also carried out on the structures of R303 and R303C33A/C102A in complex with the VHH's antigen, the LRR domain of the Listeria virulence factor InlB (Figure 5). Similar to what was observed in the unliganded VHH structures, the B‐factors in mutant R303C33A/C102A were remarkably similar to the wild type protein across the majority of the structure (Figure 5a). Similar to the unliganded structures, CDR1 in the mutant structure displayed elevated B‐factors, within the vicinity of Residues 27 and 28, while CDR2 and CDR3 display similar B‐factor values (Figure 5a). The residues in CDR1 (Residues 27 and 28) do not directly contact the antigen and instead the mobile region of the loop is facing away from the InlB interaction site (Figure 5b, Table S1). However, similar to what was described above for the unliganded structures, there was no significant difference in the mean normalized B‐factors of each CDR loop when comparing the wild type and mutant structures (Figure 5c, Table S2). Furthermore, there is no major change in the flexibility of any CDR loops upon complex formation with InlB for either R303 or R303C33A/C102A (Figure 5c).
Figure 5.
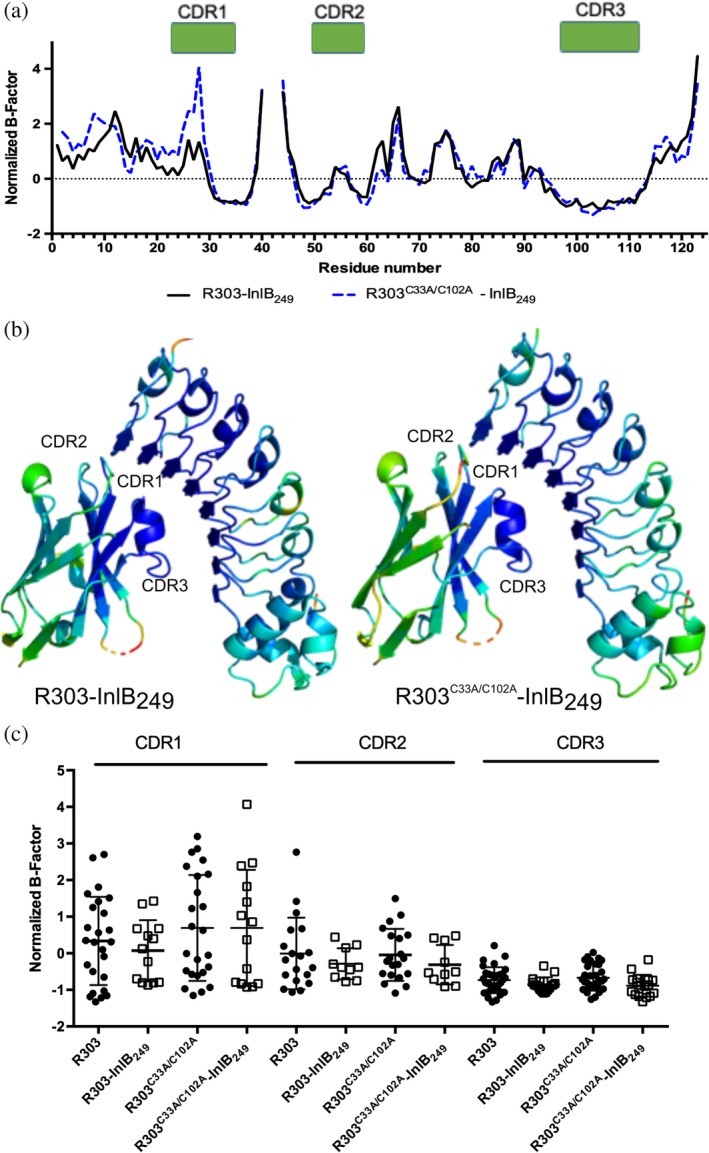
B‐factor analysis of R303C33A/C102A and R303 in complex with InlB249 (a) Plot of normalized B‐factor Z‐scores for R303C33A/C102A‐InlB249 (blue) and R303‐InlB249 (black). There is no major difference in the normalized B‐factors within the 3 CDR loops between the wild type and mutant structures. (b) R303C33A/C102A‐InlB249 (blue) and R303‐InlB249 (black). Darker blue indicates lower relative B‐factor, lighter blue to green indicates increasing B‐factors values. The C33A/C102A mutation does not dramatically alter the mobility of an of the CDR loops. (c) Box plot of normalized B‐factor distribution in the three CDR loops in the unliganded and liganded states for R303 and R303C33A/C102A. Binding to InlB in both structures does not cause a significant decrease in the mean normalized B‐factors
3. DISCUSSION
Camelid VHHs have emerged as an attractive and powerful class of biologics. VHHs are thought to offer several distinct advantages compared to the traditional monoclonal antibody format. VHH are stable, and in at least some cases display reversible folding upon thermal and chemical denaturation.28, 29, 35 Furthermore, due to unusually long CDR3 lengths, in some instances VHH are able to bind epitopes inaccessible to IgG antibodies such as enzyme active sites.36 The advantages conveyed by the VHH format are likely the consequence of camelid evolutionary adaptation to the loss of the light chain. The frequent occurrence of noncanonical disulfide bonds in camelid VHH domains begs the question as to what evolutionary function these bonds serve.
In Camelus dromedarius, the most common noncanonical disulfide bond occurs between CDR1 and CDR3, with the cysteine on CDR1 (position 33) being germline encoded.16 This observation is highly suggestive that the noncanonical disulfide bond plays an important structural and/or functional role in the HCAbs coded by this germline segment. A systematic investigation into the role of the CDR1–CDR3 disulfide bond in antigen binding and thermal stability of five camel VHH resulted in two key hypothesis regarding the functional role of the bond.19 The first hypothesis put forward is that the disulfide bond is an important contributor to HCAbs/VHH antigen affinity owing to entropic effects. The CDR3 loops found in camelid VHH are on average three residues longer than those found in murine or human antibodies.11 Given the longer length of CDR3, the noncanonical disulfide bond could influence affinity by reducing the entropic penalty associated with the immobilization of a long flexible loop. This hypothesis was supported by a landmark study carried out by Govaert et al.19 where cysteine residues in the bond were substituted with a variety of amino acid pairs. The authors observed a range of decreased binding affinity (ranging from 2 to 155 fold decrease) for three of the five VHH in the study (Table 2). However, despite the logic and elegance of this entropic effect hypothesis, the evidence presented here suggests that the effects of the disulfide bond on antigen affinity are not universal, and challenges that the effect on affinity is solely due to entropic effects.
Table 2.
Effect of amino acid substitution of the CDR1–CDR3 noncanonical disulfude bond in camel VHH
In Govaert et al.19 antigen affinity of 2 of the 5 VHH remained unaffected by removing the CDR1–CDR3 disulfide bond, and a similar effect is seen for VHH R303 (Table 2). The affinity of VHH R303 for the InlB antigen remained unaffected by removing the disulfide bond between CDR1 and CDR3 (Figure 1a), indicating that at least in this case, the bond was not a major contributor to antigen binding. Second, the influence of the disulfide bond on the flexibility and mobility of the CDR loops in R303 is minor. Despite R303 having a long CDR3 loop (Table 2), removal of the disulfide bond between CDR1 and CDR3 had no significant effect on the flexibility of the loops (Figure 4). Third, based on the structures presented here, there is no evidence that immobilization of the CDR3 loop upon antigen binding represents an entropic penalty that needs to be paid. There was no evidence in either the wild type R303 or R303C33A/C102A mutant that any of the CDR loops underwent a significant change in flexibility upon binding the InlB antigen (Figure 5). Finally, if the CDR1–CDR3 disulfide bond played an entropic role we may predict the affinity of VHHs with longer CDR3 loops to be more dramatically affected by removal of the disulfide bond. However, the available data suggests this is not the case. There was no observed correlation between CDR3 length and the magnitude of the change in affinity associated with removal of the disulfide bond (Table 2). While clearly at least in some cases the disulfide bond can influence VHH affinity for antigen (Table 2), the mechanism is not entirely clear. The determination of more VHH structures with substituted disulfide bonds may address this question. Taken together, the weight of the evidence suggests that the role the noncanonical disulfide bonds in antigen binding affinity may be rather small, and is certainly not universal. The CDR1–CDR3 noncanonical disulfide bond may potentially serve an alternative evolutionary function.
A second hypothesis put forward by Govaert et al.19 which may explain the evolutionary conservation of the CDR1–CDR3 disulfide bond is related to the biophysical stabilization of the VHH domain. Unsurprisingly, in all the VHH studied by Govaert et al.19 and R303, removal of the disulfide bond resulted in a reduction of the thermal stability of the domain (Table 2). In addition to thermal stability, the reversible refolding of R303 was impacted by the presence of the C33–C102 disulfide bond (Figure 2). Strikingly, while ∼70% of the wild‐type R303 refolded after heating and cooling, removal of the CDR1–CDR3 disulfide bond resulted in completely irreversible unfolding due to protein aggregation. This observation is consistent with a recent large‐scale study on the folding reversibility of 70 VHHs, which found that the presence of a second disulfide bond was strongly correlated with reduced aggregation and reversible folding.37 These results are suggestive that a potential driving force behind the evolution of a second noncanonical disulfide bond in dromedary VHH domains, is not antigen affinity, but rather domain stabilization and prevention of heat induced aggregation.
In the VHH domain, the CDR3 loop folds across a hydrophobic patch which in other IgG antibodies would pair with the light chain, shielding the side chains of residues 37F and 47F/G from solvent.9, 10 Even in the unfolded state, the disulfide bond between CDR1 and CDR3 would maintain the position of CDR3 across the framework which would continue to shield these residues from solvent and prevent heat induced aggregation. This would explain the propensity of R303C33A/C102A to aggregate upon heating, and the irreversible unfolding of the protein following heating, as the 37F/47G hydrophobic patch may trigger aggregation once the CDR3 loop has unfolded. This is supported not only by the observation that R303C33A/C102A is prone to heat induced aggregation while the wild type protein is not (Figure 2), but also that in the presence of a reducing agent, R303 aggregates and no longer displays reversible folding (Figure 2c).
The results presented here, by Govaert et al.19 and by Kunz et al.37 point toward the evolutionary role of the second CDR1–CDR3 disulfide bond being related to the biophysical adaptation of the VHH domain as a means of preventing heat induced aggregation in the absence of a light chain partner and an adaptation to higher temperatures. We can speculate that this may be a specific niche adaptation associated with the hot and arid habitats in which camels live. Among the Camelidae family only camels have specifically evolved to survive in arid, hot climates, while llamas and alpacas generally occupy more temperate zones. If a second noncanonical disulfide bond represented an evolutionary adaptation to higher temperatures, we would expect the bond to be far more frequent in camels. Analysis of unique VHH sequences in the abYsis antibody database38 for putative inter‐CDR noncanonical disulfide bonds revealed that the CDR1–CDR3 disulfide bond is almost exclusively unique to camels (Figure 6). Interestingly, while a second disulfide bond can occur in llama and alpaca VHH domains, the bond tends to occur between CDR2 and CDR3 rather than CDR1.14, 39, 40 In general, the presence of any additional noncanonical disulfide bond is more frequent in camels (∼30% of sequences) compared to alpacas (∼12%) and llamas (∼3.5%; Figure 6). The frequency that camel VHH possess the disulfide bond, and the presence of a germline encoded cysteine in camel VHH point to a specific niche adaptation unique to camels.
Figure 6.

Distribution of noncanonical disulfide bonds in camels (Camelus dromedarius), alpacas (Vicugna pacos), and llamas (Llama glama). VHH sequences from the abYsis database38 were analyzed for the presence of putative noncanonical disulfide bonds in CDR1–CDR3 and CDR2–CDR3
In order to survive the extreme desert climate, camels have evolved to conserve water by increasing their internal body temperature as high as 40°C.41 By storing heat in the body during the day, and only evolving it at night, water loss is prevented as there no heat dissipation through the evaporation of water.41 The higher internal body temperature of camels may have provided the initial evolutionary stimulus to maintain a germline encoded cysteine at position 33, and a second disulfide bond which reduces the heat induced aggregation propensity and increases the thermal stability of the camel VHH domain. This hypothesis is partially supported by experiments which examined the aggregation propensity of R303 and R303C33A/C102A over time at moderate temperatures (Figure 2g,h). R303C33A/C102A was found to exhibit significantly greater amounts of aggregation over time at 50°C compared to wild‐type R303, indicating that the CDR1–CDR3 disulfide bond is a factor which may contribute to the aggregation propensity of the domain at temperatures approaching (although not equal) to those observed in the camel's interior. Although certainly not conclusive, we can speculate that the hypothesis that CDR1–CDR3 disulfide bond evolved to prevent VHH domain aggregation is plausible.
The results presented here serve as a single example which point to the potential role of the CDR1–CDR3 disulfide bond found in camels. Further structural and biophysical studies on additional camel VHH are needed in order to draw a more general conclusion regarding the effect of the disulfide bond on antigen affinity and role in reversible folding of the VHH domain.
4. MATERIALS AND METHODS
4.1. Site directed mutagenesis
VHH R303 was previously cloned into the periplasmic expression vector pSJF2H.20 To delete the interloop disulfide bond between CDR1 and CDR3 in R303 a double mutant (C33A/C102A) was created using site‐directed mutagenesis (Quick Change II, site directed mutagenesis kit, Agilent Genomics). Two sets of mutagenic primers were designed to substitute the TGC codon (C33 of CDR1) to GCC (A33) and the TGT codon (C102 of CDR3) to GCC (A102) within the R303 gene. Two rounds of site directed mutagenesis were carried out on R303 according the manufacturer's instructions. Introduction of the two mutations was confirmed by sequencing (Eurofins Genomics).
4.2. Expression and purification of R303C33A/C102A and InlB249
The purification of VHH R303C33A/C102A was performed as described previously for wild‐type R303.21 Briefly, R303C33A/C102A in plasmid pSJF2H was transformed into Escherichia coli BL21 (DE3). An overnight culture was used to inoculate 2× YT media (30°C, 225 RPM), when the cells reached mid‐log phase (OD600 = 0.6) protein expression was induced with IPTG (0.4 mM) grown for 16 hr (30°C, 225 RPM). The periplasmic fraction was released by osmotic shock, and the protein purified to homogeneity using Ni‐NTA chromatography.
The purification of InlB249 (Internalin B Residues 31–249) I was performed as described previously.22 Briefly, InlB249 cloned into pET‐15‐TEV‐NESG and transformed into E. coli BL21 (DE3). Overnight cultures were used to inoculate 2× YT media (30°C, 225 RPM). At mid‐log phase (OD600 = 0.6) protein expression was induced with IPTG (0.4 mM) and then grown for 16 hr (30°C, 225 RPM). Bacteria were harvested by centrifugation (6°C, 5,000g, 10 minu). The cell pellet was suspended in TBS buffer (20 mM Tris‐HCl pH = 8, 150 mM NaCl, 1 mM PMSF) and lysed using by sonication. The cytoplasmic fraction (supernatant) was isolated by centrifugation (6°C, 10,000 rpm, 30 min) and protein were purified Ni‐NTA affinity and SEC.
4.3. Solid phase (ELISA) binding assay
R303C33A/C102A and wild‐type R303 were biotinylated using the EZ‐Link Sulfo‐NHS‐Biotinylation Kit (Thermo Scientific). A 96 well microtitre plate was coated with of InlB249 (10 ng/μl) in phosphate buffer saline (PBS) overnight at 4°C. The wells were blocked for 1 hr with bovine serum albumin (3%, in PBS). Serial dilutions (1:2 starting at 10 ng/μl) of biotinylated VHH (R303C33A/C102A, R303) was applied and incubated (1 hr). The plate was washed three times with PBS‐tween (0.05% Tween‐20). Following the wash step, streptavidin Horseradish Peroxidase was added (Fisher Scientific, MA) (1:50,000 dilution in 3% BSA 1 hr). Finally, detection was carried out by the addition of 3, 3, 5, 5‐tetramethyl benzidine (TMB; 15 min). The reaction was stopped with 0.18 M H2SO4 and the absorbance measured at 450 nm using a plate reader (BioTek, Biotek Instruments Inc., VT). After adjusting the data for background absorbance, binding curves were constructed in Graph Pad Prism 7.
4.4. Fluorescent microcopy to measure listeria invasion
The assay was carried out as reported previously.22 HeLa cells were cultured in 1X RPMI 1640 media (HyClone) containing 2.05 mM l‐Glutamine, 10% FBS, and Penicillin/Streptomycin and incubated at 37°C with 5% CO2. HeLa cells were seeded at a density of 4 × 105 cells/ml onto a microscope cover glass placed the wells of a 24‐well plate. GFP‐Listeria monocytogenes 42 were grown overnight in BHI broth. The bacteria were washed with sterile 1X PBS (pH 7.4) and labeled with biotin following the manufacturer's instructions (EZ‐Link Sulfo‐NHS‐LC‐Biotin, Thermo Scientific). The bacteria were incubated with R303 or R303C33A/C102A (100 μg/ml, 37°C, 30 min). HeLa cells were stained DAPI dilactate (4′,6‐diamidino‐2‐phenylindole, 10 μg/ml) and infected with biotinylated GFP‐Listeria at a MOI of 50:1. Following centrifugation (15 minutes, 300 RPM) the plate was incubated for 1 hr at 37°C with 5% CO2 followed by three washes RPMI 1640. Biotinylated GFP‐Listeria were detected with Streptavidin‐Dylight550 (1 mg/ml, 30 min at 37°C with 5% CO2) to. The wells were washed with RPMI 1640 and the coverslips were fixed with 4% p‐formaldehyde (30 min, 4°C). After washing three times with PBS (pH 7.4), the coverslips were removed and Fluoromount‐G (SouthernBiotech) was added to mount them onto slides. The slides were analyzed in a Leica DMI3000 B fluorescence microscope at ×63 magnification.
4.5. Thermal stability by circular dichroism spectroscopy
R303 or R303C33A/C102A were diluted to 2.5 μM in PBS pH 7.4. CD spectra were collected using a Jasco J‐815 CD spectropolarimeter between the wavelengths of 180–300 nm at room temperature. A 0.1 cm pathlength rectangular quartz cuvette was used for spectral data collection. Spectral data were analyzed on DichroWeb using the CDSSTR algorithm and reference data sets 6 and 7. CD spectra were then collected in 10°C intervals over a temperature range of 25–95°C to determine the optimal wavelength for monitoring secondary structure denaturation. The ramp rate was programmed at 5.0°C/min. CD data were then collected in triplicate at a wavelength of 202 nm in 5°C intervals over a temperature range of 25–95°C. Raw ellipticity data (mdeg) was smoothed using Jasco software, and converted to molar ellipticity ([θ]) in deg cm2/dmol using Equation (1) 43
| (1) |
where, MRW (mean residue weight) = molecular weight of the protein in Da/number of amino acids, pathlength = cell pathlength in mm, and [VHH] = concentration of VHH in mg/ml. Molar ellipticity values were then used to calculate the fraction of folded protein (FF), shown in Equation 2:
| (2) |
Where [θ F] is the molar ellipticity of the folded protein (25°C) and unfolded (95°C) states. The thermal unfolding midpoint temperature (T M, where FF = 0.5), was obtained by plotting FF against temperature and fitting with a sigmoidal Boltzmann function in Graph Pad Prism 7.
4.6. Reversible folding and aggregation monitored by SEC
The aggregation propensity of R303 and R303C33A/C102A were determined with SEC similar to previously reported experiments with VHH.30 150 μl of R303 and R303C33A/C102A at 0.5 mg/ml in PBS or PBS with 5 mM DTT were incubated at room temperature, or in a PCR thermocycler at 80°C for 10 min, followed by cooling at 4°C for 10 min. The samples were then centrifuged at 17,000g for 1 min, filtered through 0.45 μm filters, and analyzed on an Enrich SEC70 size exclusion column (Biorad). The fraction of nanobody recovered was determined by measuring the area under the curve of the heated samples over the nonheated samples.
For time dependent aggregation experiments, 50 μl aliquots of R303 and R303C33A/C102A (0.5 mg/ml, in PBS pH 7.4) were incubated at 25°C, 40°C, or 50°C for 0–72 hr, followed by cooling in an ice bath for at least 10 min. Samples were then centrifuged at 17,000g for 1 min, and injected through an Enrich SEC650 (Bio‐Rad) size exclusion column. The fraction of nanobody retained from each sample were determined by measuring the area of under the curves of the heated samples over the nonheated samples.
4.6.1. Crystallization of R303C33A/C102A and R303C33A/C102A in complex with InlB249
Purified R303C33A/C102A was dialyzed against 20 mM Tris pH 8.0 and concentrated to 8 mg/ml and screened for crystals in 96 well sitting drop plates using the PEG Rx and Index screens (Hampton Research) screens by the sitting‐drop vapor diffusion method. Crystal optimization was carried out in using hanging drop vapor diffusion in 24 well Limbro plates (Hampton Research). Optimal crystals were grown in 0.1 M Bis‐Tris pH 4.8 with 2 M Ammonium sulfate.
The complex of R303C33A/C102A and InlB249 was formed by mixing the two proteins (1:1.2 M ratio). The complex was purified by gel filtration chromatography (BioRad NGC quest system using Enrich Sec70 column). The complex was concentrated to 10 mg/ml and screened for crystals as described above. Optimal crystals of the complex grew in 0.49 M Sodium Phosphate monobasic monohydrate, 0.91 M Potassium Phosphate dibasic pH 6.8.
4.6.2. Data collection and x‐ray structure determination
Crystals were dipped in cryoprotectant (mother liquor supplemented with 25% glycerol) and flash frozen in liquid nitrogen. X‐ray data for R303C33A/C102A and R303C33A/C102A ‐ InlB249 was collected at the Canadian Light Source on beamline 08ID‐1.44 X‐ray diffraction data was processed using Xia2.45 Both structures were solved by molecular replacement using Phaser as implemented in Phenix.46 For molecular replacement of R303C33A/C102A the previously solved structure of wild type R303 was used as a search model22 (PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6DBA). For the molecular replacement of R303C33A/C102A in complex with InlB249, the structure of R303 ‐ InlB249 was used as a search model22 (PDB code: http://bioinformatics.org/firstglance/fgij//fg.htm?mol=6DBF). The structures were refined using Phenix and manual fitting of sigma A weighted 2Fo‐Fc electron density maps was carried out using Coot.47 The final model and refinement statistics are given in Table 1.
4.6.3. B‐factor analysis to measure main chain protein flxibility
As a measure of protein main chain flexibility, the B‐factors of the Cα atoms in the structures of R303, R303C33A/C102A and the two VHHs in complex with InlB. B‐factors cannot be used directly, so the values were normalized according to the procedure outlined by Parthasarathy and Murphy34 and given in the following equation:
Where, <B> is the average value of all Cα in the structure and σB is the standard deviation of the B‐values within the structure.
Supporting information
Table S1 – Summary of Polar Interactions Between VHH R303/R303C33A/C102A and InlB249
Table S2 – ANOVA Table of differences between average B‐factors in CDR Loops
ACKNOWLEDGMENTS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Research reported in manuscript was supported (100%) by the National Institute of General Medical Sciences of the National Institutes of Health under award number SC3GM112532.
We thank Dr. Roger Mackenzie, Robert Gene and Dr. Jyothi Kumaran (National Research Council, Ottawa, Canada) for the R303 plasmid. We also thank Penny for being a good dog. Part of the research described in this paper was performed using beamline 08ID‐1 at the Canadian Light Source, a national research facility of the University of Saskatchewan, which is supported by the Canada Foundation for Innovation (CFI), the Natural Sciences and Engineering Research Council (NSERC), the National Research Council (NRC), the Canadian Institutes of Health Research (CIHR), the Government of Saskatchewan, and the University of Saskatchewan.
Mendoza MN, Jian M, King MT, Brooks CL. Role of a noncanonical disulfide bond in the stability, affinity, and flexibility of a VHH specific for the Listeria virulence factor InlB. Protein Science. 2020;29:1004–1017. 10.1002/pro.3831
Funding information National Institute of General Medical Sciences, Grant/Award Number: SC3GM112532
REFERENCES
- 1. Muyldermans S. Nanobodies: Natural single‐domain antibodies. Annu Rev Biochem. 2013;82:775–797. [DOI] [PubMed] [Google Scholar]
- 2. Beghein E, Gettemans J. Nanobody technology: A versatile toolkit for microscopic imaging, protein‐protein interaction analysis, and protein function exploration. Front Immunol. 2017;8:771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bever CS, Dong JX, Vasylieva N, et al. Vhh antibodies: Emerging reagents for the analysis of environmental chemicals. Analyt Bioanalyt Chem. 2016;408:5985–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Y, Fan Z, Shao L, et al. Nanobody‐derived nanobiotechnology tool kits for diverse biomedical and biotechnology applications. Int J Nanomedicine. 2016;11:3287–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bannas P, Hambach J, Koch‐Nolte F. Nanobodies and nanobody‐based human heavy chain antibodies as antitumor therapeutics. Front Immunol. 2017;8:1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iezzi ME, Policastro L, Werbajh S, Podhajcer O, Canziani GA. Single‐domain antibodies and the promise of modular targeting in cancer imaging and treatment. Front Immunol. 2018;9:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peyvandi F, Scully M, Kremer Hovinga JA, et al. Caplacizumab for acquired thrombotic thrombocytopenic purpura. New Engl J Med. 2016;374:511–522. [DOI] [PubMed] [Google Scholar]
- 8. Wang N, Smith WF, Miller BR, et al. Conserved amino acid networks involved in antibody variable domain interactions. Proteins. 2009;76:99–114. [DOI] [PubMed] [Google Scholar]
- 9. De Genst E, Saerens D, Muyldermans S, Conrath K. Antibody repertoire development in camelids. Dev Comp Immunol. 2006;30:187–198. [DOI] [PubMed] [Google Scholar]
- 10. Riechmann L, Muyldermans S. Single domain antibodies: Comparison of camel vh and camelised human vh domains. J Immunol Methods. 1999;231:25–38. [DOI] [PubMed] [Google Scholar]
- 11. Mitchell LS, Colwell LJ. Comparative analysis of nanobody sequence and structure data. Proteins. 2018;86:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sela‐Culang I, Kunik V, Ofran Y. The structural basis of antibody‐antigen recognition. Front Immunol. 2013;4:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nguyen VK, Hamers R, Wyns L, Muyldermans S. Camel heavy‐chain antibodies: Diverse germline v(h)h and specific mechanisms enlarge the antigen‐binding repertoire. EMBO J. 2000;19:921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harmsen MM, Ruuls RC, Nijman IJ, Niewold TA, Frenken LG, de Geus B. Llama heavy‐chain v regions consist of at least four distinct subfamilies revealing novel sequence features. Mol Immunol. 2000;37:579–590. [DOI] [PubMed] [Google Scholar]
- 15. Li X, Duan X, Yang K, et al. Comparative analysis of immune repertoires between bactrian camel's conventional and heavy‐chain antibodies. PLoS One. 2016;11:e0161801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nguyen VK, Muyldermans S, Hamers R. The specific variable domain of camel heavy‐chain antibodies is encoded in the germline. J Mol Biol. 1998;275:413–418. [DOI] [PubMed] [Google Scholar]
- 17. Dumoulin M, Conrath K, Van Meirhaeghe A, et al. Single‐domain antibody fragments with high conformational stability. Protein Sci. 2002;11:500–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muyldermans S, Atarhouch T, Saldanha J, Barbosa JA, Hamers R. Sequence and structure of vh domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 1994;7:1129–1135. [DOI] [PubMed] [Google Scholar]
- 19. Govaert J, Pellis M, Deschacht N, et al. Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J Biol Chem. 2012;287:1970–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gene RW, Kumaran J, Aroche C, et al. High affinity anti‐internalin b vhh antibody fragments isolated from naturally and artificially immunized repertoires. J Immunol Methods. 2015;416:29–39. [DOI] [PubMed] [Google Scholar]
- 21. Huh I, Gene R, Kumaran J, MacKenzie CR, Brooks CL. In situ proteolysis, crystallization and preliminary x‐ray diffraction analysis of a vhh that binds listeria internalin b. Acta Cryst. 2014;F70:1532–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. King MT, Huh I, Shenai A, Brooks TM, Brooks CL. Structural basis of vhh mediated neutralization of the foodborne pathogen listeria monocytogenes . J Biol Chem. 2018;293:13626–13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. North B, Lehmann A, Dunbrack RL Jr. A new clustering of antibody cdr loop conformations. J Mol Biol. 2011;406:228–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Agerer F, Waeckerle S, Hauck CR. Microscopic quantification of bacterial invasion by a novel antibody‐independent staining method. J Microbiol Methods. 2004;59:23–32. [DOI] [PubMed] [Google Scholar]
- 25. Kubala MH, Kovtun O, Alexandrov K, Collins BM. Structural and thermodynamic analysis of the gfp:Gfp‐nanobody complex. Protein Sci. 2010;19:2389–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Betz SF. Disulfide bonds and the stability of globular proteins. Protein Sci. 1993;2:1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Goldman ER, Liu JL, Zabetakis D, Anderson GP. Enhancing stability of camelid and shark single domain antibodies: An overview. Front Immunol. 2017;8:865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ewert S, Cambillau C, Conrath K, Pluckthun A. Biophysical properties of camelid v(hh) domains compared to those of human v(h)3 domains. Biochemistry. 2002;41:3628–3636. [DOI] [PubMed] [Google Scholar]
- 29. van der Linden RH, Frenken LG, de Geus B, et al. Comparison of physical chemical properties of llama vhh antibody fragments and mouse monoclonal antibodies. Biochim Biophys Acta. 1999;1431:37–46. [DOI] [PubMed] [Google Scholar]
- 30. Dudgeon K, Rouet R, Christ D. Rapid prediction of expression and refolding yields using phage display. Protein Eng Des Sel. 2013;26:671–674. [DOI] [PubMed] [Google Scholar]
- 31. White B, Huh I, Brooks CL. Structure of a vhh isolated from a naive phage display library. BMC Res Notes. 2019;12:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Adolf‐Bryfogle J, Xu Q, North B, Lehmann A, Dunbrack RL Jr. Pyigclassify: A database of antibody cdr structural classifications. Nucleic Acids Res. 2015;43:D432–D438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun Z, Liu Q, Qu G, Feng Y, Reetz MT. Utility of b‐factors in protein science: Interpreting rigidity, flexibility, and internal motion and engineering thermostability. Chem Rev. 2019;119:1626–1665. [DOI] [PubMed] [Google Scholar]
- 34. Parthasarathy S, Murthy MR. Analysis of temperature factor distribution in high‐resolution protein structures. Protein Sci. 1997;6:2561–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perez JM, Renisio JG, Prompers JJ, et al. Thermal unfolding of a llama antibody fragment: A two‐state reversible process. Biochemistry. 2001;40:74–83. [DOI] [PubMed] [Google Scholar]
- 36. De Genst E, Silence K, Decanniere K, et al. Molecular basis for the preferential cleft recognition by dromedary heavy‐chain antibodies. Proc Natl Acad Sci U S A. 2006;103:4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kunz P, Zinner K, Mucke N, Bartoschik T, Muyldermans S, Hoheisel JD. The structural basis of nanobody unfolding reversibility and thermoresistance. Sci Rep. 2018;8:7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swindells MB, Porter CT, Couch M, et al. Abysis: Integrated antibody sequence and structure‐management, analysis, and prediction. J Mol Biol. 2017;429:356–364. [DOI] [PubMed] [Google Scholar]
- 39. Maass DR, Sepulveda J, Pernthaner A, Shoemaker CB. Alpaca (lama pacos) as a convenient source of recombinant camelid heavy chain antibodies (vhhs). J Immunol Methods. 2007;324:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vu KB, Ghahroudi MA, Wyns L, Muyldermans S. Comparison of llama vh sequences from conventional and heavy chain antibodies. Mol Immunol. 1997;34:1121–1131. [DOI] [PubMed] [Google Scholar]
- 41. Schmidt‐Nielsen K, Schmidt‐Nielsen B, Jarnum SA, Houpt TR. Body temperature of the camel and its relation to water economy. Am J Physiol. 1957;188:103–112. [DOI] [PubMed] [Google Scholar]
- 42. Ma L, Zhang G, Doyle MP. Green fluorescent protein labeling of listeria, salmonella, and Escherichia coli o157:H7 for safety‐related studies. PLoS One. 2011;6:e18083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grochulski P, Fodje MN, Gorin J, Labiuk SL, Berg R. Beamline 08id‐1, the prime beamline of the Canadian macromolecular crystallography facility. J Synch Radiat. 2011;18:681–684. [DOI] [PubMed] [Google Scholar]
- 45. Winter G, Lobley CM, Prince SM. Decision making in xia2. Acta Crystallogr. 2013;D69:1260–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: A comprehensive Python‐based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 – Summary of Polar Interactions Between VHH R303/R303C33A/C102A and InlB249
Table S2 – ANOVA Table of differences between average B‐factors in CDR Loops