

Abstract
Most genes in evolutionarily complex genomes are expressed to multiple protein isoforms, but there is not yet any simple high‐throughput approach to identify these isoforms. Using an oversimplified top‐down LC–MS/MS strategy, we detected, around the 26‐kD position of SDS‐PAGE, proteins produced from 782 genes in a Cdk4−/− mouse embryonic fibroblast cell line. Interestingly, only 213 (27.24%, about one‐fourth) of these 782 genes have their proteins with a theoretical molecular mass (TMM) 10% smaller or larger than 26 kD, that is, between 23 and 29 kD, the range set as allowed variation in SDS‐PAGE. These 213 proteins are considered as the wild type (WT). The remaining three‐fourths includes proteins from 66 (9.44%) genes with a TMM smaller than 23 kD and proteins from 503 (64.32%, nearly two‐thirds) genes with a TMM larger than 29 kD; these proteins are categorized into a larger‐group or a smaller‐group, respectively, for their appearance at a higher or lower position of SDS‐PAGE. For instance, at this 26‐kD position we detected proteins from the Rps27a, Snrpf, Hist1h4a, and Rps25 genes whose proteins' TMM is 8.6, 9.7, 11.4, and 13.7 kD, respectively, and detected proteins from the Plelc1 and Prkdc genes, whose largest isoform is 533.9 and 471.1 kD, respectively. We extrapolate that many of those proteins migrating unexpectedly in SDS‐PAGE may be isoforms besides the WT protein. Moreover, we also detected a Cdk4 protein in this Cdk4−/− cell line, thus wondering whether some of other gene‐knockout cells or organisms show similar incompleteness of the knockout.
Keywords: gene knockout, LC–MS/MS, protein isoform, proteomics, SDS‐PAGE, top‐down
1. INTRODUCTION
The chromosomal genome in evolutionarily‐higher animals stores its genetic information in an extremely compact manner. While both strands of the DNA double helix encode genes, on each strand one gene often contains other gene(s), resulting in a common situation dubbed by us as “gene(s) within a gene” or “a gene containing other gene(s)”.1, 2 This structural complexity makes it possible that genetic manipulation of a gene on one strand may mistakenly manipulate genes on the other strand as well. Moreover, manipulation of the expression of one gene with antisense, small interference (siRNA), or related techniques may also interfere with the expression of the other gene(s) within the target gene or on the opposite strand, if technical details are not well considered. The complexity occurs not only at the DNA level but also at the RNA and protein levels, since it is now well known that one gene often produces multiple mRNA variants and protein isoforms (defined as different protein forms produced by the same gene).1, 2, 3, 4 The mechanisms for the mRNA or protein multiplicity include, but are not limited to, alternative initiation or termination of transcription, alternative splicing of an RNA transcript, and alternative use of a translational start codon or stop codon of a mRNA. These complexities at the DNA, RNA, and protein levels create difficulties in estimating how many genes a genome encodes and how many protein isoforms a gene produces on average. Actually, now, in the post‐ENCODE epoch, we no longer know “what is a gene”,5, 6, 7, 8 a question that was so simple and so basic to a biologist many decades ago. One of the reasons is that each gene is no longer considered to encode a specific phenotype but is perceived to encode a full range of phenotypes, which is a newly emerging concept dubbed by Heng et al. as “fuzzy inheritance.”9, 10, 11 As another reason, those genomic loci that encode non‐coding RNAs are also considered by many peers as genes, whereas “non‐coding RNA” itself remains to be ill‐defined and is challenged by the fact that many short peptides, as short as 11 amino acids encoded by only 33 nucleotides, are known to have important biological functions.12, 13, 14, 15, 16
With the genomic complexity gradually being better known, we have started to question some routine techniques used to explore the expression and function of genes and in turn to question the conclusions from the resultant data, inspired in part by Stepanenko and Heng.17 For example, genetic manipulation to delete a gene, commonly referred to as “gene knockout”, is a widely used approach in studies of genes' functions in cells and various organisms. However, in most, if not all, of the cases, the knockout is achieved by deleting a small DNA fragment from the gene, by inserting a small fragment of DNA into the gene, or by replacing a small fragment of DNA with another DNA sequence in the gene, while most part of the target gene remains intact. This form of manipulation aims to disrupt the open reading frame (ORF) of the targeted mRNA, usually the wild type (WT), so that the mRNA cannot be translated into the target protein.1, 3 However, it remains possible that the large intact part of the gene may still express other mRNA variants and the corresponding protein isoforms. Indeed, ERα (estrogen receptor α),18, 19, 20, 21, 22 Cdk4,23 and caspase‐824 knockout mice have been suspected of expressing some mRNA variants or protein isoforms of the targeted gene detected with RT‐PCR (reverse transcription and polymerase chain reactions) or WB (western blotting), although more tangible evidence is still needed to verify this suspicion. Moreover, the deletion, insertion, or replacement of a small DNA fragment may create new non‐coding RNAs or new mRNA variants that encode new protein isoforms of the target gene, and may even create new genes, such as fusion genes that are often seen in cancer cells with similar genetic alterations. If any of these scenarios occurs in a gene‐knockout cell or organism, it is our bias to claim that the gene (and not just the targeted protein isoform) is deleted, or to attribute the observed changes in functions solely to the “loss of the gene”.
Determination of protein isoforms engendered by a particular gene, or a general estimation of protein isoforms produced by a gene on average or by the whole genome, is obviously very useful and important for the reasons described above. However, such determination or estimation is still difficult, mainly because of a lack of convenient technique to determine protein isoforms in a high throughput manner. Top‐down LC–MS/MS (liquid chromatography coupled with tandem mass spectrometry) proteomic technique is currently the standard method for the determination of protein isoforms of individual genes. However, this method possesses several weaknesses, including the requirements not only of sophisticated equipment but also of a complicated procedure. Moreover, it is not sufficiently high‐throughput. We recently developed a much more simple strategy of top‐down LC–MS/MS that allows determination of protein isoforms of genes at a given position of SDS‐PAGE (SDS‐containing polyacrylamide gel electrophoresis). With this oversimplified approach, we surprisingly found that about 90% of the human genes in some cell lines have protein isoforms other than the canonically annotated protein form, usually the WT, meaning that probably less than 10% of the human genes express only a single protein form.25, 26
In this study, we extended our work to estimate the frequency of protein isoforms produced by mouse genes and attempted to test whether a gene that is supposed to have been knocked out could still express an isoform. We took advantage of a previous finding of a putative Cdk4 protein isoform at about 26‐kD of SDS‐PAGE, which was also detected with WB in a Cdk4−/− mouse embryonic fibroblast (MEF) cell line. Our results showed that about three‐fourths of the proteins identified, including Cdk4, were unexpected at this 26‐kD position of SDS‐PAGE because they are too large or too small. Many of these unexpected proteins are likely to be an additional isoform besides the WT form.
2. MATERIALS AND METHODS
2.1. Cell line
A Cdk4−/− MEF cell line was used. This line was established by Rane et al. from their Cdk4 knockout mouse and was kindly provided to Dr. Chenguang Wang, then at the Thomas Jefferson University, Philadelphia, PA. The knockout was established by insertion of a reversely oriented Neo cassette into the Cdk4 gene.27 Our previous studies could still detect some Cdk4 mRNA with RT‐PCR23 from this cell line, although we did not check the expression of the full‐length mRNA as it was likely to have been scrambled. Our previous WB results also showed that a putative Cdk4 protein isoform at about 26‐kD was detectable in this cell line, although, as expected, the WT Cdk4 at 33‐kD was not detected.23 Another MEF cell line from a normal, that is, WT for Cdk4, mouse embryo was also used as a control for the molecular weights of the WT Cdk4 protein and the 26‐kD putative Cdk4 isoform in SDS‐PAGE.
2.2. SDS‐PAGE and excision of the gel for LC–MS/MS
As previously described,23 cells of the normal MEF line and the Cdk4−/− MEF line were cultured until the cells reached roughly 80% confluence. The cells were then washed with 1× phosphate buffered saline and harvested via scraping in a lysis buffer containing 1× Proteinase Inhibitor Cocktail (Sigma‐Aldrich, Inc, St. Louis, MO). A protein sample was routinely prepared from the cells and diluted with a gel‐loading buffer containing 2% of SDS and 2% of 2‐mercaptoethanol to the final concentration. After boiling for 4 min and then rapidly cooling on ice, the proteins were loaded into a 15% SDS‐containing polyacrylamide gel. To better separate and better detect the proteins, the gel was made with 10 × 10.5 cm glass plates included in the Hoeffer SE260 vertical slab gel system (Hoeffer Inc; http://www.hoeferinc.com/), which produced a gel 2 cm longer in the vertical direction than all gels made using the regular mini‐gel cast systems of Hoeffer and other companies. As illustrated in Figure 1, the 1st well of the gel was loaded with 60 μg of proteins from the normal MEF cells, the 2nd, 3rd, and 10th wells were loaded with a prestained protein marker containing a 33‐kD band, while the remaining 4th to 9th wells were loaded with proteins from the Cdk4−/− MEF cells (60 μg per well).
Figure 1.
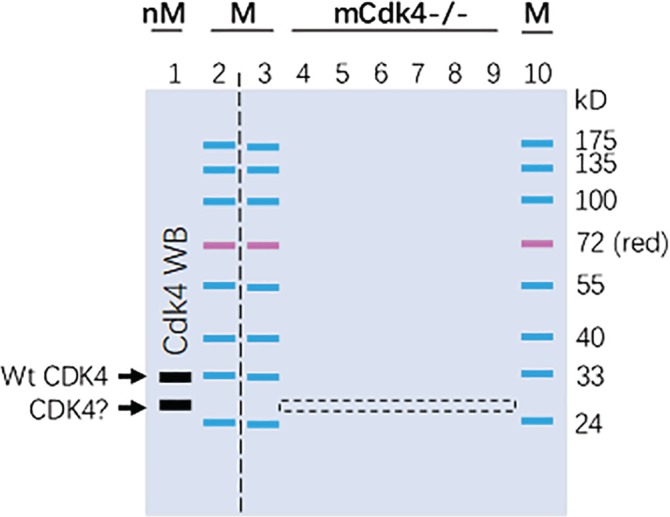
Depiction of electrophoresis and excision of the gel stripe. A 15% SDS‐containing polyacrylamide gel, which was 2‐cm longer in the vertical direction than regular mini‐gels, was loaded with a pre‐stained protein marker (M) in wells 2, 3, and 10. Proteins from normal MEF (nM) cells were loaded into well 1 and proteins from the Cdk4−/− MEF cells were loaded into wells 4–9. After protein separation via electrophoresis, the left part of the gel was cut out along the vertical dashed line and used in a quick WB procedure for detection of Cdk4. The WB resulted in multiple bands, including one at 33‐kD and another at 26‐kD (arrows). The WB membrane was then aligned to the right part of the gel to guide the excision of a 2‐mm gel stripe, shown as the dashed box, of lanes 4–9 at the 26‐kD‐band position. The proteins in this gel stripe were later extracted out during the LC–MS/MS procedure
After separation of the proteins via electrophoresis, the first two lanes were excised out, with a surgical blade and guided by a ruler along the vertical dashed line, as illustrated in Figure 1. The proteins in the 1st lane and the prestained marker in the 2nd lane were then transferred onto a PVDF membrane via electrophoresis, followed by the detection of Cdk4 proteins via a quick WB procedure using the sc‐601 primary antibody from Santa Cruz Biotech, as detailed before.23 The WB was performed in a shorter‐than‐usual time period to shorten the whole procedure, and, in the meanwhile, the remaining part of the gel was stored at 4°C. As we previously reported,23 the WB resulted in several bands on the membrane, including the WT Cdk4 protein exactly at the 33‐kD, and another one around 26‐kD, which was suggested as a putative Cdk4 isoform by WB results using different primary antibodies.23 We then aligned the WB membrane with the remaining part of the gel to determine the 26‐kD‐band position of the gel. Guided by two rulers and by estimation based on the prestained markers on lanes 3 and 10, we excised out a narrow stripe (about 2 mm in width) at the 26‐kD‐band position of the 4th‐9th lanes of the gel. The narrow gel stripe is illustrated as a dashed box in Figure 1 and was used for LC–MS/MS.
2.3. LC–MS/MS
As described before,25, 26 the excised gel stripe containing proteins from the Cdk4−/− MEF was dehydrated with escalating concentrations of acetonitrile (ACN) as per routine methods. The in‐gel proteins were reduced and alkylated with 10 mM dithiothreitol (DTT) and 55 mM iodoacetamide (IAM), followed by digestion with trypsin at 37°C for 16 hr.5 The tryptic peptides were then extracted from the gel with ACN containing 0.1% formic acid (FA), vacuum‐dried, and dissolved in 0.1% FA. The peptides were delivered onto a nano RP column (5‐μm Hypersil C18, 75 mm × 100 mm; Thermo Fisher Scientific, Waltham, MA) and eluted with escalating (50–80%) ACN for 60 min at a speed of 400 nL/min. Different fractions of the eluate were injected into a Q‐Executive mass spectrometer (Thermo Fisher Scientific, Waltham, MA) set in a positive ion mode and a data‐dependent manner with a full MS scan from 350 to 2,000 m/z. High‐collision energy dissociation (HCD) was used as the MS/MS acquisition method. Raw MS/MS data were converted into an MGF format using Proteome Discoverer 1.2 (Thermo Fisher Scientific, Waltham, MA). The exported MGF files were searched with Mascot v2.3.01 in a local server against the mouse SwissProt database. All searches were performed with a tryptic specificity allowing one missed cleavage. Carbamidomethylation was considered as fixed modification whereas oxidation (M) and Gln‐ > pyro‐Glu (N‐term Q) were considered variable modifications. The mass tolerance for MS and MS/MS was 15 ppm and 20 mmu, respectively. Proteins with false discovery rates (FDR) < 0.01 were further analyzed.
2.4. Retrieval of information on the gene number
The information about the numbers of genes and proteins for each chromosome in the human, mouse and rat was retrieved, respectively, from the following websites of the NCBI (National Center for Biotechnology Information) in February of 2019: https://www.ncbi.nlm.nih.gov/genome/?term=human+genome, https://www.ncbi.nlm.nih.gov/genome/52, https://www.ncbi.nlm.nih.gov/genome/?term=rat+genome.
In the corresponding website, there is a table that lists the gene number, protein number, pseudogene number, for each chromosome. The number of genes or proteins from each chromosome was added together to obtain the total number for the corresponding genome.
2.5. Statistical analyses
The methods used for statistical comparisons are indicated in the relevant tables, with p < .05 set for statistical significance.
3. RESULTS
3.1. How many genes are encoded by the human, mouse, or rat genome?
The total number of genes in the human, mouse, or rat genome, obtained by adding together the number of genes encoded by each chromosome, is 54,099, 45,765, or 38,882, respectively, with pseudogenes excluded (Table 1). The total number of proteins produced by the human, mouse, or rat genome is 113,138, 75,959, or 55,761, respectively (Table 1). The actual number of genes or proteins may be slightly smaller, since genes or proteins from the Y chromosome, which is very small, are also included, while some of them are their counterparts from the X chromosome. The protein‐to‐gene ratios are about 2.1 (113,138/54,099) in the human, 1.7 (75,959/45,765) in the mouse, and 1.4 (55,761/38,882) in the rat.
Table 1.
The number of genes and proteins in the human, mouse, and rat genomesa
| Chr. | Human | Mouse | Rat | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Protein | gene | Pseudogene | Protein | Gene | Pseudogene | Protein | Gene | Pseudogene | |
| 1 | 11,321 | 5,109 | 1,386 | 4,731 | 2,687 | 579 | 7,447 | 4,981 | 937 |
| 2 | 8,291 | 3,871 | 1,181 | 6,282 | 3,491 | 609 | 3,463 | 2,725 | 630 |
| 3 | 7,150 | 2,990 | 900 | 3,507 | 2,225 | 480 | 4,337 | 2,969 | 510 |
| 4 | 4,599 | 2,441 | 803 | 4,710 | 2,622 | 497 | 3,375 | 2,383 | 442 |
| 5 | 4,729 | 2,592 | 778 | 4,634 | 2,507 | 413 | 3,481 | 2,194 | 411 |
| 6 | 5,522 | 3,005 | 882 | 3,844 | 2,597 | 555 | 2,193 | 1,839 | 355 |
| 7 | 5,112 | 2,792 | 911 | 6,336 | 3,798 | 935 | 3,399 | 2,319 | 474 |
| 8 | 4,199 | 2,165 | 671 | 3,653 | 2,177 | 376 | 3,213 | 2,087 | 348 |
| 9 | 4,699 | 2,270 | 706 | 4,406 | 2,276 | 374 | 2,502 | 1,442 | 261 |
| 10 | 5,429 | 2,179 | 640 | 3,546 | 2,086 | 391 | 4,269 | 2,622 | 312 |
| 11 | 6,394 | 2,924 | 829 | 5,650 | 2,852 | 381 | 1,493 | 1,094 | 218 |
| 12 | 5,975 | 2,526 | 691 | 2,621 | 2,002 | 516 | 1,588 | 1,024 | 135 |
| 13 | 2,056 | 1,385 | 475 | 2,536 | 2,127 | 476 | 1,758 | 1,244 | 247 |
| 14 | 3,501 | 2,065 | 585 | 3,007 | 2,111 | 455 | 1,971 | 1,275 | 269 |
| 15 | 3,623 | 1,824 | 554 | 2,872 | 1,620 | 282 | 1,701 | 1,443 | 285 |
| 16 | 4,625 | 1,938 | 469 | 2,486 | 1,367 | 257 | 1,671 | 1,103 | 228 |
| 17 | 6,226 | 2,450 | 556 | 3,610 | 2,005 | 427 | 1,566 | 1,287 | 201 |
| 18 | 2,029 | 984 | 295 | 1,855 | 1,218 | 264 | 1,260 | 989 | 195 |
| 19 | 6,750 | 2,499 | 523 | 2,328 | 1,283 | 205 | 1,470 | 984 | 153 |
| 20 | 2,904 | 1,358 | 338 | No this chromosome | 1,508 | 1,124 | 267 | ||
| 21 | 1,297 | 777 | 207 | No this chromosome | No this chromosome | ||||
| 22 | 2,582 | 1,189 | 354 | No this chromosome | No this chromosome | ||||
| X | 3,801 | 2,186 | 875 | 3,010 | 2,291 | 912 | 2,054 | 1,694 | 575 |
| Y | 324 | 580 | 392 | 335 | 423 | 85 | 42 | 60 | 15 |
| Sum | 113,138 | 54,099 | 16,001 | 75,959 | 45,765 | 9,469 | 55,761 | 38,882 | 7,468 |
Abbreviation: Chr.: Chromosome.
From the NCBI data base in February 2019.
3.2. LC–MS/MS identified a peptide unique to the mouse Cdk4
LC–MS/MS identified two Cdk4 peptides from the Cdk4−/− MEF cell line. One is “ARDPHSGHFVALK,” which is unique to part of the N‐terminal region of the Cdk4 (Figure 2). The other is “IADFGLAR,” which should be the “LADFGLAR” in the mouse Cdk4 since the first amino acid (AA), “I,” in the sequence we identified has the same molecular weight as the first AA, “L,” in the mouse Cdk4 sequence (Figure 2). However, this “LADFGLAR” sequence is not unique to Cdk4 because it also exists in the mouse Cdk2, Cdk4, and Cdk5 proteins.
Figure 2.

The sequence of the WT mouse Cdk4 protein, with the two LC–MS/MS‐identified peptide sequences underlined. While the italicized sequence (ARDPHSGHFVALK) is unique to Cdk4, the other (“IADFGLAR”) should be “LADFGLAR” and is shared with Cdks 1, 2, and 5 as well
3.3. Only about one‐fourth (27.24%) of the identified proteins are expected
From the narrow gel stripe at the 26‐kD location, LC–MS/MS identified proteins produced by a total of 782 genes (Table 2). Proteins from 497 of the 782 genes contained two or more unique LC–MS/MS‐identified peptides and thus had a definite identity. Most of the 782 genes' largest protein isoforms possess a theoretical molecular mass (TMM) around 30–40 kD (Figure 3, Tables S1 and S2), but there are still many other genes with their largest proteins much greater than 40 kD or much smaller than 20 kD. For instance, the Plec1 gene has its longest protein isoform with a TMM of 533.9 kD, whereas the Rps27a gene expresses an 8.6 kD protein (Table S1 and S2). Obviously, it is unexpected that these too‐large or too‐small proteins were detected at about 26‐kD in SDS‐PAGE.
Table 2.
Categorization of genes whose protein products are identified
| TMM (kD) | Genes (%) |
|---|---|
| <23 (larger group) | 66 (9.44) |
| 23–29 (WT‐group) | 213 (27.24) |
| >29 (smaller group) | 503 (64.32) |
| Total | 782 (100) |
Figure 3.
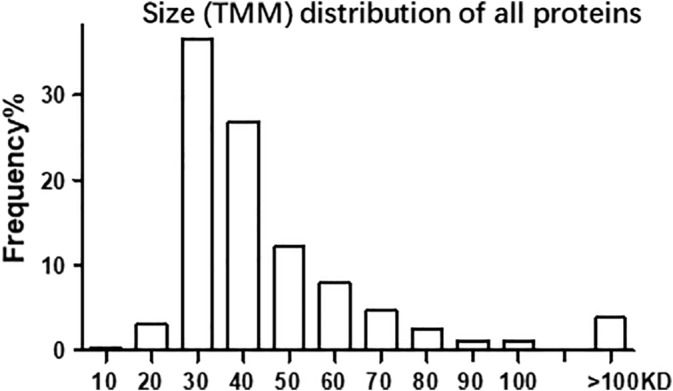
Size (TMM) distribution of the proteins CR, with the Y axis being the percentage of the genes detected and the X axis being the TMM, showing that most proteins have their TMMs around 30–40 kD
Considering that the prestained protein markers may not be accurate enough and protein migration in SDS‐PAGE can be affected by different factors, as described before,25, 26 we arbitrarily allow those proteins with a TMM in the range of 23–29 kD, which is about 10% larger or smaller than 26‐kD, to be considered the expected gene products, herein referred to as “the WT,” as described before.25, 26 Those proteins with their TMMs above this WT range are categorized into a “smaller‐group,” while those with their TMMs below this WT range are grouped into a “larger‐group,” because their positions in the gel were smaller or larger, respectively, than their WT TMMs (Table 2). Because many genes are expressed as multiple protein isoforms, the categorization is made based on the largest isoform listed in the NCBI database, which may or may not be the canonical WT. By this lax criterion, proteins from 213 (27.24%, or about one‐fourth) of the 782 genes fell into the WT‐group, proteins from 66 (9.44%) genes were assigned into the larger group, and proteins from 503 (64.32%, or nearly two‐thirds) were attributed to the smaller group (Table 2). The sum of the larger and smaller groups is 569 (73.76%), indicating that about three‐fourths of the proteins detected are unexpected at the 26‐kd position.
3.4. Lengthy proteins have large region(s) without LC–MS/MS‐identified peptides
We mapped the LC–MS/MS‐identified peptide sequences onto the largest protein isoform of four genes, that is, Plec1 (Figure 4), Flna (Figure 5), Prkdc, and Sptan1 (Figure 6), whose protein products have the largest TMMs among all proteins detected (Table S1 and S2). The largest protein isoform of Plec1, which has 4,691 AAs and a TMM of 533.9 kD, has six identified peptides. However, three of the six peptides are repeats and one of the repeats has two mismatched AAs (Figure 4). Interestingly, all three repeats are located beyond the middle of the protein (Figure 4). The largest protein isoform of the Flna gene, which has 2,647 AAs and a TMM of 281 kD, has 23 identified peptides distributed throughout the protein (Figure 5).
Figure 4.

Mapping LC/MS/MS‐identified peptides (underlined) onto the largest protein isoform of the Plec1 gene (upper panel), showing that there is a large sequence gap between any two neighboring identified peptides, such as a 1,371‐AA gap between “GGAEGELQALR” and “GFFDPNTHENLTYLQLLER” in the middle of the protein. Moreover, there are three repeated sequences identified (lower panel), from the 2948th to the 3028th, the 3607th to the 3687th, and the 4185th to the 4265th AA regions, respectively, with single AA mutations shown as the italicized letters. One of the three repeats contains two mismatched AAs (boldfaced and italicized letters) in the LC–MS/MS‐identified peptide (lower panel)
Figure 5.

Mapping the 23 LC–MS/MS‐identified peptides onto the largest protein isoform of the Flna gene. In several circumstances, one identified peptide (italicized) is consecutive to another, making the two forming a single peptide sequence. Two identified peptides (shaded with grey color) are not unique to the Plna but, instead, are shared with proteins of other gene(s). Although the 23 peptides are distributed throughout the whole protein, there are still large gaps between some identified peptides, such as the 667‐AA gap between “YGGDEIPFSPYR” and “FNEEHIPDSPFVVPVASPSGDAR”
Figure 6.

Mapping of the only LC–MS/MS‐identified peptide (underlined) onto the largest protein isoform of the Prkdc gene (top panel) and the Sptan1 gene (bottom panel), with many AAs at the N‐ and C‐terminal regions not shown
Both Plec1 and Flna proteins consistently display a lengthy undetected region between two neighboring LC–MS/MS‐identified peptides. For instance, there is a 1,371‐AA region between “GGAEGELQALR” and “GFFDPNTHENLTYLQLLER” in the Plec1 protein (Figure 4). This phenomenon is even more prominent in the largest protein isoform of the Prkdc and Sptan1 genes, because the largest Prkdc has 4,128 AAs with 471.1 kD as its TMM and the largest Sptan1 has 2,498 AAs with 284.4 kD as its TMM, but these two proteins have only one identified peptide (Figure 6).
We also mapped the LC–MS/MS‐identified peptides onto the proteins of four other genes, that is, Rps27a, Snrpf, Hist1h4a and Rps25, whose largest protein isoform is 8.6, 9.7, 11.4, and 13.7 kD, respectively (Figure 7), according to the NCBI database. While some of these very small proteins have only one peptide identified, others have two or more (Figure 7).
Figure 7.

Mapping the LC–MS/MS‐identified peptides (underlined) onto the proteins of the Rps27a, Snrpf, Hist1h4a and Rps25 genes, whose protein products have the lowest TMMs among the proteins of the genes detected in this study
3.5. A smaller CR suggests a higher possibility of lacking part(s) of the AA sequence
Since it is unlikely that a large region of a protein sequence lacks a trypsin‐digested peptide for LC–MS/MS detection, as shown in Figures 4, 5, and 6, a likely reason for the detection of the smaller group, generally speaking, is that the detected protein is either a degraded fraction or a smaller isoform lacking part(s) of the sequence. The LC–MS/MS results provide coverage rate (CR) of identified AAs (Table S2, column D), that is, the total AAs of all identified peptides as the percentage of the total AAs of the largest protein isoform listed in the NCBI database, as illustrated in Figure 8a using the 6PGL protein as an example.26 A larger CR indicates that the detected protein isoform is closer to the full‐length form, with a CR of 1 (100%), which will not occur in any real case, indicating that all AAs are LC–MS/MS identified. While the average of the CRs of the proteins of all 782 genes were 13.95%, the average CR of the WT‐group was 18.74%, which was significantly higher than 11.64%, the average CR of the smaller‐group but was not statistically different from 16.13%, the average CR of the larger group (Table 3).
Figure 8.
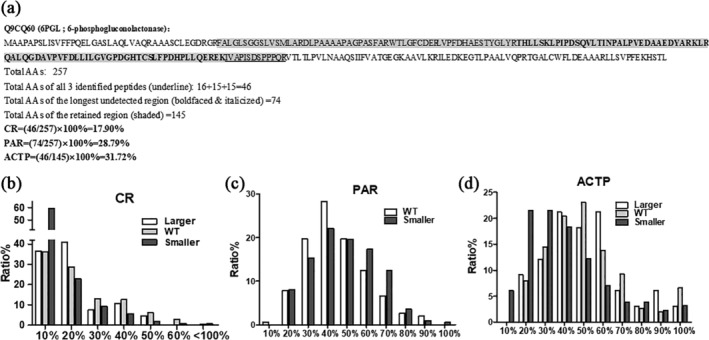
Calculation of CR, PAR, and ACTP, as well as their distributions with the Y axis being the percentage of the genes detected and the X axis being the CR, PAR, or ACTP. (a) Methods of calculation of CR, PAR, and ACTP with the 6PGL protein as the example. (b) CR distribution, showing that in all three groups the majority of the proteins detected have a CR of about 20% or less. (c) PMCUF distribution showing that in the WT‐ and smaller‐groups PMCUF tends to manifest a normal distribution, with most proteins having a PMCUF around 20–80%. (d) ACTP distribution showing that in all three groups ACTP tends to distribute normally, with most proteins having an ACTP around 20–70%
Table 3.
CR of each group
| Total | Smaller | WT | Larger | |
|---|---|---|---|---|
| Average | 13.95% | 11.64% | 18.74%a | 16.13% |
| CR > 20% | 35.01% | 17.50% | 35.21% | 22.73% |
Significantly higher than the smaller‐group (p < .05; Wilcoxon rank sum test, due to large variation).
Since most proteins had a CR of about 20% or less (Figure 8b), we calculated how many genes expressed proteins with a CR larger than 20% and found 35.01% of the 782 genes possessing this larger CR (Table 3). Interestingly, while 35.21% of the genes in the WT‐group showed a CR >20% for their proteins, only 17.50 and 22.73% of the genes in the smaller and larger groups, respectively, had a CR >20% for their proteins; which is significantly lower than the same parameter of the WT‐group (Table 3).
3.6. A larger PAR suggests a greater possibility that part(s) of the sequence may be absent
As aforementioned, each detected protein consists of LC–MS/MS‐detected and undetected peptides, and a larger region without any detected peptide indicates a greater likelihood that this region is absent in the detected protein isoform. If the number of detected peptides is coined as N, the number of undetected peptides or regions should be N + 1. For instance, if a protein has two detected peptides, like the Cdk4 (Figure 2), the number of undetected regions should be 3. For those proteins with two or more detected peptides, that is, when N ≥ 2, we mapped all detected peptides onto the largest isoform of each detected gene to determine the longest undetected region, and then calculated the ratio of the total AAs of this region to the total AAs of the protein, as illustrated with the exemplary protein in Figure 8a. The resultant parameter, dubbed as the “possibility of the absent region” (PAR), reflects the possibility for the lack of this region in the detected isoform. A larger N, that is, a larger number of detected peptides, is associated with a shorter the‐longest‐undetected‐region and in turn a smaller PAR, which indicates a smaller possibility for the lack of this region in the detected isoform. Obviously, in this study, the proteins in the larger group are too few in number to be present in the figure and are too short (<23 kD) to get a reliable PAR.
The PAR distribution shifted to the right (Figure 8c), compared with the CR distribution (Figure 8b). At the low range (<40% on the X axis), the PAR was higher in the WT‐group compared to the smaller‐group, whereas at the higher range (>60%) the opposite is true (Figure 8c), indicating that genes in the smaller group might express proteins lacking part(s) of the AA sequence. While the CR distribution of all groups peaked at about 10–20% and did not show a normal distribution (Figure 8b), PAR shifted to the right, peaked at 30–60%, and tended to show a normal distribution (Figure 8c).
3.7. A larger ACTP also indicates a higher possibility of lacking part(s) of the sequence
RNA transcription often starts from an alternative initiation site, resulting in an RNA variant with a shorter or longer 5′‐region that may be translated to a protein isoform with a shorter or longer N‐terminus.28 Transcription often terminates at an alternative site as well, resulting in an RNA variant with a shorter or longer 3′‐region that may be translated to a protein isoform with a shorter or longer C‐terminus.28 Actually, translation of a given mRNA may also use an alternative start codon or stop codon, engendering a protein isoform with a longer or shorter N‐ or C‐terminus.28 To evaluate the possibility of the occurrence of an isoform with the N‐ or C‐terminal truncation, we mapped the detected peptides onto the largest isoform. We assume that the region between the first and the last identified peptides is the most likely region to actually exist in the detected protein isoform, shown as the boldfaced and italicized region in the exemplary protein in Figure 8a. Conversely, a longer N‐ or C‐terminal region without a detected peptide implies a higher possibility that this region does not exist in the detected isoform. Therefore, we calculated the ratio of the number of total AAs of all detected peptides to the number of AAs in the region between the first and last detected peptides. We refer this ratio to as the “approximate coverage of truncated proteins” (ACTP) (Figure 8d), with an assumption that all detected peptides are from the same protein isoform. A larger number of AAs in these two undetected ends, that is, longer undetected N‐ and C‐terminuses, will result in a larger ACTP, which connotes a higher possibility of the N‐ or C‐terminal truncation, and vice versa.26
The ACTP of the WT‐ and larger groups, but not that of the smaller group, tended to show a normal distribution (Figure 8c). In general, at the lower range (<30%), the ACTP was much higher in the smaller group than in the other two groups. Conversely, the opposite was discerned in the higher range (50%–70%), similar to PAR, which suggests that there might be isoforms smaller than the WT form with a N‐ and/or C‐terminal truncation.
4. DISCUSSION
4.1. How many genes are there in the human, mouse, and rat genomes?
There have been different estimations in the past decade on the total number of genes in the human, mouse, and rat genomes, varying from about 20,000 to 17,000 genes in each of the three genomes.29, 30, 31, 32, 33, 34 All these figures are obviously much smaller than the data we obtained herein by using a simple way which, to our knowledge, is the first of its kind, for obtaining the total gene number, the total protein number, and in turn the gene‐protein ratio. While the exact reason for this huge difference remains unclear, we extrapolate that the figures reported in the literature, that is, 17,000‐20,000, are for the protein‐coding genes, whereas in the NCBI database the genes in each chromosome may include both coding and non‐coding, although this is not clearly stated in the database. We will probably have no correct estimation of the total number of genes in the near future, partly because it is unlikely that molecular biologists will have a consensus on the definitions of “gene,” “non‐coding gene,” “transcriptional‐read‐through gene,” “pseudogene,”1, 2, 35 Since many small peptides have important biological functions,12, 13, 14, 15, 16 most non‐coding genes likely also elicit functions by producing small peptides and may be later reclassified. If this is the case, the total numbers of genes we obtained herein are closer to the real figures than the 17,000–20,000 reported in the literature. However, the total numbers of proteins we obtained herein are probably much smaller than the actual figures, because the protein‐to‐gene rations we obtained (2.1 in the human, 1.7 in the mouse, and 1.4 in the rat) are too small to be congruent with the generally accepted notion that most genes, especially the human ones, produce multiple mRNA variants and protein isoforms. This putative discrepancy appeals anew for the need to establish good methods for identification of protein isoforms. Considering the novel concept of Heng's “fuzzy inheritance,”9, 10, 11 we anticipate that there should be many more proteins produced from a genome and even more protein isoforms for most gene individuals to be identified.
4.2. Gene knockout may not be complete
Most, if not all, gene‐knockout cells or organisms have been created by scrambling the ORF of the target mRNA, usually the WT form, leading to its failure of translation to protein. The scrambling is most often made by manipulation of a small DNA fragment of the target gene without affecting the most part of the gene that may, theoretically, still be able to produce some RNA variants coding or non‐coding for proteins. Suspected examples include ERα18, 19, 20, 21, 22 and caspase‐824 knockout mice, although convincing evidence such as protein sequence is still lacking. In this study, our LC–MS/MS identified a unique region of Cdk4 protein in the Cdk4−/− cells, which serves as the first tangible evidence for the incompleteness of a knockout product. Interestingly, it is the 23rd‐35th AA region of the Cdk4 that is identified, while the predominant portion of the 303‐AA Cdk4 protein, that is, from the 36th AA to the end, lacks any detected region. It is possible that this 26‐kD isoform lacks a part of the mid‐ or C‐terminal sequence of the 33‐kD WT form of Cdk4.
4.3. Many genes may express protein isoforms smaller than the WT or the canonical form
This study detected many genes whose proteins are supposed to be very large, categorized into the smaller group. For example, four detected genes are supposed to have a protein of 533.9 (Plec1), 471.1 (Prkdc), 284.4 (Sptan1), and 280 kD (Flna) in molecular weight (Table S1 and S2). Although each of these four genes may be expressed to multiple mRNA variants and thus multiple protein isoforms, none of their protein isoforms listed in the NCBI database are as small as 26 kD. The reason for their appearance at such a low position of SDS‐PAGE is unknown. A simple assumption is that what we detected is a randomly degraded fragment. However, some of these proteins have also been detected by us previously in the HEK293, MCF7, and MDA‐MB231 human cell lines at the same and other positions of SDS‐PAGE.25, 26 Such a high frequency of detection at different positions diminishes the possibility of random degradation. In the Plec1 and Flna proteins that have multiple trypsin‐digested fragments detected, there often is a large undetected region between two neighboring detected peptides, such as the region of 1,371 AAs between the “GGAEGELQALR” and the “GFFDPNTHENLTYLQLLER” in Plec1 (Figure 4). The Prkdc and Sptan1 proteins have only one peptide detected; also leaving large parts of the protein without any LC–MS/MS‐detected peptide. As explained above, a likely explanation for the detection of the smaller group, generally speaking, is that they are detected as one or more isoforms that are smaller than their corresponding canonical form, which often is the WT protein.
If two or more protein isoforms of the same gene are similar in length, despite lacking different parts of the full‐length sequence, they can appear at the same position of SDS‐PAGE and be detected simultaneously. This is because our LC–MS/MS approach uses a bottom‐up strategy after horizontally excising the narrow gel stripe, that is, using short peptide sequence(s) to predict a gene's protein product. In this regard, it remains possible that we actually detected multiple smaller isoforms of the same gene. Therefore, each of the four genes mentioned above may have more than one smaller protein isoform expressed and detected. We have actually mapped the LC–MS/MS‐detected peptides onto the largest isoform from 40 genes with the largest TMM among all genes detected and found that only a few of them have the identified peptides evenly distributed throughout the entire sequence (data not shown), similar to what we have reported previously.25, 26 For example, five heat shock proteins (Hspa1a, Hspa8, Hspd1, Hsp90aa1, and Hsp90ab1) have their identified peptides evenly distributed across the entire protein with a high CR. In contrast, for more than 80% of these 40 genes, their proteins have one or more large regions lacking a detected peptide, also similar to what we have reported previously.25, 26 It is worth mentioning that many lengthy proteins have repeated sequences, such as the Plec1 (Figure 4, lower panel), usually to reinforce some important functional domains. It is a plausible conjecture that the smaller isoforms would be the ones lacking one or more of the repeated regions.
4.4. The reason for the appearance of the larger group remains unclear
There are 66 detected genes whose proteins have a TMM smaller than 23 kD and thus, theoretically, should not appear at the 26‐kD position of SDS‐PAGE. Our previous studies on proteins from different human cell lines at this and other positions of SDS‐PAGE also detected proteins from a large number of genes in the larger group.25, 26 In general, at the low region of SDS‐PAGE, such as at the 26‐kD position, there should be a smaller number of genes identified in the larger group but a larger number of genes in the smaller group, and the opposite is true at the high region of SDS‐PAGE, as inferred before.25, 26
The protein identified with the lowest TMM in this study is Rps27a, which actually has a much longer AA sequence but a smaller Dalton value than the other three proteins presented in Figure 7. Theoretically, each of these four proteins with the lowest TMMs can migrate more sluggishly in SDS‐PAGE and appear at the 26‐kD position if they have experienced multiple posttranslational modifications, including a few glycosylations, SUMOylations, ubiqulations, S‐nitrosylations, and phosphorylations. However, considering that our previous studies have also identified a large number of genes in the larger‐group at higher positions of SDS‐PAGE, such as at the 55‐kD and 72‐kD, multiple posttranslational modifications still cannot explain the appearance of the larger group. For instance, we have previously detected histone 4 (with a TMM of 11.4 kD) and cytochrome c (with a TMM of 11.7 kD) at the 72‐kD position,25 which cannot be explained by multiple posttranslational modifications. Therefore, the true reason remains unknown for this phenomenon, that is, why many proteins with a very small TMM can be detected at a much higher position of SDS‐PAGE, and further study on it is required.
5. SUMMARY
Most published proteomic studies present only a tiny fraction of the data produced by LC–MS/MS, usually just the total number of genes or which genes that are expressed to proteins in the tissue or cells studied, while the vast majority of the data produced, as shown in the supplementary Tables S1 and S2, is basically not used. In an attempt to make the LC–MS/MS data more useful, we have developed an oversimplified top‐down procedure of LC–MS/MS and several algorithms to process the resultant data, as shown in this study. We found that only 27.24%, or about one‐fourth, of the proteins detected at about 26‐kD of SDS‐PAGE are expected from their TMMs. The remaining three‐fourths of the proteins have a TMM smaller or larger than the range of 23–29 kD, which is arbitrarily set as the allowed variation of the molecular weight for SDS‐PAGE. Considering that numerous WB studies can detect the target proteins at the positions expected from their TMMs, such as the P53 protein being detected at the 53‐kD position, we conclude that many, but certainly not all, of those proteins detected at unexpected positions are additional isoforms besides the WT or the expected form. Moreover, the Cdk4−/− MEF cell line we studied still expressed a Cdk4 protein isoform, suggesting that the knockout is not complete. We therefore suggest that, when a gene is knocked out with manipulation of a small DNA fragment, a more‐thorough examination of the expression of all RNA variants and protein isoforms is needed, and not just the targeted protein form.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHORS CONTRIBUTIONS
JYQ prepared the figures, tables and the manuscript. JZ and HH analyzed data and revised the manuscript. CGW provided the cell lines and participated in discussion. SQL performed the LC–MS/MS and participated in discussion. HH and HY wrote the point‐by‐point response document and revised the manuscript accordingly. LZ performed English editing. NZX and CFY participated in discussion and data explanations. DZL finalized the manuscript.
CONSENT FOR PUBLICATION
All authors agree on the publication.
Supporting information
Table S1 Supporting Information
Table S2: Supporting Information
ACKNOWLEDGMENTS
We would like to thank Dr. Fred Bogott at Austin Medical Center, Mayo Clinic in Austin, Minnesota, USA, for his excellent English editing of this manuscript.
Qu J, Zhang J, Zellmer L, et al. About three‐fourths of mouse proteins unexpectedly appear at a low position of SDS‐PAGE, often as additional isoforms, questioning whether all protein isoforms have been eliminated in gene‐knockout cells or organisms. Protein Science. 2020;29:978–990. 10.1002/pro.3823
Funding information National Natural Science Foundation of China, Grant/Award Number: 81660501
Contributor Information
Jiayuan Qu, Email: jiayuan.qu@ctgu.edu.cn.
Ju Zhang, Email: zhangju@big.ac.cn.
Hai Huang, Email: huanghai828@gmc.edu.cn.
Dezhong J. Liao, Email: djliao@gzy.edu.cn.
REFERENCES
- 1. Jia Y, Chen L, Ma Y, Zhang J, Xu N, Liao DJ. To know how a gene works, we need to redefine it first but then, more importantly, to let the cell itself decide how to transcribe and process its RNAs. Int J Biol Sci. 2015;11:1413–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He Y, Yuan C, Chen L, et al. Transcriptional‐readthrough RNAs reflect the phenomenon of "a gene contains gene(s)" or "gene(s) within a gene" in the human genome, and thus are not chimeric RNAs. Genes. 2018;9:E40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He Y, Yuan C, Chen L, et al. While it is not deliberate, much of today's biomedical research contains logical and technical flaws, showing a need for corrective action. Int J Med Sci. 2018;15:309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu X, Wang Y, Yang W, Guan Z, Yu W, Liao DJ. Protein multiplicity can lead to misconduct in western blotting and misinterpretation of immunohistochemical staining results, creating much conflicting data. Prog Histochem Cytochem. 2016;51:51–58. [DOI] [PubMed] [Google Scholar]
- 5. Finta C, Warner SC, Zaphiropoulos PG. Intergenic mRNAs. Minor gene products or tools of diversity? Histol Histopathol. 2002;17:677–682. [DOI] [PubMed] [Google Scholar]
- 6. Gerstein MB, Bruce C, Rozowsky JS, et al. What is a gene, post‐ENCODE? History and updated definition. Genome Res. 2007;17:669–681. [DOI] [PubMed] [Google Scholar]
- 7. Ponting CP, Belgard TG. Transcribed dark matter: Meaning or myth? Hum Mol Genet. 2010;19:R162–R168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Portin P. The elusive concept of the gene. Hereditas. 2009;146:112–117. [DOI] [PubMed] [Google Scholar]
- 9. Ye CJ, Regan S, Liu G, Alemara S, Heng HH. Understanding aneuploidy in cancer through the lens of system inheritance, fuzzy inheritance and emergence of new genome systems. Mol Cytogenet. 2018;11:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ye CJ, Stilgenbauer L, Moy A, Liu G, Heng HH. What is karyotype coding and why is genomic topology important for cancer and evolution? Front Genet. 2019;10:1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heng HH, Horne SD, Chaudhry S, et al. A postgenomic perspective on molecular cytogenetics. Curr Genomics. 2018;19:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chu Q, Ma J, Saghatelian A. Identification and characterization of sORF‐encoded polypeptides. Crit Rev Biochem Mol Biol. 2015;50:134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kondo T, Hashimoto Y, Kato K, Inagaki S, Hayashi S, Kageyama Y. Small peptide regulators of actin‐based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol. 2007;9:660–665. [DOI] [PubMed] [Google Scholar]
- 14. Kondo T, Plaza S, Zanet J, et al. Small peptides switch the transcriptional activity of Shavenbaby during drosophila embryogenesis. Science. 2010;329:336–339. [DOI] [PubMed] [Google Scholar]
- 15. Ladoukakis E, Pereira V, Magny EG, Eyre‐Walker A, Couso JP. Hundreds of putatively functional small open reading frames in drosophila. Genome Biol. 2011;12:R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Galindo MI, Pueyo JI, Fouix S, Bishop SA, Couso JP. Peptides encoded by short ORFs control development and define a new eukaryotic gene family. PLoS Biol. 2007;5:e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stepanenko AA, Heng HH. Transient and stable vector transfection: Pitfalls, off‐target effects, artifacts. Mutat Res. 2017;773:91–103. [DOI] [PubMed] [Google Scholar]
- 18. Kos M, Denger S, Reid G, Korach KS, Gannon F. Down but not out? A novel protein isoform of the estrogen receptor alpha is expressed in the estrogen receptor alpha knockout mouse. J Mol Endocrinol. 2002;29:281–286. [DOI] [PubMed] [Google Scholar]
- 19. Couse JF, Curtis SW, Washburn TF, et al. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol. 1995;9:1441–1454. [DOI] [PubMed] [Google Scholar]
- 20. Denger S, Reid G, Kos M, et al. ERalpha gene expression in human primary osteoblasts: Evidence for the expression of two receptor proteins. Mol Endocrinol. 2001;15:2064–2077. [DOI] [PubMed] [Google Scholar]
- 21. Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. [DOI] [PubMed] [Google Scholar]
- 22. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993;90:11162–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun Y, Lou X, Yang M, et al. Cyclin‐dependent kinase 4 may be expressed as multiple proteins and have functions that are independent of binding to CCND and RB and occur at the S and G 2/M phases of the cell cycle. Cell Cycle. 2013;12:3512–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakamaki K, Inoue T, Asano M, et al. Ex vivo whole‐embryo culture of caspase‐8‐deficient embryos normalize their aberrant phenotypes in the developing neural tube and heart. Cell Death Differ. 2002;9:1196–1206. [DOI] [PubMed] [Google Scholar]
- 25. Yan R, Zhang J, Zellmer L, et al. Probably less than one‐tenth of the genes produce only the wild type protein without at least one additional protein isoform in some human cancer cell lines. Oncotarget. 2017;8:82714–82727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J, Lou X, Shen H, et al. Isoforms of wild type proteins often appear as low molecular weight bands on SDS‐PAGE. Biotechnol J. 2014;9:1044–1054. [DOI] [PubMed] [Google Scholar]
- 27. Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin‐deficient diabetes and Cdk4 activation results in beta‐islet cell hyperplasia. Nat Genet. 1999;22:44–52. [DOI] [PubMed] [Google Scholar]
- 28. Jia Y, Chen L, Jia Q, Dou X, Xu N, Liao DJ. The well‐accepted notion that gene amplification contributes to increased expression still remains, after all these years, a reasonable but unproven assumption. J Carcinog. 2016;15:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. [DOI] [PubMed] [Google Scholar]
- 30. Belizario JE. The humankind genome: From genetic diversity to the origin of human diseases. Genome. 2013;56:705–716. [DOI] [PubMed] [Google Scholar]
- 31. Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. Pseudogenes: Pseudo‐functional or key regulators in health and disease? RNA. 2011;17:792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Foley JF, Phadke DP, Hardy O, et al. Whole exome sequencing in the rat. BMC Genomics. 2018;19:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pruitt KD, Harrow J, Harte RA, et al. The consensus coding sequence (CCDS) project: Identifying a common protein‐coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mudge JM, Harrow J. Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm Genome. 2015;26:366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan C, Han Y, Zellmer L, et al. It is imperative to establish a pellucid definition of chimeric RNA and to clear up a lot of confusion in the relevant research. Int J Mol Sci. 2017;18:714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Supporting Information
Table S2: Supporting Information