

Abstract
Several factors should be taken into account when it comes to the first exposure of humans to a novel vaccine.
Subject terms: Vaccines, Adjuvants, Clinical trials
Vaccines have a long history of excellent safety and a highly positive benefit/risk profile. Even so, the lack of specific guidance from regulatory agencies specifically relating to the first application of a new experimental vaccine in humans has hampered product development. Most of the regulatory guidance documents for manufacturers are too broad and sometimes only vague where vaccines are concerned. As regulators deeply involved both in the development of the European Medicines Agency's (EMA; London) new regulatory framework on risk identification and mitigation, and in assessment and authorization of clinical trial applications for biotechnological and biological products (especially vaccines), we have been repeatedly approached by companies and vaccine developers regarding regulatory issues for first-in-human clinical trials. Here, we discuss these considerations as they relate to vaccines within the context of the current EMA guideline for risk identification and mitigation for first-in-human clinical trials based on the apparently considerable uncertainty among developers. We describe how regulators apply the guideline and where we see the limitations or the need to take alternative approaches. The discussion primarily focuses on prophylactic and therapeutic vaccines against infectious diseases as this classic field of products is associated with particular uncertainty.
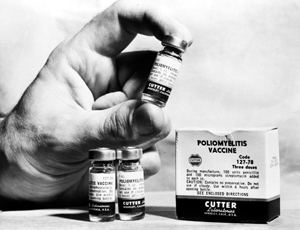
In the so-called Cutter incident in 1955, Cutter Laboratories of Berkeley, California, failed to fully inactivate a batch of polio vaccine (vials shown). This is one of the rare examples where documented adverse events were associated with the use of a vaccine in humans.
© Bettmann/CORBIS
General considerations
The EMA's Committee for Medicinal Products for Human Use (CHMP) has assembled a Guideline on Strategies to Identify and Mitigate Risks for First-in-Human Clinical Trials with Investigational Medicinal Products as a joint effort of European regulators and scientists from various disciplines1. This guideline is applicable to any new molecular entity, both chemical and biotechnological and/or biological. Its main principle, which is now also widely applied by regulators assessing clinical trial applications in Europe, is an approach of risk identification and risk mitigation. This is done by assessing the mode of action, the nature of the target and the relevance of the animal species used for testing of nonclinical safety and toxicity. These issues are particularly pertinent to the design of first-in-human clinical trials of products that have a seemingly potentially higher risk in the first administration to humans than the nth iteration/reformulation of an established product. The most important consideration is to commence testing with a conservative calculation of a safe starting dose and sequential inclusion of subjects in the trial to limit exposure.
Unfortunately, little if any specific guidance is available for first-in-human trials specific to vaccines. The guidance for industry issued by the US Food and Drug Administration (FDA) concerning dose calculation for a first-in-human clinical study2 describes in detail the initial dose finding but states explicitly that it is not pertinent for vaccines. Only general guidance concerning the principles for conduct of clinical studies is available from the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). EMA guidance specific to nonclinical and clinical evaluation of vaccines is available but also includes only limited guidance specific to first-in-human studies3,4,5.
Because vaccines resemble pathogen antigens, which usually have antigenic features distinct from physiological structures found in human tissues, the risk accompanied with administration of these products is usually considered relatively low; frequently reported adverse events in clinical trials are in most cases manageable and transient (e.g., fever and local reactogenicity). In addition, knowledge of immunological processes and the role of specific cells and mediators in this context continues to advance, facilitating our understanding of the mechanism of action of individual components of vaccines.
The overall safety of vaccines is corroborated by the fact that during decades of vaccine development and application, cases of severe damage caused by these products have been uncommon. However, rare examples of adverse events have been observed. In 1955, for example, insufficiently inactivated batches of polio vaccine caused an outbreak of polio due to the presence of wild-type poliovirus strains. This became known world wide as the Cutter incident, in which 40,000 children developed mild polio, 200 were permanently paralyzed and 10 died6. Another example is an aggravated or atypical disease following vaccination and exposure to wild-type viruses caused by a measles and respiratory syncitial virus7 or experimental severe acute respiratory syndrome (SARS)8 vaccine. Regarding live-attenuated vaccines, data suggesting elevated mortality observed in developing countries following vaccination with medium- and high-titer measles vaccines demonstrate again the need for cautious approaches when entering into early clinical trial phase9. But these examples also highlight that root causes for problems can often be identified, and principles for risk identification and mitigation can be developed.
Infections themselves can trigger immunological sequelae that can even be more harmful than the actual infection itself (e.g., rheumatic fever after infection with group A streptococci, such as strep throat or scarlet fever, or Guillain-Barré syndrome (GBS) following viral infections or infection with Campylobacter jejuni or certain other bacteria). Guillain-Barré syndrome is a rapidly progressing ascending paralysis, mediated by a cross-reactive attack of antibodies (e.g., cross-reacting against the GM1 ganglioside)10. Such knowledge is relevant to the risk assessment of a novel vaccine against an infection for the following reasons: first, vaccines often present an antigen in an artificial context (that is, as repetitive structures, such as in virus-like particles, as fragments of epitopes or as capsules); second, in many cases, vaccines are administered together with an adjuvant that enhances or modulates the immune response (see below); and third, vaccines provide an antigen dose that is both different from that seen in a natural infection and most times presented to the immune system by a different route. It may, thus, be (theoretically) possible that a vaccine could lead to clinical symptoms similar to infections that trigger downstream immunological sequelae and thus study protocols could benefit from implementing respective endpoints. This is borne out by the observation that especially in live vaccines, but also in some others, there have been sporadic reports of rheumatic fever and Guillain-Barré syndrome. At the same time, such complications are very rare and the causality is not always clear. For instance, sometimes concomitant minimal (respiratory) infections are present in a subject when an experimental vaccine is tested, but these are, of course, no reason to delay a vaccination. Thus, for the time being, there are doubts of a causal relation between vaccination and the onset of autoimmune diseases, apart from isolated cases.
Another aspect that must be taken into account is that vaccines are biological products. As such, even small changes to the established manufacturing processes may significantly alter product safety and/or efficacy. For example, simultaneous elimination of thiomersal and human serum albumin from a European tick– borne encephalitis vaccine drastically increased cases of moderate and severe fever after the first dose of primary immunization, which could only be corrected by reintroducing human serum albumin into the vaccine formulation11. These events demonstrate that the manufacturing process is an integral part of the concept and that changes in the formulation of a given vaccine may benefit from risk identification and mitigation considerations.
Finally, both novel adjuvants12 that enhance the immune response and novel routes for antigen delivery (e.g., antigen delivery based on gene transfer) will affect the perception of risks and require specific regulatory strategies. With novel adjuvant or emerging new vaccine formats, including vaccines against pathogens for which no vaccine exists so far, safety considerations have to be put on a broader scale as has previously been done for rather straightforward cases like insufficient inactivation of a live virus.
On the other hand, vaccines have an excellent safety record and most new vaccines can a priori be considered low-risk medicinal products. It needs to be emphasized that we do not have to assume that a vaccine with a new mechanism of action or a novel structure is a high-risk vaccine. Likewise, not every new medicinal product should automatically be considered a high-risk medicinal product13. The first-in-human trial is a critical turning point between preclinical studies and first human exposure and subsequent larger clinical trials in hundreds or (for many vaccines) thousands of subjects. For sponsors, relevant risk assessment for first-in-human clinical studies means careful design and conduct of studies that reduce potential risk to humans. In comparison to therapeutic proteins or other medicinal products, however, the prophylactic character and mechanism of action of vaccines warrant particular attention. Indeed, some of the concepts introduced in the aforementioned EMA guideline1 may not even be readily applicable.
First, pharmacokinetics should be considered relevant only if, for example, a vaccination approach involves either a novel or different means of delivery (the first pass effect for oral application versus the usual intramuscular route) or a novel live vaccine (where shedding rates can differ). Pharmacodynamics in vaccines is usually gauged by immunogenicity (appearance and increase of antibody titers).
Second, vaccines often include an adjuvant or are administered concomitantly with an immunomodulator that has its own impact on the overall risk assessment. As such agents can influence the behavior of a vaccine or the host's responses to a vaccine14,15, it is often important to assess their effects (including, for instance, pharmacokinetics and pharmacodynamics) both separately from the vaccine antigen (as its own entity) and in combination with the antigen.
Third, the target population for vaccine trials is usually healthy and young—often infants from six weeks of age and up, children or adolescents. This requires special diligence concerning benefit/risk assessment.
Fourth, unlike other medicinal products, efficacy measurements are often indirect; thus, the elicited immune response is the active principle of a vaccine and needs to be part of the risk assessment.
And finally, the risk profile of a vaccine may be different over time and dependent on exposure to both vaccine and pathogen infection. For vaccines, acute risks have to be distinguished from sub-acute or chronic (long-term) risks after (repeated) product administration.
Although the general criteria and considerations mentioned in the EMA guideline1 should always be taken into account, Figure 1 displays criteria that are more specific to vaccines and may be helpful for developers. The relative importance of these criteria should be decided on a case-by-case basis for each product; if developers are in doubt, they should consult with regulators (either the national agencies of EU member states or the EMA) when designing a trial.
Figure 1.
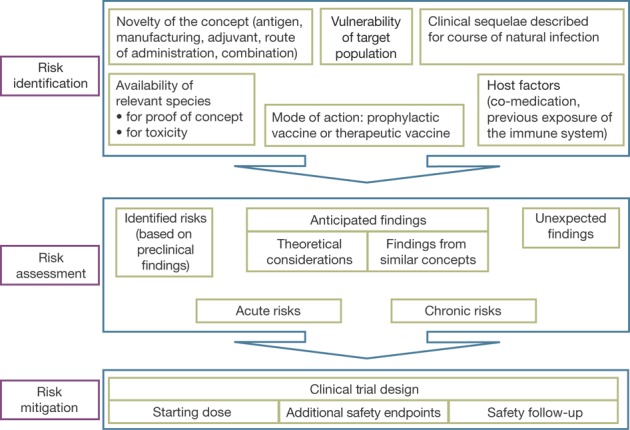
Risk assessment for a vaccine intended for first-in-human administration.
The safe starting dose: is the MABEL relevant?
The calculation of a safe starting dose is a central aspect for a first-in-human trial. The classic approach to calculate the starting dose for a first-in-human trial for a classic medicinal product (not a vaccine) is based on toxicity in the relevant animal model specifically on the no-observed-adverse-effect level (NOAEL). This approach was also chosen for the agonistic anti-CD28 monoclonal antibody TGN1412, but was apparently insufficient to prevent the highly elevated pharmacodynamic effect, a massive cytokine-release syndrome16. Thus, the EMA guideline advocates an alternative approach, that is, a calculation based on the minimal-anticipated-biological-effect level (MABEL), being the dose level at which a minimal biological effect in humans is expected by in vitro or in vivo data. It is based on the occurrence of any biological effect, not only toxicity. Thus, the MABEL approach usually results in a much lower dose than that calculated with the NOAEL approach, which relates to toxicity findings. Here, the classic paradigm of a dose-dependent effect (including toxicity) is implicitly assumed—a principle already questionable for certain biotechnological medicinal products that can exhibit distinct pharmacodynamic effects at a low dose. For vaccines, additional—or even alternative—considerations need to be made because often thresholds for eliciting an immune response exist. Thus, the principle of little dose increases in cohorts might not be applicable here regarding the toxicity (if any) of the vaccine itself and consequences of the elicited immune response. In addition, if no correlate of clinical protection yet exists for the respective vaccination or if thresholds of antibodies are different between serotypes included in a vaccine (e.g., pneumococcal vaccines), the respective dose for a MABEL would be difficult to determine.
If a similar vaccine exists, for instance, a conventional Bacillus Calmette Guérin (BCG) vaccine in relation to new BCG-based tuberculosis-vaccine developments, information about the immunological pathways and clinical effects (efficacy and safety) may be extrapolated to indicate a safe starting dose and a possible test setting for the first-in-human clinical trial. If such a 'prototype' product is not available, a safe starting dose can be achieved by dissecting the different aspects that characterize a vaccine (Fig. 2), which are discussed below.
Figure 2.
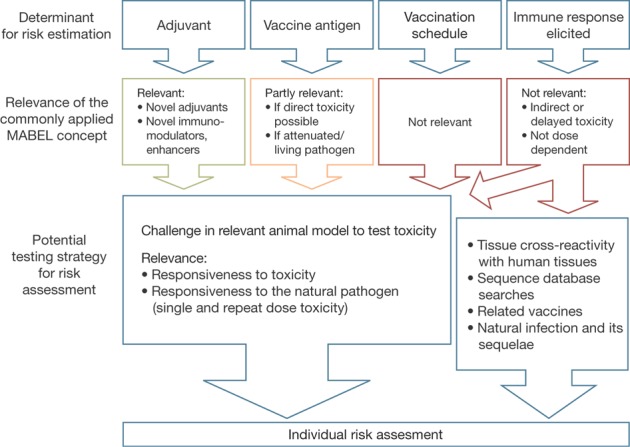
Factors to be considered for the starting dose of a vaccine for first-in-human administration.
Vaccine antigen. Traditionally, the vaccine antigen consists of a live-attenuated pathogen, an inactivated pathogen or a recombinant or chemically synthesized antigen that resembles the natural antigen of the pathogen. An attenuated vaccine strain has impaired replication competence in humans. Here, a dose escalation starting from a low dose of the vaccine can indeed be feasible when it comes to test safety of the pathogen itself. But for inactivated pathogens and synthetic antigens, this may be less feasible because the protein or polysaccharide in itself might not exert any toxicity at all. These considerations apply both to dose escalation in nonclinical safety studies and for first-in-human trials. A MABEL approach may well be feasible only for certain types of antigens and may produce misleading results and even a false feeling of safety for other types.
Concerning direct toxicity, nonclinical studies might already be of help here. If there is a direct toxicity of the antigen, for instance, this normally will be apparent in nonclinical toxicity studies.
Adjuvants and immunomodulators. Adjuvants are an important component of a vaccine and a rapidly growing number of new adjuvant systems are used to enhance the immune response either through ideal presentation of the antigen or by immunomodulating effects. They are traditionally composed of mineral salts (e.g., alum), or more advanced developments derived from microorganisms like muramyl dipeptide, monophosphoryl lipid A or trehalose dimycolate. The mechanism of action of adjuvant emulsions includes the formation of a depot at the injection site enabling the slow release of antigen and the stimulation of antibody-producing plasma cells. Other adjuvants may be particulate antigen delivery systems (that is, liposomes, polymeric microspheres, nano-beads, immunostimulating complexes and virus-like particles), polysaccharides or nucleic acid–based adjuvants17. They may consist of combinations of two or more adjuvant systems (e.g., AS04) or not even be part of the formulation at all but concomitantly administered (that is, cytokines).
Some of these adjuvants are well known or at least 'established' through their long use. For newer adjuvants, however, less exposure data are available and the ideal dose of adjuvant with a certain antigen (content) has to be sought each time a novel vaccine is developed.
The dose of a novel adjuvant may feasibly be found through a MABEL approach. For example, a dose-dependent effect might exist for adjuvants targeting Toll-like receptors. A MABEL approach could also be used for immunomodulating adjuvants like cytokines. However, a threshold effect could exist here as for some antigens or other adjuvants.
Novel adjuvants can be species specific (e.g., cytokines), posing an additional challenge to find a relevant animal model (see discussion further below). Thus, even individual testing of the adjuvant or the immunomodulator in a separate first-in-human study might become necessary. Experience gained with a specific adjuvant in another vaccine could be considered supportive data, but it cannot be excluded that the same adjuvant causes serious side effects in combination with a different antigen. In any case, a thorough risk assessment is necessary also for the adjuvant.
The elicited immune response. The elicited immune response surely represents the main 'active' principle of vaccination. The vaccine antigen (such as a protein or polysaccharide) may in itself be harmless (and cause only unspecific local reactions), but the immune response against it could be harmful. Antibodies can cross-react with physiological structures and the concept of 'molecular mimicry'18 is one of the hypotheses by which autoimmunity is explained. Antibodies, as well as CD4+ T-helper cells that are part of activating and promoting a specific immune response against an antigen, might not only detect the target antigen they are intended for (that is, the pathogen) but also cross-react in an unwanted fashion with other structures that have a similar formation. T-cell recognition is 'degenerate'19, meaning that T cells also react with structures that have less than 100% identity with the T-cell receptor's primary target.
Thus, for risk estimation of a novel vaccine candidate one needs to consider the immune response that is elicited by a vaccine as a potentially 'toxic' principle. Because activation of the immune system and the resulting immune response is not necessarily dose dependent and may be associated with particular threshold considerations, MABEL might also not be feasible here. One possible solution could be nonclinical studies. Unfortunately, for observation of a potential cross-reactivity animal toxicology data might not always be sufficiently helpful because the biological structures of animal and human organs regarding epitopes are different. Cross-reactivity of the sera of accinated animals with animal organs might not necessarily imply that the same would happen in humans and, likewise, absence of cross-reactivity or autoimmunity in animals would not imply safety in humans.
On the other hand, in vitro tissue cross-reactivity studies performed with animal sera can be helpful. This approach, in which animals are vaccinated and their sera tested for cross-reactivity with human tissue sections, is routinely carried out in nonclinical toxicology testing of monoclonal antibodies. In the case of a vaccine product, the animal species might not necessarily have to be 'relevant' (see discussion below) because the animals are used rather to obtain the antibodies that can then be tested for toxicity. In addition, for the choice of the species, one may have to take into account that unrelated species may produce cross-reactive antibodies. These might be deleted in highly related species due to a tolerance for self that is shared by related but not by unrelated species. Nevertheless, such results can be useful to create a 'worst-case scenario' for cross-reactivity and may be helpful when considering risks. It is acknowledged that cross-reactivity studies have their inherent difficulties as data may be misleading if artifacts arise due to tissue preparation and fixation procedures. Even if true binding or cross-reactivity is observed, this might not necessarily point to a safety concern as the tissue structure may not be accessible under physiological conditions in humans. A shortcoming of this approach is that it tests only the antibody response; a T-cell response cannot be tested. This is problematic as T cells might be the main driver for toxicity in humans. Even so, such findings should be regarded as potential safety signals and will be helpful in assessing risks. For a first-in-human study (and subsequent clinical studies), such signals trigger the implementation of relevant clinical safety endpoints to detect potential clinical manifestations of such bindings or confirm that it does not occur in patients. These will be aimed mostly at subclinical changes, for instance, echocardiography in case binding to human cardiac tissue sections had been observed. It is fully acknowledged that such events can be rare and that the true risk of occurrence might not even become apparent before marketing authorization and use in large numbers of people. Nevertheless, for novel vaccination, principles such as precautionary measures are helpful in risk identification and mitigation strategies.
This discussion shows that for a first-in-human trial for a novel vaccine one also needs to consider the definition of risk. The EMA guideline1 was written to detect and mitigate acute risks like cytokine release syndrome. For vaccines, such acute events derive either from an allergic reaction or are elicited by an adjuvant that triggers a skewed immune activation. The cross-binding of sera, however, would not be included in such acute risks because autoimmunity or other symptoms elicited by a real cross-binding of antibodies take a longer time. Antibodies have to be formed after vaccination and clinical manifestation normally requires a certain time following binding of those antibodies to the target organ structures. Therefore, these 'long-term' risks are hardly suitable to determine any inter-subject interval for administration in a sequential dosing concept but are rather meant to define suitable endpoints as discussed above. Another potential long-term risk is the possibility for a paradoxical enhancement of the disease (e.g., overstimulating immune cells by prolonging presentation of the pathogen antigen–antibody complexes).
The vaccination schedule. For most vaccines, single-dose administration is insufficient to establish immune protection as well as boosterable long-term persistence of the immune response. Adverse effects not triggered by a first dose might be triggered or will become detectable only during completion of the primary vaccination regimen or at the time of booster vaccination. These phenomena are known as positive re-exposure. The risk might then increase with the frequency of administration of a specific vaccine to achieve an acceptable immune response and might be particularly high for vaccines that must be administered at regular intervals. In view of these effects, the vaccination schedule also needs to be taken into consideration for the definition of the safe starting dose. As such safety issues cannot readily be predicted, a simulation of the vaccination schedule in animals should be made. In addition, appropriate safety evaluations in vaccinees and regular and extensive follow-up visits suitable to detect late effects (up to several months) must be implemented.
Quality/CMC considerations
At the time the step from animals to humans is made in drug development, the product should already be very well characterized. The potency of bacterial or viral antigens in the vaccine should be given special attention as this is a crucial factor to mediate toxicity and other adverse reactions. Therefore, specifications for potency should ideally be set sufficiently narrow. Specifications being too wide might project into false dose estimations, thus leaving room for uncertainty regarding the validity of dosing assumptions defining the starting dose. Assays measuring impurities, sterility and inactivation of biological agents have to be available at this early point in development. If possible, components (e.g., reagents, adjuvant and excipients) should be referenced (for trials in Europe) to the European Pharmacopoeia where monographs are available.
Of course, the manufacturing should be undertaken according to good manufacturing practice. Newly developed components have to be described in great detail, including chemical definition and biological structure, normally all the way down to amino acid sequence. For recombinant vaccines, as much data as possible on post-translational modifications, like addition of sugar structures (glycosylation), should be provided. Depending on the nature of a novel vaccine, similarity to human cell structures, receptors, nucleic acid or other possibly 'immunoactive' structures have to be described and evaluated with respect to (unwanted) interaction within the organism (see also discussion elsewhere in this article). If changes were made in the production process after the nonclinical studies, comparability would have to be shown between pre- and post-change product as per the relevant guideline20 to demonstrate that the nonclinical data supporting a first-in-human use can still be applied.
Nonclinical considerations
Although animals present 'good models' for a variety of human physiological functions, they also have significant limitations when it comes to species-specific aspects; diseases induced by infectious agents relevant to humans may not exist in animals or may cause different symptoms. Likewise, certain adverse reactions can be seen only in humans and some adverse events of special interest cannot be predicted or reproduced in animals (e.g., a potential impact on functions of the central nervous system, especially learning difficulties or development of speech). Thus, a 'relevant' animal model is needed that maps the respective disease to be prevented as well as organ systems influenced by the new agent. In this respect, it is important to discuss the concept of the 'relevant species' specifically for vaccines. For a vaccine as well as for certain other biologicals, there needs to be a distinction of 'relevance' in respect of susceptibility and the clinical course of infection with the pathogen (proof of concept) and in respect of reliable prediction of safety and toxicology of the vaccine in humans.
Regarding safety evaluation, the ICH S6 guideline21 defines a “relevant risk” species as one in which the test material is pharmacologically active due to the expression of the receptor or, in the case of monoclonal antibodies, an epitope. This is not feasible for a vaccine because here the medicinal product—the vaccine—itself is in most cases not the active principle (but the immune response against it is) and the target structure is the pathogen or infected cell containing the pathogen. This needs to be taken into account in the planning of the nonclinical development strategy.
When it comes to proof of concept, the relevant species might have to be defined differently. Here, the relevant species is one that is susceptible to infection with the pathogen and at best also resembles clinical features of humans suffering from infection and its subsequent resolution. Relevant animal models for most kinds of vaccine-targeted diseases exist (e.g., ferrets for influenza and chimpanzees for hepatitis A and B), but for specific scenarios investigators might have to combine different approaches to describe the human infection and the way the vaccine will prevent it. When selecting an animal model, which type of immune response is elicited in conjunction with the adjuvant, for example, the kind of T-cell response (cytotoxic T cells or T-helper cell responses), is also among the factors to be considered.
If a novel adjuvant is species specific (e.g., a cytokine), then the relevant animal model might have to be chosen based on the activity of the adjuvant in the respective animal species. On a case-by-case basis such an immunomodulator might have to be exchanged with the homolog active in the respective species. Also the immune response against a given vaccine antigen might be different in animals and in humans. Thus, extrapolation of data is difficult and often not feasible. Nevertheless, a nonclinical proof-of-concept study is usually mandatory before a first-in-human trial can be commenced because it adds valuable data to the overall concept of vaccine development and is needed to decide on the benefit/risk estimation to allow the first-in-human trial to be initiated (that is, to provide a rationale that the vaccine is likely to fulfil its purpose). A practical shortcoming can be the nonavailability of some animal models (e.g., the aforementioned chimpanzee model for animal protection reasons). A crucial point is to carefully consider physiological systems that might be or will be affected and how a vaccine could affect the response of different immunological cells that would be observed during natural infection; for example, overstimulated T cells can result in unexpected acute and chronic adverse events. Use of worst-case-scenario data from animals obtained with different doses of antigen and adjuvant and/or immunomodulator can be helpful in the estimation of the likelihood of such events to occur in humans, up to a full-blown systemic inflammatory response syndrome with its adverse impact on heart, liver, kidney and the central nervous system. For some organ systems and physiological scenarios, computer models are available that derive their accuracy from data that have been collected in all kinds of previous studies in humans of different age, gender and with co-morbidities; these can be helpful tools to estimate potential reactions only seen in human organisms22. Whether such models are useful for the development of vaccines has to be considered on a case-by-case basis and may best be discussed with regulators upfront.
Usually, only single and repeated dose toxicity studies in at least one animal species are required before first-in-human administration (repeat dose toxicity for most vaccines that are applied at least twice). For vaccines that target children and/or women of child-bearing potential, the influence on the reproductive system has to be explored. Here, different animal models might be defined as 'relevant' compared with the other nonclinical studies. For the emerging class of genetically modified biological systems, the risk of possible gene transfer into humans (or the human germ line) also needs to be quantified. Reproductive toxicity includes male and female reproductive capacity as well as the possible influence of transferred genes on the development of the embryo/fetus during pregnancy. This might indeed be an issue, given the complex changes to the maternal organism during pregnancy, including maternal-fetal exchange (hormones, antibodies and so forth). Therefore, the possible influence on fetal development (bone structure, central nervous system, organs and so forth) has to be closely surveyed as well.
Clinical trial design considerations
One central aspect of clinical trial design is the translation of potential findings from the nonclinical and in vitro studies (e.g., unexpected cross-reactivity of induced antibodies in animals with human tissue, to suitable clinical endpoints). As discussed, surveillance of subjects should be designed on a risk-based approach including acute and chronic risks (Figs. 1 and 2). Because they can affect immunological responses, several intrinsic and extrinsic factors influence the conduct and structure of a clinical trial design.
Intrinsic factors derive from subjects enrolled in the trial. They can cover, for example, concurrent diseases (e.g., HIV and malaria) and genetic polymorphisms, including major histocompatibility complex (MHC) haplotypes, receptor sensitivity or organ function. Ethnic factors, drug habits and nutritional status also directly affect the immune system.
Extrinsic factors derive from the socioeconomic background of the region where a trial takes place. Crucial factors for vaccines are the climate (that is, the ability to maintain the cold chain for the product), diagnostic and case definition practices. Drug compliance influences the trial subjects' view on multi-dose vaccinations as well as repeated visits for blood draws and adverse event checks. Some of these factors cannot be controlled or avoided (e.g., MHC haplotypes), but should nevertheless be considered in clinical trial protocol design as relevant. For example, developers elect to conduct a trial in a region where disease incidence or prevalence is high because only in this region would subjects be sufficiently motivated to comply with the trial protocol. Also crucial are local views on regulatory practice and good clinical practice as well as methodology and endpoints for the trial. This last instance, of course, influences all drug trials and is not unique for vaccines, but local ethical or religious views determine the acceptability of certain vaccines, as can be seen by the difficulty in eradicating polio, and might even be more an issue with vaccines preventing sexually transmittable diseases. To take into account these factors, the EMA has drafted a reflection paper containing examples of product groups and special extrinsic factors influencing studies23 and the ICH has issued 'frequently asked question' paper E5 (ref. 24).
Statistical methods for limiting trial size. In first-in-human studies, only a very small number of participants are enrolled to minimize risk in light of the usually—at this point—nonexisting benefit for the enrolled study subjects (if, for example, the vaccine dose when deciding to follow the MABEL approach is too low and is maybe immunogenic but not yet protective). As most first-in-human studies have dose escalation in their procedure reliable measures for proceeding to the next dose cohort have to be implemented. Besides orientation from nonclinical animal challenge studies, several statistical methods limit the number of study subjects while at the same time allowing good estimates of nontoxic (minimal toxicity dose, MTD) and beginning effect (minimal effective dose, MED) levels. This includes, for example, the standard 3+3 cohort analysis and the continual reassessment method (CRM). The CRM is usually used to estimate the maximum tolerated dose but might be used as well to define the MED and MTD when starting from a dose level estimated to be between nontoxic and a beginning effect, as previously observed in nonclinical studies25,26.
Surveillance of subjects. Safety is not restricted to 'tolerability' as this rather relates to local tolerance of the vaccine only. First, as usually only healthy participants are included in these studies, all possible control mechanisms must be applied. These include recording of routine laboratory parameters, including those specific to the expected interaction of the vaccine with the physiological environment, such as differential blood count and blood chemistry. Systematic evaluation should also include the recording of parameters in organs previously observed to be affected in animals (e.g., liver enzyme levels associated with hepatotoxicity) and those deduced from tissue cross-reactivity studies. Imaging techniques like (contrast) magnetic resonance imaging, computer tomography, ultrasound or X-ray of suspected vulnerable tissues as well as regular medical surveillance (electrocardiography or clinical examination) before, during and after the application of the new vaccine are also common. In addition, a first-in-human administration should not only be performed in a suitable hospital environment that provides the investigator with all necessary equipment, including an adjourning intensive care unit, but also cover a time span estimated to include all possible short-term adverse events and/or serious adverse events. After this period has elapsed, subjects are released from the trial center and examined as outpatients at regular intervals until the end of the expected interference induced by the vaccine (that is, long-term adverse events). Agencies often request long-term follow-up visits up to six months from the start of the trial, depending on the perceived potential risk for long-term events like autoimmunity.
If a genetically modified organism is used in a vaccine, there could be the risk of shedding (feces, urine) or direct transmission by means of a local inflammatory reaction at the injection site (e.g., smallpox or tuberculosis vaccination). Here, special environmental risk assessments are needed27, and risk estimation thus implies not only vaccinees but also the persons coming into contact with them. Where possible, the existence of individuals of vulnerable immunological status (e.g., those with immunosuppression, premature newborns, the elderly, people with atopic diseases and those with severe co-morbidities in which an infection could be life-threatening) that are in contact with trial participants should be considered in the trial protocol and before the trial commences.
Pediatric studies. In contrast with most conventional pharmaceuticals, the target population for many vaccines is infants and children. First use in a pediatric population is, therefore, a particularly critical step that again needs careful consideration with respect to additional animal studies that might potentially be required (juvenile animals), further dose reduction and different dosing schemes. In addition, studies in children regardless of age are ethically difficult if no comparator yet exists and the disease to be prevented is at the same time not life threatening. Thus, justification of the trial design has to be very thorough, covering availability of a comparator (at least established medicinal use), impact and epidemiology of the disease as well as resulting age escalation/de-escalation planned.
For the different age groups, separate studies are usually required by European Union (EU; Brussels) regulators, especially in view of the new EU Paediatric Regulation28, which entered into force in 2007. Here, the crucial point of decision is whether testing of the different subgroups should be done by age de-escalation or whether the disease to be prevented has its peak in the first few weeks and months of age and thus, the age group with the highest risk of infection as well as the maximum benefit by the vaccination should be vaccinated first. This approach should be agreed upon on a case-by-case basis involving (in Europe) the Paediatric Committee of EMA and, in general, the regulatory authorities concerned in the respective member states where the clinical trial is conducted. Vaccination of infants as the first age subgroup in the pediatric field has been agreed upon for the new live tuberculosis vaccines as infants are at the highest risk of tuberculosis in the first two years of life (thus, there is a dire need) and vaccination with the established BCG vaccine takes place shortly after birth (ideal comparator).
Guidance for this field is provided in various documents by EMA (http://www.ema.europa.eu/htms/human/paediatrics/sci_gui.htm). As in the EU, all different pediatric age groups (up to 18 years of age) will usually have to be evaluated separately in accordance with the European Paediatric Regulation; possibly only very small numbers for the individual trials will be available.
Conclusions
Most vaccines have an excellent safety record. As vaccination against infectious diseases contributes hugely to public and individual health all over the world, one needs to exercise caution when discussing risks associated with vaccines so as to avoid false and misleading signals for the public and politicians. Such a balanced view is particularly important with the emergence of new infectious agents (e.g., SARS and H1N1 influenza), the continued battle against neglected (tropical) diseases and the re-emergence of pathogens and vectors worldwide displaying increasing resistance to existing therapeutic agents. In this context, the establishment of new vaccines both against known and novel infectious diseases, as well as the improvement of established vaccines through novel techniques (e.g., genetic modification), is of the utmost importance. In addition, vaccination usually represents the cheapest and at the same time the most effective means of disease prevention worldwide.
In this context, a balanced and reasonable approach for first-in-human studies of a novel vaccine candidate is crucial to ensure safety of trial participants. The principles of the EMA guideline need to be applied in a reasonable and scientific way based on how prophylactic and therapeutic vaccines against infectious diseases function. Some principles, like the MABEL or NOAEL approaches, might require very careful adaptation to the specific needs and/or aspects of any given product, including a novel vaccine, as we discuss above. If a first-in-human trial for a vaccine does apply the MABEL strategy (e.g., for a novel adjuvant) and implements gradual dose increases, this merely represents the first step in defining the safety of administration. It must not be mistaken as 'dose finding' for immunogenicity, safety and tolerability. These are integral parts of further vaccine development to arrive at a dose that is maximally safe and immunogenic.
The discussion in this article demonstrates that the definition of a starting dose for a novel vaccine might not be straightforward; indeed, 'automatic' use of the MABEL approach might lead to misleading results. When uncertainty or doubt arises, we strongly advise manufacturers to seek discussion with regulatory agencies. This can be done either on a national level with the respective national competent authority in the EU member states (e.g., in Germany, the Paul-Ehrlich-Institut) or on a European level by using the CHMP Scientific Advice Procedure29 (http://www.ema.europa.eu/htms/human/raguidelines/sa_pa.htm.). The former approach has the advantage of a direct discussion with the competent authority later responsible for evaluating and granting the clinical trial application in the respective EU member state. On the other hand, approaching European authorities has the advantage of receiving a European position on respective issues. Regulators are increasingly open for dialog, even at very early stages of development as well as for the development of future vaccines; such a dialog is to be considered an increasingly important factor for success.
The principles discussed in this article apply primarily to prophylactic and therapeutic vaccines against infectious agents. However, many of the principles discussed here might also readily be applied to other classes of vaccines, including therapeutic 'anti-tumor vaccines'. Because of their different immunological mode of action, these products should not be grouped with traditional vaccines and thus, according to their specific mode of action and nonprophylactic timing of use, have been classified as 'immunotherapy medicinal products'.
It is important to emphasize that not every novel vaccine or adjuvant system bears a high risk and 'higher risk' might likewise imply 'more effective' concepts (e.g., enhanced immunogenicity or protection against pathogens where no functional vaccine principle exists yet). The European guideline for first-in-human trials is intended to be a step forward to develop innovative compounds more safely. It is a certainty that even this guideline and all precautionary principles will never reduce the risk to zero. Transition from nonclinical studies will always be a risk but is also a necessity to develop more efficacious medicines against human diseases.
Competing interests
The authors declare no competing financial interests.
Footnotes
The views expressed in this article are our personal views and may not be understood or quoted as being made on behalf of the EMA committees or reflecting the position of the EMA committees or one of the CHMP Working Parties.
References
- 1.European Medicines Agency. Guideline on Strategies to Identify and Mitigate Risks for First-in-Human Clinical Trials with Investigational Medicinal Products CHMP/SWP/28367/07 (EMA, London; 2007). <http://www.ema.europa.eu/pdfs/human/swp/2836707enfin.pdf>
- 2.US Food and Drug Administration (FDA). Guidance for Industry and Reviewers, Estimating the Safe Starting Dose in Clinical Trials for Therapeutics in Adult Healthy Volunteers (FDA, Washington, DC; 2002). <http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078932.pdf>
- 3.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Guideline E8: General Considerations for Clinical Trials. (ICH, Geneva; 1997). <http://www.ich.org/LOB/media/MEDIA484.pdf>
- 4.European Medicines Agency (EMA). Note for Pre-clinical Pharmacological and Toxicological Testing of Vaccines CPMP/SWP/465/95. (EMA, London; 1997). <http://www.ema.europa.eu/pdfs/human/swp/046595en.pdf>
- 5.European Medicines Agency (EMA). Guideline on Clinical Evaluation of New Vaccines CHMP/VWP/164653/05. (EMA, London; 2006). <http://www.ema.europa.eu/pdfs/human/vwp/16465305enfin.pdf>
- 6.Nathanson N, Langmuir AD. Am. J. Hyg. 1963;78:16–28. doi: 10.1093/oxfordjournals.aje.a120327. [DOI] [PubMed] [Google Scholar]
- 7.Polack FP. Pediatr. Res. 2007;62:111–115. doi: 10.1203/PDR.0b013e3180686ce0. [DOI] [PubMed] [Google Scholar]
- 8.Yang ZY, et al. Proc. Natl. Acad. Sci. USA. 2005;102:797–801. doi: 10.1073/pnas.0409065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knudsen KM, et al. Int. J. Epidemiol. 1996;25:665–673. doi: 10.1093/ije/25.3.665. [DOI] [PubMed] [Google Scholar]
- 10.Hughes RA, Hadden RD, Gregson NA, Smith KJ. J. Neuroimmunol. 1999;100:74–97. doi: 10.1016/S0165-5728(99)00195-2. [DOI] [PubMed] [Google Scholar]
- 11.Marth E, Kleinhappl B. Vaccine. 2001;20:532–537. doi: 10.1016/S0264-410X(01)00329-2. [DOI] [PubMed] [Google Scholar]
- 12.Aguilar JC, Rodriguez EG. Vaccine. 2007;25:3752–3762. doi: 10.1016/j.vaccine.2007.01.111. [DOI] [PubMed] [Google Scholar]
- 13.Schneider CK. Expert Rev. Clin. Pharmacol. 2008;1:327–331. doi: 10.1586/17512433.1.3.327. [DOI] [PubMed] [Google Scholar]
- 14.European Medicines Agency (EMA). Guideline on Adjuvants in Vaccines for Human Use EMA/CHMP/VEG/134716/2004 (EMA, London; 2005). <http://www.ema.europa.eu/pdfs/human/vwp/13471604en.pdf>
- 15.European Medicines Agency (EMA). Explanatory Note on Immunomodulators for the Guideline on Adjuvants in Vaccines for Human Use EMA/CHMP/VWP/244894/2006 (EMA, London; 2006). <http://www.ema.europa.eu/pdfs/human/vwp/24489406en.pdf>
- 16.Suntharalingam G, et al. N. Engl. J. Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 17.Plotkin S, Offit O. Vaccines. 2009. [Google Scholar]
- 18.Wucherpfennig KW. J. Autoimmun. 2001;16:293–302. doi: 10.1006/jaut.2000.0499. [DOI] [PubMed] [Google Scholar]
- 19.Gran B, Hemmer B, Vergelli M, McFarland HF, Martin R. Ann. Neurol. 1999;45:559–567. doi: 10.1002/1531-8249(199905)45:5<559::AID-ANA3>3.0.CO;2-Q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.European Medicines Agency (EMA). Guideline on Comparability of Biotechnology-Derived Medicinal Products after a Change in the Manufacturing Process—Non-Clinical and Clinical Issues EMA/CHMP/BMWP/101695/2006 (EMA, London;2007). <http://www.ema.europa.eu/pdfs/human/biosimilar/10169506enfin.pdf>
- 21.European Medicines Agency (EMA). ICH Topic S 6. Note for Guidance on Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals (CPMP/ICH/302/95). (EMA, London; 1998). <http://www.ema.europa.eu/pdfs/human/ich/030295en.pdf>
- 22.Gibson GG, Rostami-Hodjegan A. Xenobiotica. 2007;37:1013–1014. doi: 10.1080/00498250701649873. [DOI] [PubMed] [Google Scholar]
- 23.European Medicines Agency (EMA). Reflection Paper on the Extrapolation of Results from Clinical Studies Conducted Outside Europe to the EU-Population EMA/CHMP/EWP/692702/2008 (EMA, London; 2009). <http://www.ema.europa.eu/pdfs/human/ewp/69270208en.pdf>
- 24.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). Topic E 5 (R1): Ethnic Factors in the Acceptability of Foreign Clinical Data (ICH, Geneva; 2008). <http://www.ich.org/LOB/media/MEDIA481.pdf>
- 25.Neuenschwander B, Branson M, Gsponer T. Stat. Med. 2008;27:2420–2439. doi: 10.1002/sim.3230. [DOI] [PubMed] [Google Scholar]
- 26.Thall PF, Lee SJ. Int. J. Gynecol. Cancer. 2003;13:251–261. doi: 10.1046/j.1525-1438.2003.13202.x. [DOI] [PubMed] [Google Scholar]
- 27.European Medicines Agency (EMA). Guideline on Environmental Risk Assessments for Medicinal Products Consisting of, or Containing, Genetically Modified Organisms (GMOs) EMA/CHMP/BWP/473191/2006 (EMA, London; 2008). <http://www.ema.europa.eu/pdfs/human/bwp/47319106en.pdf>
- 28.European Parliament. Official J. Eur. Union27, L378/1–19 (2006).