

Abstract
SRSF7 is an essential RNA-binding protein whose misexpression promotes cancer. Here, we describe how SRSF7 maintains its protein homeostasis in murine P19 cells using an intricate negative feedback mechanism. SRSF7 binding to its premessenger RNA promotes inclusion of a poison cassette exon and transcript degradation via nonsense-mediated decay (NMD). However, elevated SRSF7 levels inhibit NMD and promote translation of two protein halves, termed Split-ORFs, from the bicistronic SRSF7-PCE transcript. The first half acts as dominant-negative isoform suppressing poison cassette exon inclusion and instead promoting the retention of flanking introns containing repeated SRSF7 binding sites. Massive SRSF7 binding to these sites and its oligomerization promote the assembly of large nuclear bodies, which sequester SRSF7 transcripts at their transcription site, preventing their export and restoring normal SRSF7 protein levels. We further show that hundreds of human and mouse NMD targets, especially RNA-binding proteins, encode potential Split-ORFs, some of which are expressed under specific cellular conditions.
Subject terms: RNA-binding proteins, RNA decay
The RNA-binding protein SRSF7 autoregulates its protein levels through an intricate negative feedback mechanism that involves translation of two distinct protein halves, termed Split-ORFs; the potential to encode Split-ORFs is also seen in other targets of nonsense-mediated decay.
Main
Control of gene expression in mammalian cells occurs at multiple levels of post-transcriptional regulation and involves 5′ capping, pre-mRNA splicing and alternative splicing, 3′ end processing, mRNA export, translation and mRNA decay. Each step is controlled by RNA-binding proteins (RBPs) that densely coat mRNAs and form large mRNA ribonucleoprotein particles (mRNPs)1. The usage of splice sites in pre-mRNAs is determined by serine- and arginine-rich (SR) proteins, a family of essential RBPs comprising 12 members (SRSF1–SRSF12)2. SR proteins contain one or two RNA recognition motifs (RRMs) and bind to specific sequences within exons, promoting their inclusion by recruiting the spliceosome to nearby splice sites. This recruitment is mediated by their arginine- and serine-rich (RS) domain, a low-complexity C-terminal region enriched in serine-arginine dipeptides that interacts with spliceosomal proteins; for example, U170K or U2AF2 (ref. 3).
SRSF7 is the only SR protein that contains, in addition to its RRM, a zinc knuckle (Zn), which contributes to its RNA-binding specificity4. This multitasking RBP regulates the inclusion of circadian and disease-relevant alternative splicing events such as FAS exon 6 (refs. 5–7), it modulates alternative polyadenylation and mRNA export and promotes translation of unspliced viral transcripts8,9. Recently, SRSF7 emerged as an oncogene that is overexpressed in various cancers and promotes the progression of colon and lung cancers10–12.
Many RBPs engage in auto-regulatory feedback loops to control their levels13, but the mechanisms that control SRSF7 protein homeostasis and the reasons for its disruption in cancer cells are not well understood. In renal cancer cells, SRSF7 is both a target and a regulator of microRNAs miR-30a-5p and miR-181a-5p (ref. 14). SRSF7 was also suggested to regulate its own transcript levels through the inclusion of an ultraconserved alternative exon, called poison cassette exon (PCE), a process referred to as unproductive splicing. The PCE contains a premature termination codon (PTC) and causes the rapid cytoplasmic degradation of the transcript by NMD15,16. SRSF7 transcript levels are also crossregulated by SRSF3, which binds to the PCE and promotes its inclusion17.
NMD is triggered during translation of PTC-containing transcripts to prevent the production of potentially deleterious truncated proteins. However, NMD gets frequently inactivated globally; for example, by viral infections, the tumor microenvironment or upon endoplasmic reticulum stress18–22. Thus, fail-safe mechanisms must be in place for RBPs that regulate their levels through unproductive splicing. Indeed, NMD alone was not sufficient to maintain protein homeostasis of the oncogenic SRSF1 (ref. 23).
Here, we describe an intricate auto-regulatory feedback mechanism for SRSF7 that involves unproductive splicing, bicistronic transcripts encoding truncated proteins (Split-ORFs), intron retention and the formation of large RNPs that assemble into phase-separated nuclear bodies. We provide evidence that Split-ORFs might contribute to auto-regulation of other SR proteins and are possibly a widespread feature among RBPs. Our findings further highlight that the retention of specific introns with repeated RBP binding sites can convert an mRNA into an architectural RNA that contributes to protein homeostasis.
Results
SRSF7 overexpression induces auto-regulation
To investigate the mechanisms of SRSF7 homeostasis, we generated cell lines overexpressing SRSF7 and examined transcript and protein expression. Bacterial artificial chromosomes (BACs) encoding C-terminally green fluorescent protein (GFP)-tagged SRSF7 (or SRSF3 as control) were integrated into diploid mouse P19 cells (Fig. 1a), and clonal cell lines with overexpression (OE) were derived by fluorescence-activated cell sorting (FACS)8. BACs enforce a sustained and homogenous OE in all cells and, given that they contain all gene-regulatory elements, can serve as genomic reporter genes that can be distinguished from their endogenous counterparts through their GFP tag.
Fig. 1. SRSF7 OE induces auto-regulation and promotes the splicing of NMD-sensitive and -resistant SRSF7 isoforms.

a, Domains and exonic organization of SRSF3 and SRSF7 BAC constructs. The mouse SRSF7 gene contains eight exons encoding the domains shown. An EGFP tag is inserted in frame at the C terminus, followed by the endogenous 3′ UTR. NXF1, nuclear export factor 1. b, WB comparing endogenous SRSF3 and SRSF7 protein levels in WT, SRSF3- and SRSF7-GFP overexpressing (OE) P19 cell lysates using SRSF3 and SRSF7 antibodies. GAPDH served as loading control. c, Quantification of seven WB experiments using FIJI normalized to GAPDH levels. Mean and error bars, s.d. *P < 0.05, **P < 0.001 (two-sided t-test). Endo, endogenous. d, Distribution of normalized significant crosslink events (Norm. X-links) of SRSF3- and SRSF7-GFP on the SRSF7 gene. e, Distribution of RNA-seq reads from WT (Ctrl) and SRSF7 OE samples on the SRSF7 gene. Splice junction read counts are given in percentage of the total junction read counts. f, Reverse transcription PCR (RT–PCR) of SRSF7 isoforms (see text for details) generated from the endogenous SRSF7 gene in WT and SRSF7 OE cells treated with CHX (100 µg ml−1) (+) or DMSO (−) using the indicated primers. The first lane for each cell line is without treatment. g, Reverse transcription qPCR analysis of SRSF7 mRNA (main), SRSF7-PCE1/4 and SRSF7-PCE isoforms in WT and SRSF7 OE cells treated with CHX (100 µg ml−1) (+) or with DMSO (−). Transcript levels were normalized to GAPDH and are shown relative to the respective DMSO control (−). Mean and error bars, s.d. n = 3 independent experiments. *P < 0.05 (two-sided t-test). h, Reverse transcription PCR of SRSF7 isoforms generated from the endogenous SRSF7 gene in fractionated SRSF7 OE cells treated with CHX (100 µg ml−1) (+) or with DMSO (−) using the indicated primers. in, input; nu, nucleus; cy, cytoplasm. Asterisks: orange, SRSF7-PCE; yellow, SRSF7-PCE1/4; red, SRSF7-I3a + b; green, SRSF7-I3b. Uncropped blot images for b and data for graphs in c and g are available as source data.
We obtained clones with eightfold OE for SRSF7 transcripts and 3.4-fold for SRSF7 protein (endogenous + transgene-derived; Fig. 1b,c and Extended Data Fig. 1a). Endogenous SRSF7 protein levels were markedly decreased (eightfold), whereas endogenous SRSF7 transcript levels showed only a modest decrease (twofold). Similar OE of SRSF3 had no apparent effect on SRSF7 protein levels, indicating that auto-regulation of SRSF7 is more efficient than its crossregulation by SRSF3 (ref. 17).
Extended Data Fig. 1. SRSF7 overexpression induces auto-regulation and promotes the splicing of NMD-sensitive and -resistant SRSF7 isoforms.

a, Quantification of four northern blot (NB) experiments using FIJI. All values were normalized for equal loading to 18S rRNA. Error bars indicate s.d. b, RT-PCR of SRSF7 isoforms (see text for details) generated exclusively from the SRSF7-GFP reporter gene in WT and SRSF7 OE cells treated with CHX (+) or DMSO (-) using the indicated primers. The first lane for each cell line is without any treatment. c, RT-PCR of SRSF7 isoforms generated exclusively from the endogenous SRSF7 gene in WT and SRSF7 OE cells with CHX (+) or DMSO (-). d, RT-PCR of SRSF7 isoforms generated from the endogenous SRSF7 gene in fractionated WT cells treated with CHX (+) or DMSO (-) using the indicated primers. e, Denaturing urea gel (7%) monitoring the success of the subcellular fractionation of WT and SRSF7 OE cells treated with DMSO or CHX.
SRSF7 binding promotes splicing of NMD-sensitive and -resistant SRSF7 isoforms
To understand the mechanism of SRSF7 auto-regulation, we examined the binding of SRSF7 protein to SRSF7 transcripts using individual-nucleotide resolution ultraviolet (UV) crosslinking and immunoprecipitation (iCLIP). We used normalized significant crosslink events (X-links, false discovery rate (FDR) < 0.05) from SRSF3 and SRSF7 iCLIP datasets of P19 cell lines without OE8. Similar to SRSF3, which promotes the inclusion of the PCE in SRSF7 transcripts17, SRSF7 showed enriched crosslinks in an extended region encompassing the PCE, its flanking introns 3a and 3b, and exons 3, 4 and 5 (Fig. 1d). Quantification revealed that SRSF7 binds ~50-fold more to SRSF7 transcripts than SRSF3 (Supplementary Table 1), indicating that SRSF7 has an unusual preference for its own transcripts.
RNA-seq followed by quantification of junction reads revealed that SRSF7 OE promotes inclusion of either the complete PCE (460 nucleotides (nt)) or a partial PCE (107 nt) in SRSF7 transcripts (Fig. 1e). Additionally, both PCE-flanking introns (3a and 3b) and intron 5 displayed increased read coverage, indicating that they are partly retained upon SRSF7 OE. Semiquantitative reverse transcription PCR and sequencing confirmed that SRSF7 OE caused the appearance of transcripts containing the entire PCE (SRSF7-PCE, orange asterisk), the partial PCE (SRSF7-PCE1/4, yellow asterisk) or the PCE in combination with both flanking introns (SRSF7-I3a+b, red asterisk) or with only intron 3b (SRSF7-I3b, green asterisk; Fig. 1f and Extended Data Fig. 1b,c). Identical isoforms were detected for endogenous and SRSF7-GFP reporter transcripts, indicating that auto-regulation operates similarly on both.
All these transcripts contain PTCs and should be susceptible to NMD. Indeed, NMD inhibition using cycloheximide (CHX) increased SRSF7-PCE and SRSF7-PCE1/4 levels in wild-type (WT) cells, confirming that they are bona fide NMD targets (Fig. 1f). However, in SRSF7 OE cells, SRSF7-PCE transcript levels were not increased by CHX treatment, which was validated by quantitative PCR (qPCR) with reverse transcription (Fig. 1g). Cytoplasmic-nuclear fractionation confirmed that both PCE-containing isoforms are exported to the cytoplasm, but whereas SRSF7-PCE1/4 is degraded by NMD, SRSF7-PCE is NMD-resistant (Fig. 1h and Extended Data Fig. 1d,e). Intron-containing isoforms SRSF7-I3a+b and SRSF7-I3b were also NMD-resistant, probably due to their nuclear retention. NMD was not globally compromised in SRSF7 OE cells since SRSF7-PCE1/4 was stabilized by CHX to the same extent in WT and SRSF7 OE cells (Fig. 1g). These results hint at a multilayered auto-regulatory mechanism, where SRSF7 OE promotes the generation of various SRSF7 transcript isoforms that are NMD-sensitive (SRSF7-PCE1/4) or NMD-resistant (SRSF7-PCE).
Translation of the SRSF7-PCE transcripts generates truncated SRSF7 proteins
Given that SRSF7-PCE transcripts are not degraded by NMD, they might be translated. Translation of SRSF7-PCE would result in a truncated protein with a predicted molecular weight of 15.7 kDa. This SRSF7 variant contains only the RNA-binding module (RRM + Zn) and a unique C terminus but lacks the RS domain and was termed SRSF7_RRM (Extended Data Fig. 2a). Indeed, western blots (WBs) using an antibody against the RRM of SRSF7 revealed a protein of ~16 kDa upon SRSF7 OE (Fig. 2a). This protein was not detectable using the antibody mAb104 for phosphorylated RS domains24. SRSF7_RRM was also detectable at low levels in WT cells and accumulated upon NMD inhibition by UPF1 knockdown (KD) (Extended Data Fig. 2b). Stringent immunoprecipitation (IP) coupled with tandem mass spectrometry (MS/MS) analyses confirmed the presence of peptides mapping to the RRM and the unique C terminus of SRSF7_RRM (Fig. 2b–d). Furthermore, ribosome profiling (Ribo-Seq) revealed the presence of ribosome-protected fragments mapping to the exon 3-PCE junction, confirming that SRSF7-PCE transcripts undergo translation and that SRSF7_RRM is produced from these transcripts (Fig. 2e, Extended Data Fig. 2c and Supplementary Table 2).
Extended Data Fig. 2. The SRSF7-PCE isoform is translated into two distinct truncated SRSF7 proteins.

a, Sequence and predicted molecular weight (MW) of the SRSF7_RRM and SRSF7_RS isoforms. SRSF7 protein domains are highlighted with the indicated colors. Hydrophobic amino acids within the RS domain are indicated in bold. The deleted 27aa stretch (see Fig. 6) is indicated in dark green. The mutated cysteine residues are indicated in red. b, SRSF7_RRM level in WT and SRSF7 OE cells upon knockdown (KD) of UPF1. The WB membrane was probed with α-SRSF7 and α-UPF1 antibodies. GAPDH was used as loading control. c, Ribo-Seq was performed from WT (Ctrl) and SRSF7 OE cells in two replicates. Scatter plot of Rlog-transformed raw Ribo-Seq reads from both replicates. d, WB analysis of WT and SRSF7 OE cells upon knockdown of UPF1. The blot was probed with mAb104 (anti-phospho-RS) and α-GFP. α-Beta-Catenin (Cttnb) was used as loading control. e, Co-IP of purified SRSF7-GFP probed with mAb104 to verify the presence of phosphorylated SRSF7_RS. PABPN1 was used to control for RNase treatment. IgG – unspecific antibody control, In – Input. f, Coomassie-stained SDS-PAGE of a stringent IP to purify the SRSF7_RS-GFP isoform for MS analysis. Cut bands are indicated in orange squares. M – marker g, Identified high-confidence peptides (FDR<0.01) mapping to the PCE, the RS domain and GFP. h, Alignment of PCE 3’ends indicate conservation of the in-frame START codon in mammals. i, iCLIP from polysomal fractions (piCLIP). Polysome profile after UV crosslinking of SRSF7-GFP to RNA in P19 cells. Indicated fractions were pooled to obtain monosomal (5+6) and polysomal (7–10) fractions. A representative rRNA gel and α-GFP WB are shown below (n=3). j, Protein-RNA complexes were immunopurified under stringent conditions from pooled fractions using α-GFP antibodies. RNA-protein complexes were undetectable when unspecific antibodies (IgG) were used.
Fig. 2. The SRSF7-PCE isoform is translated into two distinct truncated SRSF7 proteins.

a, WB analysis of WT and SRSF7 OE cells upon knockdown (KD) of UPF1. The membrane was probed with antibodies to α-SRSF7, mAb104 (antiphospho-RS) and α-UPF1. The KD efficiency is indicated below the UPF1 blot. β-catenin (Cttnb) was used as loading control. b, Coomassie-stained SDS–PAGE gel of stringent IP to purify the SRSF7_RRM isoform for MS analysis. Cut bands are indicated in orange boxes. IgG, unspecific antibody control; M, marker; In, input. c, Scheme of two truncated SRSF7 proteins encoded by the SRSF7-PCE transcript. Sequences of the unique C- and N-termini are shown above in yellow and green, respectively. d, Identified peptides (FDR < 0.05) mapping to exons 1–3 and the PCE. e, Top: distribution of Ribo-Seq reads from WT (Ctrl) and SRSF7 OE samples on the SRSF7 gene. The PCE is indicated in light blue. Bottom: distribution of Ribo-Seq reads within the PCE. Start (AUG, green Ms) and stop codons (red asterisks) as well as amino acids encoded in all three possible open reading frames are shown below. A STOP sign marks the termination codon of SRSF7_RRM. A START sign marks the putative initiation codon of SRSF7_RS. f, Top: distribution of SRSF7 piCLIP X-links on the SRSF7 gene. Bottom: distribution of SRSF7 piCLIP X-links in the PCE and PCE1/4 regions. The main binding peaks are highlighted with green arrows. Uncropped images for a are available as source data.
Ribo-Seq reads mapped throughout the PCE, indicating that translation might reinitiate downstream of the PTC (Fig. 2e). Translation initiation from the 3′ most AUG codon within the PCE would resume the original open reading frame (ORF) and generate a second polypeptide comprising the entire RS domain with 11 additional N-terminal amino acids, termed SRSF7_RS (Fig. 2c and Extended Data Fig. 2a). This polypeptide was indeed detectable in SRSF7 OE cells using both anti-GFP and mAb104 antibodies (Extended Data Fig. 2d,e, pink asterisk). MS/MS analyses after IP confirmed the presence of peptides mapping to the junction between the RS domain and the GFP tag (Extended Data Fig. 2f,g), although few peptides mapped to the RS domain itself and none to the unique 11-amino acid N terminus. This is likely due to the low abundance of this protein, small peptide sizes and the high charge and repetitive nature of RS domains. The in-frame start codon of SRSF7_RS is ultraconserved in mammals and the same protein halves would also arise from translation of the human SRSF7-PCE isoform (Extended Data Fig. 2h). Altogether, our data reveal that upon PCE inclusion, the resulting transcript encodes two SRSF7 protein halves (referred to as Split-ORFs).
Binding of SRSF7 and translation might protect SRSF7-PCE transcripts from NMD
SRSF7-PCE transcripts are NMD-resistant despite having multiple exon–exon junctions downstream of their PTC, a feature normally triggering NMD25. Translation of Split-ORF2 would ensure that translating ribosomes reach the end of the ORF, thereby removing all exon–exon junction complexes (EJCs). SRSF7 binding to the PCE might promote translation of Split-ORF2 as it shuttles between the nucleus and the cytoplasm26 and was previously shown to promote the translation of unspliced viral and reporter transcripts9,27. To test where SRSF7 binds in SRSF7-PCE transcripts when they undergo translation, we performed iCLIP on monosomal and polysomal fractions (piCLIP)26 (Fig. 2f and Extended Data Fig. 2i,j). piCLIP reads were virtually absent from coding exons of SRSF7, consistent with efficient translation of the ORF and removal of SRSF7 by translating ribosomes, but two abundant crosslink peaks were present upstream of the AUG of SRSF7_RS (Fig. 2f, green arrows, and Supplementary Table 3). This raises the possibility that SRSF7 binding promotes translation of Split-ORF2, thereby protecting SRSF7-PCE transcripts from NMD. Consistently, both SRSF7 binding sites and in-frame start codon are missing in NMD-sensitive SRSF7-PCE1/4 transcripts (Fig. 2f).
The first protein half (SRSF7_RRM) inhibits splicing by competing with SRSF7
SRSF7_RRM contains the RNA-binding module but lacks the RS domain for spliceosomal recruitment. Thus, it might act as a dominant-negative and antagonize the functions of full-length SRSF7. This hypothesis predicts that SRSF7_RRM (1) localizes to the nucleus, (2) binds to RNA with similar preference as SRSF7 and (3) is impaired in spliceosome recruitment.
Nuclear-cytoplasmic fractionation revealed that SRSF7_RRM indeed localized predominantly to the nucleus (Extended Data Fig. 3a). But, unlike full-length SRSF7-GFP, SRSF7_RRM did not accumulate in nuclear speckles (Fig. 3a and Extended Data Fig. 3b). This indicates that the unique C terminus of SRSF7_RRM acts as a nuclear localization signal but is insufficient for nuclear speckle targeting, which requires phosphorylated RS dipeptides28.
Extended Data Fig. 3. SRSF7_RRM competes with SRSF7 for binding to SRSF7-PCE transcripts and inhibits splicing of intron 3.

a, WB analysis of cytoplasmic (cyt) and nuclear (nuc) fractions of WT and SRSF7 OE cells. The blot was probed with α-SRSF7 as well as with α-Histone H3 and α-Tubulin antibodies to verify the fractionation efficiency. b, Confocal microscopy following transient expression of SRSF7_RRM-mCherry in SRSF7-GFP expressing cells. DNA was stained with Hoechst. c, WB analysis of iCLIP samples to validate the IP efficiency. Samples without antibodies served as controls. Blots were probed with α-SRSF7. d, WB showing the comparable expression of full-length SRSF7-mCherry, SRSF7_RRM-mCherry and empty mCherry plasmids in P19 WT cells. Blots were probed with α-mCherry and α-Beta-Catenin (Ctnnb) as loading control. e, RT-PCR of SRSF7 isoforms generated from the endogenous SRSF7 gene in WT cells transfected with plasmids transiently overexpressing full-length SRSF7-mCherry, SRSF7_RRM-mCherry or empty mCherry using the indicated primers.
Fig. 3. SRSF7_RRM competes with SRSF7 for binding to SRSF7-PCE transcripts and inhibits splicing of intron 3.

a, Confocal microscopy analysis of cells transiently expressing SRSF7_RRM-GFP or EGFP as control. DNA was stained with Hoechst. Nuclei are outlined with dashed lines. Scale bar, 5 µm. b, Autoradiograph of an iCLIP experiment using α-SRSF7 antibodies to purify SRSF7-GFP, SRSF7 and SRSF7_RRM from SRSF7 OE and size-matched WT samples. Crosslinked RNA was labeled with 32P. Non-crosslinked samples (-UV) and samples without antibody served as controls. Cut bands are indicated with black boxes. c, Consensus binding motifs of SRSF7-GFP, SRSF7 and SRSF7_RRM derived from an alignment of the top 20 enriched 5-mers. d, Distribution of significant crosslink events (X-links) of SRSF7-GFP, SRSF7 and SRSF7_RRM from OE and WT samples on the SRSF7 gene. Peaks at 5′ splice sites are indicated by green arrows. RNA-seq read coverage on introns 3 and 5 is shown below. e, Co-IPs of purified SRSF7-GFP and SRSF7_RRM-GFP following RNase A treatment, probed for interaction with spliceosomal factors U2AF65, U170K and PRP8. NXF1 and RNA PolII were used as controls. f, Reverse transcription qPCR analysis of PCE splicing and intron 3 retention in WT cells after transient OE of SRSF7-mCherry, SRSF7_RRM-mCherry or mCherry (Ctrl). Mean and error bars, s.d.; n = 4–6 independent experiments as specified in the source data file. *P < 0.05, **P < 0.001 (two-sided t-test). Uncropped blot images for e and data for graphs in f are available as source data.
To assess whether SRSF7_RRM binds to RNA, we performed iCLIP from WT and SRSF7 OE cells using an anti-SRSF7 antibody. IP efficiency was verified by WB, and uncrosslinked samples and unspecific antibodies served as controls (Extended Data Fig. 3c). The presence of radioactively labeled RNA-protein complexes between 15 and 35 kDa whose abundance increased upon SRSF7 OE indicated that SRSF7_RRM indeed binds to RNA (Fig. 3b). We found similar consensus binding motifs for endogenous SRSF7, SRSF7-GFP and SRSF7_RRM (GAYGAY) (Fig. 3c) and all three proteins bound SRSF7 transcripts to a similar extent (Supplementary Table 4), indicating that SRSF7_RRM has a similar RNA-binding specificity and affinity as full-length SRSF7. Moreover, all SRSF7 variants bound near the 5′ splice sites of introns 3a, 3b and 5, which all remain partly unspliced on SRSF7 OE, while other introns are efficiently spliced (Fig. 3d, green arrows). However, SRSF7_RRM lacked the strong binding peak of full-length SRSF7 (WT and OE) and SRSF7-GFP near the 3′ splice site of intron 3a (black arrow), expected to promote PCE inclusion.
To test whether SRSF7_RRM recruits the spliceosome, we performed co-IPs from cells transiently expressing SRSF7-GFP or SRSF7_RRM-GFP. The three tested spliceosomal factors U2AF65, U170K and PRP8 interacted less with SRSF7_RRM-GFP than with SRSF7-GFP (Fig. 3e), indicating impaired spliceosome recruitment. Moreover, transient OE of full-length SRSF7-mCherry promoted PCE inclusion, whereas SRSF7_RRM-mCherry impaired PCE inclusion and instead promoted retention of the flanking introns 3a and 3b (Fig. 3f and Extended Data Fig. 3d,e).
Altogether, these results indicate the existence of a negative feedback mechanism in which SRSF7 OE promotes PCE inclusion, thereby generating a splicing-incompetent SRSF7_RRM isoform. Upon accumulation, SRSF7_RRM competes with full-length SRSF7 for binding to 5′ splice sites, resulting in the retention of introns 3 and 5.
Intron-containing SRSF7 transcripts are retained in large nuclear bodies
The observed shift from PCE inclusion to intron retention indicates additional layers of regulation. Northern blots and Reverse transcription PCRs confirmed that SRSF7 transcripts with unspliced introns 3 and 5 (SRSF7-I3+5) were confined to the nucleus (Fig. 1h, Extended Data Fig. 4a–c). RNA fluorescence in situ hybridization (RNA FISH) using probes specific for introns 3 and 5 detected one to three large foci per nucleus that colocalized with SRSF7-GFP accumulations (Fig. 4a,b and Extended Data Fig. 4d).
Extended Data Fig. 4. Intron-containing SRSF7 transcripts are retained in large nuclear bodies.

a, Northern blot with total RNA isolated from WT and SRSF7 OE samples probed for SRSF7 transcripts. A probe for 18S served as loading control. b, RT-PCR of intron 5 amplified from endogenous and reporter SRSF7 genes in fractionated WT and SRSF7 OE cells using the indicated primers. c, RT-PCR of intron 5 amplified from the endogenous SRSF7 gene in fractionated WT cells treated with CHX (100 µg/mL) (+) or with DMSO (-) using the indicated primers. Exon- and intron-specific primers for Beta-actin (BA) served to control for fractionation efficiency. d, RNA FISH in SRSF7 OE cells using probes specific for introns 3 and 5. DNA was stained with Hoechst. SRSF7 bodies are labeled with arrowheads. Scale bar, 5 µm. e, P19 cells expressing the paraspeckle markers PSPC1-GFP and NONO-GFP. Scalebar – 5 µm. f, RNA FISH in SRSF7 OE cells using probes specific for introns 3 and Malat1. Left, SRSF7 bodies in magenta are outlined and analyzed for co-localization with Malat1 speckles. Orange arrowheads, no colocalization; purple arrowheads - co-localization. Scalebar – 10 µm. Right: Pie chart showing the percentage of Malat1 co-localization analysing 150 bodies. g, RNA FISH in SRSF7 OE cells treated for 5 minutes with 10% 1,6 HD or 2,5-HD using probes specific for introns 3 and 5. Treatment with DMSO is shown as additional control. DNA was stained with Hoechst. SRSF7 bodies are labeled with arrowheads. Scale bar – 5 µm.
Fig. 4. Intron-containing SRSF7 mRNAs are retained in large nuclear bodies.

a, Top: RNA FISH in SRSF7-GFP (OE) cells using a probe specific for intron 5. Bottom: quantification of body number per cell from 100 cells from three independent experiments. b, Top: RNA FISH in SRSF7- and SRSF3-GFP (OE) cells using a probe specific for intron 5. This image is representative of three independent experiments. Bottom: line scan of two example cells showing colocalization with SRSF7-GFP. No FISH signal for intron 5 was detected in the SRSF3 OE line. c, RNA FISH in SRSF7-GFP (OE) cells treated for 2 h with Actinomycin D (ActD) using probes specific for intron 3 and intron 5. Cells treated with DMSO or hybridization buffer containing no probe are shown as controls. d, RNA FISH in SRSF7-GFP (OE) cells using probes specific for intron 3 and poly(A)+ RNA. e, RNA FISH in SRSF7-GFP (OE) cells treated for 5 min with 10% 1,6-HD or 2,5-HD using probes specific for introns 3 and 5. DMSO treatment is shown as additional control. In all figures, DNA was stained with Hoechst. SRSF7 bodies are indicated with arrowheads. Scale bars, 5 µm. Data for graphs in a are available as source data.
The number of observed foci indicates that SRSF7-I3+5 transcripts accumulate at transcription sites of exogenous and endogenous SRSF7 alleles. Accordingly, treatment with actinomycin D completely abrogated the formation of SRSF7 foci (Fig. 4c), while SRSF7 relocalized to perinucleolar caps, similar to RBPs that assemble paraspeckles29. SRSF7 is found in paraspeckles and nuclear speckles30; however, SRSF7 bodies are unlikely to constitute a subpopulation of those nuclear bodies, since (1) pluripotent P19 cells do not contain any paraspeckles (Extended Data Fig. 4e), and (2) nearly half of SRSF7 bodies do not colocalize with the nuclear speckle marker Malat1 (Extended Data Fig. 4f).
Accumulation of SRSF7 transcripts at their transcription sites could be due to perturbed cleavage and polyadenylation. RNA-seq, northern blot and 3′ RACE analyses indicated that SRSF7 transcripts have longer 3′ untranslated regions (UTRs) in OE cells due to enhanced use of a distal poly(A) site, but they were otherwise properly processed at their 3′ ends (Fig. 1e and Extended Data Fig. 4a, data not shown). RNA FISH using oligo(dT) probes revealed that SRSF7 foci contained substantial amounts of polyadenylated transcripts (Fig. 4d and Extended Data Fig. 4g). Since fully spliced SRSF7 mRNAs were also retained in the nucleus (Fig. 1h and Extended Data Fig. 1d), these observations indicate that SRSF7 OE causes the transient sequestration of fully spliced and intron-containing, polyadenylated SRSF7 transcripts in a new type of nuclear body.
Nuclear bodies often assemble through the binding of RBPs containing intrinsically disordered regions to scaffolding RNAs, and their oligomerization induces a liquid–liquid phase separation31. To assess whether SRSF7 bodies are phase-separated nuclear bodies, we treated SRSF7 OE cells with 10% 1,6-HD, which disrupts weak hydrophobic interactions32–34, and performed RNA FISH. Indeed, 1,6-HD treatment efficiently dispersed SRSF7 bodies into numerous smaller foci that still colocalized with SRSF7-GFP. The control compound 2,5-HD did not affect the integrity of SRSF7 bodies (Fig. 4e and Extended Data Fig. 4h). This indicates that SRSF7 bodies are assembled from many SRSF7-containing RNPs that are held together by weak hydrophobic interactions, similar to many other nuclear bodies35.
SRSF7 promotes the formation of higher-order assemblies in vitro
We hypothesized that SRSF7 body formation is driven by the SRSF7 protein based on the following observations: (1) the PCE and intron 3 together contain ~80 regularly spaced SRSF7 binding sites, and SRSF7 binds massively within this region (Fig. 1d), (2) SRSF7 colocalizes with SRSF7 bodies (Fig. 4b), and (3) SRSF7 contains domains that enable its simultaneous binding to SRSF7 transcripts (RRM-Zn domain) and to other SRSF7 proteins (RS domain). Co-IPs confirmed that SRSF7 oligomerizes via its RS domain as only full-length SRSF7, but not SRSF7_RRM, coimmunoprecipitated with SRSF7-GFP in the presence of RNase A (Fig. 5a). SRSF7_RRM coimmunoprecipitated with SRSF7-GFP in the absence of RNase A, strongly indicating that SRSF7_RRM binds to the same transcripts as SRSF7-GFP and SRSF7.
Fig. 5. SRSF7-GFP promotes the formation of higher-order assemblies in vitro.
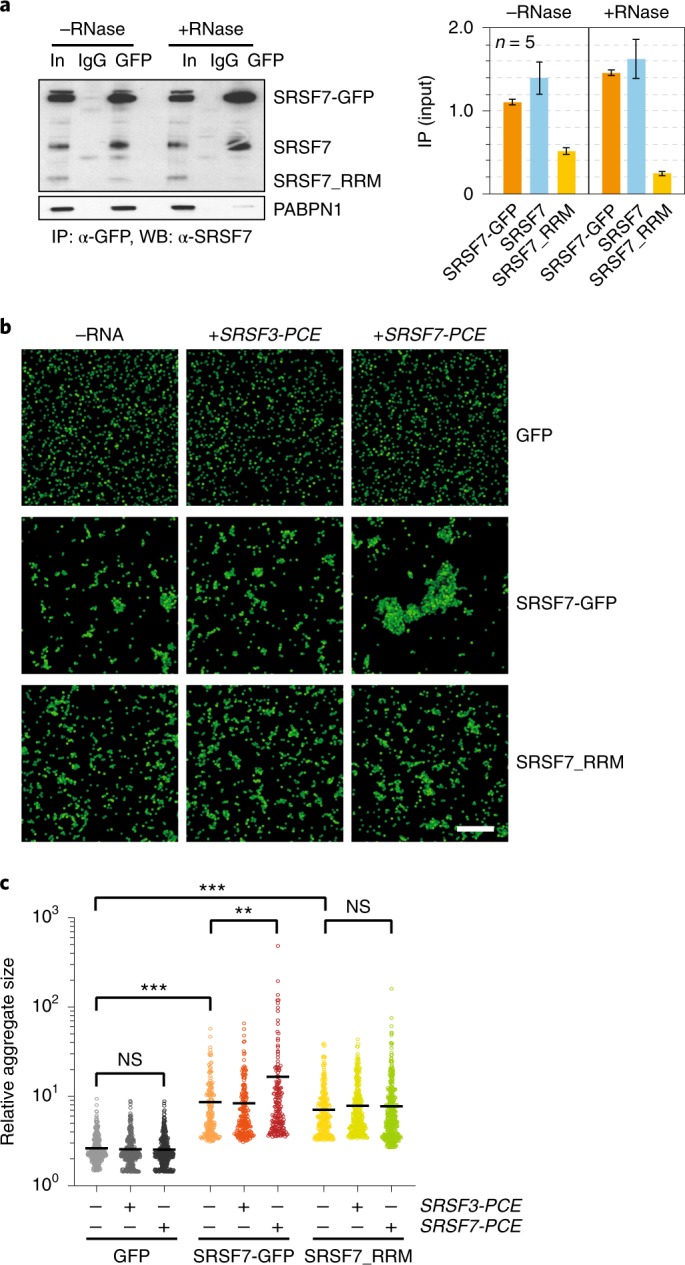
a, Left: co-IP of purified SRSF7-GFP probed with α-SRSF7 to verify the interaction with endogenous SRSF7 and SRSF7_RRM. PABPN1 was used to control for RNase treatment. Right: quantification of five Co-IP experiments with and without RNase treatment. Mean and error bars, s.e.m.; n = 5 independent experiments. b, SRSF7-GFP and SRSF7-PCE transcripts promote the formation of higher-order assemblies in vitro. Bead aggregation assays were performed with magnetic beads coupled to α-GFP antibodies that immunoprecipitated GFP, SRSF7-GFP or SRSF7_RRM-GFP from P19 cell lysates. Their aggregation propensity was tested in the absence or presence of transcripts corresponding to the PCE of SRSF7 or SRSF3. Shown is a representative confocal micrograph of fluorescent single beads and aggregates at ×40 magnification from three independent experiments. Scale bar, 50 µm. c, Quantification of beads per aggregate using FIJI (Analyze particles). Shown is the top 25th percentile of aggregate areas normalized to the area of one bead. This experiment is representative of three independent experiments. Center, median. ***P < 0.0002; **P < 0.002 (two-tailed Mann–Whitney test); NS, not significant. Uncropped blot images for a and data for graphs in a and c are available as source data.
To assess whether binding of SRSF7 proteins to SRSF7 transcripts induces higher-order assemblies, we performed in vitro bead aggregation assays34. Using magnetic beads coupled with anti-GFP antibodies, we first immunoprecipitated SRSF7-GFP, SRSF7_RRM-GFP or GFP alone from cell lysates and measured the size of bead aggregates observed by fluorescence microscopy (Fig. 5b,c and Extended Data Fig. 5a). SRSF7-GFP and SRSF7_RRM-GFP formed larger bead aggregates compared to GFP alone, likely caused by simultaneous binding to RNA molecules within the lysates (Fig. 5c). Addition of in vitro-transcribed SRSF7 PCE RNA that contains ~34 predicted SRSF7 binding sites (Extended Data Fig. 5b) produced super-aggregates of beads coated with SRSF7-GFP but not SRSF7_RRM-GFP, indicating that oligomerization via the RS domain drives aggregation. Notably, addition of SRSF3 PCE RNA, which has a similar length (460 nt) and GC content (51 versus 45%) but contains few SRSF7 binding sites, did not cause super-aggregates (Extended Data Fig. 5b,c). Together, these results indicate that both sequence-specific RNA binding and oligomerization of SRSF7 are required to promote high-order assembly of mRNPs in vitro.
Extended Data Fig. 5. SRSF7-GFP promotes the formation of higher-order assemblies in vitro.
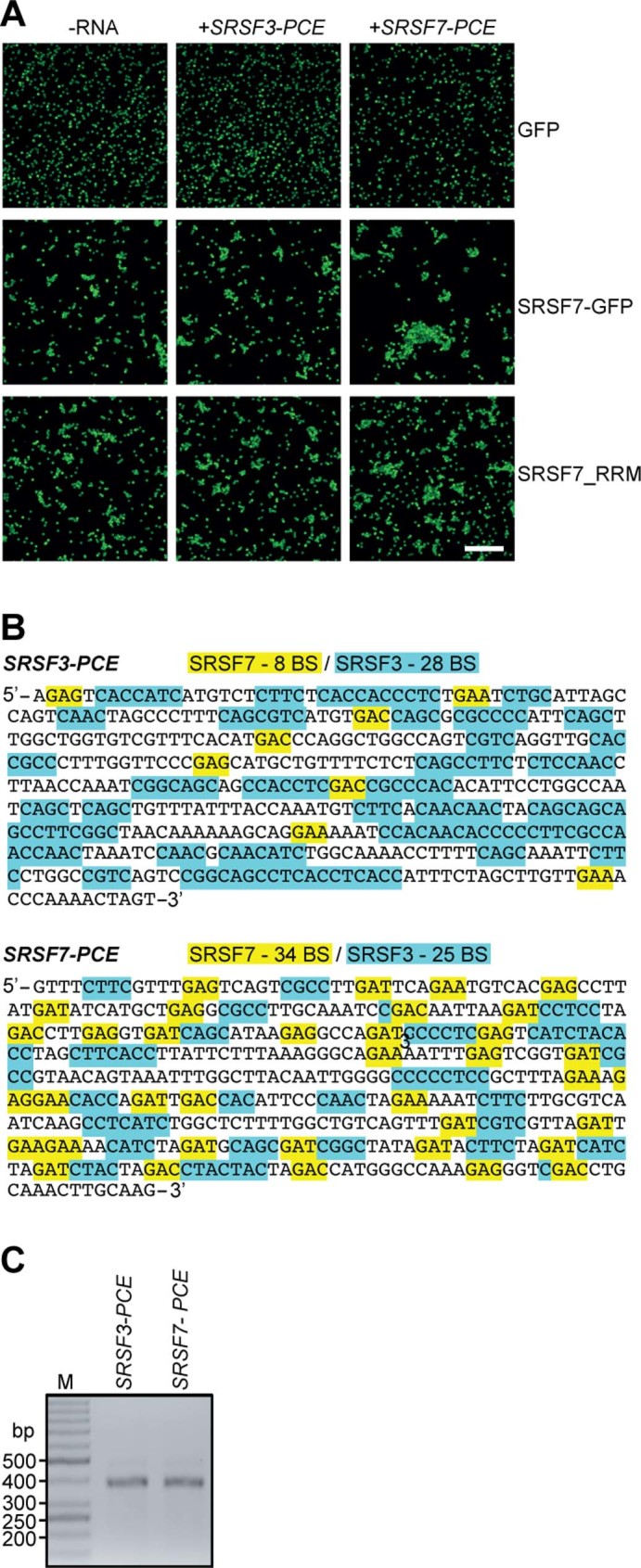
a, Bead aggregation assay. Representative confocal micrographs of fluorescent single beads and aggregates at 40x magnification. Scalebar – 50 µm. b, Sequences of the PCEs of SRSF3 and SRSF7, which have a similar length and GC content but differ in the number of SRSF7-binding sites (BS, highlighted in yellow). SRSF3-binding sites are highlighted in blue. C, Representative agarose gel of purified in vitro transcribed SRSF3 and SRSF7 PCE transcripts.
RNA binding and oligomerization of SRSF7 drive the formation of SRSF7 bodies in vivo
To confirm that RNA binding and oligomerization of SRSF7 are required for SRSF7 body formation in vivo, we generated two SRSF7 mutants using BAC recombineering36 (Fig. 6a). We altered the RNA-binding specificity of SRSF7 by mutations in the zinc knuckle domain (Cys106Ala, Cys109Ala; SRSF7_mutZn)4 and the oligomerization propensity by deleting three of the four RSRSXSXR repeats (X, hydrophobic amino acid) within its RS domain (SRSF7_Δ27aa, Extended Data Fig. 6a)37. Co-IPs confirmed that oligomerization (interaction with endogenous SRSF7) was reduced for SRSF7_Δ27aa but not for SRSF7_mutZn (Extended Data Fig. 6b,c). Alternative 5′ splice site usage produces four distinct SRSF7 isoforms in P19 cells containing all four repeats (full-length) or lacking three repeats (Δ27aa), one repeat (Δ11aa) or only the last three amino acids of exon 5 (ΔYFQ), indicating that the mutated SRSF7 variants can naturally occur in cells (Fig. 6b, Extended Data Fig. 6a and Supplementary Table 5).
Fig. 6. Translation of Split-ORF2, RNA-binding and oligomerization of SRSF7 contribute to the formation of SRSF7 bodies in vivo.

a, Mutated SRSF7 BAC constructs. See text for details. b, Reverse transcription PCR of SRSF7 isoforms generated by alternative 5′ splice site usage of intron 5 in WT and SRSF7 OE cells. c, WB comparing protein levels of endogenous SRSF7, SRSF7-GFP and SRSF7_RRM in WT, SRSF7 OE, SRSF7_Δ27aa and SRSF7_mutZn cells. β-Catenin (Cttnb) served as loading control. d, RNA FISH with SRSF7-GFP, SRSF7_Δaa and SRSF7_mutZn cells using a probe specific for intron 3. DNA was stained with Hoechst. SRSF7 bodies are indicated with arrowheads. Scale bar, 10 µm. e, Mean signal intensity of SRSF7 bodies quantified from 100 cells. f, Size (longer axis, in µm) of SRSF7 bodies quantified from 100 cells from three independent experiments. Box plots indicate median, first and third quartiles (box); whiskers show 1.5× interquartile range; outliers were omitted. **P < 0.0005; *P < 0.05 (Wilcoxon rank-sum tests). g, Consensus binding motifs of SRSF7-GFP (OE), SRSF7_Δ27aa and SRSF7_mutZn derived from an alignment of the top 20 enriched 5-mers. h, The conserved in-frame START codon in the PCE was mutated to AAA. i, WB comparing protein levels of endogenous SRSF7, SRSF7-GFP, SRSF7_RS-GFP (pink asterisk) and SRSF7-RRM in WT, SRSF7 OE and two different SRSF7-ΔAUG clones. β-Catenin (Cttnb) served as loading control. j, RNA FISH with SRSF7 OE and ΔAUG cells using a probe specific for intron 5. DNA was stained with Hoechst. Scale bar, 5 µm. k, Size of SRSF7 bodies (in µm2) quantified from 100 cells from three independent experiments. Box plot indicates median, first and third quartiles (box), whiskers show 1.5× interquartile range. Open circles and squares, outliers. Uncropped blot images for c and i and data for graphs in e,f,k are available as source data.
Extended Data Fig. 6. Translation of Split-ORF2, RNA-binding and oligomerization of SRSF7 contribute to the formation of SRSF7 bodies in vivo.

a, Intron 5 undergoes alternative 5’ splice site usage, which gives rise to four different SRSF7 isoforms lacking different portions of the four RSRSXSXR repeats (X – hydrophobic aa, highlighted in orange). b, Co-IP of purified SRSF7-GFP, SRSF7_Δ27aa-GFP and SRSF7_mutZn-GFP probed with α-SRSF7 to verify oligomerization with endogenous SRSF7. α-PABPN1 was used as control for RNase treatment. IgG – unspecific antibody control, In – Input. c, Quantification of (n=3) Co-IP experiments. Shown is the interaction of endogenous SRSF7 with SRSF7-GFP, SRSF7_Δ27aa or SRSF7_mutZn normalized to the respective baits. Two-sided T-test, **p-value < 0.01. Error bars s.d. d, Northern blot to compare the levels of intron-containing transcripts SRSF7-I3 (red asterisk) and SRSF7-I3+5 (purple asterisk) in SRSF7-GFP, SRSF7_Δ27aa or SRSF7_mutZn cells. e, RT-PCR of SRSF7 isoforms generated from reporter and endogenous SRSF7 genes in WT, SRSF7 OE, SRSF7_Δ27aa and SRSF7_mutZn P19 cells with (+) and without (-) CHX using the indicated primers. f, Same as in E), except that primers specific for both endogenous and reporter SRSF7 genes were used. g, RT-qPCR quantification of SRSF7-PCE 1/4 and SRSF7-PCE transcript level after CHX treatment in SRSF7-GFP, SRSF7_Δ27aa and SRSF7_mutZn cells normalized to the respective DMSO controls (n=4). Two-sided T-test, *p-value < 0.05. Error bars s.d. h, Line scans of example cells to assess co-localization of SRSF7 bodies with SRSF7-GFP, SRSF7_Δ27aa and SRSF7_mutZn. i, Autoradiograph of an iCLIP experiment using α-GFP antibodies to purify GFP-tagged SRSF7, SRSF7_Δ27aa and SRSF7_mutZn. Crosslinked RNA was labeled with 32P. Non-crosslinked samples (-UV) served as controls. Cut bands are indicated in grey squares. j, Venn diagram displaying the overlap in RNA targets. K, Comparison of crosslink preferences of SRSF7-GFP and SRSF7_mutZn around 5’ splice sites. L, Mean signal intensities of SRSF7 bodies quantified from 100 cells. Box plot indicates median, first and third quartiles (box), whiskers show 1.5× interquartile range.
Auto-regulation of endogenous SRSF7 protein is less efficient in SRSF7_Δ27aa cells compared to SRSF7 OE (Fig. 6c), although both produced similar levels of GFP-tagged proteins, NMD-resistant SRSF7-PCE isoforms, truncated SRSF7_RRM protein and SRSF7-I3+5 transcripts (Fig. 6c and Extended Data Fig. 6d,g). However, SRSF7_Δ27aa formed smaller SRSF7 bodies and intron 3 showed reduced colocalization with SRSF7_Δ27aa protein compared to SRSF7 (Fig. 6d–f and Extended Data Fig. 6h).
In SRSF7_mutZn cells, SRSF7 auto-regulation was more severely compromised. Endogenous SRSF7 protein levels were not reduced, SRSF7_RRM protein was hardly detectable and SRSF7-PCE transcripts were NMD-sensitive (Fig. 6c and Extended Data Fig. 6e,g). Very few SRSF7-I3+5 isoforms were detectable, and SRSF7 bodies were drastically diminished and showed poor colocalization with SRSF7-mutZn protein (Fig. 6d–f and Extended Data Fig. 6d–h). To verify the altered RNA-binding specificity of SRSF7_mutZn, we performed iCLIP (Extended Data Fig. 6i and Supplementary Table 6). SRSF7 and SRSF7_mutZn showed 84% overlap in bound target transcripts, but SRSF7_mutZn crosslinked more to introns and less to exons (Extended Data Fig. 6j,k). The binding motif of SRSF7_mutZn changed from purine-rich GAYGAY triplets to pyrimidine-rich CNYC motifs, similar to the SRSF3 binding motif (Fig. 6g), indicating that its RNA-binding specificity is now determined exclusively by its RRM, which is highly similar to the RRM of SRSF3 (ref. 38). Altogether, these data exclude the possibility that high levels of SRSF7 mRNA simply overwhelm the NMD machinery and confirm that sequence-specific binding and oligomerization of SRSF7 are required for body formation and auto-regulation in vivo.
Translation of Split-ORF2 prevents NMD and contributes to SRSF7 homeostasis
Based on our results, we speculated that translation initiation at the AUG codon of Split-ORF2 would allow ribosomes to remove all NMD-triggering EJCs, rendering SRSF7-PCE transcripts NMD-resistant and allowing for accumulation of SRSF7_RRM encoded by Split-ORF1. To test this hypothesis, we used BAC recombineering to mutate the in-frame AUG (Fig. 6h). Indeed, the ΔAUG mutant showed strongly reduced levels of SRSF7_RS-GFP and SRSF7_RRM and impaired SRSF7 body formation (Fig. 6i–k and Extended Data Fig. 6l). These data indicate that the conserved in-frame START codon of Split-ORF2 is required for the accumulation of SRSF7_RRM encoded by Split-ORF1 and SRSF7 body formation.
Genome-wide identification of Split-ORFs in human and mouse NMD targets
To test whether Split-ORFs exist in other transcripts, we designed a computational pipeline to search within annotated NMD transcripts from mouse and human genes (Extended Data Fig. 7a, see Methods). We found 2,723 human and 1,423 mouse genes that encoded 4,473 and 1,859 NMD transcripts with Split-ORFs, respectively (Fig. 7a, Extended Data Fig. 7b and Supplementary Table 7) with a mean length of 879 and 854 nt, respectively (293 and 285 amino acids, Fig. 7b). Notably, 475 NMD transcripts encode Split-ORFs in both species (Fig. 7c). Split-ORF genes were enriched for the gene ontology terms ‘RNA binding’ and ‘mRNA processing’, and among the significantly enriched protein domains were known RNA-binding domains, such as zinc-finger and RRM domains and WD40 repeats (Fig. 7d and Supplementary Tables 8 and 9), indicating that many RBPs have the potential to express Split-ORFs. Two scenarios for the generation of Split-ORFs were common: the inclusion of an alternative exon, for example an ultraconserved PCE that provides a STOP and a downstream in-frame START codon, or the skipping of an alternative exon that generates a frameshift and a PTC in close proximity. In the latter case, a downstream in-frame START codon already present in the original ORF would be used.
Extended Data Fig. 7. Genome-wide identification of putative Split-ORFs in annotated human and mouse NMD targets.

a, Scheme of the computational pipeline. b, Numbers of identified Split-ORFs per NMD target (mouse). c, Protein sequences of Split-ORFs for SRSF6, SRSF5 and TIAL1. d, Quantification of Ribo-Seq reads (mouse hearts and MHV infection) within unique NMD exons of selected RBPs.
Fig. 7. Genome-wide identification of putative Split-ORFs in annotated human and mouse NMD targets.

a, Numbers of identified Split-ORFs per NMD target (human). b, Length of identified Split-ORFs (4,473 human and 1,859 mouse). Box plot indicates median, first and third quartiles (box); whiskers show 1.5× interquartile range; black dots, outliers. c, Overlap of orthologous NMD targets with putative Split-ORFs from mouse and human. d, Gene ontology (GO) term enrichment of NMD targets with identified Split-ORFs (human). BH, Benjamini–Hochberg. e, WB of lysates from WT or SRSF5 and SRSF6 overexpressing P19 cells probed with the indicated antibodies. Asterisks: pink, ORF2-GFP; orange, ORF1. f, Examples of identified RBPs that bind to their own transcripts within a conserved PCE, whose inclusion generates NMD-transcript isoforms that encode two Split-ORFs. g, Distribution of Ribo-Seq reads within unique NMD exons of mouse SRSF7 and human SRSF6 genes. MHVA59, murine coronavirus; CM, cardiomyocytes. Uncropped blot images for e and data for graphs in b are available as source data.
Interesting examples of RBP-encoding genes that generate Split-ORFs include several SR proteins (SRSF5, SRSF6, SRSF9 and SRSF10) as well as BCLAF1, HNRNPL, MAGOHB, PRPF39, RBM39, DDX3X, TIAL1 and U2AF1L4 (Supplementary Table 7). To test whether these RBPs express Split-ORFs under some conditions, we followed three different strategies. First, we generated P19 cell lines overexpressing either SRSF5- or SRSF6-GFP. WB using specific antibodies revealed that the levels of endogenous proteins were strongly decreased in the OE samples, indicative of auto-regulation (Fig. 7e). In both cases additional protein bands appeared whose sizes would fit the predicted Split-ORFs (Extended Data Fig. 7c). Second, we inspected binding of RBPs to their own NMD transcripts using available eCLIP and iCLIP data39–41. In several cases, such as BCLAF1, TIAL1, SRSF5, SRSF6 and HNRNPL, a conserved PCE that generates Split-ORFs was massively bound by the encoded RBP (Fig. 7f, data not shown). This indicates that this NMD isoform is made, stably detectable and possibly involved in an auto-regulatory feedback mechanism. Third, to assess whether some of the predicted Split-ORFs are translated in a physiological context, we interrogated publicly available Ribo-Seq data from mouse hearts42, 17Cl-1 cells infected with murine coronavirus, human cardiomyocytes42, A549 cells infected with Dengue virus and human embryonic kidney 293 cells43. We selected RBP-encoding transcripts where insertion of a unique NMD exon gives rise to Split-ORFs and counted Ribo-Seq footprint reads mapping to these exons. We identified 29 RBPs with at least two footprint reads within their Split-ORF-encoding NMD exon in mouse and 19 RBPs in human datasets (Fig. 7g, Extended Data Fig. 7d and Supplementary Table 10). Among those, SRSF5, SRSF6, SRSF7, BCLAF1, HNRNPL, PRPF39 and TIAL1 were found in both species. Our data indicate that PCE location, sequence and Split-ORF coding potential are conserved between mouse and human and widespread among multi-domain RBPs. Split-ORF translation would represent an intriguing mechanism to rescue transcripts from NMD and thereby generate distinct protein isoforms or contribute to the regulation of RBP expression. The individual mechanisms and functions require further studies.
Discussion
We report the discovery of an intricate auto-regulatory feedback loop ensuring protein homeostasis of the essential splicing factor SRSF7. At low levels, SRSF7 binds the SRSF7 pre-mRNA in both exons flanking the PCE and promotes its skipping, thereby producing functional SRSF7 protein (Fig. 8, upper panel). Upon transient OE, SRSF7 binds to splice sites within the PCE and promotes its inclusion. PTC-containing SRSF7-PCE transcripts perform two different functions: normally they are NMD-sensitive and rapidly degraded in the cytoplasm, thereby decreasing the levels of functional SRSF7 protein (Fig. 8, middle panel). However, sustained SRSF7 OE, for example after oncogenic transformation, protects SRSF7-PCE transcripts from NMD. Under such conditions, they become bicistronic mRNAs encoding two SRSF7 protein halves, here termed Split-ORFs. Split-ORF1 is translated into SRSF7_RRM, which acts as a dominant-negative form of SRSF7 (Fig. 8, lower panel). It retains its RNA-binding specificity and nuclear localization but cannot recruit the spliceosome due to its missing RS domain. SRSF7_RRM can easily outcompete full-length SRSF7 as it is not stored in nuclear speckles and therefore readily available. Within SRSF7 pre-mRNA, it inhibits PCE inclusion and promotes retention of introns 3a, 3b and 5. The intron-containing SRSF7-I3+5 transcripts are sequestered in the nucleus and act as architectural RNAs (arcRNAs). Massive binding of SRSF7 and its oligomerization assemble large nuclear bodies at SRSF7 transcription sites (Fig. 8, lower panel), which sequester intron-retained but likely also fully spliced SRSF7 mRNAs. This results in a reduction of translatable SRSF7 transcripts in the cytoplasm and functional SRSF7 protein in the nucleus and ultimately restores normal SRSF7 levels.
Fig. 8. Model of SRSF7 auto-regulation.

Top: at low levels, SRSF7 promotes PCE skipping, producing a functional SRSF7 protein. Middle: upon transient OE, SRSF7 binds to splice sites within the PCE and promotes its inclusion. SRSF7-PCE transcripts are rapidly degraded by NMD in the cytoplasm, and the levels of functional SRSF7 protein decreases. Bottom: sustained SRSF7 OE protects bicistronic SRSF7-PCE transcripts from NMD and translation produces two SRSF7 protein halves (SRSF7_RRM and SRSF7_RS). SRSF7_RRM acts as a dominant-negative form and outcompetes full-length SRSF7, inhibiting PCE inclusion and promoting retention of introns 3a, 3b and 5 instead. Intron-containing SRSF7-I3+5 transcripts now act as arcRNAs, which assemble nuclear bodies at SRSF7 transcription sites via massive binding of SRSF7 and its oligomerization. SRSF7 bodies sequester intron-retained and likely fully spliced SRSF7 mRNAs, resulting in the reduction of functional SRSF7 protein in the nucleus and translatable SRSF7 transcripts in the cytoplasm, ultimately restoring normal SRSF7 levels.
Expression of arcRNAs and sequestration of specific RBPs in nuclear bodies contribute to the regulation of gene expression under stress conditions, oncogenic transformation and disease44–46. So far, all known arcRNAs are long noncoding RNAs (lncRNAs). SRSF7-I3+5 transcripts satisfy all arcRNA requirements defined in a previous study47 and represent the first arcRNA generated from a protein-coding transcript through regulated intron retention. In cancers where SRSF7 is overexpressed, SRSF7 arcRNAs might promote oncogenesis through sequestering other RBPs with similar binding motifs.
ArcRNAs are usually bound by RBPs that contain low-complexity domains, which enable them to oligomerize and bridge multiple RNPs47. Similarly, SRSF7 binds to about 80 regularly spaced binding sites within intron 3 and the PCE. Oligomerization between SRSF7 proteins bound to distinct SRSF7-I3+5 transcripts likely triggers the formation of large nuclear bodies that undergo phase separation35. In line with this, SRSF7 bodies consist of multiple SRSF7-containing RNPs that are held together by weak hydrophobic interactions. Their assembly depends on transcription and their size correlates with the levels of SRSF7-I3+5 and the oligomerization capacity of SRSF7. Differences in the number of hydrophobic RSRSXSXR repeats in SRSF7 isoforms indicate that the oligomerization properties of SRSF7 might be regulated by alternative splicing.
Our experiments further indicate that translation occurs downstream of the PTC in NMD-resistant SRSF7-PCE transcripts and that this contributes to auto-regulation. Ribosomes might fail to terminate at the PTC48 or reinitiate at downstream AUGs49. Alternatively, leaky scanning, ribosome shunting or initiation at an internal ribosomal entry site might cause ribosomes to skip the normal translation start site and initiate at the AUG within the PCE50,51. The detection of an N-terminally truncated SRSF7 peptide (SRSF7_RS-GFP), the conserved START codon in a strong Kozak context (ACCAUGG) and the presence of Ribo-Seq reads at the AUG (Figs. 2e and 7g) indicate that translation initiation might occur downstream of the PTC. Translation of Split-ORF2 resumes the original reading frame and would allow ribosomes to remove all EJCs, thereby rendering SRSF7-PCE transcripts NMD-resistant and allowing for bulk translation of Split-ORF1. Indeed, translational read-through and reinitiation have been shown to rescue transcripts from NMD52–54. Patients with nonsense mutations in genes causing several diseases have been shown to express NMD-resistant PTC-containing transcripts. In all cases, reinitiation at an AUG downstream of the PTC caused the expression of an N-terminally truncated protein, which delayed the symptoms or acted as a dominant-negative variant55–58. Ribosome profiling in human hearts further revealed that translation frequently occurs downstream of PTCs, which might prevent the degradation of transcripts with nonsense mutations42.
Inclusion of the PCE into SRSF7 transcripts precisely separates the RNA-binding module of SRSF7 from its protein-interaction platform. If PCE inclusion only served to introduce PTCs and create unstable NMD transcripts, PCEs could be inserted anywhere in pre-mRNAs. However, a similar gene organization is found in SRSF5 and SRSF6, where PCE inclusion also generates Split-ORFs that separate RRM and RS domains. Moreover, the PCE of SRSF7 is 99% conserved between mouse and human, which is significantly higher than any coding exon of SRSF7 (ref. 15). Regulation of PCE inclusion and translation initiation would require high conservation only around the splice sites, the PTC and the downstream AUG, not within the middle region. However, our data indicate that sequence-specific binding of SRSF7 along the entire PCE is required for proper auto-regulation of SRSF7. Thus, the similar PCE organization for other SR proteins and the ultraconservation of PCE length, sequence and location indicate additional roles, such as the assembly of nuclear bodies.
We provide evidence that the auto-regulatory feedback mechanism of SRSF7 operates under physiological conditions: (1) endogenous genes and transgenes produce the same SRSF7 isoforms and both appear to assemble nuclear bodies. (2) SRSF7 binds strongly to the PCE and intron 3 in WT cells (ref. 16, this study). (3) Introns 3 and 5 are retained in cancer cell lines that overexpress SRSF7 (MCF-7, A549; data not shown). (4) Ribo-Seq reads are detectable in the PCE in mouse hearts, upon virus infection, in human cardiomyocytes and in A549 cells. (5) SRSF7_RRM is detectable in WT cells and accumulates upon UPF1 depletion. Altogether, this indicates that our proposed mechanism operates at low levels in unperturbed cells but is amplified upon persistent OE of SRSF7 or other cellular conditions.
What could be the advantage of such a complex feedback loop with multiple NMD-independent routes? NMD can be globally impaired, for example in cancer cells, requiring NMD-independent regulatory mechanisms to ensure protein homeostasis of critical regulators. Extensive NMD might also result in large amounts of potentially harmful degradation products, which could induce transcriptional compensation of related genes, as recently shown in mouse ES cells59. Moreover, the reversible sequestration of functional SRSF7 transcripts and SRSF7 proteins in nuclear bodies provides a fast way to adjust protein levels when cellular conditions change. The nuclear-retained SRSF7-I3+5 isoforms might be spliced posttranscriptionally to increase protein expression when conditions become more favorable60,61. Ultimately, this regulatory mechanism could also represent an antiviral defense, since several viruses inactivate NMD and hijack SRSF7 for processing and translating their transcripts9,62–65. In line with this, Ribo-Seq reads are present on the PCE in cells infected with coronavirus (Fig. 7g). The accumulation of truncated SRSF7_RRM during viral infection might slow down the production of new viruses.
We discovered that hundreds of annotated NMD target transcripts encode Split-ORFs, among which RBPs and ATP-binding proteins were enriched. Split-ORFs often precisely separate RNA-binding domains from the rest of the protein, offering a simple way to generate dominant-negative protein isoforms or to increase protein diversity. We confirmed the expression of Split-ORFs for other SR proteins by WB and identified Ribo-Seq reads within Split-ORF-encoding NMD exons of ~30 RBP transcripts. This indicates that multiple Split-ORFs are expressed and might contribute to the regulation of gene expression under certain physiological conditions.
Methods
Generation and maintenance of stable BAC P19 cell lines
Murine P19 cells (ECACC 95102107) were purchased from Sigma-Aldrich and grown in DMEM (1×) GlutaMAX medium (ThermoFisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum (ThermoFisher Scientific) and 100 U ml−1 penicillin-streptomycin (ThermoFisher Scientific) on 10-cm cell culture dishes coated with phosphate-buffered saline (PBS, Sigma) containing 0.1% gelatin (Sigma) under humidified 5% CO2 at 37 °C. BACs harboring the complete mouse SRSF7 gene with an inserted GFP tag were mutated by BAC recombineering according to ref. 36 with slight modifications. Mutated BAC DNA was isolated from Escherichia coli DH10 cells using the Nucleobond Xtra Midi EF kit (Macherey-Nagel) and transfected into wild-type P19 cells using Effectene (Qiagen). Stable clonal cell lines were obtained after selection with 500 µg ml−1 Geneticin (G418, ThermoFisher Scientific) and FACS sorting or limited dilution. Cells were tested regularly for mycoplasma.
iCLIP library preparation
Approximately 1 × 107 P19 cells were irradiated once with 150 mJ cm−2 UV light (254 nm) on ice and iCLIP was performed according to ref. 66 with slight modifications. Briefly, crosslinked RNA was digested to lengths of 80–200 nucleotides using RNase I (Ambion) and RNA-protein complexes were immunopurified using Dynabeads Protein G (ThermoFisher Scientific, 10004D) coupled with a goat anti-GFP antibody (provided by D. Drechsel, MPI-CBG) or a rabbit anti-SRSF7 antibody (Assay Biotech C18943). Isolated and purified RNA fragments were ligated to preadenylated DNA 3′ adapters (Integrated DNA Technologies) and reverse transcribed using barcoded RT primers (Supplementary Table 12) and Invitrogen Superscript IV (ThermoFisher Scientific). Complementary DNA fragments were size-selected, circularized using CircLigase II (Epicentre/Lucigen) and relinearized by BamHI HF (New England Biolabs). The final cDNA libraries containing 5′ and 3′ adapters were amplified using AccuPrime SuperMix I (ThermoFisher Scientific) and sequenced on an Illumina HiSeq2000 instrument (single-end 75-nucleotide reads, 20 million reads per replicate).
Ribo-Seq and RNA-seq library preparation
For Ribo-Seq, cells were pretreated with 100 µg ml−1 CHX (Sigma) in culture medium (see above) for 1 min at room temperature and washed with ice-cold PBS containing 100 µg ml−1 CHX. PBS was quickly removed and culture dishes were immediately flash frozen on dry ice. Ribosome profiling experiments were performed as described in ref. 67. The final Ribo-Seq libraries were amplified using Phusion polymerase (New England Biolabs) and sequenced on an Illumina HiSeq2000 instrument (single-end 75-nucleotide reads, 20 million reads per replicate).
For RNA-seq, total RNA was subjected to poly(A)+ selection and Illumina library preparation following standard procedures. RNA-seq libraries were sequenced on an Illumina NEXTSeq500 instrument (single-end 75-nt reads, 50 million reads per replicate).
Analysis of iCLIP, piCLIP, Ribo-Seq and RNA-seq data
Analysis of iCLIP and piCLIP sequencing data was done using the iCount package (http://icount.biolab.si). Briefly, adapters and barcodes were removed from all reads before mapping to the mouse mm9 genome assembly (Ensembl59 annotation) using the Bowtie aligner (v.0.12.7). To determine protein-RNA contact sites, all uniquely mapping reads were used, PCR duplicates were removed and crosslink events (X-links) were extracted (first nucleotide of the read). To determine statistically significant X-links, an FDR < 0.05) was calculated using normalized numbers of input X-links and randomized within cotranscribed regions41,68,69. To obtain comparable numbers of significant binding sites, replicates that correlated well were pooled according to their overall number of X-links.
For motif searching, a z-score analysis for enriched k-mers was performed as described previously41. Sequences surrounding significant X-links were extended in both directions by 30 nucleotides (windows: −30 to −5 nt and 5–30 nt). All occurring k-mers within the evaluated interval were counted and weighted. Then, a control dataset was generated by randomly shuffling 100 times significant X-links within the same genes and a z-score was calculated relative to the randomized genomic positions. The top 15 k-mers were aligned to determine the in vivo binding consensus motif. Sequence logos were produced using WebLogo (http://weblogo.berkeley.edu/logo.cgi).
For quantification of significant X-links in genes, significant X-links were counted into transcript regions using mm9 transcript coordinates (Ensembl59) using the iCount annotate and segment functions, respectively (https://github.com/tomazc/iCount).
RNA-seq reads were trimmed (Cutadapt) and mapped to the mouse genome (mm10 assembly) using STAR70. Aligned reads were counted into genic regions (HTSeq) and normalized to the library size. Ribo-Seq reads were trimmed (Cutadapt), reads mapping to ribosomal RNA were removed and the remaining reads were mapped either to the mouse genome (mm10) using STAR70 or separately to the SRSF7-PCE isoform using Bowtie2 (ref. 71). Sorted .bam files were visualized using the integrated genomics viewer (http://software.broadinstitute.org/software/igv/). Splice junctions on the SRSF7 gene were visualized and counted using the Sashimi plot function.
SRSF7 IP, sample preparation and MS analysis
Truncated and full-length SRSF7 isoforms were purified from WT and OE cells by stringent IPs as follows. Briefly, cells grown on 14-cm culture dishes were washed in ice-cold PBS and pelleted. For stringent IPs 100 µl beads (Dynabeads Protein G, ThermoFisher Scientific, 10004D) were washed twice with 800 µl lysis buffer (100 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 50 mM Tris, pH 7.4) and resuspended in 200 µl lysis buffer. Beads were incubated with 5 µl rabbit IgG α-SRSF7 (Assay Biotech C18943) or 12 µg goat IgG α-GFP antibodies (provided by D. Drechsel, MPI-CBG) on a rotating wheel at 4 °C for 1 h. Rabbit or goat IgGs (Sigma) served as specificity controls. Beads were washed once with 800 µl high-salt buffer (1 M NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM EDTA, 50 mM Tris, pH 7.4), twice with 800 µl lysis buffer and suspended in cell lysates prepared as follows. Cell pellets were resuspended in 1 ml ice-cold lysis buffer supplemented with cOmplete Protease Inhibitor Cocktail (Sigma) and 10 mM β-glycerophosphate (Fluka BioChemica) and sonicated on ice for 30 s (3 pulses of 10 s at 20-s intervals) at 20% amplitude (Branson W-450 D, ThermoFisher Scientific). Lysates were treated with Invitrogen TURBO DNase (ThermoFisher Scientific) for 5 min at 37 °C and were cleared by centrifugation (17,000g, 10 min, 4 °C). Beads were incubated with cell lysates for 1.5 h at 4 °C on a rotating wheel, washed three times with 800 µl high-salt buffer, twice with 800 µl lysis buffer and resuspended in 25 µl Laemmli buffer. Samples were heated at 95 °C for 3 min and beads were separated from eluates on a magnetic rack for 1 min. Eluates were subjected SDS–PAGE, and gels were stained overnight in a colloidal Coomassie solution (0.08% Coomassie Brilliant Blue G250, 10% citric acid, 8% ammonium sulfate, 20% methanol) and destained with distilled water. Protein bands and size-matched empty controls were cut and digested with Trypsin/LysC, LysC or Chymotrypsin (Promega) overnight and analyzed by liquid chromatography–MS.
Plasmids and transfections
The proofreading thermostable ALLin RPH Polymerase (highQu) was used for the amplification of PCR fragments for cloning. Primers are listed in Supplementary Table 12. PCR fragments were subcloned in the pGEM-T Easy Vector (Promega), digested with NheI and KpnI (New England Biolabs) and after purification ligated into the expression vectors pEGFP-N1 (Clontech) or pmCherry-N1 (Clontech). Plasmid DNA was verified by sequencing and used for transfection. WT and SRSF7 OE cells were transfected for 24 h using JetPRIME Transfection Reagent (Polyplus) or Lipofectamine 2000 (Invitrogen) according to the manufacturers’ instructions.
Co-IP, western blotting and antibodies
For Co-IPs, approximately 5 × 107 cells were lysed in NET-2 buffer (150 mM NaCl, 0.05% NP-40, 50 mM Tris, pH 7.5) supplemented with cOmplete Protease Inhibitor Cocktail (Sigma) and 10 mM β-glycerophosphate (Fluka BioChemica) and sonicated on ice for 30 s (see above). Cleared cell lysates were treated with 100 µg ml−1 RNase A for 20 min at 21 °C or left without. 0.2% of lysate served as input. Next, 10 µg of goat IgG (Sigma) or goat α-GFP (provided by D. Drechsel, MPI-CBG) were preincubated with Gammabind G Sepharose beads (GE Healthcare) for 2 h at 4 °C and were then was mixed with equal amounts of untreated or RNase A-treated lysates for 1.5 h at 4 °C. Beads were washed, and coprecipitated proteins were eluted with 2× Laemmli buffer. For Western blotting, proteins were resolved on NuPAGE 4–12% Bis-Tris Gels (ThermoFisher Scientific), blotted onto nylon membranes (EMD Millipore) and probed with the following antibodies: rabbit α–SRSF7 (Assay Biotech C18943), goat α–GFP (MPI-CBG), goat α–NXF1 (Santa Cruz Biotechnology, Inc.), rabbit α–PABPN1 (Abcam), mouse α–glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Biotechnology, Inc.), mouse mAb104 (CRL_2067; ATCC), rabbit α–beta-catenin (Abcam), mouse α–PRP8 (Santa Cruz Biotechnology, Inc.), rabbit α–U170k (Sigma), mouse α–U2AF65 (Santa Cruz Biotechnology, Inc.), rabbit α–UPF1 (Abcam), mouse α–PolII (Cell Signalling), rabbit α–tubulin (Abcam), rabbit α–histone H3 (Abcam), rabbit α–SRSF5 (Merck Millipore), rabbit α–SRSF6 (LSBio) and mouse α–SRSF3 (Sigma-Aldrich). Quantifications were performed using FIJI. Values were normalized to input and bait.
RNA FISH
Custom probes for Malat1, intron 3 and intron 5 were designed using the Stellaris FISH probe designer (www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer) and purchased from Biosearch Technologies. Probes from intron 3 and Malat1 were coupled with Quasar 570 fluorophores, and probes for intron 5 and poly(A)-tails (T20) were coupled with Quasar 670 fluorophores. FISH was performed as recommended by the manufacturer with minor changes. Briefly, cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 20 min at room temperature, washed and permeabilized in 70% (v/v) ethanol for 1 h at 4 °C. FISH probes (~20 nt) were diluted in Stellaris RNA FISH Hybridization Buffer (intron 3/5 at 1:50 and Poly(A)+ at 1:100) and incubated with the cells at 37 °C for 4–16 h. DNA was counterstained with Hoechst 34580 (Sigma) in Wash Buffer A (1:4,000) at 37 °C for 15 min. After washing, cells were dried and mounted on slides using ProLong Diamond Antifade Mountant (ThermoFisher Scientific) and stored at 4 °C until imaging.
Fluorescent bead aggregation assay
HeLa cells were transfected with plasmids expressing GFP, SRSF7-GFP and SRSF7_RRM-GFP using JetOPTIMUS Transfection Reagent (Polyplus) and were subsequently cultured for 20 h. Cells were lysed in NET-2 buffer supplemented with cOmplete Protease Inhibitor Cocktail (Sigma) and 10 mM β-glycerophosphate (Fluka BioChemica) and sonicated on ice for 30 s (see above). 25 µl Dynabeads Protein G (ThermoFisher Scientific, 10004D) were coupled with 2.5 µg goat α–GFP (provided by D. Drechsel, MPI-CBG) in NET-2 buffer while rotating for 2 h at 4 °C. Unbound antibody was removed by washing with NET-2 and high-salt buffer. Cleared lysates were added to the beads and incubated with rotation for 2 h at 4 °C. After washing with NET-2 buffer, the beads were resuspended in PBS and split into three tubes for addition of in vitro transcribed RNAs. RNAs were in vitro transcribed with HiScribe T7 Quick High Yield RNA Synthesis Kit (New England Biolabs), purified, added to the beads at a final concentration of 100 ng µl−1 and incubated with rotation for 10 min at 4 °C. Bead suspensions were diluted 1:10 in PBS, transferred to an eight-well glass chamber slide (Sarstedt) and imaged using a confocal laser microscope (Zeiss LSM780) as Z-stacks. FIJI was used to quantify the sizes of fluorescence bead aggregates using maximum projection and the ‘analyze particles’ option.
Microscope image acquisition and quantification
Images were acquired using a confocal laser-scanning microscope (LSM780; ZEISS) with a Plan-Apochromat ×63 1.4 numerical aperture oil differential interference contrast objective equipped with two photomultiplier tubes and a gallium arsenite phosphate (GaAsP-PMT) detector system. Fluorescence signal was detected with an Argon laser (GFP, 488 nm excitation, Qasar 570–561 nm excitation and Qasar 670–647 nm excitation). Line scans were performed using the line scan and fluorescence measure from ZEN 2012 (black edition v.8.0.5.273; Zeiss) software using the Profile definition tool (arrow) (manually pointed through the target signal) and the results in distance (x axis) in pixels to intensity (y axis) were depicted in graphs.
Fiji was used to process and analyze all acquired images72. Pictures were cropped with the Image crop function and scale bars were added. For the quantification of number, size and fluorescence intensity of SRSF7 bodies, fields with similar numbers of cells were captured using Z-stacks until at least 100 cells were obtained. The BioVoxxel plugin was used to generate a sum of slides image (Sum) from the Intron 3, Intron 5 or Hoechst channel Z-stacks. Intron 3/5 and Hoechst images were merged and the number of bodies per nucleus was counted using the three-dimensional object counter. To obtain body area and fluorescence intensity the Particle analyzer plug in was first used to derive the regions of interest (ROI) from the bodies and subsequently area and fluorescence was quantified using the Area and Integrated density value (mean gray value per pixel × area), respectively. Area and fluorescence values were plotted with GraphPad Prism. For Malat1 colocalization slices from the Intron 5 Z-stacks and MALAT1 channels were merged and at least 100 bodies were investigated for colocalization. For representative images, the ROI from SRSF7 bodies were generated as described above and transferred to the MALAT1 channel. Presence or absence of Malat1 fluorescence within the body ROI was quantified and plotted with GraphPad Prism.
RNA isolation, reverse transcription, reverse transcription PCR and qPCR
Cells were collected and resuspended in TRIzol (ThermoFisher Scientific). RNA was extracted according to the manufacturer’s instructions, treated with TURBO DNase (ThermoFisher Scientific) for 30 min to remove genomic DNA contamination and subsequently purified by ethanol precipitation. For RNA-seq, 2 µg of total RNA were subjected to poly(A)+ selection and library generation using the TRUSeq ILL kit (Illumina). Libraries were sequenced on an Illumina NextSeq instrument (single-end 75 nucleotide reads, 50 million reads, three replicates per condition). For cDNA generation, 1 µg of RNA was reverse transcribed into cDNA using Superscript III and a 1:1 mix of random hexamers and oligo(dT) primers according to the manufacturer’s instructions (ThermoFisher Scientific). Reverse transcription PCRs were performed with Phusion HF Taq DNA Polymerase and qPCRs with the ORA SEE qPCR Green ROX L kit (highQu). All primers are listed in Supplementary Table 12.
Knockdown of UPF1
EsiRNAs specific for UPF1 were designed and generated as described previously29,73. For knockdowns, 5 × 104 P19 cells were seeded in six-well plates, grown until 25% confluency and 2 µg esiRNAs were transfected per well using Lipofectamine 2000 (Life Technologies). An esiRNA against GFP was used as control. Cells were collected after 48 h.
Inhibitor treatments
To block NMD, P19 cells were treated with 100 µg ml−1 CHX for 2 h before collection. To block transcription by RNA Pol II, P19 cells were treated with 5 µg ml−1 actinomycin D (ActD) for 2 h before fixation. A similar concentration of DMSO was added to control cells. To disrupt phase separation, P19 cells were treated with 10% 1,6-hexanediol (1,6-HD) for 5 min before fixation. A similar treatment with 10% 2,5-HD was used as control.
Polysome profiling for PiCLIP
Approximately 2 × 107 P19 cells were treated with 100 µg ml−1 CHX for 5 min to stabilize translating ribosomes and were subsequently irradiated with 150 mJ cm−2 UV light (254 nm) in PBS containing 100 µg ml−1 CHX. The cells were collected by trypsinization and lysed. Cell extracts were fractionated over linear 15–45% sucrose density gradients (prepared in 10 mM HEPES at pH 7.2, 150 mM KCH3CO2, 5 mM MgCl2) by centrifugation at 270,000g for 120 min. Fractions (1 ml each) were collected from the top to the bottom of the gradient and analyzed for absorption at 260 nm and on agarose gels. For RNA analysis, 300 μl of each fraction were extracted with Trizol (ThermoFisher Scientific) and separated on 1.5% agarose gels. For protein analysis, 20 μl of each fraction were mixed with 5× Laemmli buffer, separated on a 4–12% NuPAGE gradient gel (Life Technologies), and analyzed by WB using an α-GFP antibody. For each replicate, fractions 5–6 were pooled for the monosomal fraction and 7–10 for the polysomal fraction. The pooled fractions were separated in two and diluted in iCLIP lysis buffer to a final volume of 50 ml. One half was incubated with Protein G Dynabeads (ThermoFisher Scientific) coupled with a goat α-GFP antibody (D. Drechsel, MPI-CBG) and the other half with goat IgG (Sigma-Aldrich) as control. After IP, iCLIP was performed according to ref. 66. The final cDNA libraries were amplified using Phusion HF Master Mix and sequenced on an Illumina HiSeq2000 instrument (single-end 75-nt reads, 15 million reads per replicate, three replicates).
Subcellular fractionation
Subcellular fractionation was performed using a fractionation kit for Protein and RNA (Abcam) according to the manufacturer’s recommendations with slight modifications. Buffer A was supplemented with 1 U ml−1 RNaseOut for RNA samples and with 1× protease inhibitor and 10 mM β-glycerophosphate (Fluka BioChemica) for protein samples.
Northern blot
We mixed 2–6 µg RNA with 2× RNA sample buffer (Thermo Scientific), preheated it at 60 °C for 10 min and separated it on a denaturing agarose gel (1–3%) supplemented to 5% formaldehyde and 1× MOPS buffer. The RNA integrity was verified by staining total RNA with SYBR Gold (Thermo Scientific) and determined by the 28S:18S rRNA ratio. Blotting was performed overnight in 20× SSC buffer on a 0.1 µm charged nylon membrane (GE Healthcare). After RNA crosslinking (at 120 mJ cm−2), the membranes were preincubated with ULTRA Hybridization buffer (Ambion) for 30 min at 42 °C. RNA probe synthesis was performed by PCR and DIG-dUTP (Roche) incorporation. Denatured probes (95 °C for 5 min) were added to the membranes in hybridization buffer and rotated for 14–24 h at 42 °C. The membranes were washed two times with 2× SSC + 0.1% SDS for 5 min at 42 °C and twice with 0.1× SSC + 0.1% SDS for 15 min at 42 °C. After swirling the membrane in washing buffer (0.3% Tween20 in Maleic acid, pH 7.5), the membrane was blocked (1% blocking reagent in maleic acid, pH 7.5) (Roche) for 30 min at room temperature. To detect the RNA probe, the membrane was incubated with Anti-DIG fab fragment (0.005% in blocking solution) (Roche) for 30 min. After washing two times 15 min with washing buffer, the membrane was incubated with detection solution (100 mM Tris, 150 mM NaCl, 50 mM MgCl2, pH 9.5) (Roche). Then, 0.07% CDP Star in detection buffer was added and chemiluminescence was recorded by a ChemiDoc (Bio-Rad) in High Sensitivity mode.
Split-ORF pipeline, cross-species and Ribo-Seq analyses
A Split-ORF pipeline was developed for the prediction of ORFs resulting from the translation of NMD transcripts. For this, all annotated NMD transcripts were obtained from the Ensembl database (v.95) using Biomart resulting in 6,996 mouse and 16,141 human sequences. First, NMD transcripts were translated in silico from the forward strand only allowing three possible reading frames. Second, all peptides with a minimal length of 50 amino acids were aligned with BlastP using local alignments74 against all proteins of the respective species using Ensembl proteins (v.95). All alignments with an E value ≤10 were retained. Third, all NMD transcripts were kept, which generate at least two peptides that align to the same protein sequence. Predicted ORFs were required to align with at least 50% of their sequence length and with at least 90% sequence identity to the Ensembl proteins. Both filters avoided short spurious ORF matches that were false positives. Fourth, after filtering, NMD transcripts with at least two matching ORFs were tested for encoded protein domains, by overlapping the ORF locations with annotated PFAM domains in Ensembl proteins75. Only PFAM domains that overlapped completely with matching ORFs were considered. To derive orthologous human and mouse genes we used the Biomart (Ensembl v.95). Fisher’s Exact test was used to compute enrichment of a PFAM domain type using all genes with NMD transcripts as background. P values were corrected using FDR in R and an FDR ≤ 0.05 was considered significant. Gene-ontology enrichment of the identified NMD gene sets were conducted using the functional annotation tool of the DAVID webserver (https://david.ncifcrf.gov). Enriched categories with a corrected P ≤ 0.001 (Benjamini–Hochberg) were considered significant.
The Split-ORF prediction pipeline is implemented using python, available under https://github.com/SchulzLab/SplitOrfs.
To count Ribo-Seq reads within unique NMD exons in RBP genes, we downloaded four Ribo-Seq datasets from mouse (SRA IDs: ERR3367717, ERR3367718, ERX1264382) and four from human (SRA IDS: SRR9332878, SRX3849510, ERX3391949, ERX339195) and aligned them using STAR. The junction index was built using the reference genome sequence and Ensembl gene annotations (v.95). Genomic regions of unique NMD exons were intersected with BAM alignments using bedtools76, counting only reads with a minimal alignment overlap of 20% of the read length with the unique NMD exon.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41594-020-0385-9.
Supplementary information
Supplementary Tables 1–6 and Supplementary Table 8.
Human NMD targets of RBPs with encoded Split-ORFs.
Protein domains within Split-ORFs (human and mouse).
Ribo-Seq read counts in unique NMD exons of RBPs (human and mouse).
Acknowledgements
We thank J. Ule and T. Curk for access to the iCOUNT server, A. Dahl for advice and sequencing of the RNA-seq, iCLIP and piCLIP libraries, V. Botti for establishing the piCLIP protocol, I. Poser (MPI-CBG, Dresden) for the BACs encoding NONO- and PSPC1-GFP, R. Rosenkranz for generating stable P19 clonal lines, J. Wöhnert, M. Feldbrügge and D. Staiger for important discussions and mentoring, M. Seiler for NLS predictions, K. Zarnack for excellent comments on the manuscript and all members the Müller-McNicoll laboratory for support. We are grateful for funding from the Deutsche Forschungsgemeinschaft (grant nos. CEF-MC, ECCPS, MU 3915/2-1 and SFB902-2, B13 to M.M.M., CPI to M.H.S. and SFB815-3, Z01 to I.W.).
Extended data
Source data
Uncropped western blots
Statistical source data
Uncropped western blots
Uncropped western blot co-IPs
Statistical source data
Statistical source data
Uncropped western blots
Statistical source data
Uncropped western blots
Statistical source data
Statistical source data
Uncropped western blots
Author contributions
M.M.M., F.M. and M.H.S. designed the experiments and pipelines. F.M., V.K., N.B., B.L.A., M.S., S.L. and C.O.F.M. performed the experiments, I.R.d.l.M., M.H.S., F.M., B.L.A., C.O.F.M. and M.M.M. performed the analyses. Figures were prepared by F.M., V.K., N.B., B.L.A., C.O.F.M. and M.M.M. The manuscript was written by F.M. and M.M.M.
Data availability
All deep sequencing data have been deposited at Gene Expression Omnibus under the accession number GSE142802. The MS data and a detailed method description have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) with the dataset identifiers PXD016871 and PXD016884. Source data for Figs. 1 b,c,g, 2a, 3 e,f, 4a, 5a,c, 6 c,e,f,i,k and 7b,e are available online.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Anke Sparmann was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Vanessa Königs, Camila de Oliveira Freitas Machado, Benjamin Arnold, Nicole Blümel.
Contributor Information
Francois McNicoll, Email: mcnicoll@bio.uni-frankfurt.de.
Michaela Müller-McNicoll, Email: mueller-mcnicoll@bio.uni-frankfurt.de.
Extended data
is available for this paper at 10.1038/s41594-020-0385-9.
Supplementary information
is available for this paper at 10.1038/s41594-020-0385-9.
References
- 1.Müller-McNicoll M, Neugebauer KM. How cells get the message: dynamic assembly and function of mRNA-protein complexes. Nat. Rev. Genet. 2013;14:275–287. doi: 10.1038/nrg3434. [DOI] [PubMed] [Google Scholar]
- 2.Manley JL, Krainer AR. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins) Genes Dev. 2010;24:1073–1074. doi: 10.1101/gad.1934910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Änkö ML. Regulation of gene expression programmes by serine-arginine rich splicing factors. Semin. Cell Dev. Biol. 2014;32:11–21. doi: 10.1016/j.semcdb.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Cavaloc Y, Bourgeois CF, Kister L, Stevenin J. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA. 1999;5:468–483. doi: 10.1017/S1355838299981967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao L, Wang J, Wang Y, Andreadis A. SR protein 9G8 modulates splicing of tau exon 10 via its proximal downstream intron, a clustering region for frontotemporal dementia mutations. Mol. Cell Neurosci. 2007;34:48–58. doi: 10.1016/j.mcn.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldammer G, et al. Characterization of cis-acting elements that control oscillating alternative splicing. RNA Biol. 2018;15:1081–1092. doi: 10.1080/15476286.2018.1502587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tejedor JR, Papasaikas P, Valcarcel J. Genome-wide identification of Fas/CD95 alternative splicing regulators reveals links with iron homeostasis. Mol. Cell. 2015;57:23–38. doi: 10.1016/j.molcel.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 8.Müller-McNicoll M, et al. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 2016;30:553–566. doi: 10.1101/gad.276477.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swartz JE, Bor YC, Misawa Y, Rekosh D, Hammarskjold ML. The shuttling SR protein 9G8 plays a role in translation of unspliced mRNA containing a constitutive transport element. J. Biol. Chem. 2007;282:19844–19853. doi: 10.1074/jbc.M701660200. [DOI] [PubMed] [Google Scholar]
- 10.Fu Y, Wang Y. SRSF7 knockdown promotes apoptosis of colon and lung cancer cells. Oncol Lett. 2018;15:5545–5552. doi: 10.3892/ol.2018.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park WC, et al. Comparative expression patterns and diagnostic efficacies of SR splicing factors and HNRNPA1 in gastric and colorectal cancer. BMC Cancer. 2016;16:358. doi: 10.1186/s12885-016-2387-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saijo S, et al. Serine/arginine-rich splicing factor 7 regulates p21-dependent growth arrest in colon cancer cells. J. Med. Invest. 2016;63:219–226. doi: 10.2152/jmi.63.219. [DOI] [PubMed] [Google Scholar]
- 13.Müller-McNicoll M, Rossbach O, Hui J, Medenbach J. Auto-regulatory feedback by RNA-binding proteins. J. Mol. Cell Biol. 2019;11:930–939. doi: 10.1093/jmcb/mjz043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boguslawska J, et al. microRNAs target SRSF7 splicing factor to modulate the expression of osteopontin splice variants in renal cancer cells. Gene. 2016;595:142–149. doi: 10.1016/j.gene.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 15.Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007;446:926–929. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- 16.Pervouchine D, et al. Integrative transcriptomic analysis suggests new autoregulatory splicing events coupled with nonsense-mediated mRNA decay. Nucleic Acids Res. 2019;47:5293–5306. doi: 10.1093/nar/gkz193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Änkö ML, et al. The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol. 2012;13:R17. doi: 10.1186/gb-2012-13-3-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balistreri G, Bognanni C, Muhlemann O. Virus escape and manipulation of cellular nonsense-mediated mRNA decay. Viruses. 2017;9:24. doi: 10.3390/v9010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fiorini F, et al. HTLV-1 Tax plugs and freezes UPF1 helicase leading to nonsense-mediated mRNA decay inhibition. Nat. Commun. 2018;9:431. doi: 10.1038/s41467-017-02793-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol. Cell Biol. 2008;28:3729–3741. doi: 10.1128/MCB.02284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Vuong JK, Zhang M, Stork C, Zheng S. Inhibition of nonsense-mediated RNA decay by ER stress. RNA. 2017;23:378–394. doi: 10.1261/rna.058040.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D, et al. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol. Cell Biol. 2011;31:3670–3680. doi: 10.1128/MCB.05704-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun S, Zhang Z, Sinha R, Karni R, Krainer AR. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat. Struct. Mol. Biol. 2010;17:306–312. doi: 10.1038/nsmb.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahler AM, Neugebauer KM, Stolk JA, Roth MB. Human SR proteins and isolation of a cDNA encoding SRp75. Mol. Cell Biol. 1993;13:4023–4028. doi: 10.1128/MCB.13.7.4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YK, Maquat LE. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA. 2019;25:407–422. doi: 10.1261/rna.070136.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botti V, et al. Cellular differentiation state modulates the mRNA export activity of SR proteins. J. Cell Biol. 2017;216:1993–2009. doi: 10.1083/jcb.201610051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mo S, Ji X, Fu XD. Unique role of SRSF2 in transcription activation and diverse functions of the SR and hnRNP proteins in gene expression regulation. Transcription. 2013;4:251–259. doi: 10.4161/trns.26932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lai MC, Lin RI, Tarn WY. Transportin-SR2 mediates nuclear import of phosphorylated SR proteins. PNAS. 2001;98:10154–10159. doi: 10.1073/pnas.181354098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fox AH, Lamond AI. Paraspeckles. Cold Spring Harb. Perspect. Biol. 2010;2:a000687. doi: 10.1101/cshperspect.a000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.An H, Tan JT, Shelkovnikova TA. Stress granules regulate stress-induced paraspeckle assembly. J. Cell Biol. 2019;218:4127–4140. doi: 10.1083/jcb.201904098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maharana S, et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science. 2018;360:918–921. doi: 10.1126/science.aar7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kroschwald S, et al. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible RNP granules. ife. 2015;4:e06807. doi: 10.7554/eLife.06807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin YH, Forman-Kay JD, Chan HS. Sequence-specific polyampholyte phase separation in membraneless organelles. Phys. Rev. Lett. 2016;117:178101. doi: 10.1103/PhysRevLett.117.178101. [DOI] [PubMed] [Google Scholar]
- 34.Yamazaki T, et al. Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell. 2018;70:1038–1053 e1037. doi: 10.1016/j.molcel.2018.05.019. [DOI] [PubMed] [Google Scholar]
- 35.Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science357, 10.1126/science.aaf4382 (2017). [DOI] [PubMed]
- 36.Wang H, et al. Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res. 2014;42:e37. doi: 10.1093/nar/gkt1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popielarz M, Cavaloc Y, Mattei MG, Gattoni R, Stevenin J. The gene encoding human splicing factor 9G8. Structure, chromosomal localization, and expression of alternatively processed transcripts. J. Biol. Chem. 1995;270:17830–17835. doi: 10.1074/jbc.270.30.17830. [DOI] [PubMed] [Google Scholar]
- 38.Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip. Rev. RNA. 2012;3:1–12. doi: 10.1002/wrna.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krchnakova Z, et al. Splicing of long non-coding RNAs primarily depends on polypyrimidine tract and 5′ splice-site sequences due to weak interactions with SR proteins. Nucleic Acids Res. 2019;47:911–928. doi: 10.1093/nar/gky1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Nostrand EL, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP) Nat. Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, et al. iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol. 2010;8:e1000530. doi: 10.1371/journal.pbio.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Heesch S, et al. The translational landscape of the human heart. Cell. 2019;178:242–260 e229. doi: 10.1016/j.cell.2019.05.010. [DOI] [PubMed] [Google Scholar]
- 43.Clamer M, et al. Active ribosome profiling with RiboLace. Cell Rep. 2018;25:1097–1108 e1095. doi: 10.1016/j.celrep.2018.09.084. [DOI] [PubMed] [Google Scholar]
- 44.Goenka A, et al. Human satellite-III non-coding RNAs modulate heat-shock-induced transcriptional repression. J. Cell Sci. 2016;129:3541–3552. doi: 10.1242/jcs.189803. [DOI] [PubMed] [Google Scholar]
- 45.Pettersson OJ, Aagaard L, Jensen TG, Damgaard CK. Molecular mechanisms in DM1—a focus on foci. Nucleic Acids Res. 2015;43:2433–2441. doi: 10.1093/nar/gkv029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yap K, et al. A short tandem repeat-enriched RNA assembles a nuclear compartment to control alternative splicing and promote cell survival. Mol. Cell. 2018;72:525–540 e513. doi: 10.1016/j.molcel.2018.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chujo T, Yamazaki T, Hirose T. Architectural RNAs (arcRNAs): a class of long noncoding RNAs that function as the scaffold of nuclear bodies. Biochim. Biophys. Acta. 2016;1859:139–146. doi: 10.1016/j.bbagrm.2015.05.007. [DOI] [PubMed] [Google Scholar]
- 48.Arribere JA, et al. Translation readthrough mitigation. Nature. 2016;534:719–723. doi: 10.1038/nature18308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gunisova S, Hronova V, Mohammad MP, Hinnebusch AG, Valasek LS. Please do not recycle! translation reinitiation in microbes and higher eukaryotes. FEMS Microbiol. Rev. 2018;42:165–192. doi: 10.1093/femsre/fux059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haimov O, Sinvani H, Dikstein R. Cap-dependent, scanning-free translation initiation mechanisms. Biochim. Biophys. Acta. 2015;1849:1313–1318. doi: 10.1016/j.bbagrm.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 51.Hinnebusch AG. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011;75:434–467. doi: 10.1128/MMBR.00008-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cohen S, et al. Nonsense mutation-dependent reinitiation of translation in mammalian cells. Nucleic Acids Res. 2019;47:6330–6338. doi: 10.1093/nar/gkz319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neu-Yilik G, et al. Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA. 2011;17:843–854. doi: 10.1261/rna.2401811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, Maquat LE. Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J. 1997;16:826–833. doi: 10.1093/emboj/16.4.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lopez-Granados E, et al. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum. Mutat. 2008;29:861–868. doi: 10.1002/humu.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paulsen M, et al. Evidence that translation reinitiation leads to a partially functional Menkes protein containing two copper-binding sites. Am. J. Hum. Genet. 2006;79:214–229. doi: 10.1086/505407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stalke A, et al. Homozygous frame shift variant in ATP7B exon 1 leads to bypass of nonsense-mediated mRNA decay and to a protein capable of copper export. Eur. J. Hum. Genet. 2019;27:879–887. doi: 10.1038/s41431-019-0345-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stump MR, Gong Q, Packer JD, Zhou Z. Early LQT2 nonsense mutation generates N-terminally truncated hERG channels with altered gating properties by the reinitiation of translation. J. Mol. Cell Cardiol. 2012;53:725–733. doi: 10.1016/j.yjmcc.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.El-Brolosy MA, et al. Genetic compensation triggered by mutant mRNA degradation. Nature. 2019;568:193–197. doi: 10.1038/s41586-019-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boutz PL, Bhutkar A, Sharp PA. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015;29:63–80. doi: 10.1101/gad.247361.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ninomiya K, Kataoka N, Hagiwara M. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J. Cell Biol. 2011;195:27–40. doi: 10.1083/jcb.201107093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Escudero-Paunetto L, Li L, Hernandez FP, Sandri-Goldin RM. SR proteins SRp20 and 9G8 contribute to efficient export of herpes simplex virus 1 mRNAs. Virology. 2010;401:155–164. doi: 10.1016/j.virol.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jacquenet S, Decimo D, Muriaux D, Darlix JL. Dual effect of the SR proteins ASF/SF2, SC35 and 9G8 on HIV-1 RNA splicing and virion production. Retrovirology. 2005;2:33. doi: 10.1186/1742-4690-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maciolek NL, McNally MT. Serine/arginine-rich proteins contribute to negative regulator of splicing element-stimulated polyadenylation in rous sarcoma virus. J. Virol. 2007;81:11208–11217. doi: 10.1128/JVI.00919-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Valente ST, Gilmartin GM, Venkatarama K, Arriagada G, Goff SP. HIV-1 mRNA 3′ end processing is distinctively regulated by eIF3f, CDK11, and splice factor 9G8. Mol. Cell. 2009;36:279–289. doi: 10.1016/j.molcel.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huppertz I, et al. iCLIP: protein-RNA interactions at nucleotide resolution. Methods. 2014;65:274–287. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.König J, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yeo GW, et al. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Surendranath V, Theis M, Habermann BH, Buchholz F. Designing efficient and specific endoribonuclease-prepared siRNAs. Meth. Mol. Biol. 2013;942:193–204. doi: 10.1007/978-1-62703-119-6_11. [DOI] [PubMed] [Google Scholar]
- 74.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinf. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Finn RD, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables 1–6 and Supplementary Table 8.
Human NMD targets of RBPs with encoded Split-ORFs.
Protein domains within Split-ORFs (human and mouse).
Ribo-Seq read counts in unique NMD exons of RBPs (human and mouse).
Data Availability Statement
All deep sequencing data have been deposited at Gene Expression Omnibus under the accession number GSE142802. The MS data and a detailed method description have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (http://www.ebi.ac.uk/pride) with the dataset identifiers PXD016871 and PXD016884. Source data for Figs. 1 b,c,g, 2a, 3 e,f, 4a, 5a,c, 6 c,e,f,i,k and 7b,e are available online.