

Abstract
Asthma is the most common inflammatory disease of the lungs. The prevalence of asthma is increasing in many parts of the world that have adopted aspects of the Western lifestyle, and the disease poses a substantial global health and economic burden. Asthma involves both the large-conducting and the small-conducting airways, and is characterized by a combination of inflammation and structural remodelling that might begin in utero. Disease progression occurs in the context of a developmental background in which the postnatal acquisition of asthma is strongly linked with allergic sensitization. Most asthma cases follow a variable course, involving viral-induced wheezing and allergen sensitization, that is associated with various underlying mechanisms (or endotypes) that can differ between individuals. Each set of endotypes, in turn, produces specific asthma characteristics that evolve across the lifecourse of the patient. Strong genetic and environmental drivers of asthma interconnect through novel epigenetic mechanisms that operate prenatally and throughout childhood. Asthma can spontaneously remit or begin de novo in adulthood, and the factors that lead to the emergence and regression of asthma, irrespective of age, are poorly understood. Nonetheless, there is mounting evidence that supports a primary role for structural changes in the airways with asthma acquisition, on which altered innate immune mechanisms and microbiota interactions are superimposed. On the basis of the identification of new causative pathways, the subphenotyping of asthma across the lifecourse of patients is paving the way for more-personalized and precise pathway-specific approaches for the prevention and treatment of asthma, creating the real possibility of total prevention and cure for this chronic inflammatory disease.
Supplementary information
The online version of this article (doi:10.1038/nrdp.2015.25) contains supplementary material, which is available to authorized users.
Subject terms: Asthma, Respiratory tract diseases, Allergy, Inflammation
Asthma is a chronic inflammatory disease of the airways. Here, Holgate and colleagues outline the need for a better mechanistic understanding of the origins and heterogeneity of asthma, and discuss how this is leading a move towards more-personalized and targeted treatments.
Supplementary information
The online version of this article (doi:10.1038/nrdp.2015.25) contains supplementary material, which is available to authorized users.
Introduction
The 2015 Global Strategy for Asthma Management and Prevention by the Global Initiative for Asthma (GINA) defined asthma as a heterogeneous disease characterized by chronic airway inflammation and variable remodelling that results in a range of clinical presentations, treatment responses and natural history across the lifecourse of the patient1. Asthma involves a history of respiratory symptoms — including wheeze, shortness of breath, chest tightness and cough — that vary over time and in intensity, variable expiratory airflow limitation and airway hyper-responsiveness to a range of stimuli, such as exercise and inhaled irritants. At the population level, a subset of individuals with asthma exhibit an accelerated decline in lung function over their lifetime2, which, in severe chronic disease, manifests as fixed airflow obstruction. This decline is especially prominent in late-onset asthma3. The origin and severity of asthma are driven by strong genetic and environmental factors. Although most cases of asthma begin in childhood in association with IgE-dependent sensitization to common environmental allergens4, asthma can also emerge later in life. Adult-onset asthma often occurs in the absence of allergy but can be accompanied by intolerance to NSAIDs, rhinosinusitis and nasal polyps5. Intolerance to NSAIDs most likely results from reduced production of the anti-bronchoconstrictor prostaglandin E2 under conditions of inflammation.
Asthma is often accompanied by co-morbidities including multi-organ allergies, such as allergic rhinitis, conjunctivitis, atopic dermatitis and food allergy, as well as non-allergic disorders, such as obesity, gastro-oesophageal reflux and psychiatric conditions6. Asthma is subject to periods of rapid deterioration (or exacerbations) that are provoked by viral infection and exposure to allergens, air pollutants and certain drugs such as aspirin and other NSAIDs7. In addition, certain types of asthma can enter spontaneous remission (that is, patients become symptom-free), such as during late childhood and adolescence8, and can respond to allergen-specific immunotherapy through the acquisition of immunological tolerance9.
In both adults and children, asthma has been traditionally classified by either symptom severity or the extent of disease control achieved using a stepwise management process, in which patients are grouped into one of four or five categories that are used to determine treatment requirements with controller drugs. These drugs include inhaled corticosteroids (ICSs), long-acting β2-adrenergic receptor agonists (LABAs), long-acting muscarinic antagonists, leukotriene receptor antagonists (LTRAs) and, for the most severe disease, the IgE-specific monoclonal antibody omalizumab1. Although this stepwise approach has improved the management of asthma and reduced dependency on inhaled short-acting bronchodilators (SABAs) for symptom relief, none of these treatments have been shown to alter the natural history of the disease10,11. Cluster and other non-hierarchical analyses have identified subtypes of asthma associated with differing causal pathways, natural histories and responses to interventions12 (Fig. 1). In this Primer, we discuss the epidemiology, origins, pathophysiology, diagnosis and treatment of asthma with specific reference to disease heterogeneity and stratification according to causal pathways, and show how this is shaping a new personalized or stratified approach to treatment.
Figure 1. Selected asthma subphenotypes.
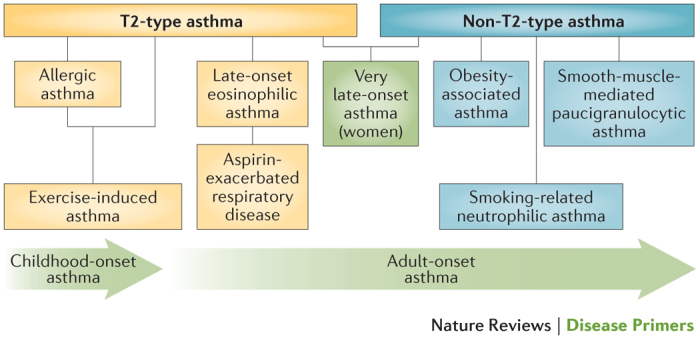
New subphenotypes and associated causal pathways, or endotypes, of asthma are being discovered through the application of non-hierarchical statistical analyses of clinical, physiological and laboratory characteristics. Figure from Ref. 163, Nature Publishing Group.
Epidemiology
There are >300 million people in the world who are affected by asthma, making it one of the most common chronic diseases13. Although the prevalence of asthma is greatest in countries with a high gross domestic product, the disease is recognized worldwide. In the lowest-income and most rural countries14, the prevalence of asthma tends to be ≤1%, far lower than the 10% usually seen in developed western countries (Fig. 2). Within populations of a given gross domestic product, the prevalence of asthma follows an urban–rural gradient and a weak latitudinal gradient, that is, there is greater disease prevalence with greater distance from the equator and asthma is more common in urban areas14. Despite the low prevalence of asthma in low-income and middle-income countries, underdiagnosis and misdiagnosis together with inadequate treatment in these regions leads to considerable, and potentially avoidable, disease morbidity and mortality.
Figure 2. Changing trends in the prevalence of asthma according to gross domestic product.
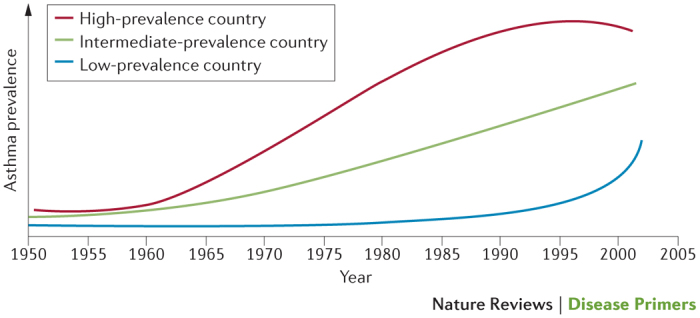
The prevalence of asthma has plateaued in recent decades in high-prevalence countries, which have a high gross domestic product (GDP). Conversely, there has been a steep increase in the prevalence of asthma in low-prevalence and intermediate-prevalence countries, which have low-ranking and middle-ranking GDPs, respectively. These steep increases have occurred alongside acquisition aspects of the western lifestyle in these countries254. Reprinted from Bulletin of World Health Organisation, 83, Bousquet, J., Bousquet, P. J., Godard, P. & Dyres, J.-P., The public health implications of asthma, 548–554, Copyright (2005).
The prevalence of asthma has increased in many parts of the world over the past few decades, and, until recently, asthma prevalence was increasing on a year-by-year basis in developed western countries (Fig. 2). The cause of the epidemic that began in the late 1970s is unclear, but the rise in asthma prevalence is consistent with a rise in other immune-mediated diseases, such as type 1 diabetes mellitus, inflammatory bowel disease and multiple sclerosis. Recent epidemiological research has focused on changes in maternal diet during pregnancy — particularly on the levels of micronutrients such as ω-3 fatty acids, folate and vitamin D (the latter two modifying methylation), and hence fetal programming — along with the gut and airway microbiota, prematurity and maternal paracetamol use during pregnancy15.
Immunological factors, age and sex all influence the development of asthma. The disease is closely linked to the presence of immediate hypersensitivity, and 50% of children who are diagnosed with asthma by 3 years of age and 80% of those diagnosed by the time they are 6 years of age are atopic — that is, they are genetically predisposed to allergic hypersensitivity16. The prevalence of wheezing exceeds the prevalence of asthma in children up to 6 years of age. This observation suggests that factors other than asthma, such as physician diagnostic bias and lower respiratory tract infections, can drive the onset and persistence of wheezing. Asthma is not constant across the lifecourse of the patient, and patients can experience periods of remission and the onset of new asthma17. Whereas asthma is more common in boys than girls in early childhood, throughout puberty and early adulthood, boys experience asthma remission at a higher rate than in girls. In addition, girls acquire asthma more often than boys in this age period. Consequently, the sex ratio of asthma during childhood reverses in adolescence and in young adulthood16,18,19 (Fig. 3). The reasons for this variability in asthma across the lifecourse of the patient are not clear, but evidence is mounting in support of a key role for hormones in this process. Furthermore, asthma remission during adolescence is associated with lower initial airway hyper-responsiveness and greater gain in the function of small airways compared with asthma that begins after childhood18. The reasons for this variability between early-onset and late-onset asthma are probably complex, but differing environmental exposures (exposome) — including those that occur in occupational settings — are thought to be important20.
Figure 3. Proportions of children and adolescents with asthma in the Isle of Wight birth cohort.
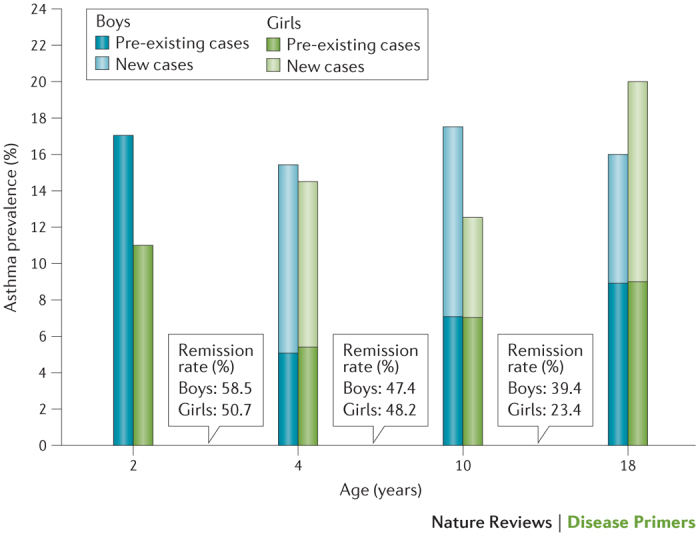
Bars represent the proportion of either boys or girls at particular ages who have asthma, with light-shaded sections representing the proportion of new cases that did not carry over from the previous age group (positive transition); the boxes between bars indicate the percentage of children who grew out of asthma in the intervening years (negative transition or remission). Data from Ref. 18.
The heterogeneity of asthma can pose challenges for epidemiological research. Cross-sectional studies of asthma can be difficult to interpret given that both recall bias and the fact that most patients with asthma will have had a varied disease course. As such, at any one time, individuals at different stages of their disease and with different pathophysiological mechanisms for their asthma might be affected. The exception to this is severe asthma, which usually has an onset in early childhood, is associated with multi-allergen sensitization21 and persists across the lifecourse22. In addition, new asthma in older adults tends to be more severe from the onset than asthma that develops in younger age groups23. Finally, asthma might co-occur with chronic obstructive pulmonary disease (COPD) to cause asthma COPD overlap syndrome1. The prevalence of this overlap is substantial.
Mechanisms/pathophysiology
Pathophysiology
Airway inflammation. Airway inflammation is a prominent feature of asthma (Fig. 4). T2-type inflammation occurs in >80% of children and in the majority of adults with asthma in association with sensitization to environmental allergens, such as those from dust mites, fungi, pets and pollens24–26. This sensitization is often associated with other clinical manifestations of atopy such as atopic dermatitis (eczema), allergic rhinoconjunctivitis and food allergy. The inflammatory infiltrate that accompanies T helper 2 (TH2) lymphocyte responses is mainly composed of eosinophils but also includes mast cells, basophils, neutrophils, monocytes and macrophages27. Cellular activation and release of inflammatory mediators in asthma is evidenced by mast cell degranulation and eosinophil vacuolation. The majority of mucosal mast cells in mild-to-moderate allergic-type asthma are of the TH2 cell-dependant tryptase-expressing type (MCT)28. In the more intractable forms of asthma, mast cells containing both tryptase and chymase (MCTC) predominate, which are more dependent on stem cell factor (also known as KIT ligand) for their survival than are MCT cells29,30.
Figure 4. Histopathology of the asthmatic airway.
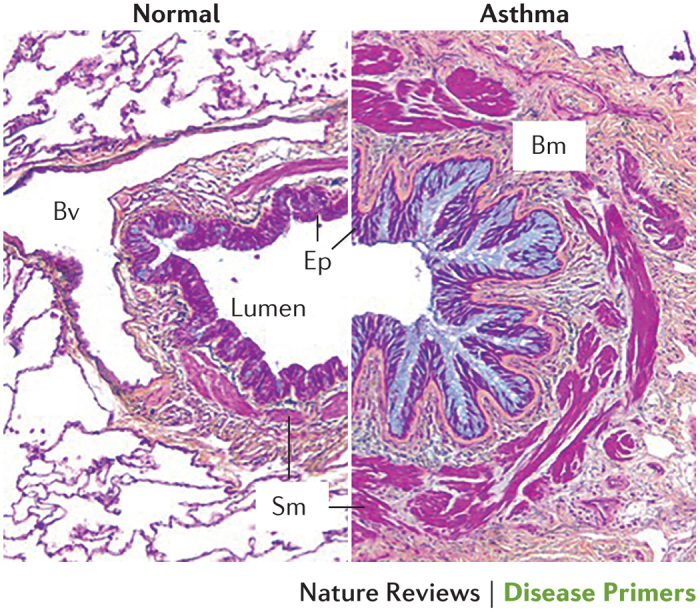
Cross section of a severe asthmatic airway (right) compared with a normal airway (left). Asthma involves mucosal inflammation that most frequently consists of activated eosinophils, mast cells and T lymphocytes within the context of a remodelled airway with mucous metaplasia, an increase in smooth muscle (Sm), fibrosis and angiogenesis. Bm, basement membrane; Bv, blood vessel; Ep, epithelium. Republished with permission of Dove Medical Press, from Clinical update on the use of biomarkers of airway inflammation in the management of asthma. Wadsworth, S., Sin, D. & Dorscheid, D., 4, 2011; permission conveyed through Copyright Clearance Center, Inc.
The principal role of T cells in asthmatic airways is in controlling the inflammatory cell profile31. Whereas the activity of TH2 CD4+ lymphocytes predominate in classic allergic-type asthma32, roles for a range of other T cells in different asthma subtypes have been described, including the association of TH1 cells33 and TH17 cells34 with neutrophilic asthma (Fig. 5). In eosinophilic allergic asthma35, and potentially non-allergic asthma36, the initiation of T2-type immune responses occurs through secretion of the epithelial cell-derived cytokines IL-25, IL-33 and thymic stromal lymphopoietin (TSLP). These cytokines induce a new innate lymphoid subset (nuocytes, a type of group 2 innate lymphoid cells (ILC2s)) to produce the T2-type cytokines IL-5, IL-9 and IL-13 (Ref. 37) (Fig. 6).
Figure 5. Involvement of various T cell subtypes in asthma pathogenesis and asthma endotypes.

Dendritic cells (DCs) and the thymic epithelium together trigger the immune response that drives the development of asthma. In response to a combination of signals transmitted by cytokines and direct contact, the DCs and thymic epithelium promote the differentiation of an array of different leukocyte subsets (inner, yellow circle). Before differentiation, these subsets either augment or protect the airways from inflammatory responses linked to asthma, whereas following differentiation, they generate cytokines that promote the development of asthma. These cytokines influence various different cell types and the attendant inflammatory responses to drive allergic airway inflammation and airway hyper-responsiveness. GM-CSF, granulocyte–macrophage colony-stimulating factor; ILC2, group 2 innate lymphoid cell; iTReg, inhibitory regulatory T; NKT, natural killer T; TGFβ, transforming growth factor-β TH, T helper; TNF, tumour necrosis factor. Figure from Ref. 31, Nature Publishing Group.
Figure 6. Allergen sensitization of the airways during the induction of allergic-type asthma.

Infection (bacterial and viral) and pollutants perturb the airway epithelium, which leads to initial danger signalling and activation of innate signalling receptors. This signalling causes airway epithelial cells (ECs) to secrete chemokines and leads to trafficking of immature dendritic cells (DCs) to the mucosal epithelium. These DCs respond to danger signals through pattern recognition receptors (PRRs), which leads to their maturation into competent antigen-presenting myeloid-type DCs. Allergen detection and processing by these activated DCs is mediated by the extension of cellular processes into the airways or by the capture of allergens that have breached the epithelium. Allergen-loaded DCs then drive T cell differentiation by migrating to local lymph nodes where they interact with naive T cells (TN) via the T cell receptor (TCR), major histocompatibility complex (MHC) class II and co-stimulatory molecules. DC activation and T helper 2 (TH2) cell maturation and migration into the mucosa are influenced by additional epithelial-derived cytokines and chemokines, including IL-25, IL-33, CC-chemokine ligand 17 (CCL17) and CCL22. CCR, CC-chemokine receptor; GM-CSF, granulocyte–macrophage colony-stimulating factor; mDC, mucosal DC; TNF, tumour necrosis factor; TSLP, thymic stromal lymphopoietin. Figure from Ref. 31, Nature Publishing Group.
Allergen sensitization also requires an interaction between specialized antigen-presenting airway dendritic cells (DCs) and T cells. This mechanism involves processing of allergen into small peptides and the selective major histocompatibility complex (MHC) class II presentation of these processed peptides to the T cell receptors of naive T cells31 (Fig. 6). Effective allergen signalling also requires co-stimulatory interactions between DCs and T cells38 that take place in local lymphoid collections39, resulting in T cell differentiation into TH2-type T cells. These TH2-type T cells secrete the pro-allergic cytokines, IL-3, IL-4, IL-5, IL-9, IL-13 and granulocyte–macrophage colony-stimulating factor (GM-CSF), which in turn leads to the IgE, mast cell and eosinophilic responses that are characteristic of allergic asthma40 (Fig. 5). Many of the asthma-related allergens — such as those from dust mite, cockroach, animal and fungal sources — exhibit enzymatic properties that enable them to penetrate the epithelial barrier and directly interact with mucosal DCs41 (Fig. 6). During this time, quiescent DCs transform to express an array of cell adhesion and co-stimulatory molecules. These molecules are recognized by naive T cells, which interact with DCs to create an immunological synapse that facilitates allergen presentation42. Whereas a minority of allergen-specific TH2 cells migrate to the B cell follicle to initiate immunoglobulin class switching from IgM to IgE43, others relocate to the airway mucosa, under the influence of chemoattractants, to elicit the T2-type inflammatory response and the associated coordinated secretion of pro-allergic cytokines44.
Once sensitized, further exposure of the airways to allergen results in a mast-cell-driven early-type bronchoconstrictor response (EAR) that lasts for 5–90 minutes and involves IgE-dependent release of histamine, prostaglandin D2 and the leukotriene C4 (LTC4), which is subsequently converted to LTD4 and LTE4 (Ref. 45) (Fig. 5). The EAR is followed by a late-phase response (LAR) that evolves over 3–12 hours, and is linked to infiltration and activation of leukocytes (especially eosinophils) with further LTC4 generation, TH2 cytokine release from mast cells and T cells46 and an increase in airway responsiveness47. Although EAR and LAR have been studied in humans and animal models to dissect the allergic mechanisms of asthma, these two responses are not accompanied by the chronic persistent inflammation that characterizes most cases of asthma. Consequently, EAR and LAR do not provide an adequate mechanistic setting within which to investigate exacerbations other than those triggered by allergen exposure48.
Although much of the focus in asthma pathophysiology has been on positive drivers of inflammation, the defective resolution of inflammation is emerging as a mechanism that might also be involved in asthma. The failure to adequately downregulate the inflammatory response could result in the prolonged survival of mast cells and eosinophils as a result of the cytokine milieu of the asthmatic airway. In addition, an important new paradigm in asthma pathophysiology is the potential role of lipoxins and resolvins as mediators of the endogenous resolution of inflammation. For instance, lipoxin A4 can induce apoptosis of eosinophils and decrease the activity of ILC2s and natural killer lymphocytes, and the production of lipoxin A4 is reduced in asthmatic airways49.
Airway remodelling. In asthma, the airway wall thickens in proportion to disease severity and duration50,51. This remodelling involves an increase in airway smooth muscle, thickening of the subepithelial reticular lamina, matrix deposition throughout the airway wall, angiogenesis, neuronal proliferation and epithelial mucous metaplasia — a process that involves the appearance of mucous cells in new areas of the airways and increased production of mucus (Fig. 4). These events are thought to underlie airway hyper-responsiveness, whereas mucus forms plugs that can extend into the small airways and lead to air trapping and hyperinflation52. In addition, epithelial goblet cell metaplasia results from the actions of IL-4, IL-9 and IL-13, as well as from the secretion of growth factors such as members of the epidermal growth factor family, which cause epithelial cell stress and injury53.
Epithelial damage results from the separation of columnar cells from basal cells. This can be detected by staining of sputum from patients with asthma, showing detached columnar cells as Creola bodies. Thickening of the subepithelial basement membrane is confined to the reticular lamina and results from deposition of ‘repair-type’ collagens I, III, V and VI together with periostin, tenascin, osteopontin and fibronectin54–56. Subepithelial collagen is produced by a sheath of myofibroblasts that lie beneath the epithelium. An epithelial–mesenchymal trophic unit, located between the epithelial and smooth muscle layers of the airway, is established in response to epithelial cell injury57 (Fig. 7). Within the epithelial–mesenchymal trophic unit, the epithelium is a potent source of growth factors, including functionally active periostin33, platelet-derived growth factors58, fibroblast growth factors and members of the transforming growth factor-β (TGFβ) family. In addition, the epithelium is a source of members of the epidermal growth factor family59 — which are capable of driving both fibrosis and smooth muscle proliferation — as well as neurotrophins60 and angiogenic factors, such as vascular endothelial growth factors61. Together, these growth factors promote the neuronal and microvascular proliferation that accompany airway remodelling62. Enhanced growth factor production occurs as a direct consequence of epithelial injury and delayed repair, and in this respect resembles a chronic wound scenario63,64. Finally, migration of subepithelial microvascular pericytes65 and proliferation of fibroblasts followed by their differentiation into myofibroblasts contribute to mucosal fibrosis, muscle hyperplasia54,66 and the reduction in distance between airway smooth muscle cells and the epithelium67.
Figure 7. Epithelial–mesenchymal trophic unit in asthma.
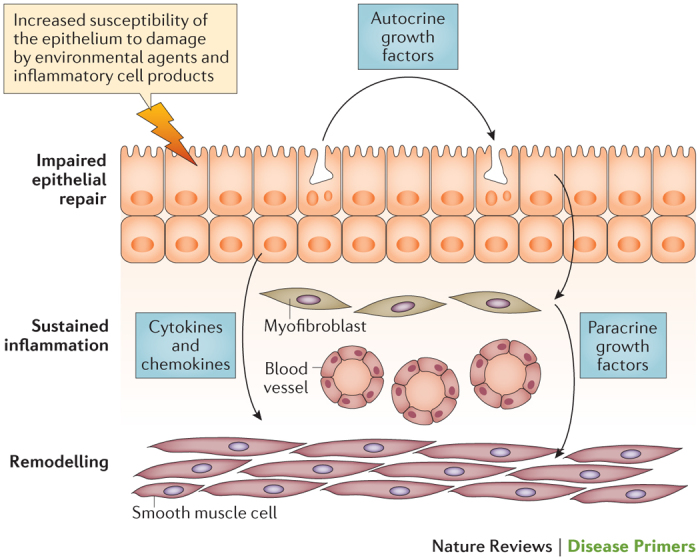
In chronic moderate-to-severe asthma, the behaviour of the epithelium resembles that in chronic wound scenarios: it is more susceptible to environmental and viral injury than usual and exhibits impaired repair. In addition, the epithelium no longer undergoes healing by ‘primary intention’ but instead undergoes ‘secondary intention’ — a process that involves the production of growth factors that drive remodelling responses in the underlying airway wall. Similarly, the damaged and stimulated epithelium generates growth factors that contribute to goblet cell metaplasia. The augmented communication between the epithelium and the underlying mesenchyme resembles the activation of the epithelium–mesenchymal trophic unit that drives airway morphogenesis in the developing fetal lung. The resulting cytokine milieu also provides a favourable environment for sustaining chronic inflammation. This research was originally published in Clin. Sci. (Lond.). Holgate, S. T., Arshad, H. S., Roberts, G. C., Howarth, P. H., Thurner, P. & Davies, D. E., A new look at the pathogenesis of asthma. Clin. Sci. (Lond.). 2009; 118: 439–450 © Portland Press.
Disease onset
Childhood viral illness and lung function. A key trigger for the onset of asthma in children is severe wheezing in early life in response to viral infections, especially respiratory infection with syncytial virus (RSV) or rhinovirus. A second trigger is the emergence and then persistence of a T2-type allergic immune response in the airways (Fig. 8). In the first 2 years of life, all children become infected with RSV and rhinovirus68, so the question is not whether infection is a causal factor in the onset of asthma, but whether there is an underlying developmental defect of the lungs and/or the innate immune system that confers asthma susceptibility.
Figure 8. Asthma as a developmental disease.

Asthma is caused by failure of the respiratory and immune systems to develop normally. This schematic represents asthma risk factors that operate at different stages of life. Asthma risk at birth is influence by genetic predispositions, impaired lung function and delayed immune maturation. Postnatal risk factors that increase asthma risk include reduced lung growth resulting in low lung function, the timing of acquisition of specific components of the pulmonary microbiota, repeated episodes of viral upper respiratory tract infections (URTIs) that spread to the lower airway and result in severe lower respiratory tract infections (LRTIs), maturational deficiencies in the innate and adaptive immune systems that increase the risk of severe LRTIs and favour primary allergic sensitization and repeated allergen exposure, resulting in persistent airway inflammation. The maternal gastrointestinal tract (GIT) microbiota is thought to influence priming of the fetal immune system, and the postnatal development of the infant GIT microbiota is influenced by early-life exposures. Although each individual pathway increases the risk of asthma, the major risk is produced when a child progresses through multiple risk pathways simultaneously.
For instance, an important risk factor for persistent asthma is low lung function, and most longitudinal cohort studies have found a deficit in lung function in children with asthma when lung function was first measured69–71. Lung function is partially influenced by genetics, and inherited factors might contribute to low lung function in those with asthma72,73. Exposure to environmental toxicants, such as cigarette smoke and ambient air pollution during pregnancy, is associated with reduced lung function at birth74–76 and an increased risk of subsequent asthma (Fig. 8). The debate over whether early-life infections damage the lungs (viral-induced effect) or whether such infections unmask vulnerable individuals (susceptible host) to cause asthma has not yet been settled. Severe viral infections that require hospitalization, especially infection with adenoviruses and RSV, can damage the developing lung and lead to recurrent respiratory problems including recurrent wheeze in childhood77,78. Longitudinal birth cohort studies have shown that wheezing in early life associated with rhinovirus infection is a risk factor for both subsequent asthma and lower lung function in childhood compared with infants who did not show rhinovirus-related wheezing79–81. However, none of these studies provide definitive evidence to determine whether these postnatal exposures increase the risk for asthma by limiting lung growth during early childhood.
Another key factor in the relationship between viral infection and developing asthma is the existence of impaired antiviral innate immunity in the airway epithelium. This impairment might include deficiency in mounting a robust type 1 (IFNβ) and type 3 (IFNλ) protective response when confronted by common respiratory viruses, such as rhinovirus, RSV, influenza virus, coronaviruses and adenoviruses82,83, and will be discussed in further detail in relation to viral exacerbations of asthma.
Prenatal and postnatal risk factors. Prenatal risk factors for the development of asthma include ethnicity, low socioeconomic status, stress, caesarean section and maternal tobacco smoking, whereas postnatal risk factors include the levels of endotoxins and allergens within the home, viral and bacterial infection, air pollution, antibiotic use, paracetamol exposure and obesity84. For example, prematurity confers a fourfold increase in the risk of developing asthma85, representing the largest effect of any known epidemiological risk factor for this disease86. In addition, increased airway responsiveness is present at birth. Given that this phenotype is known to be associated with prematurity and low birth weight, this physiological marker of asthma susceptibility is thus present in at-risk babies before any viral illness74,85,87. As such, airway responsiveness is an important determinant of the response to viral infections early in life and of ultimate asthma risk. However, with the exception of smoking, the identification of these risk factors has failed to translate into implementable public health policy.
Microbiota and the ‘hygiene hypothesis’. Epidemiologists have also had difficulty in integrating knowledge about putative risk factors into a comprehensive theory about the developmental origins of the disease and the relationship of asthma to patient susceptibility to infection. One such attempt is the ‘hygiene hypothesis’, which was developed almost 25 years ago. This hypothesis posited that respiratory infections ‘protected’ against asthma and allergies by ‘educating’ the immature infant immune system88. However, although the potential protective effect of microorganisms on the development of allergy is an attractive idea, the mechanisms that underlie the negative association between microorganisms and allergy acquisition have proven to be complex and elusive89. Recent interest has focused on vitamin D90 and the developing infant gut microbiota91, both of which might influence developmental asthma susceptibility and immune function through epigenetic mechanisms92,93. Evidence that supports a role for the maternal gastrointestinal microbiota in asthma development is also emerging. The maternal microbiota might affect the developing fetal immune system during pregnancy and influence respiratory health during infancy94. The infant gastrointestinal microbiota develops postnatally and is influenced by factors linked to asthma risk, such as mode of delivery, infant feeding practices, antibiotic exposure and exposure to siblings and pets. Whether the infant gastrointestinal microbiota is the ‘missing link’ between early-life environmental exposures and asthma risk is yet to be determined.
Bacterial pathogens. Along with a potential involvement of the intestinal microbiota in asthma, there is also increasing recognition that the presence of microorganisms in the respiratory tract, including the upper airways, and the way that the immune system responds to these, is likely to have an effect on respiratory health95 (Fig. 8). The presence of bacterial pathogens per se is not necessarily associated with disease risk, suggesting that an effective mechanism must normally operate to protect against trans-epithelial invasion in situations where local homeostasis is disturbed and barrier function is compromised, such as during viral infections. Immune recognition of invading bacteria has the potential to play such a part. For example, longitudinal cohort studies have determined that the development of IgG1 (T1-type immunity) and IgE (T2-type immunity) antibodies against bacterial antigens might limit tissue-damaging inflammatory responses to bacterial invasion across the respiratory epithelium96. These data indicate that a delayed postnatal rise in serum titres of IgG1 against Haemophilus influenzae and Streptococcus pneumoniae in children with a family history of atopy is associated with increased risk of sensitization to perennial aeroallergens and of persistent asthma at 5 years of age97,98. In this context, the likely role of specific IgG1 is to accelerate phagocytic clearance of microorganisms that breach the mucosal barrier, and thus limit potential tissue damage99. The presence of potentially pathogenic bacteria in the lower airways is likely to generate a vigorous inflammatory response associated with substantial lower respiratory symptoms, and might result in unbalanced T1-type immunity that increases the risk of developing asthma.
T2-type immunity against respiratory mucosal dwelling bacteria such as H. influenzae and S. pneunomiae, detectable as bacterial-specific serum IgE, is present in teenagers regardless of their atopic status. The strength of this immunity is inversely associated with asthma risk100. This bacterial-specific IgE is probably a surrogate marker for underlying populations of bacterial-specific TH2 memory cells that secrete IL-4 and IL-13, which are pluripotent cytokines that probably have a role in downregulating bacterial-induced macrophage activation and thus limiting local tissue damage96. Exaggerated and unbalanced T2-type immunity is also probably associated with the eosinophilic inflammation and tissue damage observed in chronic asthma. Thus, deficiencies in the normal immune recognition of bacterial incursion across the respiratory epithelium, especially at times of respiratory viral infection that spreads to the lower airways, probably results in an unbalanced immune response that translates into an increased risk of subsequently developing asthma.
Airway modelling and remodelling. Although histological evidence of airway inflammation and modelling (or remodelling) in infants and young children with recurrent wheeze or asthma is limited, what little evidence there is suggests that recurrent wheeze is associated with thickening of the epithelial reticular basement membrane and T2-type airway inflammation101. These changes relate to symptom severity, the need for anti-asthma medication102 and long-term evidence of airway inflammation103. It is noteworthy that thickening of the reticular lamina only begins with the onset of asthma, and in infants this thickening is not a feature of viral wheezing per se104. Perplexing questions are how and when the airways smooth muscle bulk increases and what is the relationship of these increases to the development of inflammation. There is evidence in support of the notion that smooth muscle hyperplasia accompanies early-onset childhood asthma104. However, studies in older children with asthma, cystic fibrosis or non-cystic fibrosis bronchiectasis have shown that the increases in smooth muscle mass in these children were similar to those observed in children without a lung disease105. This similarity suggests that a proportion of changes in smooth muscle mass might be related to chronic inflammation generally rather than being disease-specific. However, the remarkable hyper-responsive behaviour of asthmatic smooth muscle does suggest that there are highly specific changes in morphology linked with muscle function, such as an increase in the expression of oxidant pathways that changes the behaviour of muscle fibre contractility106. Recent data from challenge tests in adults with atopic asthma indicate that bronchoconstriction per se, regardless of the mechanism inducing it, increases the secretion of pro-fibrotic cytokines and the deposition of subepithelial collagen107. Thus, in addition to chronic inflammation and responses to tissue injury, altered mechanotransduction might be a further cause of airway wall remodelling. In this setting, remodelling might be promoted by induction of factors in the epithelium such as resistin-like-β (RELMβ), which accentuates collagen deposition108, members of the plasminogen activator system and chitinase-3-like protein 1 (CHI3L1; also known as YKL40)109,110.
Disease chronicity and exacerbations
Through a continuing cycle of epithelial injury and repair, chronic inflammation and airway remodelling occur in parallel to create the disease chronicity that is characteristic of asthma57,111. Superimposed on this chronicity is the acute worsening of asthma (also termed asthma exacerbation), which is most often driven by common respiratory viruses, allergen exposure and air pollutants. One possible explanation for why the asthmatic airways are so susceptible to usually innocuous viruses such as rhinovirus is that the epithelium, which undergoes repeated damage and repair, also exhibits an impaired innate immune response that involves a reduced capacity for induction of the protective interferons IFNβ and IFNλ in response to viruses. This defect is linked to the chronic wound status of the epithelium112, and inhaled IFNβ has recently been shown to attenuate viral exacerbations in moderate-to-severe asthma by restoring defective antiviral innate immunity113.
Remission of asthma
Up to one-third of children with asthma become disease free in young adulthood, with boys outgrowing asthma more frequently than girls18,19,114 (Fig. 3). Up to one-third of those who become asthma-free as young adults remain in remission throughout their adult life — that is, they remain symptom-free and have normal lung function in the absence of medication use. However, given that some evidence of airway inflammation remains in these individuals, it is debatable whether their lack of obvious symptoms indicates complete recovery as opposed to signifying clinical remission only18,115. Sensitization to small-particle allergens from animals with fur, especially from cats, is a risk factor for the persistence of asthma into adulthood. These allergens, such as major allergen I polypeptide chain 1 (allergen Fel dI) found in the saliva of cats116, can penetrate the small airways.
Complete remission from asthma (cure) can also occur, but in this case, airway hyper-responsiveness also resolves117–119. Although complete remission of asthma is accompanied by the normalization of lung function, which is usually measured by spirometry as forced expiratory volume in 1 second (FEV1), the small airways, which are known to contribute to asthma, have not been well studied in this context. In addition, bronchial biopsy studies have revealed that airway remodelling, defined by thickening of the reticular lamina of the epithelial basement membrane, persists in adults who experience complete remission of asthma, and these individuals also show some residual increase in the number of eosinophils compared with those who do not have asthma. However, the function of these residual eosinophils is important to note. In contrast to the eosinophils of patients with asthma, those present in individuals in complete remission are neither ‘activated’ nor are they easily attracted to the airway lumen in the absence of ICSs or by indirect stimuli such as inhaled adenosine120. These differences indicate that eosinophil activation and associated mediator secretion, rather than the mere presence of these inflammatory cells, is linked to the persistence of asthma.
Diagnosis, screening and prevention
Risk factors
Environmental allergens. Children sensitized to aeroallergens early in childhood are more likely than their non-sensitized peers to experience persistent asthma later in life. In addition, seasonal allergic rhinitis and exposure to indoor allergens are associated with asthma development, severity and morbidity121. In particular, the relationship between asthma and exposure to dust mites is especially strong121–123, with >50% of children and adolescents with asthma sensitized to dust mites, whereas generally <20% of children without asthma are sensitized to dust mites.
Consequently, to prevent asthma onset, allergen avoidance is strongly recommended in cases of existing respiratory allergies. However, the role of allergen avoidance in preventing allergic asthma is poor, unless it is undertaken as part of a complex and multifaceted prevention effort124,125. The disappointing outcomes of indoor allergen reduction programmes either in pregnancy and early life (referred to as primary prevention) or once asthma is established (termed secondary prevention) are difficult to explain126–128. One potential reason for the failure of such efforts is that they might have simultaneously reduced exposure to protective factors, such as microbial products, which shape innate immunity by interacting with pattern recognition receptors, such as Toll-like receptors129. Indeed, microbial products present in indoor environments are protective against allergen sensitization if exposure occurs prenatally or in the first few years of life130,131. The mechanisms that underlie this protection might involve altered signalling through Toll-like receptors in the placenta and induction of tolerogenic regulatory T (TReg) cells in infants89,132.
Viral infection. Childhood viral infections, especially with rhinovirus and RSV, are associated with the development of asthma (Fig. 8) and are the most common cause of asthma exacerbations (Fig. 9). RSV is a major cause of bronchiolitis in the first year of life and an independent predictor of recurrent wheeze and early-childhood asthma133. With the advent of modern diagnostic technologies, rhinovirus-induced wheezing has also been recognized as a potential predictor of asthma development134. Several longitudinal and birth-cohort studies provide evidence that respiratory viral infections, particularly with rhinovirus that led to wheezing events during the first 3 years of life, are strong predictors of later asthma development80,81. A relative lack of production of type 1 and type 3 interferons together with the release of the T2-type alarmin cytokines TSLP, IL-25 and IL-33 by the airway epithelium provides a link between epithelial injury, enhanced allergen sensitization and the acquisition of asthma82,83,135 (Fig. 6). Initiation of asthma also involves preferential expression of susceptibility genes in different regions of the airway. These genes include protocadherin 1 (PCDH1) and cadherin 3 (CDH3), which are preferentially expressed in the airway epithelium136,137, and genes that are preferentially expressed in airway mesenchymal cells including ADAM33 (which encodes disintegrin and metalloproteinase domain-containing protein 33) (Ref. 138).
Figure 9. Contribution of risk factors to the development and/or exacerbation of asthma.
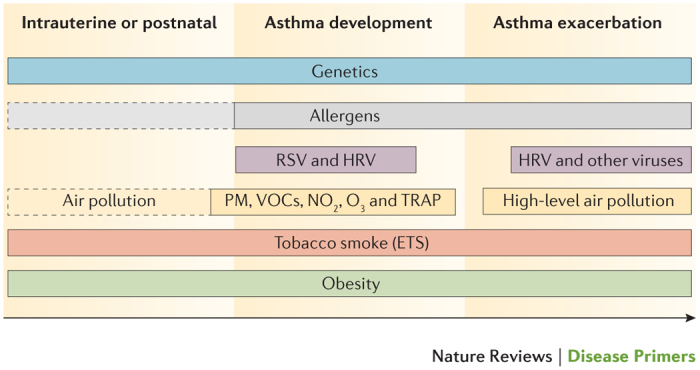
Solid outline indicates that the role of these risk factors has been firmly established. Dotted outline indicates that there are controversial and/or preliminary results for the contribution of these risk factors. ETS, environmental tobacco smoke; HRV, human rhinovirus; NO2, nitric dioxide; O3, ozone; PM, particulate matter; RSV, respiratory syncytial virus; TRAP, traffic-related air pollution; VOCs, volatile organic compounds.
Air pollution. Indoor and outdoor air pollution, which have increased alongside urbanization and population growth, are important contributing factors to the development and exacerbation of asthma, particularly in the developing world139,140 (Fig. 9). The pollutants implicated in asthma are nitrogen dioxide, ozone, volatile organic compounds, particulate matter (PM10 and PM2.5) and traffic-related air pollution, which contains fresh vehicle exhaust pollutants and non-combustion-derived particles. These pollutants induce oxidative stress and epithelial damage to initiate or augment airway inflammation141,142 and reduce inhibitory TReg function143. Although controversial, such pathways are being linked to the initiation of asthma in those who otherwise would not experience the disease, as all of these pathways could contribute to enhanced respiratory sensitization to aeroallergens144 and increased airway hyper-responsiveness and remodelling145. It has even been suggested that prenatal exposure to pollutants might contribute to postnatal asthma146. One possible mechanism in which pollutants could induce asthma involves interaction with genetic risk factors such as polymorphisms in antioxidant genes, for instance in those that encode various glutathione S-transferases, and in the TNF promoter147. In addition, pollutants might influence gene function through epigenetic pathways by affecting promoter methylation and histone acetylation status148.
Tobacco smoke. Exposure to tobacco smoke, which comprises a complex mixture of many volatile organic compounds and nitrogen dioxide, is an independent risk factor for the development of asthma149,150 and acts, in part, by augmenting T2-type responses151 (Fig. 9). Even grandmaternal smoking during pregnancy increases the risk for asthma for the second generation152, thereby further incriminating intrauterine epigenetic mechanisms in asthma causation.
Obesity. Obesity during childhood is strongly associated with the incidence and severity of asthma153, and maternal obesity and high gestational weight gain are associated with an increased risk of childhood asthma, especially in non-asthmatic mothers154 (Fig. 9). A combination of mechanical factors and shared causal metabolic, hormonal and low-grade inflammatory pathways might help explain these associations. For instance, the obesity-associated chronic inflammatory response is defined by increased levels of inflammatory cytokines, such as TNF, IL-1 and IL-6, produced by macrophages and adipokines, such as leptin, chemerin and adiponectin, produced by adipocytes that inhibit cell proliferation and cause tissue damage. Leptin is a sentinel mediator of differentiation of lipofibroblasts and controls pulmonary surfactant synthesis in fetal lungs155. In mice, leptin infusion enhances airway hyper-responsiveness, increases the levels of IgE following allergen challenge156 and increases airway levels of IL-6 (Ref. 157). A low-grade maternal inflammatory response with increased levels of leptin also contributes to postnatal risk of asthma158. However, the exact mechanism linking obesity and asthma still remains to be fully clarified.
Diagnosis
Asthma is among the most common chronic diseases in the developed and developing world, but its diagnosis can be difficult. Although symptoms including wheeze, chest tightness and shortness of breath are often considered essential features of asthma in humans, the adage ‘all that is asthma does not wheeze and all that wheezes is not asthma’ holds true. Epidemiological studies that rely on ‘doctor diagnosis’ of asthma overestimate the true disease prevalence owing to misclassification159. A diagnosis of asthma usually begins when a child or adult presents with a range of spontaneous respiratory symptoms including recurrent cough and nocturnal awakening, along with symptoms triggered by external stimuli, such as allergens, viral infections, exercise and cold air. In adults, a history of asthma or recurrent ‘bronchitic episodes’ in childhood holds an important clue to diagnosis. However, a diagnosis of asthma should not be made on clinical characteristics alone. The definition of asthma requires a combination of appropriate clinical symptoms in association with documented reversible airflow limitation and/or airway hyper-responsiveness160. Although reversibility of airway obstruction and hyper-responsiveness are considered hallmarks of asthma, the sensitivity and specificity of the diagnositic criteria to identify these symptoms are poorly defined. Guidelines have added the important extra dimension of airway inflammation to the diagnosis of asthma, which is measured by eosinophil counts in sputum or blood and/or increased fractional exhaled nitric oxide (FeNO)161,162.
Physiologically determined abnormalities, such as reduced spirometry, are also of value in establishing an asthma diagnosis early in the course of the condition. Patients should be tested on a spirometer that is equipped with population normal values and, ideally, one that generates a flow-volume loop that can be evaluated for both inspiratory and expiratory effort. FEV1, forced vital capacity (FVC) and the FEV1/FVC ratio should be reported alongside reversibility of lung function with an inhaled SABA. Symptomatic asthma is often associated with a predicted FEV1 of <80% and an age-adjusted FEV1/FVC of <75%. Testing should be repeated after inhalation of a SABA to establish reversibility of airway obstruction, a hallmark of asthma, although as noted, both bronchodilator reversibility and peak expiratory flow (PEF) variability have poor sensitivity and specificity for the diagnosis of asthma. By convention, a diagnosis of asthma requires at least a 12% improvement in FEV1 over baseline and a total improvement of at least 200 ml. As asthma is frequently highly variable, normal spirometry results do not exclude the disease. Additional diagnostic aids include repeat testing over time and diurnal PEF monitoring using a portable PEF meter. If spirometry results remain normal, bronchial provocation testing with inhaled methacholine or mannitol should be considered to establish if airway hyper-responsiveness exists as another characteristic feature of asthma, although some variability in responses can be seen. To perform this test, the patient inhales increasing concentrations of the challenge substance until there is a ≥20% fall in the FEV1 from the saline control value. Each challenge agent has a threshold concentration for the fall that identifies asthma. Exercise testing or, as an alternative, eucapnic hyperventilation, which mimics the volume of air exchanged during exercise, is another method for uncovering hyper-responsiveness and is especially useful in diagnosing asthma in children.
The documentation of asthma-related airway inflammation is an important recent development in asthma diagnosis and is especially useful for ruling out asthma, as many diseases can produce asthma-like symptoms and provide positive results in the tests described above. Recent UK National Institute for Health and Care Excellence (NICE) guidelines for asthma diagnosis highlighted an urgent need to mainstream the use of inflammatory biomarkers, such as FeNO and sputum eosinophil counts, for reliable diagnosis of T2-type asthma162 (Fig. 10). If diagnosis is still questionable, then a practical approach is to treat the patient with medications that are appropriate to their level of severity defined by national or international asthma guidelines. If the patient's symptoms become markedly better in response to this treatment, then asthma is the likely cause. Possible other causes of wheezing and asthma-like symptoms should always be considered in the face of a poor response to a trial of treatment, and can include COPD, upper airway obstruction and laryngospasm in adults and viral-associated infection in children.
Figure 10. Biomarkers for the assessment of T2-type asthma.
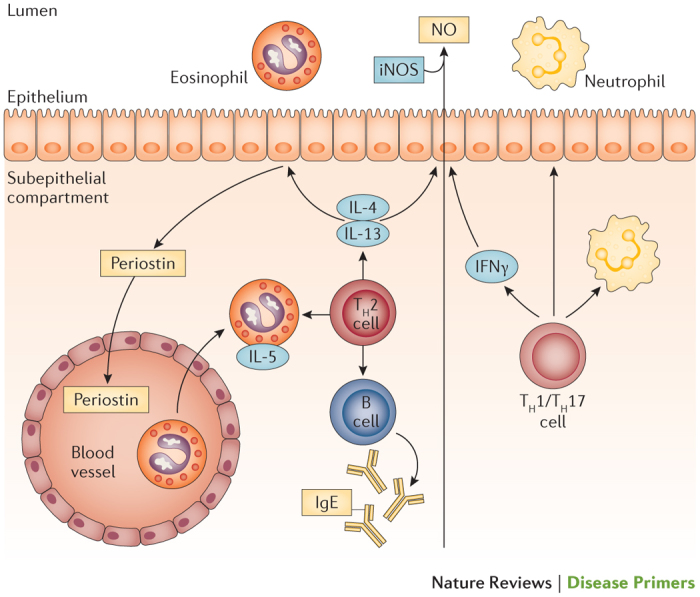
This molecular phenotype, or endotype, of asthma can be identified using biomarkers. These include already established biomarkers (blue boxes) and markers currently under clinical evaluation (yellow boxes). iNOS, inducible nitric oxide synthase; NO, nitric oxide; TH, T helper.
Biomarkers
Many different ‘subphenotypes’ of asthma with differing characteristics are becoming increasingly recognized163 (Fig. 1). In terms of disease classification, perhaps the most important distinction to make is whether the patient has evidence of an eosinophilic T2-type inflammatory process. This is important for disease management, as evidence is emerging that responses to ICSs and biologic therapies that target IgE, the IL-4–IL-13 pathway and IL-5 are all greater in patients with evidence of T2-type inflammation than in patients without evidence of T2-type inflammation32,164,165. A characteristic feature of this immune activation profile is the appearance of eosinophils in the blood and induced sputum as well as increased FeNO. ICS treatment monitored by sputum eosinophil testing is highly effective166,167, and both sputum and blood eosinophil counts are being used to effectively identify patients who might be responsive to biologic therapies that target IL-4, IL-13 and IL-5 (Refs 168,169). Measurement of sputum and blood eosinophilia is, unfortunately, not widely implemented162.
Nitric oxide produced by inducible nitric oxide synthase in the bronchial epithelium increases in response to IL-4 and IL-13, is a marker for T2-type inflammation and is highly corticosteroid sensitive170,171. Although measurement of the FeNO requires a specific analyser, the test itself is easy to perform, is reproducible and can be measured as a point-of-care biomarker with instant results. Elevated FeNO increases the likelihood of an asthma diagnosis involving T2-type inflammation and can be used as a predictor of and to follow therapeutic responses to biologics that are targeted at IgE, the IL-4 receptor, IL-13 and IL-5 (Refs 172–174) (Fig. 10).
Expression of periostin, an extracellular matrix protein, is induced by IL-4 and IL-13 in airway epithelial cells and lung fibroblasts175, and periostin is secreted as a soluble peptide from the basolateral surface from which it gains access to the circulation176. Periostin functions as a ligand for αVβ3 and αVβ5 integrins to promote adhesion and migration of epithelial cells and aids in the crosslinkage of submucosal collagen177. As a T2-type immunity biomarker, reduced serum levels of periostin predict the clinical efficacy of biologics targeting the IL-4–IL-13 pathway and discriminate patients with high numbers of eosinophils in their airways178,179. As a high proportion of patients expressing T2-type airway inflammation are atopic, assessment of allergen-specific IgE in the serum provides information on patient-specific allergic triggers180.
Perhaps the most crucial use of these biomarkers will be to identify various molecular phenotypes of asthma, in particular severe asthma, where biologic agents are likely to be targeted. To date, the biomarkers identified are all linked to T2-type inflammatory phenotypes, which might either predict or be responsive to these T2-type-targeted therapies. Blood eosinophil counts of ∼150 per microlitre seem to both predict responses to IL-5-targeted therapies and fall in response to these treatments181. At present, it is unclear which biomarker will best predict response to IL-4–IL-13 pathway-targeted therapies, as all have shown some predictive ability. However, for IL-4–IL-13 pathway-targeted therapies, whereas blood eosinophil counts predict response, these counts do not decline in response to therapy164,173. It remains unclear whether any current biomarker, alone or in combination with a second or third biomarker, will identify patients who respond better to IL-5-specific as compared with IL-4–IL-13 pathway-specific or IgE-specific approaches. These biomarkers are linked closely to disease pathways, with less relation to treatment responses. As such, their inhibition by specific targeted biologic approaches and use to produce improved clinically meaningful outcomes might eventually translate what are currently molecular phenotypes into endotypes — which are definitively defined biological processes that underlie particular subgroups of the disease.
Most recently, a cross-sectional study of patients with asthma of varying severity and endobronchial tissue gene expression analysis has revealed three major patient clusters: TH2-high, TH17-high, and TH2/TH17-low182. In individual patient samples, TH2-high and TH17-high patterns were mutually exclusive and their gene signatures were inversely correlated and differentially regulated by IL-13 and IL-17A. In a mouse model of allergen-induced lung inflammation, IL-4–IL-13 blockade caused an increase in TH17 cells and neutrophilic inflammation, whereas neutralization of IL-13 and IL-17 protected mice from eosinophilia, mucus metaplasia and airway hyper-responsiveness as well as causing an attenuation of neutrophilic inflammation. The authors conclude that combination therapy targeting both pathways may maximize therapeutic efficacy across a patient population comprising both TH2 and TH17 endotypes.
A crucial question is whether intervening on selective pathways prevents or reverses airway wall remodelling to influence the natural history of asthma. Although there is very limited evidence in support of the notion that anti-asthma therapy influences remodelling processes, a recent report showing that IgE blockade with omalizumab downregulates bronchial smooth muscle proteins in severe asthma provides justification for some optimism in the use of targeted biologics183.
Prevention
Allergen avoidance. Although prevention of asthma, especially asthma associated with allergy, should be straightforward it has in fact proven difficult. Specific allergen avoidance and strategies with the aim to reduce asthma have produced disappointing results. This outcome could be taken to mean that allergic sensitization does not contribute to asthma, or it might also signify that the process of allergic sensitization in general, rather than sensitization to a specific allergen, is key to disease expression. For example, comprehensive (multifaceted) allergen avoidance that includes breastfeeding while the mother is on a low allergen diet or given an extensively hydrolysed formula in conjunction with dust mite reduction strategies in the first year of life seems to be most effective in preventing asthma onset in individuals who are genetically at risk of asthma, with protection extending to 18 years of age124. However, as this study was single-blinded in design, more studies are needed to confirm the benefits of multifaceted allergen avoidance.
Dietary measures. Breastfeeding is one of the best-studied asthma prevention measures. Whereas initial studies have provided some evidence for a reduction of the incidence of both allergy and childhood asthma184,185 as a result of breastfeeding, these findings have recently been challenged186,187. Nonetheless, on the basis of the overwhelming general health benefits of breastfeeding, many guidelines still recommend exclusive breastfeeding during the first months of life188,189. Vitamin D levels might also influence asthma. A large trial aimed at testing whether vitamin D supplementation for pregnant women prevents their children from developing asthma has recently concluded, but its results are yet to be reported190. Finally, dietary supplementation with fish oil — an important source of long-chain polyunsaturated fatty acids — has received attention on account of the immune-modulatory activities of the altered and less-active eicosanoid derivatives that are metabolically produced instead of those derived from arachidonic acid. However, despite the theoretical advantages of fish oil supplementation, attempts to use fish oil to prevent asthma have been unsuccessful191,192.
Exposure to microorganisms. Microbial exposures have an essential role in priming immune responses, particularly early in life. Sources of exposure include certain allergy-protective and asthma-protective lifestyle conditions, such as traditional farms that contain environmental microorganisms — for example, high levels of Gram-positive and Gram-negative bacteria and of fungi193 — and biodiverse environments found, for example, in rural areas. By contrast, urbanization is associated with both low biodiversity and an increase in the prevalence of asthma. Although epidemiological and experimental studies strongly support the concept that a decrease in exposure to diverse microorganisms causes increases in asthma prevalence89,132, this now needs to be translated into a clinical application for asthma prevention. Interventions that have aroused recent interest are the use of probiotics, which are live bacteria found in foods such as yoghurt or taken as supplements, and prebiotics, which are specialized plant fibres that promote the growth of ‘good bacteria’ already resident in the colon, especially in those living in rural environments with livestock farming. There have been a range of studies in which the gut microbiota has been changed by probiotics or prebiotics. For instance, prenatal and postnatal administration of certain probiotics decreases the risk of allergen sensitization194. However, although there have been claims there are some benefits of using probiotics to prevent atopic dermatitis195, results in asthma prevention have so far been disappointing196,197.
Finally, prevention strategies that can address other asthma risk factors include avoidance of active and passive tobacco smoke exposure, interventions that target obesity and achievement of a balanced diet comprising adequate micronutrients such as vitamin D. However, for these strategies to have their greatest effect, major public health measures that begin early in life and run as lifelong programmes are needed198.
Management
Over the past decade, understanding of asthma has changed considerably, with better insights into its heterogeneity. Consequently, treatment has changed from being based on a ‘one-size-fits-all’ model to a more patient-centred or personalized approach199. Central to this development is supported self-management and provision of an individualized written asthma action plan, referred to in guidelines as the co-management of asthma200. This approach ensures that interventions are tailored to the needs of patients, such as prioritizing minimal absences from school and work, creation of a supportive partnership with their doctor, family support and use of the appropriate drug regimen and inhaler devices. Such co-management plans should provide the best quality of life (QOL) through minimizing disease symptoms and abolishing disease exacerbations201. As far as is possible, drug treatment should only be initiated after removal of the stimuli known to exacerbate the disease (Box 1).
This management situation becomes more complex in children, especially in those <5 years of age, as many children who present with asthma symptoms have one of the various wheezing syndromes that, compared with asthma, are less likely to persist into adult life and do not require treatment with ICSs202. Unfortunately, the clinician has few tools available to determine which children are likely to have or develop persistent asthma and therefore benefit from early treatment.
In adults, GINA guidelines, which were updated in 2015, have provided sound recommendations to be used worldwide1,203. However, health services and treatment facilities, including the availability of drugs, differ between countries, and these disparities need to be taken into account when constructing action plans for asthma204.
Adults
Standard asthma treatment. Asthma guidelines such as those provided by GINA1, the US National Asthma Education and Prevention Program205 and the British Thoracic Society206 classify asthma by severity and extent of disease control. This classification is done in 4 or 5 steps that are used to assess the use of controller drugs, such as inhaled ICSs, LABAs, LTRAs and, for the most severe disease, the IgE-specific monoclonal antibody omalizumab (Fig. 11). By starting ICSs as early as step 1 of the management plan and scaling controller therapy up in a stepwise manner, dependency on inhaled SABAs for symptom relief has been reduced, exacerbations prevented, asthma-related mortality reduced and QOL improved in the majority of patients with asthma. Combination ICS–LABA therapy in moderate-to-severe asthma has achieved considerable success, as has omalizumab in severe allergic asthma. Moreover, maintenance and rescue therapy with the combination of ICS and the rapidly acting LABA formoterol has shown beneficial effects when used as maintenance and reliever treatment in adult patients with asthma207. In those patients who remain symptomatic despite taking an ICS–LABA combination, adding a long-acting anti-muscarinic antagonist is beneficial208. Inhaled drugs are most frequently used on demand or twice daily. Novel ultra-long-acting bronchodilators have been developed to be used once daily209 but their effectiveness is yet to be fully evaluated.
Figure 11. Global Initiative for Asthma-based steps for treatment of asthma at all stages of severity.

The Global Initiative for Asthma (GINA) recommends that asthma is classified and treated according to 5 steps that reflect symptom severity and patient characteristics. The relative widths of each step reflect the number of patients who receive each treatment. In particular, dose–response studies demonstrate that the majority of patients with asthma should be able to be treated with low-dose inhaled corticosteroids (ICSs; step 2). The relative heights of each step reflect disease severity, with treatment in step 5 reserved for those with the most severe disease. *For children 6–11 years of age, theophylline is not recommended and the preferred step 3 treatment with is medium-dose ICSs. ‡Tiotropium by soft-mist inhaler is indicated as add-on treatment for patients with a history of exacerbations; it is not indicated in patients <18 years of age. §For patients prescribed inhaled beclomethasone dipropionate/formoterol or budesonid/formoterol maintenance and reliever therapy. LABA, long-acting β2-adrenergic receptor agonist; LTRA, leukotriene receptor antagonist; OCS, oral corticosteroid; SABA, short-acting β2-receptor agonist. Adapted from the Global Strategy for Asthma Management and Prevention 2015, © Global Initiative for Asthma (GINA) all rights reserved. Available from http://www.ginasthma.org.
Despite the availability of a wide range of controller and reliever therapies, uncontrolled asthma remains a challenge. This problem reflects the need for new therapeutic options, especially as the number of deaths due to asthma has been reduced drastically since the introduction of anti-inflammatory treatments but, importantly, significant asthma-related mortality persists. Figure 12 shows that treatment should be based on both current control of a patient's asthma and reduction of future asthma risk. A recent meta-analysis compared methods for preventing severe disease exacerbations210 and found that, for this purpose, both combined maintenance and reliever treatment and combined fixed-dose treatment with ICSs and a LABA performed equally well and were ranked first for effectiveness. All other treatment strategies that used ICSs with another agent, whether by single or separate inhalers, tended to perform better than low-dose ICSs, but the difference failed to achieve significance. In patients sensitized to a single allergen, for instance allergen Fel dI from cats or pectate lyase 1 (also known as antigen Amb a I) from ragweed, subcutaneous allergen immunotherapy is an option211. The effect of sublingual immunotherapy on asthma is still being evaluated212.
Figure 12. Overall goals of asthma management.

Asthma management should aim to both control the current symptoms of asthma and reduce the risk of future adverse outcomes for patients. Adapted with permission from Ref. 256, Elsevier.
Before deciding whether to start a new treatment or change treatment, health practitioners should reconsider whether there are any factors that are modifiable. These include incorrect use of an inhaler, which needs repeat instruction especially in cases of uncontrolled asthma, active smoking or passive smoke exposure, obesity, rhinitis, sinusitis, diabetes mellitus, gastro-oesophageal reflux, anxiety and depression, low vitamin D status, limited language proficiency, absence of specialty care, lack of an asthma action plan and lack of self-management education. Special consideration also needs to be given to managing asthma in pregnancy213.
Improvement in the treatment of asthma in adults requires that, in the future, study designs take into account a range of considerations. For example, they need to ensure that their findings have applicability for various settings and patient populations and that they have a patient-centred focus, such as that taken in pragmatic comparative effectiveness studies214. Nonetheless, although there is an ongoing need for further research in some areas, the evidence base in support of patient-centred and co-management approaches to asthma is strong215.
Novel therapeutic approaches. Cluster analyses have identified subtypes of asthma, one of which is severe asthma that is unresponsive to ICS and oral corticosteroid treatment12,23,216. Despite extensive research, the underlying pathobiology of severe asthma has not been established. Nonetheless the aetiology of severe asthma is likely to be heterogeneous rather than based on a single unifying process (Fig. 1). As a result of increased awareness of this heterogeneity, the identification of appropriate biomarkers in individual patients with asthma is becoming crucial in guiding the use of therapies that target the specific causative pathways or endotypes of asthma217–220. Individual T2-type cytokine biologics that target the IL-4–IL-13, IL-5 and TSLP pathways and IgE itself are promising treatment options for severe disease, especially when used with biomarkers such as sputum and blood eosinophil numbers, FeNO, serum periostin levels and total IgE levels to differentiate ‘responders’ from ‘non-responders’. Blockade of a single pathway has clear clinical benefits and few associated adverse effects. However, whereas this strategy leads to improvement in some asthma outcomes, such as a reduction in asthma exacerbations produced with IL-5-specific therapy, outcomes for other measures, such as lung function and hyper-responsiveness, are less favourable. As a result, single pathway blockade has only partial efficacy169. Additional research is needed to define how such agents should be used in a stratified or personalized approach to management. The same is true for pharmacogenomics, in which single-nucleotide polymorphisms in genes contribute to treatment responsiveness. The proportion of phenotypic variability accounted for by genetic variation (heritability) is estimated to be 28.5% for bronchodilator responsiveness and 50% for lung function221. For example, the Arg16 allele of the gene encoding the β2-adrenergic receptor has been shown to associate with worsening of asthma when children with the disease receive continuous SABA or LABA monotherapy222,223. This and other variants of the β2-adrenergic receptor have different effects on the disease in different ethnic populations224. Polymorphisms also influence responsiveness to ICSs and LTRAs and deserve further study225.
Bronchial thermoplasty, delivered by the Alair™ System, is a treatment for severe asthma that was approved by the US FDA in 2010. This treatment involves the delivery of controlled, therapeutic radiofrequency energy to the airway wall to heat the tissue and thereby reduce the amount of smooth muscle in the airway wall226. This procedure causes epithelial injury followed by regeneration of the epithelium, blood vessels, mucosa and nerves but not airway smooth muscle227,228. Indeed, there is preliminary evidence that this procedure is able to reduce airway smooth muscle mass in patients with severe asthma228. Although bronchial thermoplasty is an exciting development for managing some types of severe asthma, its place in management guidelines is still being evaluated229,230.
Children
Although early treatment reduces asthma symptoms in those at high risk of developing persistent asthma, there is no evidence that early treatment decreases the risk of subsequent asthma or alters its natural history10,11,202,231,232. In addition, with the exceptions of treatment with omalizumab for children >12 years of age who have severe atopic asthma that is not controlled by maximal conventional therapy233 and treatment with oral montelukast (a cysteinyl LTRA) for exercise-induced asthma234, so far, there is little evidence that ‘phenotype-specific’ therapy is helpful in children235. Treatment of asthma in school-aged children is perhaps the most straightforward, with most patients responding to simple first-line therapy that forms the basis of most asthma management plans. If first-line therapy is not successful, there is no single ‘best’ add-on therapy for all children. Thus, if the first add-on is not sufficient, it should be stopped and an alternative therapy of a different drug class tried in its place236.
The diagnosis and treatment of asthma in infants and preschool-aged children is much more problematic than it is for older children. As outlined earlier, although most severe childhood asthma starts early in life, the majority of infants and young children who have wheezing episodes do not progress to persistent asthma237. A further problem arises with the apparent lack of clinical or physiological response to bronchodilators in infants and young children with established asthma-related risk factors that predict long-term outcomes238. The aim of asthma management at all ages is to achieve good clinical control for as long as possible. Young children with persistent symptoms should be managed in a similar manner to that of older children with persistent symptoms. As pointed out in the GINA guidelines, for children ≤5 years of age1,169, a combination of increased daytime cough, daytime wheeze and night-time use of an inhaled SABA is a strong predictor of acute exacerbations in these children. Inhaled medications are the cornerstone of asthma management in younger children; however, substantial importance must be placed on the ability of the child to use the inhalation device. The use of ICSs as first-line management for children with persistent symptoms is supported by placebo-controlled clinical trials239. In contrast with adults, there is no evidence for the use of combination ICS–LABA therapy in this age group, with the GINA guidelines specifically advising against this in children ≤5 years of age.
The treatment of intermittent, usually viral-induced, symptoms in young children is more controversial. As discussed earlier, the use of ICSs or the LTRA montelukast as maintenance therapy does not reduce the frequency of acute intermittent symptoms in young children. Montelukast but not ICSs169,239,240 or oral corticosteroids241 given as a short-course treatment at the onset of symptoms can reduce the severity and duration of asthma symptoms, but do not reduce the need for additional medication or hospitalization. There is no justification for using a LTRA in infants169,242. Many young children can be treated with SABAs alone to relieve symptoms provided that symptoms are troublesome enough to warrant any treatment.
Box 1: Asthma triggers.
Wherever possible, avoidance of triggers should be part of every patient's written asthma action plan. Common triggers include253:
Inflammatory factors
Allergens
Respiratory infections
Work
Irritants
Exercise
Cold air
Temperature change
Strong odours
Stress and emotions
Others
Tobacco
Medication
Food additives
Pollutants
Gastric reflux
Quality of life
For the vast majority of patients with asthma, life expectancy should not differ from the general population. Thus, treatment of asthma is primarily focused on improving the day-to-day symptoms of the patient, preventing exacerbations and generally improving their QOL (Fig. 12). Indeed, GINA1 identifies asthma control as the main outcome of asthma management. Asthma control incorporates level of symptoms, number and severity of exacerbations, as well as lung function. The effect of these factors on patients' activities and daily living is generally referred to as a patient's QOL. To address this more holistic approach, patient-reported outcome measures (PROMs; defined in Box 2) are becoming more attractive in the assessment of treatment success. These are typically short, self-completed questionnaires, most commonly used to measure the health status or health-related QOL of a patient before and after an intervention. The concept that PROMs are necessary to assess the impact of asthma treatment has contributed to their increasing use in clinical practice243,244. The PROMs generally used for the assessment of asthma are the asthma QOL questionnaire (AQLQ), the paediatric version of the AQLQ (PAQLQ), the Asthma Control Test (ACT) and the Asthma Control Questionnaire (ACQ)245. The ACT and ACQ focus on symptoms and functional status rather than how these affect the patient's personal perceptions of the effect of asthma on their day-to-day QOL. However, these outcomes are intimately related as studies show strong correlations between the level of asthma control and QOL, even when controlling for disease severity246.
QOL in patients with asthma is likely to be affected by various factors and co-morbidities associated with asthma, including asthma control (even when controlling for asthma severity), duration of disease and the concurrent need for chronic therapies, frequent exacerbations, obesity or weight gain and psychosocial factors241,247,248. In addition, sex and socioeconomic factors, as well as co-morbidities such as gastro-oesophageal reflux and rhinoinusitis also probably have a role in determining QOL in patients with asthma. These co-morbidities are particularly prevalent in more-severe asthma6. Patients with more-severe asthma experience an increase in symptom severity, more frequent and severe exacerbations and more co-morbidity, all of which contribute to an overall poor QOL.
Treatment with ICSs, usually with SABAs or ICSs in combination with a LABA, montelukast or omalizumab have all been reported to improve QOL, with varying effects on asthma control, when compared with a placebo. Interestingly, not all therapies affect these parameters in the same manner. For instance, although IgE-specific treatment improves exacerbations in patients with more-severe asthma, the mean improvement in QOL as measured by the AQLQ, while substantial, does not achieve the 0.5 points threshold change for clinical significance on the 7-point scale of this questionnaire249. The intensity of the intervention also seems to influence the magnitude of improvement in the AQLQ. This improvement is demonstrated by the invasive treatment bronchial thermoplasty; however, the difference in improvement between active and sham thermoplasty also did not achieve clinical significance250. Finally, it should be recognized that the mere label of asthma as a chronic disease, with lifelong required treatment and daily confrontation with the disease, even though controlled to some extent, might affect the QOL of an individual with asthma considerably, irrespective of disease severity251. Hence, all efforts should be made to prevent disease chronicity.
Box 2: Definition of a patient-reported outcome measure.
A patient-reported outcome measure (PROM) is reported directly by a patient or their carer. It is a measure used to assess the outcome and/or the effect that a particular treatment for a long-term condition has on the quality of life of a patient. These reports might include how the condition affects health-related quality of life, a patient's perceptions of their health or functional status in relation to their long-term condition and the effect that treatment or care strategies have on the quality of life of the patient244.
Outlook
Although substantial progress has been made in our understanding of asthma mechanisms and epidemiology, these are only just being translated into novel approaches for disease management. However, the prevention of asthma in at-risk infants and cure of the fully developed disease seem distant goals. All of our current therapies are still based on damping down airway inflammation and on relieving airway obstruction. To make further progress and recognizing that the majority of the disease begins early in life, we need to understand what the genetic, prenatal, maternal and early-life environmental factors are that initiate asthma or at least render particular infants more susceptible to it. A most promising field connecting these two drivers is the uncovering of gene functions by environmental exposures through epigenetics. There are already novel data about the relationship of the epigenome to maternal smoking; similar epigenetic studies looking at the plethora of chemicals that humans are now exposed to, such as air pollutants and endocrine disruptors, as well as diet might cast new light on preterm susceptibility. In terms of established asthma and disease severity, novel mechanisms are emerging to explain the way that cells adapt to environmental cues. These mechanisms include changes in DNA methylation, histone modifications and regulation of transcription and translation by non-coding RNAs and, when coupled with recent advances in lung delivery of oligonucleotides and small molecules, are opening up new opportunities for intervention219,252.
An important question stemming from lifecourse epidemiology is what limits the development of asthma in high-risk infants and, in particular, what role the maternal gut microbiota has in priming fetal immune development, predisposition to atopy and allergy and preventing viral-induced respiratory wheezing from progressing into subsequent asthma? Of equal importance is the infant upper airway and lung microbiota and understanding how different microbial signalling pathways modify innate immunity towards protection against or susceptibility to asthma. The role of metabolic pathways relating to diet, micronutrients, obesity and the microbiota and how all these interact with environmental exposures are also likely to be of great relevance.
From a mechanistic standpoint, the pathobiology of asthma is in large part restricted to the central and peripheral conducting airways, including inflammation and the structural changes referred to as airway modelling or remodelling, depending on the age of the patient. This has implications for lung development in utero and its association with large and small airway dysfunction and hyper-responsiveness that might already be present at birth in those likely to develop asthma. As the majority of asthma cases, at least in childhood, adopt a T2-type immune pattern, a key question is what drives the development of this immunological pathway as opposed to others. Adaptive immunity is just that. It has to be initiated by some other event (or events). Where does the initial defect (or defects) lie? Have we overemphasized altered immunity towards the allergic phenotype at the expense of abnormalities of the airway structural cells, such as epithelial cells, fibroblasts, nerves, microvasculature and smooth muscle, along with the modelling or remodelling components? Given that these airway components of adult asthma increase in response to mechanotransductive as well as inflammatory stimuli, is it possible that repeated wheeze in infancy can similarly initiate asthma in susceptible individuals?
Bronchial thermoplasty is an example of a disruptive technology that looks promising but needs further study to identify which patients benefit most from this invasive intervention. There is early evidence that this procedure targets smooth muscle, possibly suggesting that to overcome asthma more attention needs to be given to influencing modelling or remodelling pathways as well as halting inflammation. A crucial question that still needs answering is whether there is a mutual interdependency of airway inflammation (T2 type) and the onset of smooth muscle and other remodelling characteristics of asthma. If so, how and when does this occur? If interventions were able to alter the airway structural components (the ‘soil’) in which the T2-type inflammatory response (the ‘seed’) takes hold at the initiation of asthma, would we stand a better chance of preventing or even curing established disease?
Lifecourse epidemiology also indicates that severe asthma in early childhood continues throughout life. Low lung function at birth is a primary risk factor for asthma and, therefore, research into the causes of this both at birth and in early life is needed to uncover potentially modifiable exposures. Understanding the heterogeneity of severe asthma and using modern biomarker and statistical tools are opening up the field into asthma subtypes. This is a key focus of current research as differing causal pathways (endotypes) might be involved in different subtypes of asthma. New diagnostic tests to identify these subtypes and specific treatments, especially biologics, which target particular pathways represent new directions for asthma research where the focus is on identifying treatable disease traits. Thus, the proliferation of biologics now being trialled in severe asthma, although at present these only target components of the T2-type pathway (including IgE, IL-4, IL-13, IL-5 and TSLP), herald a new era in highly targeted, pathway-directed therapy that will require companion diagnostics to make them cost effective. Progress is also being made on the diagnostics front, with sputum and blood eosinophil counts, FeNO and levels of IgE and serum periostin all emerging as candidates. It now seems crucial that, even with current biomarkers, great attempts are made to place inflammation at the forefront of asthma diagnosis and management as a treatable trait that complements PROMs, symptoms and lung function. Implementation research is required to embed such a change so that laboratories and practices routinely offer such tests. Targeted therapy will increase in use as new causative endotypes are discovered, but even with current anti-inflammatory therapy (ICSs and LTRAs), biomarker monitoring needs to become an integral part of assessing the patients' treatment response.
As in adults, the availability of these novel, mostly biologic, therapies offers new opportunities for managing asthma in children. If intervention on the T2-type pathway took place early in life, could the natural history of asthma be influenced or the disease cured? Only clinical trials dedicated to asthma in children will answer this key question. There are already encouraging results for IgE-specific and allergen immunotherapy, with the possibility of halting the ‘allergic march’. If only we knew more about what happens when children lose their asthma in adolescence, and likewise how adults can lose their asthma, then new tractable therapeutic pathways might emerge.
Turning to asthma in adults, we need better treatments for asthma exacerbations that are linked in large part to viral infections. Death from asthma is almost always avoidable as it is usually owing to undertreatment and lack of adherence to the use of anti-inflammatory controller medications. It is clear that better integrative management, based on chronic diseases education for patients, family doctors, nurses and pharmacists, and possibly supported by shared decision making and smartphone or tablet computer applications for the delivery of asthma self-management programmes, is required.
At a more mechanistic level, increased understanding of the underlying cellular and molecular events that predispose individuals with asthma to exacerbations is required. This should involve further study not only on viral infection but also on the role of air pollution and other environmental exposures, including stress in exacerbating asthma symptoms. In addition, although late-onset asthma remains enigmatic, asthma subphenotyping has identified several different phenotypic clusters linked to different causal pathways. Very little progress has been made to date on understanding how these types of asthma begin, although, as in children, there seems to be a link to an initial or persistent viral infection. There is an urgent need to understand more about this role of viral infection, the interactions with sex hormones, including why and how women and girls are more affected by asthma over the lifecourse, and the different inflammatory asthma profiles, including T2 type, T17 type and T2/T17-low type.
The future for asthma treatment very much looks as if it is moving along the personalized (P4 or stratified) approach to health care. If successful, this approach of targeting the right intervention to the right patient at the right time will create new opportunities for both prevention and cure.
PowerPoint slides
Author Contributions
Introduction (S.T.H.); Epidemiology (S.T.W.); Mechanisms/pathophysiology (P.D.S. and S.T.H.); Diagnosis, screening and prevention (D.S.P., H.R., S.T.H. and S.W.); Management (D.S.P., P.D.S. and S.T.H.); Quality of life (D.S.P.); Outlook (D.S.P., H.R., P.D.S., S.T.H., S.T.W. and S.W.); Overview of Primer (S.T.H.).
Competing interests
S.T.H. is a non-executive director and a consultant for Synairgen, and a consultant for AstraZeneca and Novartis. He is in receipt of a UK Medical Research Council (MRC) programme grant and receives salary support as an MRC clinical professor. S.W. has served as a consultant for Aerocrine, GlaxoSmithKline and AstraZeneca. She has received research funding (paid to her institution) from GlaxoSmithKline, AstraZeneca, Genentech and Sanofi-Aventis. She has also received research support from the NIH National Heart, Lung and Blood Institute (NHLBI) and the National Institute of Allergy and Infectious Diseases (NIAID), which provides her salary support. D.S.P. has received an unrestricted educational grant for research (paid to the University of Groningen) from AstraZeneca. Her travel to the European Respiratory Society (ERS) and/or the American Thoracic Society (ATS) meetings has been partially funded by AstraZeneca, Chiesi, GlaxoSmithKline and Takeda. Fees for consultancies were given to the University of Groningen by AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Takeda and Teva. Her travel and lectures in China have been funded by Chiesi. S.T.W. is funded by the NIH NHLBI and is a consultant for the TENOR study for Novartis. H.R. has received research support from the German Research Foundation (DFG), the Federal Ministry of Education and Research (BMBF), the European Union, Land Hessen, the German Academic Exchange Service (DAAD), ALK, Stiftung Pathobiochemie, Ernst-Wendt-Stiftung, Mead Johnson Nutritional and Beckman Coulter. He has also received speakers honoraria from Allergopharma, Novartis, Thermo Fisher Scientific, Danone, Mead Johnson Nutritional and Bencard Allergie. H.R. also serves as a consultant for Bencard Allergie and Sterna Biologicals (for which he is also a cofounder). P.D.S. declares no competing interests.
References
- 1.Global Initiative for Asthma. Global strategy for asthma management and prevention GINA[online], (2015).
- 2.Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. N. Engl. J. Med. 1998;339:1194–1200. doi: 10.1056/NEJM199810223391703. [DOI] [PubMed] [Google Scholar]
- 3.Porsbjerg C, Lange P, Ulrik CS. Lung function impairment increases with age of diagnosis in adult onset asthma. Respir. Med. 2015;109:821–827. doi: 10.1016/j.rmed.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Custovic A. To what extent is allergen exposure a risk factor for the development of allergic disease? Clin. Exp. Allergy. 2015;45:54–62. doi: 10.1111/cea.12450. [DOI] [PubMed] [Google Scholar]
- 5.Amelink M. Severe adult-onset asthma: a distinct phenotype. J. Allergy Clin. Immunol. 2013;132:336–341. doi: 10.1016/j.jaci.2013.04.052. [DOI] [PubMed] [Google Scholar]
- 6.Ledford DK, Lockey RF. Asthma and comorbidities. Curr. Opin. Allergy Clin. Immunol. 2013;13:78–86. doi: 10.1097/ACI.0b013e32835c16b6. [DOI] [PubMed] [Google Scholar]
- 7.Custovic A. EAACI position statement on asthma exacerbations and severe asthma. Allergy. 2013;68:1520–1531. doi: 10.1111/all.12275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu L. Natural progression of childhood asthma symptoms and strong influence of sex and puberty. Ann. Am. Thorac. Soc. 2014;11:939–944. doi: 10.1513/AnnalsATS.201402-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soyka MB, van de Veen W, Holzmann D, Akdis M, Akdis CA. Scientific foundations of allergen-specific immunotherapy for allergic disease. Chest. 2014;146:1347–1357. doi: 10.1378/chest.14-0049. [DOI] [PubMed] [Google Scholar]
- 10.Murray CS, Woodcock A, Langley SJ, Morris J, Custovic A. Secondary prevention of asthma by the use of Inhaled Fluticasone propionate in Wheezy INfants (IFWIN): double-blind, randomised, controlled study. Lancet. 2006;368:754–762. doi: 10.1016/S0140-6736(06)69285-4. [DOI] [PubMed] [Google Scholar]
- 11.Murray CS. Can inhaled corticosteroids influence the natural history of asthma? Curr. Opin. Allergy Clin. Immunol. 2008;8:77–81. doi: 10.1097/ACI.0b013e3282f41769. [DOI] [PubMed] [Google Scholar]
- 12.Moore WC. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 2010;181:315–323. doi: 10.1164/rccm.200906-0896OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.World Health Organization. Global surveillance, prevention and control of chronic respiratory diseases: a comprehensive approach. WHO[online], (2007).
- 14.Wang D. Cross-sectional epidemiological survey of asthma in Jinan, China. Respirology. 2013;18:313–322. doi: 10.1111/resp.12005. [DOI] [PubMed] [Google Scholar]
- 15.Jackson DJ, Hartert TV, Martinez FD, Weiss ST, Fahy JV. Asthma: NHLBI workshop on the primary prevention of chronic lung diseases. Ann. Am. Thorac. Soc. 2014;11:S139–S145. doi: 10.1513/AnnalsATS.201312-448LD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yunginger JW. A community-based study of the epidemiology of asthma. Incidence rates. Am. Rev. Respir. Dis. 1992;146:888–894. doi: 10.1164/ajrccm/146.4.888. [DOI] [PubMed] [Google Scholar]
- 17.Butland BK, Strachan DP. Asthma onset and relapse in adult life: the British 1958 birth cohort study. Ann. Allergy Asthma Immunol. 2007;98:337–343. doi: 10.1016/S1081-1206(10)60879-4. [DOI] [PubMed] [Google Scholar]
- 18.Arshad SH. Pathophysiological characterization of asthma transitions across adolescence. Respir. Res. 2014;15:153. doi: 10.1186/s12931-014-0153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szefler SJ. Advances in pediatric asthma in 2014: moving toward a population health perspective. J. Allergy Clin. Immunol. 2015;135:644–652. doi: 10.1016/j.jaci.2014.12.1921. [DOI] [PubMed] [Google Scholar]
- 20.Dumas O. Occupational irritants and asthma: an Estonian cross-sectional study of 34,000 adults. Eur. Respir. J. 2014;44:647–656. doi: 10.1183/09031936.00172213. [DOI] [PubMed] [Google Scholar]
- 21.Simpson A. Beyond atopy: multiple patterns of sensitization in relation to asthma in a birth cohort study. Am. J. Respir. Crit. Care Med. 2010;181:1200–1206. doi: 10.1164/rccm.200907-1101OC. [DOI] [PubMed] [Google Scholar]
- 22.Tai A. Outcomes of childhood asthma to the age of 50 years. J. Allergy Clin. Immunol. 2014;133:1572–1578.e3. doi: 10.1016/j.jaci.2013.12.1033. [DOI] [PubMed] [Google Scholar]
- 23.Wu W. Unsupervised phenotyping of Severe Asthma Research Program participants using expanded lung data. J. Allergy Clin. Immunol. 2014;133:1280–1288. doi: 10.1016/j.jaci.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sporik R, Holgate ST, Platts-Mills TA, Cogswell JJ. Exposure to house-dust mite allergen (Der p I) and the development of asthma in childhood — a prospective study. N. Engl. J. Med. 1990;323:502–507. doi: 10.1056/NEJM199008233230802. [DOI] [PubMed] [Google Scholar]
- 25.Permaul P. Allergens in urban schools and homes of children with asthma. Pediatr. Allergy Immunol. 2012;23:543–549. doi: 10.1111/j.1399-3038.2012.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olivieri M. Risk factors for new-onset cat sensitization among adults: a population-based international cohort study. J. Allergy Clin. Immunol. 2012;129:420–425. doi: 10.1016/j.jaci.2011.10.044. [DOI] [PubMed] [Google Scholar]
- 27.Djukanovic R. Mucosal inflammation in asthma. Am. Rev. Respir. Dis. 1990;142:434–457. doi: 10.1164/ajrccm/142.2.434. [DOI] [PubMed] [Google Scholar]
- 28.Fajt ML, Wenzel SE. Mast cells, their subtypes, and relation to asthma phenotypes. Ann. Am. Thorac. Soc. 2013;10:S158–S164. doi: 10.1513/AnnalsATS.201303-064AW. [DOI] [PubMed] [Google Scholar]
- 29.Brightling CE. Mast-cell infiltration of airway smooth muscle in asthma. N. Engl. J. Med. 2002;346:1699–1705. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- 30.Balzar S. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 2011;183:299–309. doi: 10.1164/rccm.201002-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holgate ST. Innate and adaptive immune responses in asthma. Nat. Med. 2012;18:673–683. doi: 10.1038/nm.2731. [DOI] [PubMed] [Google Scholar]
- 32.Fahy JV. Type 2 inflammation in asthma — present in most, absent in many. Nat. Rev. Immunol. 2015;15:57–65. doi: 10.1038/nri3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Truyen E. Evaluation of airway inflammation by quantitative Th1/Th2 cytokine mRNA measurement in sputum of asthma patients. Thorax. 2006;61:202–208. doi: 10.1136/thx.2005.052399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chesné J. IL-17 in severe asthma. Where do we stand? Am. J. Respir. Crit. Care Med. 2014;190:1094–1101. doi: 10.1164/rccm.201405-0859PP. [DOI] [PubMed] [Google Scholar]
- 35.Neill DR. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat. Immunol. 2010;11:577–584. doi: 10.1038/ni.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Gonzalez I, Steer CA, Takei F. Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol. 2015;36:189–195. doi: 10.1016/j.it.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Jain VV, Perkins DL, Finn PW. Costimulation and allergic responses: immune and bioinformatic analyses. Pharmacol. Ther. 2008;117:385–392. doi: 10.1016/j.pharmthera.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 39.Lambrecht BN, Hammad H. The immunology of asthma. Nat. Immunol. 2014;16:45–56. doi: 10.1038/ni.3049. [DOI] [PubMed] [Google Scholar]
- 40.Kay AB. The role of T lymphocytes in asthma. Chem. Immunol. Allergy. 2006;91:59–75. doi: 10.1159/000090230. [DOI] [PubMed] [Google Scholar]
- 41.Jacquet A. Interactions of airway epithelium with protease allergens in the allergic response. Clin. Exp. Allergy. 2011;41:305–311. doi: 10.1111/j.1365-2222.2010.03661.x. [DOI] [PubMed] [Google Scholar]
- 42.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–424. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Kemeny DM. The role of the T follicular helper cells in allergic disease. Cell. Mol. Immunol. 2012;9:386–389. doi: 10.1038/cmi.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perros F, Hoogsteden HC, Coyle AJ, Lambrecht BN, Hammad H. Blockade of CCR4 in a humanized model of asthma reveals a critical role for DC-derived CCL17 and CCL22 in attracting Th2 cells and inducing airway inflammation. Allergy. 2009;64:995–1002. doi: 10.1111/j.1398-9995.2009.02095.x. [DOI] [PubMed] [Google Scholar]
- 45.Bradding P, Walls AF, Holgate ST. The role of the mast cell in the pathophysiology of asthma. J. Allergy Clin. Immunol. 2006;117:1277–1284. doi: 10.1016/j.jaci.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 46.Ma LL, O'Byrne PM. The pharmacological modulation of allergen-induced asthma. Inflammopharmacology. 2013;21:113–124. doi: 10.1007/s10787-012-0155-3. [DOI] [PubMed] [Google Scholar]
- 47.Hargreave FE, Dolovich J, O'Byrne PM, Ramsdale EH, Daniel EE. The origin of airway hyperresponsiveness. J. Allergy Clin. Immunol. 1986;78:825–832. doi: 10.1016/0091-6749(86)90226-5. [DOI] [PubMed] [Google Scholar]
- 48.Diamant Z. Inhaled allergen bronchoprovocation tests. J. Allergy Clin. Immunol. 2013;132:1045–1055.e6. doi: 10.1016/j.jaci.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 49.Barnig C. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci. Transl. Med. 2013;5:174ra26. doi: 10.1126/scitranslmed.3004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barbers RG. Near fatal asthma: clinical and airway biopsy characteristics. Pulm. Med. 2012;2012:829608. doi: 10.1155/2012/829608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J. Allergy Clin. Immunol. 2011;128:451–462. doi: 10.1016/j.jaci.2011.04.047. [DOI] [PubMed] [Google Scholar]
- 52.Bjermer L. The role of small airway disease in asthma. Curr. Opin. Pulm. Med. 2014;20:23–30. doi: 10.1097/MCP.0000000000000018. [DOI] [PubMed] [Google Scholar]
- 53.Boucherat O, Boczkowski J, Jeannotte L, Delacourt C. Cellular and molecular mechanisms of goblet cell metaplasia in the respiratory airways. Exp. Lung Res. 2013;39:207–216. doi: 10.3109/01902148.2013.791733. [DOI] [PubMed] [Google Scholar]
- 54.Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. 1989;1:520–524. doi: 10.1016/S0140-6736(89)90067-6. [DOI] [PubMed] [Google Scholar]
- 55.Kanemitsu Y. Osteopontin and periostin are associated with a 20-year decline of pulmonary function in patients with asthma. Am. J. Respir. Crit. Care Med. 2014;190:472–474. doi: 10.1164/rccm.201403-0562LE. [DOI] [PubMed] [Google Scholar]
- 56.Liesker JJW. Reticular basement membrane in asthma and COPD: similar thickness, yet different composition. Int. J. Chron. Obstruct. Pulmon. Dis. 2009;4:127–135. doi: 10.2147/copd.s4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holgate ST. Epithelial–mesenchymal communication in the pathogenesis of chronic asthma. Proc. Am. Thorac. Soc. 2004;1:93–98. doi: 10.1513/pats.2306034. [DOI] [PubMed] [Google Scholar]
- 58.Chanez P. Platelet-derived growth factor in asthma. Allergy. 1995;50:878–883. doi: 10.1111/j.1398-9995.1995.tb02493.x. [DOI] [PubMed] [Google Scholar]
- 59.Puddicombe SM. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000;14:1362–1374. doi: 10.1096/fasebj.14.10.1362. [DOI] [PubMed] [Google Scholar]
- 60.Olgart Höglund C. Nerve growth factor levels and localisation in human asthmatic bronchi. Eur. Respir. J. 2002;20:1110–1116. doi: 10.1183/09031936.02.00205402. [DOI] [PubMed] [Google Scholar]
- 61.Lopez-Guisa JM. Airway epithelial cells from asthmatic children differentially express proremodeling factors. J. Allergy Clin. Immunol. 2012;129:990.e6–997.e6. doi: 10.1016/j.jaci.2011.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harkness LM, Ashton AW, Burgess JK. Asthma is not only an airway disease, but also a vascular disease. Pharmacol. Ther. 2015;148:17–33. doi: 10.1016/j.pharmthera.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 63.Puddicombe SM. Increased expression of p21waf cyclin-dependent kinase inhibitor in asthmatic bronchial epithelium. Am. J. Respir. Cell. Mol. Biol. 2003;28:61–68. doi: 10.1165/rcmb.4715. [DOI] [PubMed] [Google Scholar]
- 64.Alcala SE. Mitotic asynchrony induces transforming growth factor-β1 secretion from airway epithelium. Am. J. Respir. Cell. Mol. Biol. 2014;51:363–369. doi: 10.1165/rcmb.2013-0396OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson JR. Pericytes contribute to airway remodeling in a mouse model of chronic allergic asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015;308:L658–L671. doi: 10.1152/ajplung.00286.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Descalzi D. Importance of fibroblasts–myofibroblasts in asthma-induced airway remodeling. Recent Pat. Inflamm. Allergy Drug Discov. 2007;1:237–241. doi: 10.2174/187221307782418847. [DOI] [PubMed] [Google Scholar]
- 67.Pepe C. Differences in airway remodeling between subjects with severe and moderate asthma. J. Allergy Clin. Immunol. 2005;116:544–549. doi: 10.1016/j.jaci.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 68.Glezen WP, Taber LH, Frank AL, Kasel JA. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 1986;140:543–546. doi: 10.1001/archpedi.1986.02140200053026. [DOI] [PubMed] [Google Scholar]
- 69.Turner S. A longitudinal study of lung function from 1 month to 18 years of age. Thorax. 2014;69:1015–1020. doi: 10.1136/thoraxjnl-2013-204931. [DOI] [PubMed] [Google Scholar]
- 70.Morgan WJ. Outcome of asthma and wheezing in the first 6 years of life: follow-up through adolescence. Am. J. Respir. Crit. Care Med. 2005;172:1253–1258. doi: 10.1164/rccm.200504-525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robertson CF. Long-term outcome of childhood asthma. Med. J. Aust. 2002;177:S42–S44. doi: 10.5694/j.1326-5377.2002.tb04813.x. [DOI] [PubMed] [Google Scholar]
- 72.Van Putte-Katier N. Relationship between parental lung function and their children's lung function early in life. Eur. Respir. J. 2011;38:664–671. doi: 10.1183/09031936.00034210. [DOI] [PubMed] [Google Scholar]
- 73.Soler Artigas M. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat. Genet. 2011;43:1082–1090. doi: 10.1038/ng.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Young S. The influence of a family history of asthma and parental smoking on airway responsiveness in early infancy. N. Engl. J. Med. 1991;324:1168–1173. doi: 10.1056/NEJM199104253241704. [DOI] [PubMed] [Google Scholar]
- 75.Latzin P, Röösli M, Huss A, Kuehni CE, Frey U. Air pollution during pregnancy and lung function in newborns: a birth cohort study. Eur. Respir. J. 2009;33:594–603. doi: 10.1183/09031936.00084008. [DOI] [PubMed] [Google Scholar]
- 76.Stick SM, Burton PR, Gurrin L, Sly PD, LeSouëf PN. Effects of maternal smoking during pregnancy and a family history of asthma on respiratory function in newborn infants. Lancet. 1996;348:1060–1064. doi: 10.1016/S0140-6736(96)04446-7. [DOI] [PubMed] [Google Scholar]
- 77.Sly PD, Soto-Quiros ME, Landau LI, Hudson I, Newton-John H. Factors predisposing to abnormal pulmonary function after adenovirus type 7 pneumonia. Arch. Dis. Child. 1984;59:935–939. doi: 10.1136/adc.59.10.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sly PD, Hibbert ME. Childhood asthma following hospitalization with acute viral bronchiolitis in infancy. Pediatr. Pulmonol. 1989;7:153–158. doi: 10.1002/ppul.1950070307. [DOI] [PubMed] [Google Scholar]
- 79.Guilbert TW. Decreased lung function after preschool wheezing rhinovirus illnesses in children at risk to develop asthma. J. Allergy Clin. Immunol. 2011;128:532–538.e10. doi: 10.1016/j.jaci.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jackson DJ. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008;178:667–672. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jackson DJ. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am. J. Respir. Crit. Care Med. 2012;185:281–285. doi: 10.1164/rccm.201104-0660OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wark PAB. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Edwards MR. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol. 2013;6:797–806. doi: 10.1038/mi.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Belsky DW, Sears MR. The potential to predict the course of childhood asthma. Expert Rev. Respir. Med. 2014;8:137–141. doi: 10.1586/17476348.2014.879826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chan KN. Airway responsiveness in low birthweight children and their mothers. Arch. Dis. Child. 1988;63:905–910. doi: 10.1136/adc.63.8.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sonnenschein-van der Voort AM. Preterm birth, infant weight gain, and childhood asthma risk: a meta-analysis of 147,000 European children. J. Allergy Clin. Immunol. 2014;133:1317–1329. doi: 10.1016/j.jaci.2013.12.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kennedy JD. Lung function outcome in children of premature birth. J. Paediatr. Child Health. 1999;35:516–521. doi: 10.1046/j.1440-1754.1999.00422.x. [DOI] [PubMed] [Google Scholar]
- 88.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Renz H. Gene–environment interactions in chronic inflammatory disease. Nat. Immunol. 2011;12:273–277. doi: 10.1038/ni0411-273. [DOI] [PubMed] [Google Scholar]
- 90.Litonjua AA, Weiss ST. Is vitamin D deficiency to blame for the asthma epidemic? J. Allergy Clin. Immunol. 2007;120:1031–1035. doi: 10.1016/j.jaci.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 91.Weiss ST. Bacterial components plus vitamin D: the ultimate solution to the asthma (autoimmune disease) epidemic? J. Allergy Clin. Immunol. 2011;127:1128–1130. doi: 10.1016/j.jaci.2011.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sundar IK, Rahman I. Vitamin D and susceptibility of chronic lung diseases: role of epigenetics. Front. Pharmacol. 2011;2:50. doi: 10.3389/fphar.2011.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shaheen SO, Adcock IM. The developmental origins of asthma: does epigenetics hold the key? Am. J. Respir. Crit. Care Med. 2009;180:690–691. doi: 10.1164/rccm.200906-0893ED. [DOI] [PubMed] [Google Scholar]
- 94.Huang YJ, Boushey HA. The microbiome in asthma. J. Allergy Clin. Immunol. 2015;135:25–30. doi: 10.1016/j.jaci.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Holt PG, Strickland DH, Hales BJ, Sly PD. Defective respiratory tract immune surveillance in asthma: a primary causal factor in disease onset and progression. Chest. 2014;145:370–378. doi: 10.1378/chest.13-1341. [DOI] [PubMed] [Google Scholar]
- 96.Holt PG. Prevention — what is the most promising approach? Pediatr. Allergy Immunol. 2014;25:12–14. doi: 10.1111/pai.12194. [DOI] [PubMed] [Google Scholar]
- 97.Hales BJ. Antibacterial antibody responses associated with the development of asthma in house dust mite-sensitised and non-sensitised children. Thorax. 2012;67:321–327. doi: 10.1136/thoraxjnl-2011-200650. [DOI] [PubMed] [Google Scholar]
- 98.Hales BJ. Differences in the antibody response to a mucosal bacterial antigen between allergic and non-allergic subjects. Thorax. 2008;63:221–227. doi: 10.1136/thx.2006.069492. [DOI] [PubMed] [Google Scholar]
- 99.Holt PG, Sly PD. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat. Med. 2012;18:726–735. doi: 10.1038/nm.2768. [DOI] [PubMed] [Google Scholar]
- 100.Hollams EM. Th2-associated immunity to bacteria in teenagers and susceptibility to asthma. Eur. Respir. J. 2010;36:509–516. doi: 10.1183/09031936.00184109. [DOI] [PubMed] [Google Scholar]
- 101.Saglani S. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am. J. Respir. Crit. Care Med. 2007;176:858–864. doi: 10.1164/rccm.200702-212OC. [DOI] [PubMed] [Google Scholar]
- 102.Malmström K. Lung function, airway remodelling and inflammation in symptomatic infants: outcome at 3 years. Thorax. 2011;66:157–162. doi: 10.1136/thx.2010.139246. [DOI] [PubMed] [Google Scholar]
- 103.Sonnappa S, Bastardo CM, Saglani S, Bush A, Aurora P. Relationship between past airway pathology and current lung function in preschool wheezers. Eur. Respir. J. 2011;38:1431–1436. doi: 10.1183/09031936.00164910. [DOI] [PubMed] [Google Scholar]
- 104.O'Reilly R. Increased airway smooth muscle in preschool wheezers who have asthma at school age. J. Allergy Clin. Immunol. 2013;131:1024–1032.e16. doi: 10.1016/j.jaci.2012.08.044. [DOI] [PubMed] [Google Scholar]
- 105.Regamey N. Increased airway smooth muscle mass in children with asthma, cystic fibrosis, and non-cystic fibrosis bronchiectasis. Am. J. Respir. Crit. Care Med. 2008;177:837–843. doi: 10.1164/rccm.200707-977OC. [DOI] [PubMed] [Google Scholar]
- 106.An SS, Fredberg JJ. Biophysical basis for airway hyperresponsiveness. Can. J. Physiol. Pharmacol. 2007;85:700–714. doi: 10.1139/Y07-059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Grainge CL. Effect of bronchoconstriction on airway remodeling in asthma. N. Engl. J. Med. 2011;364:2006–2015. doi: 10.1056/NEJMoa1014350. [DOI] [PubMed] [Google Scholar]
- 108.Grainge C. Resistin-like molecule-β is induced following bronchoconstriction of asthmatic airways. Respirology. 2012;17:1094–1100. doi: 10.1111/j.1440-1843.2012.02215.x. [DOI] [PubMed] [Google Scholar]
- 109.Chu EK. Induction of the plasminogen activator system by mechanical stimulation of human bronchial epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2006;35:628–638. doi: 10.1165/rcmb.2006-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Park J-A, Drazen JM, Tschumperlin DJ. The chitinase-like protein YKL-40 is secreted by airway epithelial cells at base line and in response to compressive mechanical stress. J. Biol. Chem. 2010;285:29817–29825. doi: 10.1074/jbc.M110.103416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Holgate ST. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 2011;242:205–219. doi: 10.1111/j.1600-065X.2011.01030.x. [DOI] [PubMed] [Google Scholar]
- 112.Bedke N. Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response. PLoS ONE. 2012;7:e44580. doi: 10.1371/journal.pone.0044580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Djukanovic R. The effect of inhaled IFN-β on worsening of asthma symptoms caused by viral infections. A randomized trial. Am. J. Respir. Crit. Care Med. 2014;190:145–154. doi: 10.1164/rccm.201312-2235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vink NM, Postma DS, Schouten JP, Rosmalen JG, Boezen HM. Gender differences in asthma development and remission during transition through puberty: the TRacking Adolescents' Individual Lives Survey (TRAILS) study. J. Allergy Clin. Immunol. 2010;126:498–504.e6. doi: 10.1016/j.jaci.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 115.Van den Toorn LM. Airway inflammation is present during clinical remission of atopic asthma. Am. J. Respir. Crit. Care Med. 2001;164:2107–2113. doi: 10.1164/ajrccm.164.11.2006165. [DOI] [PubMed] [Google Scholar]
- 116.Wood RA, Laheri AN, Eggleston PA. The aerodynamic characteristics of cat allergen. Clin. Exp. Allergy. 1993;23:733–739. doi: 10.1111/j.1365-2222.1993.tb00360.x. [DOI] [PubMed] [Google Scholar]
- 117.Vonk JM. Childhood factors associated with asthma remission after 30 year follow up. Thorax. 2004;59:925–929. doi: 10.1136/thx.2003.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Broekema M. Airway eosinophilia in remission and progression of asthma: accumulation with a fast decline of FEV1. Respir. Med. 2010;104:1254–1262. doi: 10.1016/j.rmed.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 119.Broekema M. Persisting remodeling and less airway wall eosinophil activation in complete remission of asthma. Am. J. Respir. Crit. Care Med. 2011;183:310–316. doi: 10.1164/rccm.201003-0494OC. [DOI] [PubMed] [Google Scholar]
- 120.Volbeda F. Can AMP induce sputum eosinophils, even in subjects with complete asthma remission? Respir. Res. 2010;11:106. doi: 10.1186/1465-9921-11-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shaaban R. Rhinitis and onset of asthma: a longitudinal population-based study. Lancet. 2008;372:1049–1057. doi: 10.1016/S0140-6736(08)61446-4. [DOI] [PubMed] [Google Scholar]
- 122.Platts-Mills T, Vaughan J, Squillace S, Woodfolk J, Sporik R. Sensitisation, asthma, and a modified Th2 response in children exposed to cat allergen: a population-based cross-sectional study. Lancet. 2001;357:752–756. doi: 10.1016/S0140-6736(00)04168-4. [DOI] [PubMed] [Google Scholar]
- 123.Torrent M. Early-life allergen exposure and atopy, asthma, and wheeze up to 6 years of age. Am. J. Respir. Crit. Care Med. 2007;176:446–453. doi: 10.1164/rccm.200607-916OC. [DOI] [PubMed] [Google Scholar]
- 124.Scott M. Multifaceted allergen avoidance during infancy reduces asthma during childhood with the effect persisting until age 18 years. Thorax. 2012;67:1046–1051. doi: 10.1136/thoraxjnl-2012-202150. [DOI] [PubMed] [Google Scholar]
- 125.Kovac K, Dodig S, Tjesic-Drinkovic D, Raos M. Correlation between asthma severity and serum IgE in asthmatic children sensitized to Dermatophagoides pteronyssinus. Arch. Med. Res. 2007;38:99–105. doi: 10.1016/j.arcmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 126.Adgate JL, Ramachandran G, Cho SJ, Ryan AD, Grengs J. Allergen levels in inner city homes: baseline concentrations and evaluation of intervention effectiveness. J. Expo. Sci. Environ. Epidemiol. 2008;18:430–440. doi: 10.1038/sj.jes.7500638. [DOI] [PubMed] [Google Scholar]
- 127.MacDonald C, Sternberg A, Hunter PR. A systematic review and meta-analysis of interventions used to reduce exposure to house dust and their effect on the development and severity of asthma. Environ. Health Perspect. 2007;115:1691–1695. doi: 10.1289/ehp.10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Van Schayck OC, Maas T, Kaper J, Knottnerus AJ, Sheikh A. Is there any role for allergen avoidance in the primary prevention of childhood asthma? J. Allergy Clin. Immunol. 2007;119:1323–1328. doi: 10.1016/j.jaci.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 129.Braun-Fahrländer C. Environmental exposure to endotoxin and its relation to asthma in school-age children. N. Engl. J. Med. 2002;347:869–877. doi: 10.1056/NEJMoa020057. [DOI] [PubMed] [Google Scholar]
- 130.Ege MJ. Exposure to environmental microorganisms and childhood asthma. N. Engl. J. Med. 2011;364:701–709. doi: 10.1056/NEJMoa1007302. [DOI] [PubMed] [Google Scholar]
- 131.Heederik D, von Mutius E. Does diversity of environmental microbial exposure matter for the occurrence of allergy and asthma? J. Allergy Clin. Immunol. 2012;130:44–50. doi: 10.1016/j.jaci.2012.01.067. [DOI] [PubMed] [Google Scholar]
- 132.Renz H, Brandtzaeg P, Hornef M. The impact of perinatal immune development on mucosal homeostasis and chronic inflammation. Nat. Rev. Immunol. 2011;12:9–23. doi: 10.1038/nri3112. [DOI] [PubMed] [Google Scholar]
- 133.Carroll KN. Season of infant bronchiolitis and estimates of subsequent risk and burden of early childhood asthma. J. Allergy Clin. Immunol. 2009;123:964–966. doi: 10.1016/j.jaci.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jackson DJ, Lemanske RF. The role of respiratory virus infections in childhood asthma inception. Immunol. Allergy Clin. North Am. 2010;30:513–522. doi: 10.1016/j.iac.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Durrani SR. Innate immune responses to rhinovirus are reduced by the high-affinity IgE receptor in allergic asthmatic children. J. Allergy Clin. Immunol. 2012;130:489–495. doi: 10.1016/j.jaci.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mortensen LJ, Kreiner-Møller E, Hakonarson H, Bønnelykke K, Bisgaard H. The PCDH1 gene and asthma in early childhood. Eur. Respir. J. 2014;43:792–800. doi: 10.1183/09031936.00021613. [DOI] [PubMed] [Google Scholar]
- 137.Bønnelykke K. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 2014;46:51–55. doi: 10.1038/ng.2830. [DOI] [PubMed] [Google Scholar]
- 138.Klaassen EM. An ADAM33 polymorphism associates with progression of preschool wheeze into childhood asthma: a prospective case–control study with replication in a birth cohort study. PLoS ONE. 2015;10:e0119349. doi: 10.1371/journal.pone.0119349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huang S-K, Zhang Q, Qiu Z, Chung KF. Mechanistic impact of outdoor air pollution on asthma and allergic diseases. J. Thorac. Dis. 2015;7:23–33. doi: 10.3978/j.issn.2072-1439.2014.12.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Guarnieri M, Balmes JR. Outdoor air pollution and asthma. Lancet. 2014;383:1581–1592. doi: 10.1016/S0140-6736(14)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chan JK. Combustion-derived flame generated ultrafine soot generates reactive oxygen species and activates Nrf2 antioxidants differently in neonatal and adult rat lungs. Part. Fibre Toxicol. 2013;10:34. doi: 10.1186/1743-8977-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang P, Thevenot P, Saravia J, Ahlert T, Cormier SA. Radical-containing particles activate dendritic cells and enhance Th17 inflammation in a mouse model of asthma. Am. J. Respir. Cell. Mol. Biol. 2011;45:977–983. doi: 10.1165/rcmb.2011-0001OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nadeau K. Ambient air pollution impairs regulatory T-cell function in asthma. J. Allergy Clin. Immunol. 2010;126:845–852.e10. doi: 10.1016/j.jaci.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 144.Gowers AM. Does outdoor air pollution induce new cases of asthma? Biological plausibility and evidence; a review. Respirology. 2012;17:887–898. doi: 10.1111/j.1440-1843.2012.02195.x. [DOI] [PubMed] [Google Scholar]
- 145.Goodman JE, Chandalia JK, Thakali S, Seeley M. Meta-analysis of nitrogen dioxide exposure and airway hyper-responsiveness in asthmatics. Crit. Rev. Toxicol. 2009;39:719–742. doi: 10.3109/10408440903283641. [DOI] [PubMed] [Google Scholar]
- 146.Gauderman WJ. Association of improved air quality with lung development in children. N. Engl. J. Med. 2015;372:905–913. doi: 10.1056/NEJMoa1414123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang IA, Fong KM, Zimmerman PV, Holgate ST, Holloway JW. Genetic susceptibility to the respiratory effects of air pollution. Postgrad. Med. J. 2009;85:428–436. doi: 10.1136/thx.2007.079426. [DOI] [PubMed] [Google Scholar]
- 148.Esposito S. Possible molecular mechanisms linking air pollution and asthma in children. BMC Pulm. Med. 2014;14:31. doi: 10.1186/1471-2466-14-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vork KL, Broadwin RL, Blaisdell RJ. Developing asthma in childhood from exposure to secondhand tobacco smoke: insights from a meta-regression. Environ. Health Perspect. 2007;115:1394–1400. doi: 10.1289/ehp.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Burke H. Prenatal and passive smoke exposure and incidence of asthma and wheeze: systematic review and meta-analysis. Pediatrics. 2012;129:735–744. doi: 10.1542/peds.2011-2196. [DOI] [PubMed] [Google Scholar]
- 151.Feleszko W. Parental tobacco smoking is associated with augmented IL-13 secretion in children with allergic asthma. J. Allergy Clin. Immunol. 2006;117:97–102. doi: 10.1016/j.jaci.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 152.Li Y-F, Langholz B, Salam MT, Gilliland FD. Maternal and grandmaternal smoking patterns are associated with early childhood asthma. Chest. 2005;127:1232–1241. doi: 10.1378/chest.127.4.1232. [DOI] [PubMed] [Google Scholar]
- 153.Mebrahtu TF, Feltbower RG, Greenwood DC, Parslow RC. Childhood body mass index and wheezing disorders: a systematic review and meta-analysis. Pediatr. Allergy Immunol. 2015;26:62–72. doi: 10.1111/pai.12321. [DOI] [PubMed] [Google Scholar]
- 154.Forno E, Young OM, Kumar R, Simhan H, Celedon JC. Maternal obesity in pregnancy, gestational weight gain, and risk of childhood asthma. Pediatrics. 2014;134:e535–e546. doi: 10.1542/peds.2014-0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Torday JS. Leptin mediates the parathyroid hormone-related protein paracrine stimulation of fetal lung maturation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;282:L405–L410. doi: 10.1152/ajplung.2002.282.3.L405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shore SA. Effect of leptin on allergic airway responses in mice. J. Allergy Clin. Immunol. 2005;115:103–109. doi: 10.1016/j.jaci.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 157.McLachlan CR. Adiposity, asthma, and airway inflammation. J. Allergy Clin. Immunol. 2007;119:634–639. doi: 10.1016/j.jaci.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 158.Kattan M. Asthma control, adiposity, and adipokines among inner-city adolescents. J. Allergy Clin. Immunol. 2010;125:584–592. doi: 10.1016/j.jaci.2010.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Aaron SD. Overdiagnosis of asthma in obese and nonobese adults. CMAJ. 2008;179:1121–1131. doi: 10.1503/cmaj.081332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.[No authors listed.] Statement on standards for the diagnosis and care of patients with chronic obstructive pulmonary disease (COPD) and asthma. Am. Rev. Respir. Dis.136, 225–244 (1987). [DOI] [PubMed]
- 161.National Institute for Health and Care Excellence. Asthma: diagnosis and monitoring of asthma in adults, children and young people. NICE[online], (2015). [PubMed]
- 162.Pavord ID, Bush A, Holgate S. Asthma diagnosis: addressing the challenges. Lancet Respir. Med. 2015;3:339–341. doi: 10.1016/S2213-2600(15)00056-9. [DOI] [PubMed] [Google Scholar]
- 163.Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat. Med. 2012;18:716–725. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 164.Corren J. Lebrikizumab treatment in adults with asthma. N. Engl. J. Med. 2011;365:1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 165.Woodruff PG. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Green RH. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet. 2002;360:1715–1721. doi: 10.1016/S0140-6736(02)11679-5. [DOI] [PubMed] [Google Scholar]
- 167.Jayaram L. Determining asthma treatment by monitoring sputum cell counts: effect on exacerbations. Eur. Respir. J. 2006;27:483–494. doi: 10.1183/09031936.06.00137704. [DOI] [PubMed] [Google Scholar]
- 168.Bel EH. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N. Engl. J. Med. 2014;371:1189–1197. doi: 10.1056/NEJMoa1403291. [DOI] [PubMed] [Google Scholar]
- 169.Pavord ID. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–659. doi: 10.1016/S0140-6736(12)60988-X. [DOI] [PubMed] [Google Scholar]
- 170.Guo FH. Interferon γ and interleukin 4 stimulate prolonged expression of inducible nitric oxide synthase in human airway epithelium through synthesis of soluble mediators. J. Clin. Invest. 1997;100:829–838. doi: 10.1172/JCI119598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Modena BD. Gene expression in relation to exhaled nitric oxide identifies novel asthma phenotypes with unique biomolecular pathways. Am. J. Respir. Crit. Care Med. 2014;190:1363–1372. doi: 10.1164/rccm.201406-1099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Smith AD. Diagnosing asthma: comparisons between exhaled nitric oxide measurements and conventional tests. Am. J. Respir. Crit. Care Med. 2004;169:473–478. doi: 10.1164/rccm.200310-1376OC. [DOI] [PubMed] [Google Scholar]
- 173.Wenzel S. Dupilumab in persistent asthma with elevated eosinophil levels. N. Engl. J. Med. 2013;368:2455–2466. doi: 10.1056/NEJMoa1304048. [DOI] [PubMed] [Google Scholar]
- 174.Hanania NA. Exploring the effects of omalizumab in allergic asthma: an analysis of biomarkers in the EXTRA study. Am. J. Respir. Crit. Care Med. 2013;187:804–811. doi: 10.1164/rccm.201208-1414OC. [DOI] [PubMed] [Google Scholar]
- 175.Takayama G. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J. Allergy Clin. Immunol. 2006;118:98–104. doi: 10.1016/j.jaci.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 176.Sidhu SS. Roles of epithelial cell-derived periostin in TGF-β activation, collagen production, and collagen gel elasticity in asthma. Proc. Natl Acad. Sci. USA. 2010;107:14170–14175. doi: 10.1073/pnas.1009426107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Conway SJ. The role of periostin in tissue remodeling across health and disease. Cell. Mol. Life Sci. 2014;71:1279–1288. doi: 10.1007/s00018-013-1494-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jia G. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J. Allergy Clin. Immunol. 2012;130:647–654.e10. doi: 10.1016/j.jaci.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Izuhara K. Periostin in allergic inflammation. Allergol. Int. 2014;63:143–151. doi: 10.2332/allergolint.13-RAI-0663. [DOI] [PubMed] [Google Scholar]
- 180.Schulman ES, Pohlig C. Rationale for specific allergen testing of patients with asthma in the clinical pulmonary office setting. Chest. 2015;147:251–258. doi: 10.1378/chest.12-0072. [DOI] [PubMed] [Google Scholar]
- 181.Ortega HG. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 2014;371:1198–1207. doi: 10.1056/NEJMoa1403290. [DOI] [PubMed] [Google Scholar]
- 182.Choy DF. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci. Transl. Med. 2015;7:301ra129. doi: 10.1126/scitranslmed.aab3142. [DOI] [PubMed] [Google Scholar]
- 183.Mauri P. Proteomics of bronchial biopsies: galectin-3 as a predictive biomarker of airway remodelling modulation in omalizumab-treated severe asthma patients. Immunol. Lett. 2014;162:2–10. doi: 10.1016/j.imlet.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 184.Duncan JM, Sears MR. Breastfeeding and allergies: time for a change in paradigm? Curr. Opin. Allergy Clin. Immunol. 2008;8:398–405. doi: 10.1097/ACI.0b013e32830d82ed. [DOI] [PubMed] [Google Scholar]
- 185.Kramer MS. Breastfeeding and allergy: the evidence. Ann. Nutr. Metab. 2011;59:S20–S26. doi: 10.1159/000334148. [DOI] [PubMed] [Google Scholar]
- 186.Oddy WH, Peat JK, de Klerk NH. Maternal asthma, infant feeding, and the risk of asthma in childhood. J. Allergy Clin. Immunol. 2002;110:65–67. doi: 10.1067/mai.2002.125296. [DOI] [PubMed] [Google Scholar]
- 187.Sears MR. Long-term relation between breastfeeding and development of atopy and asthma in children and young adults: a longitudinal study. Lancet. 2002;360:901–907. doi: 10.1016/S0140-6736(02)11025-7. [DOI] [PubMed] [Google Scholar]
- 188.Dogaru CM, Nyffenegger D, Pescatore AM, Spycher BD, Kuehni CE. Breastfeeding and childhood asthma: systematic review and meta-analysis. Am. J. Epidemiol. 2014;179:1153–1167. doi: 10.1093/aje/kwu072. [DOI] [PubMed] [Google Scholar]
- 189.Ramos GF. Cost-effectiveness of primary prevention of paediatric asthma: a decision-analytic model. Eur. J. Health Econ. 2014;15:869–883. doi: 10.1007/s10198-013-0532-x. [DOI] [PubMed] [Google Scholar]
- 190.Litonjua AA. The Vitamin D Antenatal Asthma Reduction Trial (VDAART): rationale, design, and methods of a randomized, controlled trial of vitamin D supplementation in pregnancy for the primary prevention of asthma and allergies in children. Contemp. Clin. Trials. 2014;38:37–50. doi: 10.1016/j.cct.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yang H, Xun P, He K. Fish and fish oil intake in relation to risk of asthma: a systematic review and meta-analysis. PLoS ONE. 2013;8:e80048. doi: 10.1371/journal.pone.0080048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Anandan C, Nurmatov U, Sheikh A. Omega 3 and 6 oils for primary prevention of allergic disease: systematic review and meta-analysis. Allergy. 2009;64:840–848. doi: 10.1111/j.1398-9995.2009.02042.x. [DOI] [PubMed] [Google Scholar]
- 193.Von Mutius E, Vercelli D. Farm living: effects on childhood asthma and allergy. Nat. Rev. Immunol. 2010;10:861–868. doi: 10.1038/nri2871. [DOI] [PubMed] [Google Scholar]
- 194.Elazab N. Probiotic administration in early life, atopy, and asthma: a meta-analysis of clinical trials. Pediatrics. 2013;132:e666–e676. doi: 10.1542/peds.2013-0246. [DOI] [PubMed] [Google Scholar]
- 195.West CE, m M-L, Hernell O. Probiotics in primary prevention of allergic disease — follow-up at 8–9 years of age. Allergy. 2013;68:1015–1020. doi: 10.1111/all.12191. [DOI] [PubMed] [Google Scholar]
- 196.Bengmark S. Gut microbiota, immune development and function. Pharmacol. Res. 2013;69:87–113. doi: 10.1016/j.phrs.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 197.Nieto A. Allergy and asthma prevention 2014. Pediatr. Allergy Immunol. 2014;25:516–533. doi: 10.1111/pai.12305. [DOI] [PubMed] [Google Scholar]
- 198.Heine RG. Preventing atopy and allergic disease. Nestle Nutr. Inst. Workshop Ser. 2014;78:141–153. doi: 10.1159/000354954. [DOI] [PubMed] [Google Scholar]
- 199.Tamblyn R. Evaluating the impact of an integrated computer-based decision support with person-centered analytics for the management of asthma in primary care: a randomized controlled trial. J. Am. Med. Inform. Assoc. 2015;22:773–783. doi: 10.1093/jamia/ocu009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Griffiths C, Levy ML. Preventing avoidable asthma deaths. Practitioner. 2014;258:27–31. [PubMed] [Google Scholar]
- 201.Toelle BG, Ram FS. Written individualised management plans for asthma in children and adults. Cochrane Database Syst. Rev. 2004;2:CD002171. doi: 10.1002/14651858.CD002171.pub2. [DOI] [PubMed] [Google Scholar]
- 202.Depner M. Clinical and epidemiologic phenotypes of childhood asthma. Am. J. Respir. Crit. Care Med. 2014;189:129–138. doi: 10.1164/rccm.201307-1198OC. [DOI] [PubMed] [Google Scholar]
- 203.Pedersen SE. Global strategy for the diagnosis and management of asthma in children 5 years and younger. Pediatr. Pulmonol. 2011;46:1–17. doi: 10.1002/ppul.21321. [DOI] [PubMed] [Google Scholar]
- 204.Roberts NJ. The development and comprehensibility of a pictorial asthma action plan. Patient Educ. Couns. 2009;74:12–18. doi: 10.1016/j.pec.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 205.National Heart Lung and Blood Advisory Council Asthma Expert Working Group. Draft needs assessment report for potential update of the expert panel report-3 (2007): guidelines for the diagnosis and management of asthma January 2015. NIH[online], (2015).
- 206.British Thoracic Society & Scottish Intercollegiate Guidelines Network British guideline on the management of asthma. Thorax. 2014;69:S1–S192. [Google Scholar]
- 207.Papi A. Beclometasone-formoterol as maintenance and reliever treatment in patients with asthma: a double-blind, randomised controlled trial. Lancet Respir. Med. 2013;1:23–31. doi: 10.1016/S2213-2600(13)70012-2. [DOI] [PubMed] [Google Scholar]
- 208.Lipworth BJ. Emerging role of long acting muscarinic antagonists for asthma. Br. J. Clin. Pharmacol. 2014;77:55–62. doi: 10.1111/bcp.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Matera MG, Cazzola M. Ultra-long-acting beta2-adrenoceptor agonists: an emerging therapeutic option for asthma and COPD? Drugs. 2007;67:503–515. doi: 10.2165/00003495-200767040-00002. [DOI] [PubMed] [Google Scholar]
- 210.Loymans RJB. omparative effectiveness of long term drug treatment strategies to prevent asthma exacerbations: network meta-analysis. BMJ. 2014;348:g3009. doi: 10.1136/bmj.g3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Abramson, M. J., Puy, R. M. & Weiner, J. M. Injection allergen immunotherapy for asthma. Cochrane Database Syst. Rev.8, CD001186 (2010). [DOI] [PubMed]
- 212.Mosbech H. Standardized quality (SQ) house dust mite sublingual immunotherapy tablet (ALK) reduces inhaled corticosteroid use while maintaining asthma control: a randomized, double-blind, placebo-controlled trial. J. Allergy Clin. Immunol. 2014;134:568–575.e7. doi: 10.1016/j.jaci.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 213.Kelly W, Massoumi A, Lazarus A. Asthma in pregnancy: physiology, diagnosis, and management. Postgrad. Med. 2015;127:349–358. doi: 10.1080/00325481.2015.1016386. [DOI] [PubMed] [Google Scholar]
- 214.Price D. Reassessing the evidence hierarchy in asthma: evaluating comparative effectiveness. Curr. Allergy Asthma Rep. 2011;11:526–538. doi: 10.1007/s11882-011-0222-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Qamar N, Pappalardo AA, Arora VM, Press VG. Patient-centered care and its effect on outcomes in the treatment of asthma. Patient Relat. Outcome Meas. 2011;2:81–109. doi: 10.2147/PROM.S12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hinks TSC. Innate and adaptive T cells in asthmatic patients: relationship to severity and disease mechanisms. J. Allergy Clin. Immunol. 2015;136:323–333. doi: 10.1016/j.jaci.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Holgate ST. Stratified approaches to the treatment of asthma. Br. J. Clin. Pharmacol. 2013;76:277–291. doi: 10.1111/bcp.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Campo P. Phenotypes and endotypes of uncontrolled severe asthma: new treatments. J. Investig. Allergol. Clin. Immunol. 2013;23:76–88. [PubMed] [Google Scholar]
- 219.Krug N. Allergen-induced asthmatic responses modified by a GATA3-specific DNAzyme. N. Engl. J. Med. 2015;372:1987–1995. doi: 10.1056/NEJMoa1411776. [DOI] [PubMed] [Google Scholar]
- 220.Hambly N, Nair P. Monoclonal antibodies for the treatment of refractory asthma. Curr. Opin. Pulm. Med. 2014;20:87–94. doi: 10.1097/MCP.0000000000000007. [DOI] [PubMed] [Google Scholar]
- 221.Lima JJ. Do genetic polymorphisms alter patient response to inhaled bronchodilators? Expert Opin. Drug Metab. Toxicol. 2014;10:1231–1240. doi: 10.1517/17425255.2014.939956. [DOI] [PubMed] [Google Scholar]
- 222.Zuurhout MJL. Arg16 ADRB2 genotype increases the risk of asthma exacerbation in children with a reported use of long-acting β2-agonists: results of the PACMAN cohort. Pharmacogenomics. 2013;14:1965–1971. doi: 10.2217/pgs.13.200. [DOI] [PubMed] [Google Scholar]
- 223.Manoharan A. β2-adrenergic receptor Gly16Arg polymorphism and impaired asthma control in corticosteroid-treated asthmatic adults. Ann. Allergy. Asthma Immunol. 2015;114:421–423. doi: 10.1016/j.anai.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 224.Ortega VE. Effect of rare variants in ADRB2 on risk of severe exacerbations and symptom control during longacting β agonist treatment in a multiethnic asthma population: a genetic study. Lancet Respir. Med. 2014;2:204–213. doi: 10.1016/S2213-2600(13)70289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Meyers DA, Bleecker ER, Holloway JW, Holgate ST. Asthma genetics and personalised medicine. Lancet Respir. Med. 2014;2:405–415. doi: 10.1016/S2213-2600(14)70012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Miller RJ, Murgu SD. Interventional pulmonology for asthma and emphysema: bronchial thermoplasty and bronchoscopic lung volume reduction. Semin. Respir. Crit. Care Med. 2014;35:655–670. doi: 10.1055/s-0034-1395939. [DOI] [PubMed] [Google Scholar]
- 227.Castro M, Cox G, Wechsler ME, Niven RM. Bronchial thermoplasty: ready for prime time — the evidence is there! Chest. 2015;147:e73–e74. doi: 10.1378/chest.14-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Pretolani M. Reduction of airway smooth muscle mass by bronchial thermoplasty in patients with severe asthma. Am. J. Respir. Crit. Care Med. 2014;190:1452–1454. doi: 10.1164/rccm.201407-1374LE. [DOI] [PubMed] [Google Scholar]
- 229.Sheshadri A, McKenzie M, Castro M. Critical review of bronchial thermoplasty: where should it fit into asthma therapy? Curr. Allergy Asthma Rep. 2014;14:470. doi: 10.1007/s11882-014-0470-4. [DOI] [PubMed] [Google Scholar]
- 230.Iyer VN, Lim KG. Bronchial thermoplasty: reappraising the evidence (or lack thereof) Chest. 2014;146:17–21. doi: 10.1378/chest.14-0536. [DOI] [PubMed] [Google Scholar]
- 231.Ducharme FM, Tse SM, Chauhan B. Diagnosis, management, and prognosis of preschool wheeze. Lancet. 2014;383:1593–1604. doi: 10.1016/S0140-6736(14)60615-2. [DOI] [PubMed] [Google Scholar]
- 232.Guilbert TW. Long-term inhaled corticosteroids in preschool children at high risk for asthma. N. Engl. J. Med. 2006;354:1985–1997. doi: 10.1056/NEJMoa051378. [DOI] [PubMed] [Google Scholar]
- 233.Busse WW. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N. Engl. J. Med. 2011;364:1005–1015. doi: 10.1056/NEJMoa1009705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wasfi YS. Onset and duration of attenuation of exercise-induced bronchoconstriction in children by single-dose of montelukast. Allergy Asthma Proc. 2011;32:453–459. doi: 10.2500/aap.2011.32.3482. [DOI] [PubMed] [Google Scholar]
- 235.Schultz A, Brand PL. Phenotype-directed treatment of pre-school-aged children with recurrent wheeze. J. Paediatr. Child Health. 2012;48:E73–E78. doi: 10.1111/j.1440-1754.2011.02123.x. [DOI] [PubMed] [Google Scholar]
- 236.Lemanske RF. Step-up therapy for children with uncontrolled asthma receiving inhaled corticosteroids. N. Engl. J. Med. 2010;362:975–985. doi: 10.1056/NEJMoa1001278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Martinez FD. The connection between early life wheezing and subsequent asthma: the viral march. Allergol. Immunopathol. (Madr.) 2009;37:249–251. doi: 10.1016/j.aller.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 238.Martinez FD, Vercelli D. Asthma. Lancet. 2013;382:1360–1372. doi: 10.1016/S0140-6736(13)61536-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Van Asperen PP, Mellis CM, Sly PD, Robertson CF. Evidence-based asthma management in children — what's new? Med. J. Aust. 2011;194:383–384. doi: 10.5694/j.1326-5377.2011.tb03025.x. [DOI] [PubMed] [Google Scholar]
- 240.Zeiger RS. Daily or intermittent budesonide in preschool children with recurrent wheezing. N. Engl. J. Med. 2011;365:1990–2001. doi: 10.1056/NEJMoa1104647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Beigelman A. Do oral corticosteroids reduce the severity of acute lower respiratory tract illnesses in preschool children with recurrent wheezing? J. Allergy Clin. Immunol. 2013;131:1518–1525. doi: 10.1016/j.jaci.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Pelkonen AS. The effect of montelukast on respiratory symptoms and lung function in wheezy infants. Eur. Respir. J. 2013;41:664–670. doi: 10.1183/09031936.00173411. [DOI] [PubMed] [Google Scholar]
- 243.Wechsler ME. Getting control of uncontrolled asthma. Am. J. Med. 2014;127:1049–1059. doi: 10.1016/j.amjmed.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 244.Worth A. Patient-reported outcome measures for asthma: a systematic review. NPJ Prim. Care Respir. Med. 2014;24:14020. doi: 10.1038/npjpcrm.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Juniper, E. Measurement of health-related quality of life and asthma control. Quoltech[online],
- 246.Chen H. Asthma control, severity, and quality of life: quantifying the effect of uncontrolled disease. J. Allergy Clin. Immunol. 2007;120:396–402. doi: 10.1016/j.jaci.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 247.Lurie A, Marsala C, Hartley S, Bouchon-Meunier B, Dusser D. Patients' perception of asthma severity. Respir. Med. 2007;101:2145–2152. doi: 10.1016/j.rmed.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 248.Haselkorn T. Effect of weight change on asthma-related health outcomes in patients with severe or difficult-to-treat asthma. Respir. Med. 2009;103:274–283. doi: 10.1016/j.rmed.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 249.Hanania NA. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann. Intern. Med. 2011;154:573–582. doi: 10.7326/0003-4819-154-9-201105030-00002. [DOI] [PubMed] [Google Scholar]
- 250.Castro M. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am. J. Respir. Crit. Care Med. 2010;181:116–124. doi: 10.1164/rccm.200903-0354OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Earnshaw VA, Quinn DM. The impact of stigma in healthcare on people living with chronic illnesses. J. Health Psychol. 2012;17:157–168. doi: 10.1177/1359105311414952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Comer BS, Ba M, Singer CA, Gerthoffer WT. Epigenetic targets for novel therapies of lung diseases. Pharmacol. Ther. 2015;147:91–110. doi: 10.1016/j.pharmthera.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Boutin H, Boulet L-P. L'asthme au Quotidien. Press Universite Laval; 2005. [Google Scholar]
- 254.Bousquet J, Bousquet PJ, Godard P, Dyres J-P. The public health implications of asthma. Bull. World Health Organ. 2005;93:548–554. [PMC free article] [PubMed] [Google Scholar]
- 255.Holgate ST. A new look at the pathogenesis of asthma. Clin. Sci. 2009;118:439–450. doi: 10.1042/CS20090474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Bateman ED. Overall asthma control: the relationship between current control and future risk. J. Allergy Clin. Immunol. 2010;125:600–608.e6. doi: 10.1016/j.jaci.2009.11.033. [DOI] [PubMed] [Google Scholar]