

Key Points
The skin, together with other epithelial-cell interfaces with a hostile environment, supports a range of passive and active immune defence mechanisms.
Cutaneous immune responses serve as a model for the study of interactions between innate and acquired immune mechanisms.
Adaptive immune surveillance addresses the logistical challenge of targeting naive, effector and memory T cells to their respective sites of function by using distinct homing mechanisms at different stages of the immune response, termed primary, secondary and tertiary immune surveillance.
Primary immune surveillance involves the process by which tissue dendritic cells are induced to engulf foreign particles, undergo maturation and emigrate through the afferent lymphatics to the local draining lymph node, where they encounter naive T cells recruited from the peripheral circulation. This greatly increases the efficiency with which naive T cells are exposed to antigens presented by professional antigen-presenting cells.
Secondary immune surveillance involves the production and distribution of antigen-specific effector memory T cells that express homing receptors that direct their migration back to the tissue draining the lymph node where activation occurred and their participation in tissue-based immune responses. The persistence of memory T cells with both antigen and tissue specificity also protects against possible future encounters with the same pathogen, by providing a population of antigen-specific effector cells pre-targeted to the site where exposure to that pathogen might most probably recur.
Tertiary immune surveillance involves the production of central memory and effector cells potentially directed to lymph nodes and tissues other than the site of primary exposure, providing broad coverage in the event that the pathogen is encountered through a different route.
These concepts have implications for the understanding of both inflammatory skin disorders and the development of antitumour and antipathogen vaccine strategies.
Abstract
The skin, as the primary interface between the body and the environment, provides the first line of defence against a broad array of microbial pathogens and trauma. In addition to its properties as a physical barrier, the skin has many active defence mechanisms. In this review, we discuss the interaction between the innate and adaptive immune systems in the skin as a model for immune function at epithelial-cell interfaces with the environment. How these mechanisms account for the robust nature of cutaneous immune surveillance and how their dysregulation drives the pathogenesis of inflammatory skin disorders and skin-based tumours are the subjects of this review.
Main
We live in a hostile environment, surrounded by microbial pathogens and subject to a range of physical and chemical insults. To survive in this environment, vertebrates have evolved complex immune systems. A key element of this defence is the deployment of rapid response elements at the most probable sites of attack, which are the epithelial-cell boundaries between the body and the environment in the skin, gut and lungs.
As the body's largest and most exposed interface with the environment, the skin has a central role in host defence. Before the relatively recent discovery of the immunological defences of skin, the cutaneous interface was viewed as a passive barrier between the host and the hostile environment. In the past few decades, however, it has become apparent that the mechanical aspects of epidermal defence are reinforced by a versatile and robust system of immune surveillance1 (Fig. 1). The crucial role of immune surveillance in maintaining homeostasis is evident from the marked increase in the frequency and severity of cutaneous malignancies and infections when immune function is limited, for example in patients with genetic and acquired immunodeficiency disorders and in those receiving immunosuppressive therapy after organ transplantation2,3. The regulation of skin defence mechanisms is also crucial, as inappropriate or misdirected immune activity is implicated in the pathogenesis of a large variety of acquired inflammatory skin disorders, including psoriasis, atopic and allergic contact dermatitis, lichen planus, alopecia areata and vitiligo4,5,6,7,8,9,10. The role of immune dysfunction in these conditions is emphasized by their response to immunosuppressive therapeutic interventions11,12,13,14.
Figure 1. Immune-response elements in non-inflamed skin.
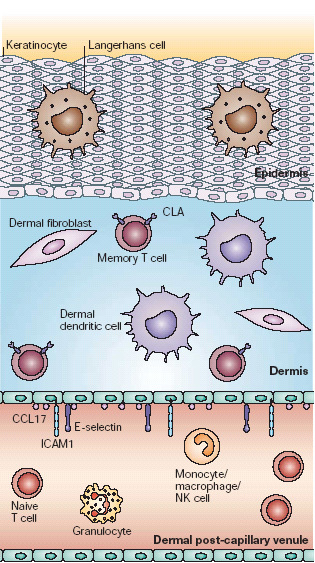
Human skin is composed of three distinct compartments relevant to its immune functions. First, the epidermis is composed of keratinized epithelial cells and functions as both a physical barrier and an early warning system. Immune cells resident in the epidermis include specialized dendritic cells (DCs) known as Langerhans cells and intraepithelial lymphocytes. Second, the dermis is mainly composed of connective tissue produced by dermal fibroblasts. Immune system cells resident in non-inflamed dermis include dermal DCs, mast cells and a small number of cutaneous lymphocyte antigen (CLA)-positive memory T cells. Third, dermal post-capillary venules constitutively express low levels of E-selectin, CC-chemokine ligand 17 (CCL17) and intercellular adhesion molecule 1 (ICAM1). These support the margination and baseline emigration of CLA+ memory T cells into non-inflamed skin. CLA− T cells, including both naive cells and memory/effector cells that are targeted to other tissues, as well as granulocytes and other immune cells, lack the appropriate receptors to attach to dermal vessels and emigrate into non-inflamed skin.
Understanding the mechanisms of immune surveillance in the skin and tissue-specific immune responses also has important implications for the rational design of vaccines. To promote protective immunity, an immunization protocol must elicit not only an antigen-specific immune response, but also an effective memory response that will provide long-lived protection at the most probable sites of invasion. This applies equally to immunization against infectious organisms, which in most cases invade through the skin or the epithelia of the gastrointestinal or respiratory systems, and to the elicitation of immune responses against tumours. As we discuss, the route and means of adjuvant stimulation that is used can affect the effectiveness and utility of specific vaccine strategies.
In this review, we discuss these issues in the context of recent advances in our understanding of cutaneous immune mechanisms, highlighting the interaction of innate and adaptive immune systems in the induction and maintenance of effective cutaneous immune surveillance.
Innate immune surveillance
Central to our model of cutaneous immune surveillance are the cells resident in the skin, which function as sentinels for DANGER SIGNALS, including invasion by microorganisms. Keratinocytes and LANGERHANS CELLS in the epidermis, as well as dermal mast cells, dendritic cells (DCs) and macrophages, provide an early warning system by releasing stored and inducible ANTIMICROBIAL PEPTIDES, chemotactic proteins and cytokines15,16,17,18,19 (Fig. 2). Keratinocyes are important and often under-appreciated participants in cutaneous immune responses. They produce large quantities of interleukin-1α (IL-1α), tumour-necrosis factor (TNF) and antimicrobial peptides such as β-defensins in response to various stimuli, including kinetic and thermal trauma, ultraviolet radiation, cytokines and neuropeptides15,20,21,22. IL-1α (and IL-1β from epidermal Langerhans cells), in turn, acts as a potent stimulator of local immune function23. Keratinocytes also produce a large number of chemokines and other immunoregulatory cytokines in response to stimulation16,24,25,26,27. These products have various important effects on resident innate immune cells in the skin, such as mast cells, DCs and macrophages, resulting in the upregulation of expression of other inducible mediators and recruitment of additional immune cells from the blood28. The induction of local inflammation through IL-1, however, depends on the balance of agonists (IL-1α, IL-1β, caspase-1 and IL-1 receptor 1; IL-1R1) and antagonists (IL-1Ra and IL-1R2) that are active in this pathway15,16,23. Each of these molecules can be produced by keratinocytes under various conditions, as well as by other cells that are resident in the skin, making it difficult to predict the effects of specific interventions. New members of the IL-1 family continue to be identified, adding to the complexity of regulation of cutaneous inflammation29.
Figure 2. Innate immune mechanisms in the skin.
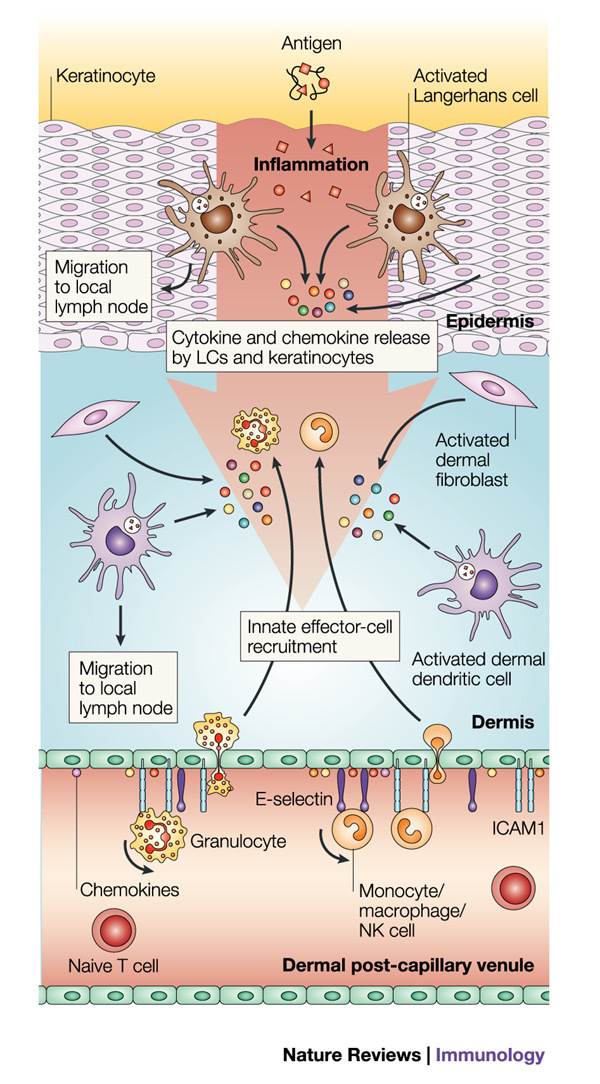
Epithelial-cell injury or pathogen invasion leads to the release of primary cytokines and the activation of both skin cells (keratinocytes and fibroblasts) and resident innate immune cells (Langerhans cells (LCs), dermal dendritic cells (DCs) and mast cells), stimulating downstream activation cascades. Activated Langerhans cells and dermal DCs are stimulated to mature and emigrate from the tissue to the draining lymph node, carrying antigen for presentation to naive and memory T cells. The cytokines and chemokines produced in response to this activation cascade act on the local endothelia through nuclear factor-κB (NF-κB)-mediated pathways to upregulate the expression of adhesion molecules, including E-selectin, P-selectin and intercellular adhesion molecule 1 (ICAM1), and direct the recruitment of additional innate immune components according to the specific signals that are generated — for example, neutrophils, eosinophils and natural killer (NK) cells. CCL17, CC-chemokine ligand 17.
Both the epithelial barrier cells and resident innate immune cells in the skin express pattern-recognition receptors that recognize specific pathogen components and can trigger downstream activation cascades. An important subset of these receptors belong to the Toll-like receptor (TLR) family30,31,32,33, which bind pathogen-associated molecular pattern molecules such as lipopolysaccharide34, bacterial lipoproteins35,36, flagellin37, yeast mannans38 and unmethylated CpG DNA motifs39,40. TLR expression is variable and might identify subsets of innate immune cells, such as DCs, with specific functions41,42,43. DC activation through TLRs results in increased production of pro-inflammatory cytokines and antimicrobial peptides, increased nitric oxide synthesis and enhanced bacterial killing, as well as increased antigen presentation30. The binding of TLR ligands is associated with the recruitment of intracellular adaptor proteins similar to those used by IL-1R and subsequent activation of the JUN N-terminal kinase (JNK) and nuclear factor-κB (NF-κB) signalling pathways30. The NF-κB signalling pathway is seen as a key link between the innate and adaptive immune systems. In the skin, NF-κB regulates the expression of numerous genes that are involved in the initiation of the inflammatory response, including adhesion molecules, chemokines and cytokines (such as IL-1 and TNF), matrix metalloproteases, nitric oxide synthase and enzymes that control PROSTANOID synthesis44. Beyond the direct effects of these compounds on pathogens and abnormal cells, products of the innate immune response direct the recruitment of additional leukocytes to the site of activation. In humans, genes regulated by NF-κB include the endothelial adhesion molecules E-selectin and P-selectin, intercellular adhesion molecule 1 (ICAM1), vascular cell-adhesion molecule 1 (VCAM1), and various chemokines and cytokines45. Collectively, these molecules are considered to be both necessary and sufficient for initiation of the leukocyte adhesion–extravasation cascade that recruits circulating leukocytes from the periphery46. These include antigen non-specific leukocytes, such as neutrophils and natural killer cells, as well as key components of the adaptive immune system, such as effector T cells (Fig. 3).
Figure 3. Adaptive immune responses in the skin.
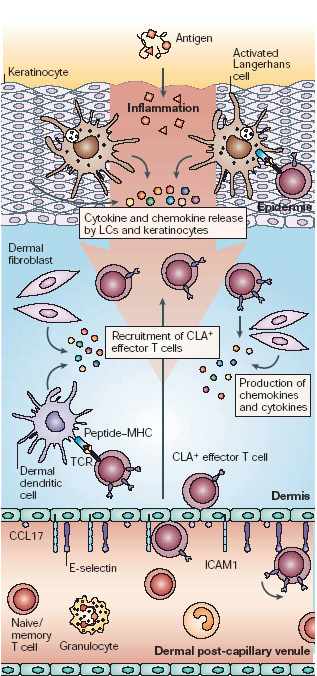
Circulating cutaneous lymphocyte antigen (CLA)-positive T cells represent a library of memory T cells with T-cell receptors (TCRs) specific for antigens previously encountered in the skin. Cytokines released by keratinocytes, fibroblasts and resident antigen-presenting cells stimulate the upregulation of expression of E-selectin and intercellular adhesion molecule 1 (ICAM1) through nuclear factor-κB (NF-κB)-mediated activation pathways. Production and presentation of T-cell-specific chemokines, such as CC-chemokine ligand 17 (CCL17), CCL22 and CCL27, on the local endothelium results in the recruitment of CLA+ T cells in an antigen non-specific manner. T cells entering the tissue that encounter their specific antigen presented by local macrophages or dendritic cells will be activated to proliferate and carry out their specific functions. Those that do not encounter their cognate antigen, which might be most of the cells that are recruited, will enter the lymphatics and return to the general circulation. LC, Langerhans cell.
Mast cells are another crucial component of the cutaneous immune response apparatus. Mast cells have been shown to release different patterns of cytokines and bioactive compounds, including leukotrienes, IL-1β, IL-4, IL-5, IL-6, IL-13, TNF and granulocyte–macrophage colony-stimulating factor (GM-CSF), in response to various TLR ligands47,48,49. These and other mast-cell products have an important role in both the initiation and modulation of innate immune responses and the generation of adaptive immune responses.
Adaptive immune surveillance
The adaptive immune system, based on T cells and B cells that express antigen-specific receptors, provides vertebrate organisms with a broader and more flexible repertoire of responses to pathogens, and a means for providing memory of past encounters. Adaptive immune surveillance addresses the logistical challenge faced by the immune system in getting the right T cell to the right place at the right time. At the skin interface, this process can be viewed as operating at three levels, which we term primary, secondary and tertiary immune surveillance (Fig. 4). Primary immune surveillance incorporates the mechanisms for bringing environmental antigens that are encountered in the skin, professional antigen-presenting cells (APCs) and naive T cells together in the specialized microenvironment of skin-draining lymph nodes. Secondary immune surveillance, in turn, involves the production and distribution of antigen-specific effector memory T cells expressing homing receptors that direct their migration to the tissue where antigen was encountered. Tertiary immune surveillance encompasses the long-term elements of the acquired immune response, including the production of central memory and effector cells that are potentially directed to tissues other than the site of primary exposure. Each of these modes of immune surveillance is a strategy used by the immune system to improve the odds that each T cell will find the antigen for which its T-cell receptor (TCRs) is specific and develop effective responses: first, by increasing the efficiency with which naive T cells are exposed to antigens; second, by targeting the effector response to the most appropriate tissue site; and third, by expanding coverage to other tissues.
Figure 4. Immune-surveillance mechanisms in the skin.
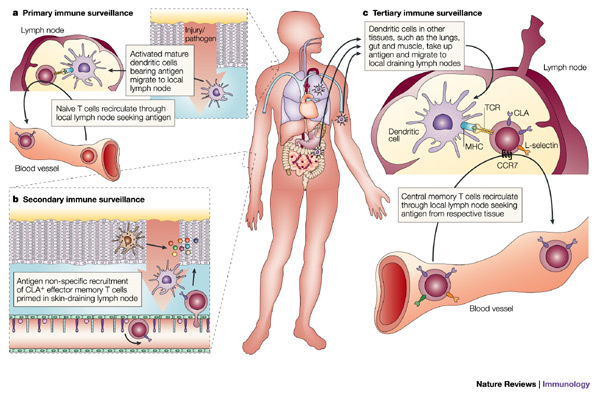
a | Primary immune surveillance is the process by which the innate immune response ensures the effective engagement of the adaptive immune response. Antigens encountered in the skin are carried by activated dendritic cells through the afferent lymphatics to the draining lymph nodes, and presented to naive and central memory T cells circulating through the node. This increases the likelihood of encountering T cells that express the appropriate T-cell receptor (TCR). T cells that encounter their cognate antigen proliferate and differentiate into effector cells expressing homing receptors for the tissue served by that node. b | Secondary immune surveillance provides a mechanism for ensuring rapid and effective local adaptive immune responses to previously encountered antigens. Tissue inflammation results in the upregulation of expression of adhesion molecules and presentation of specific chemokines on the local endothelium. Effector memory T cells that express the appropriate counter-receptors are recruited in an antigen non-specific manner. Those cells that encounter their cognate antigen presented by local antigen-presenting cells (APCs) participate in the local inflammatory response, whereas those that do not return to the general circulation. c | Tertiary immune surveillance represents a mechanism by which the immune system can hedge its bets, providing enhanced adaptive immune responses to antigens encountered in tissues distinct from those in which they were previously encountered. Central memory T cells produced in skin-draining lymph nodes express L-selectin and CC-chemokine receptor 7 (CCR7), which allows them to recirculate through lymph nodes throughout the body, where they can provide enhanced responses to antigen encountered through a different environmental interface. CLA, cutaneous lymphocyte antigen.
Primary immune surveillance. Activated DCs, whether derived from epidermal Langerhans cells or dermal DCs50,51, are professional APCs with the capacity to present antigens efficiently and to affect the maturation of naive T cells to a memory/effector phenotype52. At sites of injury or pathogen invasion in the skin, these cells become activated through innate mechanisms, including pattern-recognition receptors (such as TLRs) and exposure to the pro-inflammatory cytokines (such as IL-1 and TNF) that are released in response to tissue injury or infection. Activated APCs rapidly engulf foreign particles and undergo maturation as they emigrate through the afferent lymphatics to the local skin-draining lymph nodes52,53. This maturation process enhances antigen processing and upregulates the expression of MHC molecules and co-stimulatory molecules, including CD80 and CD86 (Ref. 54).
The function of the local draining lymph nodes is to promote frequent and supervised contact between antigens that are derived from a specific segment of the skin (carried by DCs that have migrated through afferent lymphatics) and the adaptive immune system (T cells entering the lymph node through high endothelial venules). Naive T cells that encounter their cognate antigen presented by an activated and mature DC will undergo proliferation and clonal expansion, produce autocrine growth factors and differentiate into memory/effector T cells.
Secondary immune surveillance. When an antigen is encountered in a specific tissue, such as the skin, the activation of T cells in the local draining lymph nodes results in the production of antigen-specific effector cells that express homing receptors for that site. In this way, the immune response is preferentially targeted back to the site of the initial infection or stimulation. T cells recruited to sites of inflammation in the skin will encounter a range of inflammatory mediators triggered by innate immune mechanisms, as well as activated dermal DCs and inflammatory dendritic epidermal cells (IDECs) that can present antigen and provide co-stimulatory signals to T cells that express appropriate counter-receptors55,56.
With regard to the recruitment of T cells to the skin, the earliest step in this process is the tethering and rolling of T cells on E-selectin and/or P-selectin expressed by dermal post-capillary venules. Skin-homing T cells can be identified by expression of the cell-surface carbohydrate epitope cutaneous lymphocyte antigen (CLA), which binds E-selectin. CLA is expressed by ∼30% of circulating memory T cells and is virtually absent on naive T cells57. T cells found in inflammatory skin lesions are mainly CD45RO+CLA+, whereas few T cells that accumulate in inflammatory sites other than the skin express CLA57,58. CLA is reproducibly found on most T cells present in cutaneous lymphocytic infiltrates of almost all skin diseases, including psoriasis, atopic dermatitis, allergic contact dermatitis, erythema multiforme, cutaneous GRAFT-VERSUS-HOST DISEASE (GVHD) and cutaneous T-cell lymphoma (CTCL)57,58,59,60,61,62,63,64. In biopsies of CTCL, both malignant T cells and those that respond to the presence of tumour cells in the skin are CLA+. CLA also seems to be a good marker of malignant CTCL cells in the peripheral blood of some patients with Sezary syndrome65.
The factors that induce CLA expression by T cells are less well understood, but seem to be related to the specialized environments present in secondary lymphoid tissue. That is, the microenvironment in skin-draining lymph nodes promotes the expression of CLA by newly activated effector T cells, whereas that of Peyer's patches, for example, favours the expression of α4β7 (a gut-homing receptor) by new effector T cells66,67,68. A large volume of circumstantial evidence supports this model. For example, circulating memory T cells specific for nickel or house-dust mite in allergic or atopic individuals, respectively, express high levels of CLA62, presumably because these antigens were encountered through the skin. Similarly, circulating CD8+ effector T cells specific for skin-associated viruses express CLA, whereas those specific for non-cutaneous viruses do not69. By contrast, T cells specific for rotavirus are CLA−, but express high levels of α4β7 (Ref. 70) as the immune system encounters this virus through the gut. More recently, mouse studies indicate that DCs derived from Peyer's patches can preferentially induce the expression of α4β7 by newly generated effector cells in vitro67,68.
Studies of CLA induction in vitro have indicated that expression is enhanced by CD3 activation in the presence of IL-12 and is not restricted to functional and phenotypic T-cell subsets71,72. However, the factors that regulate the induction of expression in vivo and the maintenance of expression by resting circulating cells have not been determined.
Although CLA and α4β7 mediate specific tethering and rolling steps in distinct tissue vascular beds, the activation of these rolling cells also proceeds in a tissue-specific manner. Several chemokines and their receptors are associated with skin-homing T cells73,74,75, including CC-chemokine receptor 4 (CCR4) and its ligands CCL17 (thymus and activation-regulated chemokine, TARC) and CCL22 (macrophage-derived chemokine, MDC). Constitutive and inducible expression of CCL17 on the luminal aspect of post-capillary venules in the skin has been shown, and CLA+ cells typically co-express CCR4. CCR4–CCL17 interactions can lead to the arrest of rolling T cells if they are provided an integrin ligand. CCL27 (cutaneous T-cell-attracting chemokine, CTACK) has also been implicated in skin homing. This chemokine, preferentially produced by epidermal keratinocytes, binds to CCR10 and is chemotactic for T cells in vitro76,77,78. CCR10 is expressed by a subset of CLA+ T cells only, and its role in inducing the arrest of T cells on post-capillary venules in the skin has not been shown. Other work indicates that CCR6 might be important for skin homing79, though the expression of this chemokine receptor is more variable. In most situations, it seems that skin-homing memory cells that express CLA, CCR4 and leukocyte function-associated antigen 1 (LFA1) accumulate in the skin, where E-selectin, CCL17 and ICAM1 are constitutively and inducibly expressed on post-capillary venules. What role other receptor–ligand pairs will have in specific conditions remains to be determined. Cytokines produced by T cells that are recruited to sites of inflammation can influence the content of the ongoing infiltrate by modifying the balance of chemokines produced. For example, interferon-γ (IFN-γ) can induce keratinocytes to produce a range of products, including CXCL10 (IFN-inducible protein 10, IP-10), CXCL9 (monokine induced by IFN-γ, MIG) and CXCL11 (IFN-inducible T-cell α-chemoattractant, ITAC), which act to recruit T cells that express the chemokine receptor CXCR3 (Ref. 80).
Many pathogens have evolved to use tissue-specific routes of entry. The persistence of memory T cells with both antigen and tissue specificity in the peripheral circulation prepares the immune system for possible future interaction with the same pathogen, by providing a population of antigen-specific effector cells pre-targeted to the site where exposure to that pathogen would be most likely to recur.
Although skin-homing T cells are a kind of rapid deployment corps that can be called up to inflamed tissues, there is also evidence for constitutive homing of such T cells to the skin. T cells recovered from non-inflamed skin express high levels of CLA and CCR4 as well as other chemokine receptors81,82. Even in the absence of inflammation, leukocytes are observed to tether and roll constitutively on low levels of selectin expressed in dermal post-capillary venules83,84. These cells can be thought of as continuously scanning the endothelial-cell surfaces of their target tissue for activation signals and are poised to respond to the slightest hint of danger. Constitutive expression of E-selectin on cutaneous microvessels has been described in both humans and mice, as has expression of CCL17 and ICAM1 (Refs 85–87). Using these sequential interactions, an indeterminate fraction of these T cells continuously enter the skin and traffic through it, seeking antigen-dependent activation. Antigen-specific T cells can also be detected in the uninflamed skin of patients with atopic dermatitis88. It is unclear whether T cells that home constitutively to the skin are responding to subclinical levels of inflammation or if alternative mechanisms exist that support constitutive expression of endothelial homing components. It is important to note that while they are in the skin, these cells can be thought of as 'resident' T cells; how long they reside in the skin is unknown at present.
Tertiary immune surveillance. Although a given pathogen is most likely to be re-encountered at the same epithelial-cell interface as it was originally engaged, this cannot be guaranteed. Among the T-cell subpopulations produced after an initial antigen encounter are a population of antigen-specific memory cells, known as central memory T cells, that retain expression of CD62 ligand (CD62L) and CCR7, and the ability to circulate through lymph nodes89. These cells can then emigrate from the lymph node in which they were originally produced to lymph nodes throughout the body (including those draining non-cutaneous epithelial-cell interfaces), where they may encounter DCs expressing the same cognate antigen. In this way, the immune system hedges its bets, ensuring a more rapid and effective response even if the next encounter occurs at a different interface.
Although the original description of central memory cells suggested that they could home to lymph nodes only, it has become clear that some T cells can express both central memory and tissue-homing receptors. For example, cells that express CLA, L-selectin, CCR4 and CCR7 are well represented in peripheral blood90. One interesting question that awaits investigation is whether central memory T cells that are generated in a skin-draining lymph node and resident in a different tissue lymph node (for example, gut or respiratory system lymph node) will, if exposed to antigen, give rise to new effectors of a skin-homing phenotype or effectors that home to the current source tissue, or both.
As seen from this discussion, innate immune-surveillance mechanisms drive the development of adaptive immune responses — that is, injury, inflammation and other danger signals facilitate T-cell development and entry into tissues. Memory T cells and innate immune effector cells can be thought to enter tissues not because they 'see' antigen, but because the local endothelium expresses appropriate counter-receptors and chemoattractants. Only after they have exited the blood can they respond to their antigen that is productively presented. This has important implications for the aetiopathology of inflammatory skin diseases.
Regulatory T cells. Although the mechanisms described earlier highlight the activation and recruitment of effector T cells, it is clear that REGULATORY T CELLS also have an important, though less well characterized, role in dampening exaggerated cutaneous immune responses, as well as in the maintenance of immune tolerance to innocuous self or exogenous antigens91,92. Recent reports have indicated that regulatory T-cell subsets might traffic to the skin using pathways that are similar to those used by effector cells73. An imbalance in effector/regulatory T-cell recruitment or functions might be a crucial factor in the development of inflammatory skin lesions. Conversely, for those conditions in which the antigen (self or exogenous) can be identified, induction of regulatory T cells to specific antigens could provide a powerful mechanism for inducing specific tolerance93.
Clinical implications
T-cell-mediated inflammatory skin diseases are extraordinarily common. Also, new therapies for disease have led to new T-cell-mediated skin diseases, notably GVHD after therapeutic allogeneic bone-marrow transplantation. If these diseases are viewed from our current perspective of cutaneous immune surveillance, insights emerge that are useful to understanding their clinical and biological behaviour.
The evidence described earlier indicates that the cutaneous immune-surveillance system responds to any cutaneous injury that produces danger signals as if it was potentially infectious. Both innate and adaptive immunity are mobilized, and their activities are synergistic. Inappropriate adaptive immunity can be driven by non-specific activation of innate immune pathways, for example, chronic trauma due to scratching of the skin. This in turn can lead to autoinflammatory feedback loops through the recruitment and activation of leukocytes independent of antigen-specific help. Different populations of cells accumulate in specific disease states, reflecting the patterns of expression of vascular adhesion molecules and chemoattractant cytokines induced by the balance of stimuli in that target organ (Table 1). This has potential significance for the immunopathology of diseases in organs other than the skin. Our continued understanding of mechanisms of cutaneous immune surveillance will almost certainly provide important insights into immune surveillance and diseases at other environmental epithelial-cell interfaces, including the gut, lungs, oropharyngeal and genital mucosa.
Table 1.
T cells in inflammatory skin disease
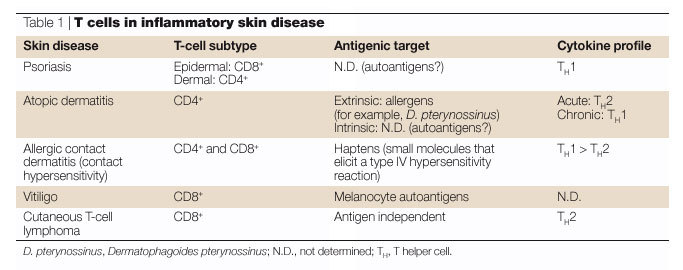
Psoriasis. Psoriasis, which affects ∼1–2% of adults worldwide, is characterized by the formation of erythematous cutaneous plaques covered with scale. Histologically, psoriatic plaques show keratinocyte hyperproliferation and both neutrophil infiltration of the upper epidermis and an infiltrate in the dermis and epidermis replete with T cells, DCs and macrophages.
Psoriasis has an obligate immunological component; therapies directed against T-cell activation and function are highly effective in this disease, and the disease can be initiated in xenograft models by activated T cells94. Increasingly, it is being understood as an autoimmune disease, although the autoantigen(s) has not been identified95. The T cells in psoriatic lesions are CLA+ and produce a type 1 cytokine profile. CD8+ T cells in particular have been identified in the epidermis and are thought to have a key role in disease expression.
So far, nearly all successful therapeutic interventions for psoriasis target T cells. These include corticosteroids, methotrexate, cyclosporine and ultraviolet light (with or without the photosensitizer 8-methoxy psoralens)96. More recently, immunobiological therapy has come to the forefront in this disease, with reports of polarizing therapies such as IL-4, IL-10 and IL-11, which inhibit T HELPER 1 (TH1) CELLS and/or enhance TH2-CELL functions, showing promise97,98,99. Other biological agents that inhibit T-cell recruitment or activation, including alefacept (an LFA3– immunoglobulin fusion protein), efalizumab (a humanized antibody specific for CD11a) and cytotoxic T lymphocyte antigen 4 (CTLA4)–immunoglobulin fusion protein have shown efficacy in clinical trials; with alefacept and efalizumab recently winning FDA approval100. Biological agents that inhibit TNF are also quite effective — etanercept is a p75 TNF receptor–IgG Fc fusion protein and infliximab is a humanized monoclonal antibody specific for TNF. Both of these compounds have been shown to be effective in the treatment of psoriasis101. Interestingly, short-term treatment with an antibody specific for E-selectin was recently shown to be ineffective in psoriasis, indicating that once lesions are established, blocking T-cell rolling on E-selectin is insufficient to block disease activity102. By contrast, Efomycin M — a small molecule inhibitor of selectin binding — has recently been shown to be effective in animal models of psoriasis and might indicate a further role for inhibition of selectins in the treatment of human psoriasis103.
The interplay of innate and adaptive immune responses in psoriasis is seen in the Koebner phenomenon, in which physical trauma provokes the development of a psoriatic lesion. As discussed earlier, skin trauma leads to the release of innate immune activators, such as IL-1 and TNF, and results in the upregulation of E-selectin and ICAM1 expression on local dermal post-capillary venules. This leads to the activation of resident T cells and the recruitment of CLA+ T cells from the blood, including the presumed subpopulation that is specific for psoriatic autoantigens. Innate immune activators also induce the maturation of DCs in the dermis and epidermis, enhancing their activity as APCs, and encouraging the activation and proliferation of the recruited T cells and the development of a psoriatic plaque.
Atopic dermatitis. Atopic dermatitis is a common disease that affects people of all age groups worldwide104,105. The prevalence has been reported to vary between 7% and 17% for children, and in 60% or more of these individuals the disease can persist into adulthood104,106,107,108,109.
Atopy is the hereditary predisposition to allergy or hypersensitivity, with the term atopic dermatitis used to describe a group of skin diseases associated with atopic conditions (allergic rhinitis, allergic keratoconjunctivitis, asthma and eczema) that might be seen in all age groups. Clinically, atopic dermatitis is characterized by the development of erythematous, exudative lesions in skin folds that are associated with intense itching. Histopathological sections show perivascular infiltration of the dermis and epidermis by lymphocytes, monocytes and macrophages.
Acute atopic dermatitis is mediated by T cells specific for environmental antigens, although there are subgroups of atopic dermatitis that might have different mechanisms of triggering and maintaining inflammation (for example, extrinsic/allergic atopic dermatitis versus intrinsic/non-allergic atopic dermatitis)6,110,111. The house-dust mite Dermatophagoides pterynossinus is a common source of extrinsic antigen, and T cells specific for this antigen can be identified in lesional and non-lesional skin of selected individuals112. Antigen presentation is enhanced by the presence of high-affinity IgE receptors on Langerhans cells, which internalize antibody–antigen complexes, process antigen and present it to T cells within evolving lesions. Interestingly, as atopic dermatitis lesions become more chronic, the cytokine profile they exhibit shifts from TH2 to TH1 type113. The mechanism behind this switch is incompletely understood.
The interplay between innate and acquired immunity emerges in this disease also. It is well established that scratching of pruritic non-lesional skin can lead to the emergence of new lesions. Presumably this occurs by the trauma of scratching, as in the Koebner response described earlier. A second link comes at the level of bacterial colonization and superinfection. Staphylococcus aureus is readily cultured from atopic skin, particularly lesional skin, and it might be that stimulation by bacterial superantigens or the activation of TLRs on resident skin cells leads to chronic release of primary cytokines. Recent studies have shown that atopic epidermis, unlike psoriatic epidermis, does not produce the antibacterial peptides β-defensin and cathelicidin in response to such infection114, and that IL-4 blocks the induction of these antimicrobial peptides from keratinocytes115. So, a product of TH2 cells blocks one pathway of innate immune activation, leading to bacterial overgrowth and the induction of another innate immune pathway. This, in turn, facilitates the continued activation of the adaptive immune system, including the recruitment and activation of atopic TH2 cells, and perpetuation of the lesion.
Allergic contact dermatitis. Allergic contact dermatitis (ACD), also referred to as contact hypersensitivity, is a T-cell-mediated type IV DELAYED-TYPE HYPERSENSITIVITY reaction to specific environmental antigens and is manifested by varying degrees of erythema, spongiosis (epidermal oedema) and vesiculation. Most contact allergens are themselves irritants (for example, uroshiol or poison ivy), and they therefore provide both antigen and danger signals when they contact skin. The pathophysiology of this disorder is multifactorial, but is characterized by the infiltration and activation of both CD4+ and CD8+ T cells91,116. A general model in which allergen-specific type 1 CD4+ and CD8+ T cells act as effectors and type 2 CD4+ T cells act as regulatory elements is supported by investigations in animal models117. Accumulation of eosinophils and enhanced production of IgE can also be seen118. ACD requires a sensitization phase of 1–2 weeks after exposure, in which small molecule components of the active agent bind to endogenous proteins and act as haptens to induce the activation and proliferation of antigen-specific T cells, which then mature into skin-homing effector memory cells. Subsequent epicutaneous exposures result in symptoms that progress over hours to days and reflect activation of resident antigen-specific effector T cells as well as their further local accumulation from the blood. This is followed by accumulation of CD4+ T cells that produce TH2-type cytokines (for example, IL-5 and IL-13) in chronic and resolving lesions. Recent studies have implicated IL-10, produced by CD4+CD25+ regulatory T cells, as a key factor in the down-modulation of allergic responses in the skin92,119.
Cutaneous graft-versus-host disease. Acute cutaneous GVHD describes a distinctive syndrome of dermatitis, developing within 100 days of allogeneic haematopoietic-cell transplantation120. Chronic cutaneous GVHD describes a more indolent syndrome that develops after day 100. Development of GVHD depends on the transfer of immunologically competent cells, such as mature T cells included in bone-marrow transplants or resident in solid organ transplants, the presence of alloantigens on host tissues that can stimulate the graft cells and the lack of an effective host immunological response to the graft. Acute cutaneous GVHD is characterized initially by a rash or by a generalized redness of the skin and desquamation. Chronic cutaneous GVHD can lead to areas of thickened skin or sclerodermatous changes that sometimes cause contractures and limitation of joint mobility. The predilection of GVHD for the skin and the gastrointestinal tract has led to speculation that it is mediated in these respective tissues by antigen-specific T cells with distinct skin- or gut-homing properties1,64. This hypothesis has not been formally tested.
Cutaneous T-cell lymphoma. We and others have proposed that CTCLs are malignancies of skin-homing T cells1,121. The most common form of this uncommon disease — mycosis fungoides — is characterized by patches and plaques on the skin, often in non-sun-exposed areas, which can resemble eczematous dermatitis. Histopathological features of CTCL include the clustering of malignant T cells in the epidermis, often around Langerhans cells. There is evidence that expression of homing molecules determines the anatomic localization of these cells81. Tumour cells that are CLA and CCR4 positive but lack expression of either L-selectin or CCR7 can be found in the skin, whereas cells that express both L-selectin and CCR7 are associated with lymph-node involvement. The CTCL cells almost invariably produce TH2-type cytokines122. Recently, evidence has emerged that CTCL is associated with marked disruption of the T-cell repertoire, indicating that it might be a systemic disease rather than simply a clonal malignancy of skin-homing T cells123.
Vitiligo. Vitiligo is characterized by complete or partial depigmentation of the epidermis. It is an acquired progressive disorder in which some or all of the melanocytes that reside in the interfollicular epidermis and, occasionally, in the hair follicles are selectively destroyed124. Vitiligo is relatively frequent, occurring in 1–2% of the population. CD8+ T cells specific for antigens that are uniquely expressed by melanocytes are frequently seen in these patients, leading to the suggestion that vitiligo is a T-cell-mediated autoimmune disorder125. Interestingly, vitiligo is most prominent in areas that are subject to minor trauma, providing another disease-related link between innate and acquired immunity in inflammatory skin diseases.
Other inflammatory skin disorders might also be mediated or modulated by these mechanisms. Although there are few data present in the literature, further investigations might identify a role for dysregulation of leukocyte homing in the pathogenesis of these and other skin conditions. It is not possible to discuss the full range of implications in this review.
Vaccine development
The concepts of immune surveillance and tissue-specific homing have important implications for the rational design of vaccines, as highlighted by the example of smallpox (Box 1). Not only must the antigen be administered in a manner that leads to DC maturation and migration to lymph nodes (danger signals or adjuvant effects), but also the route of administration might have marked qualitative and quantitative effects on the desired protective immune response. Such considerations are taking on a broader scope and purpose, with interest in the development of vaccines against tumours, HIV and emerging infectious agents responsible for diseases such as Lyme disease, West Nile virus disease and severe acute respiratory syndrome (SARS). Even long-standing immunization protocols, such as those established more than 40 years ago for smallpox, are under active investigation for improvements that might reduce complications while maintaining effectiveness. The findings outlined earlier indicate that vaccination through the skin (intradermal) will be most efficient at stimulating skin-homing effector cells, whereas alternative routes (for example, oral and intramuscular) will most efficiently generate effector memory T cells that are directed towards other sites.
Although aggressive stimulation with adjuvants might bypass the anatomically specific elements of the immune response by driving broad production of central memory cells, it might be preferable in some cases to limit responses to a desired site to avoid potential complications in other tissues.
Tumour vaccines. Our knowledge of leukocyte homing and immune-surveillance mechanisms also has implications for the field of antitumour vaccines. Despite considerable work in this area, the clinical success of antitumor vaccinations has been limited so far. One reason for this could be that the methods chosen for immunization are insufficient or inappropriate for the tumour of interest. Malignant melanoma is a cancer of melanocytes, or pigment cells, that reside in the epidermis and hair follicles and produce melanin. There is convincing evidence that malignant melanoma can evoke humoral and cellular immune responses in some patients. The radial growth phase of primary melanoma, associated with slow and superficial growth without prominent dermal invasion, is regularly associated with a marked dermal lymphocytic reaction, sometimes resulting in partial tumour destruction126,127,128. Clonal expansion of T cells occurs in regressing primary melanoma, and lymphocytes explanted from such lesions are cytotoxic in vitro to autologous melanoma cells129,130,131,132. Although a rapid lymphocytic infiltrate in the vertical growth phase (where rapid growth and prominent dermal invasion occur) of primary melanoma occurs less frequently, this response is correlated with prolonged survival and a reduced incidence of metastatic disease133,134,135.
Many strategies to enhance antimelanoma immunity are under investigation at present, based on whole tumour cells or defined tumour antigens136,137,138,139,140,141,142,143,144. In the development of such protocols, relatively little attention has been paid to the route of vaccination used145,146,147. We would predict that immunization through the skin would generate a skin-homing effector T-cell response, but might not be expected to target metastatic tumours in the lungs or gastrointestinal tract efficiently. Under normal circumstances, antimelanoma T-cell responses might first be expected to develop in the local skin-draining lymph nodes and should lead to the generation of skin-homing memory effector cells. Indeed, IL-2 therapy (which expands and activates pre-existing memory effector cell populations) and DC vaccine therapies result in more rapid responses to the cutaneous metastases of melanoma than to metastases elsewhere148. For immunization with melanoma-antigen-pulsed DCs, if they are injected into the skin, they could traffic through afferent lymphatics to draining lymph nodes, generating skin-homing memory effector cells. Injected intravenously, however, their migration patterns remain largely unknown. Protocols to enhance DC migration to peripheral lymph nodes are under investigation149. It is also important to consider the effects of vaccination strategies on DC activation, as antigen presentation by immature DCs has been shown to stimulate antigen-specific inhibition of effector T-cell functions93,150.
Conclusions
Investigation of leukocyte trafficking to the skin has provided insight into the role of primary, secondary and tertiary immune surveillance in normal cutaneous immune function and in the development of inflammatory skin diseases. The few disorders that we have discussed in detail are only a subset of clinically important T-cell-mediated skin diseases, which also include drug eruptions, alopecia areata, lichen planus and many others. The number and diversity of these diseases are testament to how many things can potentially go wrong in a complex system such as cutaneous immune surveillance. At the same time, it is extraordinary that the cutaneous immune system works as well as it does most of the time. The concept that exaggerated or inappropriate activity of an important immune-surveillance mechanism can cause organ-specific diseases might extend to inflammatory bowel disease and asthma, which occur at two other epithelial-cell interfaces with the environment. The challenge will be to design therapies that target the elements of cutaneous immune surveillance that are overactive in specific diseases of the skin or other organs, while leaving intact those functions that are central to survival in a hostile world filled with opportunistic pathogens.
Box 1 | Vaccinating against smallpox in atopic patients.
Smallpox (variola major) typically enters the host through the oropharynx, invades the mucosal epithelium, and migrates to regional lymph nodes, and then to the spleen, the bone marrow and other lymph nodes, where viral replication occurs151. After an incubation period of 12–14 days, virus enters the blood within leukocytes, which seed the skin and produce the characteristic skin lesions (pox), whereas most other tissues are spared. The fact that virus seems to travel in leukocytes that specifically exit blood vessels in the papillary dermis indicates that variola virus preferentially associates with leukocytes that can home to skin; alternatively, it might be that only skin tissues can support the subsequent replication steps that are required for lesion formation.
Protective vaccination with vaccinia virus depends on delivery of the virus to the epidermis by a technique known as scarification, leading to an epidermal 'pox' reaction — a cutaneous T-cell-mediated delayed-type hypersensitivity reaction presumably involving vaccinia-virus-specific skin-homing T cells. Both subcutaneous and intramuscular vaccinations fail to provoke a pox reaction and do not effectively incite neutralizing antibodies or vaccinia-virus-specific cytotoxic T cells152.
Patients with either active or quiescent atopic dermatitis are at risk after immunization for the development of eczema vaccinatum, which results from an inability of the host to control the spread of virus from the inoculation site, and is associated with substantial morbidity and mortality107,153. We hypothesize that atopic individuals have defects in both innate and acquired immune responses to vaccinia virus. Atopic patients preferentially generate T helper 2 (TH2)-cell responses to antigens encountered through the skin, and increased levels of the TH2-type cytokine interleukin-4 (IL-4) can be detected in both affected and unaffected skin154. Ectromelia (mousepox) virus genetically engineered to produce IL-4 results in a lethal disease in mice that are normally resistant to unmodified ectromelia, indicating a role for this cytokine in restricting immune responses to pox viruses155. Production of TH1-type cytokines, such as interferon-γ, and cytotoxic T-cell functions are also impaired in patients with atopic dermatitis91,156. T-cell homing might be dysfunctional in these patients as well. In mice, the generation of TH1-type, but not TH2-type, cytokines, is associated with a skin-homing phenotype157. The presence of skin-homing TH2 cells in atopic patients might represent an uncoupling of this association158. Surprisingly, even innate immunity might be impaired in these patients114. The production of IL-18 is increased in atopic dermatitis159, and keratinocytes from patients with atopic dermatitis produce a different array of cytokines and chemokines than do keratinocytes from non-atopic individuals160,161,162.
Acknowledgements
This work is supported by the National Institutes of Health.
Glossary
- DANGER SIGNALS
Cell-wall components and other products of pathogens that alert the innate immune system to the presence of potentially harmful invaders, usually by interacting with Toll-like receptors and other pattern-recognition receptors that are expressed by tissue cells and dendritic cells, for example.
- LANGERHANS CELLS
Immature bone-marrow-derived dendritic cells that reside for long periods of time in the epidermis. They contain Langerin and Birbeck granules, express E-cadherin and bind to contiguous keratinocytes. Stimulation through Toll-like receptors or other danger-signal receptors causes them to migrate to the draining lymph node and mature into highly efficient antigen-presenting cells.
- ANTIMICROBIAL PEPTIDES
Evolutionarily conserved peptides that directly bind to and interact with cell surfaces of bacteria and fungi, usually inducing the formation of pores, leading to the death of target cells.
- PROSTANOID
A member of the broad family of arachidonic-acid-derived inflammatory mediators.
- GRAFT-VERSUS-HOST DISEASE
(GVHD). Tissue damage in a recipient of allogeneic transplanted tissue (usually a bone-marrow transplant) that results from the activity of donor cytotoxic T lymphocytes that recognize the recipient's tissue as foreign. GVHD varies markedly in severity, but can be life threatening in severe cases. Typically, damage to the skin and gut mucosa leads to clinical manifestations.
- REGULATORY T CELL
A type of T cell that can inhibit effector-T-cell activation in an antigen-specific manner.
- T HELPER 1 CELL
(TH1). A type of T cell that, through the production of interferon-γ, interleukin-10 and other cytokines, can stimulate cellular immunity against viral and bacterial pathogens.
- T HELPER 2 CELL
(TH2). A type of T cell that, through the production of interleukin-4 (IL-4), IL-13 and other cytokines, can help B cells to produce IgE and other antibodies and, through the secretion of IL-5, IL-3 and others, can promote increased numbers of eosinophils, basophils and mast cells.
- DELAYED-TYPE HYPERSENSITIVITY
A cellular immune response to antigen that develops over 24–72 hours with the infiltration of T cells and monocytes, and depends on the production of T helper-1-specific cytokines.
Biographies
Thomas Kupper is the Thomas B. Fitzpatrick Professor of Dermatology at Harvard Medical School, Boston, USA, Chairman of the Department of Dermatology at Brigham and Women's Hospital, and Director of the Center for Cutaneous Oncology at the Dana-Farber Cancer Institute, Director of the Harvard Skin Disease Research Center, and Principal Investigator on the Skin Cancer SPORE grant (National Cancer Institute). He is also Principal Investigator of the Harvard Dermatology Training grant and is Principal Investigator on a MERIT award from the National Institute of Allergy and Infectious Diseases.
Robert C. Fuhlbrigge is an Assistant Professor of Pediatrics at Harvard Medical School, Associate Director for Research in the Department of Dermatology at Brigham and Women's Hospital and Co-Chair of the Leukocyte Migration Core of the Harvard Skin Disease Research Center. Kupper and Fuhlbrigge have collaborated for the past five years on a range of research areas relevant to skin disease, including the immunobiology of skin-homing T cells, dendritic-cell homing and trafficking, interleukin-1 and other innate immune mediators in the skin, T-cell trafficking molecules in cutaneous T-cell lymphoma, and organ-specific lymphocyte trafficking in graft-versus-host disease.
Related links
DATABASES
LocusLink
FURTHER INFORMATION
Competing interests
The authors declare no competing financial interests.
References
- 1.Robert C, Kupper TS. Inflammatory skin diseases, T cells, and immune surveillance. N. Engl. J. Med. 1999;341:1817–1828. doi: 10.1056/NEJM199912093412407. [DOI] [PubMed] [Google Scholar]
- 2.Uthayakumar S, Nandwani R, Drinkwater T, Nayagam AT, Darley CR. The prevalence of skin disease in HIV infection and its relationship to the degree of immunosuppression. Br. J. Dermatol. 1997;137:595–598. doi: 10.1111/j.1365-2133.1997.tb03793.x. [DOI] [PubMed] [Google Scholar]
- 3.Lugo-Janer G, Sánchez JL, Santiago-Delpin E. Prevalence and clinical spectrum of skin diseases in kidney transplant recipients. J. Am. Acad. Dermatol. 1991;24:410–414. doi: 10.1016/0190-9622(91)70061-6. [DOI] [PubMed] [Google Scholar]
- 4.Kadunce DP, Krueger GG. Pathogenesis of psoriasis. Dermatol. Clin. 1995;13:723–737. doi: 10.1016/S0733-8635(18)30037-8. [DOI] [PubMed] [Google Scholar]
- 5.Leung DY. Atopic dermatitis: immunobiology and treatment with immune modulators. Clin. Exp. Immunol. 1997;107(Suppl. 1):25–30. [PubMed] [Google Scholar]
- 6.Galli E, et al. Atopic dermatitis: molecular mechanisms, clinical aspects and new therapeutical approaches. Curr. Mol. Med. 2003;3:127–138. doi: 10.2174/1566524033361564. [DOI] [PubMed] [Google Scholar]
- 7.González FJ, et al. Participation of T lymphocytes in cutaneous allergic reactions to drugs. Clin. Exp. Allergy. 1998;28(Suppl. 4):3–6. [PubMed] [Google Scholar]
- 8.Porter SR, Kirby A, Olsen I, Barrett W. Immunologic aspects of dermal and oral lichen planus: a review. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 1997;83:358–366. doi: 10.1016/S1079-2104(97)90244-4. [DOI] [PubMed] [Google Scholar]
- 9.McDonagh AJ, Messenger AG. The pathogenesis of alopecia areata. Dermatol. Clin. 1996;14:661–670. doi: 10.1016/S0733-8635(05)70392-2. [DOI] [PubMed] [Google Scholar]
- 10.Boh EE, Millikan LE. Vesiculobullous diseases with prominent immunologic features. JAMA. 1992;268:2893–2898. doi: 10.1001/jama.1992.03490200145016. [DOI] [PubMed] [Google Scholar]
- 11.Wong E, Camp RDR, Greaves MW. The responses of normal and psoriatic skin to single and multiple topical applications of leukotriene B4. J. Invest. Dermatol. 1985;84:421–423. doi: 10.1111/1523-1747.ep12265520. [DOI] [PubMed] [Google Scholar]
- 12.Krueger JG, Wolfe JT, Nabeya RT. Successful ultraviolet B treatment of psoriasis is accompanied by a reversal of keratinocyte pathology and by selective depletion of intraepidermal T cells. J. Exp. Med. 1995;182:2057–2068. doi: 10.1084/jem.182.6.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gottlieb SL, Gilleaudeau P, Johnson R. Response of psoriasis to be lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nature Med. 1995;1:442–447. doi: 10.1038/nm0595-442. [DOI] [PubMed] [Google Scholar]
- 14.Lim KK, et al. Cyclosporine in the treatment of dermatologic disease: an update. Mayo Clin. Proc. 1996;71:1182–1191. doi: 10.4065/71.12.1182. [DOI] [PubMed] [Google Scholar]
- 15.Kupper TS, Groves RW. The interleukin-1 axis and cutaneous inflammation. J. Invest. Dermatol. 1995;105:62S–66S. doi: 10.1038/jid.1995.13. [DOI] [PubMed] [Google Scholar]
- 16.Murphy JE, Robert C, Kupper TS. Interleukin-1 and cutaneous inflammation: a crucial link between innate and acquired immunity. J. Invest. Dermatol. 2000;114:602–608. doi: 10.1046/j.1523-1747.2000.00917.x. [DOI] [PubMed] [Google Scholar]
- 17.Heufler C, et al. Interleukin 7 is produced by murine and human keratinocytes. J. Exp. Med. 1993;178:1109–1114. doi: 10.1084/jem.178.3.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang D, Chertov O, Oppenheim JJ. The role of mammalian antimicrobial peptides and proteins in awakening of innate host defences and adaptive immunity. Cell. Mol. Life Sci. 2001;58:978–989. doi: 10.1007/PL00000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallo RL, Murakami M, Ohtake T, Zaiou M. Biology and clinical relevance of naturally occurring antimicrobial peptides. J. Allergy Clin. Immunol. 2002;110:823–831. doi: 10.1067/mai.2002.129801. [DOI] [PubMed] [Google Scholar]
- 20.Kupper TS, Chua AO, Flood P, McGuire J, Gubler U. Interleukin 1 gene expression in cultured human keratinocytes is augmented by ultraviolet irradiation. J. Clin. Invest. 1987;80:430–436. doi: 10.1172/JCI113090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood LC, Elias PM, Calhoun C. Barrier disruption stimulates interleukin-1α expression and release from a pre-formed pool in murine epidermis. J. Invest. Dermatol. 1996;106:397–403. doi: 10.1111/1523-1747.ep12343392. [DOI] [PubMed] [Google Scholar]
- 22.Luger TA, Scholzen T, Grabbe S. The role of α-melanocyte-stimulating hormone in cutaneous biology. J. Investig. Dermatol. Symp. Proc. 1997;2:87–93. doi: 10.1038/jidsymp.1997.17. [DOI] [PubMed] [Google Scholar]
- 23.Dinarello CA. Interleukin-1, interleukin-1 receptors and interleukin-1 receptor antagonist. Int. Rev. Immunol. 1998;16:457–499. doi: 10.3109/08830189809043005. [DOI] [PubMed] [Google Scholar]
- 24.Fuhlbrigge RC, Kupper TS. Allergic Skin Disease. 2000. pp. 29–52. [Google Scholar]
- 25.Takashima A, Bergstresser PR. Cytokine-mediated communication by keratinocytes and Langerhans cells with dendritic epidermal T cells. Semin. Immunol. 1996;8:333–339. doi: 10.1006/smim.1996.0044. [DOI] [PubMed] [Google Scholar]
- 26.Dahl M. Clinical Immunodermatology. 1996. [Google Scholar]
- 27.Stoll S, et al. Production of IL-18 (IFN-γ-inducing factor) messenger RNA and functional protein by murine keratinocytes. J. Immunol. 1997;159:298–302. [PubMed] [Google Scholar]
- 28.Steinhoff M, Brzoska T, Luger TA. Keratinocytes in epidermal immune responses. Curr. Opin. Allergy Clin. Immunol. 2001;1:469–476. doi: 10.1097/01.all.0000011062.60720.e3. [DOI] [PubMed] [Google Scholar]
- 29.Kumar S, et al. Identification and initial characterization of four novel members of the interleukin-1 family. J. Biol. Chem. 2000;275:10308–10314. doi: 10.1074/jbc.275.14.10308. [DOI] [PubMed] [Google Scholar]
- 30.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 31.Sieling PA, Modlin RL. Toll-like receptors: mammalian 'taste receptors' for a smorgasbord of microbial invaders. Curr. Opin. Microbiol. 2002;5:70–75. doi: 10.1016/S1369-5274(02)00288-6. [DOI] [PubMed] [Google Scholar]
- 32.Medzhitov R. Toll-like receptors and innate immunity. Nature Rev. Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 33.Barton GM, Medzhitov R. Control of adaptive immune responses by Toll-like receptors. Curr. Opin. Immunol. 2002;14:380–383. doi: 10.1016/S0952-7915(02)00343-6. [DOI] [PubMed] [Google Scholar]
- 34.Yang RB, et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature. 1998;395:284–288. doi: 10.1038/26239. [DOI] [PubMed] [Google Scholar]
- 35.Brightbill HD, et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–736. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 36.Aliprantis AO, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi F, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 38.Tada H, et al. Saccharomyces cerevisiae- and Candida albicans-derived mannan induced production of tumor necrosis factor α by human monocytes in a CD14- and Toll-like receptor 4-dependent manner. Microbiol. Immunol. 2002;46:503–512. doi: 10.1111/j.1348-0421.2002.tb02727.x. [DOI] [PubMed] [Google Scholar]
- 39.Hemmi H, et al. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 40.Wagner H. Interactions between bacterial CpG-DNA and TLR9 bridge innate and adaptive immunity. Curr. Opin. Microbiol. 2002;5:62–69. doi: 10.1016/S1369-5274(02)00287-4. [DOI] [PubMed] [Google Scholar]
- 41.Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 2001;31:3388–3393. doi: 10.1002/1521-4141(200111)31:11<3388::AID-IMMU3388>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 42.Kadowaki N, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krug A, et al. Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur. J. Immunol. 2001;31:3026–3037. doi: 10.1002/1521-4141(2001010)31:10<3026::AID-IMMU3026>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 44.Medzhitov R, Janeway CA. Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/S0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 45.Barnes PJ. Nuclear factor-κB. Int. J. Biochem. Cell Biol. 1997;29:867–870. doi: 10.1016/S1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- 46.von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N. Engl. J. Med. 2000;343:1020–1034. doi: 10.1056/NEJM200010053431407. [DOI] [PubMed] [Google Scholar]
- 47.Supajatura V, et al. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J. Clin. Invest. 2002;109:1351–1359. doi: 10.1172/JCI0214704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marshall JS, McCurdy JD, Olynych T. Toll-like receptor-mediated activation of mast cells: implications for allergic disease? Int. Arch. Allergy Immunol. 2003;132:87–97. doi: 10.1159/000073709. [DOI] [PubMed] [Google Scholar]
- 49.McCurdy JD, Olynych TJ, Maher LH, Marshall JS. Cutting edge: distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J. Immunol. 2003;170:1625–1629. doi: 10.4049/jimmunol.170.4.1625. [DOI] [PubMed] [Google Scholar]
- 50.Inaba K, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lenz A, Heine M, Schuler G, Romani N. Human and murine dermis contain dendritic cells. J. Clin. Invest. 1993;92:2587–2596. doi: 10.1172/JCI116873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 53.Hart DNJ. Dendritic cells: unique leukocyte populations which control the primary immune response. Blood. 1997;90:3245–3287. [PubMed] [Google Scholar]
- 54.Pierre P, et al. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature. 1997;388:787–792. doi: 10.1038/42039. [DOI] [PubMed] [Google Scholar]
- 55.Schuller E, et al. In situ expression of the co-stimulatory molecules CD80 and CD86 on langerhans cells and inflammatory dendritic epidermal cells (IDEC) in atopic dermatitis. Arch. Dermatol. Res. 2001;293:448–454. doi: 10.1007/s004030100263. [DOI] [PubMed] [Google Scholar]
- 56.Wollenberg A, Kraft S, Hanau D, Bieber T. Immunomorphological and ultrastructural characterization of Langerhans cells and a novel, inflammatory dendritic epidermal cell (IDEC) population in lesional skin of atopic eczema. J. Invest. Dermatol. 1996;106:446–453. doi: 10.1111/1523-1747.ep12343596. [DOI] [PubMed] [Google Scholar]
- 57.Picker LJ, Michie SA, Rott LS, Butcher EC. A unique phenotype of skin-associated lymphocytes in human. Preferential expression of the HECA-452 epitope by benign and malignant T cells at cutaneous sites. Am. J. Pathol. 1990;136:1053–1068. [PMC free article] [PubMed] [Google Scholar]
- 58.Pitzalis C, et al. Cutaneous lymphocyte antigen-positive T lymphocytes preferentially migrate to the skin but not to the joint in psoriatic arthritis. Arthritis Rheum. 1996;39:137–145. doi: 10.1002/art.1780390118. [DOI] [PubMed] [Google Scholar]
- 59.Sigmundsdottir H, Gudjonsson JE, Jonsdottir I, Ludviksson BR, Valdimarsson H. The frequency of CLA+ CD8+ T cells in the blood of psoriasis patients correlates closely with the severity of their disease. Clin. Exp. Immunol. 2001;126:365–369. doi: 10.1046/j.1365-2249.2001.01688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noorduyn L, et al. Differential expression of the HECA-452 antigen (cutaneous lymphocyte associated antigen, CLA) in cutaneous and non-cutaneous T-cell lymphomas. Histopathology. 1992;21:59–64. doi: 10.1111/j.1365-2559.1992.tb00343.x. [DOI] [PubMed] [Google Scholar]
- 61.Santamaria Babi LF, Perez Soler MT, Hauser C, Blaser K. Skin-homing T cells in human cutaneous allergic inflammation. Immunol. Res. 1995;14:317–324. doi: 10.1007/BF02935627. [DOI] [PubMed] [Google Scholar]
- 62.Santamaria Babi LF, et al. Circulating allergen-reactive T cells from patients with atopic dermatitis and allergic contact dermatitis express the skin-selective homing receptor, the cutaneous lymphocyte-associated antigen. J. Exp. Med. 1995;181:1935–1940. doi: 10.1084/jem.181.5.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanaka Y, et al. Distinct phenotype of leukemic T cells with various tissue tropisms. J. Immunol. 1997;158:3822–3829. [PubMed] [Google Scholar]
- 64.Davis RE, Smoller BR. T lymphocytes expressing HECA-452 epitope are present in cutaneous acute graft-versus-host disease and erythema multiforme, but not in acute graft-versus-host disease in gut organs. Am. J. Pathol. 1992;141:691–698. [PMC free article] [PubMed] [Google Scholar]
- 65.Rook AH, Heald P. The immunopathogenesis of cutaneous T-cell lymphoma. Hematol. Oncol. Clin. North Am. 1995;9:997–1010. doi: 10.1016/S0889-8588(18)30054-6. [DOI] [PubMed] [Google Scholar]
- 66.Berlin C, et al. α4β7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–195. doi: 10.1016/0092-8674(93)90305-A. [DOI] [PubMed] [Google Scholar]
- 67.Mora JR, et al. Selective imprinting of gut-homing T cells by Peyer's patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- 68.Dudda JC, Simon JC, Martin S. Dendritic cell immunization route determines CD8+ T cell trafficking to inflamed skin: role for tissue microenvironment and dendritic cells in establishment of T cell-homing subsets. J. Immunol. 2004;172:857–863. doi: 10.4049/jimmunol.172.2.857. [DOI] [PubMed] [Google Scholar]
- 69.Koelle DM, et al. Expression of cutaneous lymphocyte-associated antigen by CD8+ T cells specific for a skin-tropic virus. J. Clin. Invest. 2002;110:537–548. doi: 10.1172/JCI0215537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rott LS, et al. Expression of mucosal homing receptor α4β7 by circulating CD4+ cells with memory for intestinal rotavirus. J. Clin. Invest. 1997;100:1204–1208. doi: 10.1172/JCI119633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armerding D, Kupper TS. Functional cutaneous lymphocyte antigen can be induced in essentially all peripheral blood T lymphocytes. Int. Arch. Allergy Immunol. 1999;119:212–222. doi: 10.1159/000024197. [DOI] [PubMed] [Google Scholar]
- 72.Akdis M, Klunker S, Schliz M, Blaser K, Akdis CA. Expression of cutaneous lymphocyte-associated antigen on human CD4+ and CD8+ TH2 cells. Eur. J. Immunol. 2000;30:3533–3541. doi: 10.1002/1521-4141(2000012)30:12<3533::AID-IMMU3533>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 73.Colantonio L, Iellem A, Sinigaglia F, D'Ambrosio D. Skin-homing CLA+ T cells and regulatory CD25+ T cells represent major subsets of human peripheral blood memory T cells migrating in response to CCL1/I-309. Eur. J. Immunol. 2002;32:3506–3514. doi: 10.1002/1521-4141(200212)32:12<3506::AID-IMMU3506>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 74.Biedermann T, et al. Targeting CLA/E-selectin interactions prevents CCR4-mediated recruitment of human TH2 memory cells to human skin in vivo. Eur. J. Immunol. 2002;32:3171–3180. doi: 10.1002/1521-4141(200211)32:11<3171::AID-IMMU3171>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 75.Hudak S, et al. Immune surveillance and effector functions of CCR10+ skin homing T cells. J. Immunol. 2002;169:1189–1196. doi: 10.4049/jimmunol.169.3.1189. [DOI] [PubMed] [Google Scholar]
- 76.Reiss Y, Proudfoot AE, Power CA, Campbell JJ, Butcher EC. CC chemokine receptor (CCR)4 and the CCR10 ligand cutaneous T cell-attracting chemokine (CTACK) in lymphocyte trafficking to inflamed skin. J. Exp. Med. 2001;194:1541–1547. doi: 10.1084/jem.194.10.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morales J, et al. CTACK, a skin-associated chemokine that preferentially attracts skin-homing memory T cells. Proc. Natl Acad. Sci. USA. 1999;96:14470–14475. doi: 10.1073/pnas.96.25.14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Homey B, et al. CCL27–CCR10 interactions regulate T cell-mediated skin inflammation. Nature Med. 2002;8:157–165. doi: 10.1038/nm0202-157. [DOI] [PubMed] [Google Scholar]
- 79.Fitzhugh DJ, Naik S, Caughman SW, Hwang ST. Cutting edge: C-C chemokine receptor 6 is essential for arrest of a subset of memory T cells on activated dermal microvascular endothelial cells under physiologic flow conditions in vitro. J. Immunol. 2000;165:6677–6681. doi: 10.4049/jimmunol.165.12.6677. [DOI] [PubMed] [Google Scholar]
- 80.Klunker S, et al. A second step of chemotaxis after transendothelial migration: keratinocytes undergoing apoptosis release IFN-γ-inducible protein 10, monokine induced by IFN-γ, and IFN-γ-inducible α-chemoattractant for T cell chemotaxis toward epidermis in atopic dermatitis. J. Immunol. 2003;171:1078–1084. doi: 10.4049/jimmunol.171.2.1078. [DOI] [PubMed] [Google Scholar]
- 81.Ferenczi K, Fuhlbrigge RC, Pinkus J, Pinkus GS, Kupper TS. Increased CCR4 expression in cutaneous T cell lymphoma. J. Invest. Dermatol. 2002;119:1405–1410. doi: 10.1046/j.1523-1747.2002.19610.x. [DOI] [PubMed] [Google Scholar]
- 82.Kunkel EJ, et al. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am. J. Pathol. 2002;160:347–355. doi: 10.1016/S0002-9440(10)64378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Janssen GH, Tangelder GJ, Oude Egbrink MG, Reneman RS. Spontaneous leukocyte rolling in venules in untraumatized skin of conscious and anesthetized animals. Am. J. Physiol. 1994;267:H1199–H1204. doi: 10.1152/ajpheart.1994.267.3.H1199. [DOI] [PubMed] [Google Scholar]
- 84.Robert C, et al. Interaction of dendritic cells with skin endothelium: a new perspective on immunosurveillance. J. Exp. Med. 1999;189:627–636. doi: 10.1084/jem.189.4.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chong BF, Murphy J-E, Kupper TS, Fuhlbrigge RC. E-selectin, thymus- and activation-regulated chemokine/CCL17, and intercellular adhesion molecule-1 are constitutively coexpressed in dermal microvessels: a foundation for a cutaneous immunosurveillance system. J. Immunol. 2004;172:1575–1581. doi: 10.4049/jimmunol.172.3.1575. [DOI] [PubMed] [Google Scholar]
- 86.Weninger W, et al. Specialized contributions by α(1,3)-fucosyltransferase-IV and FucT-VII during leukocyte rolling in dermal microvessels. Immunity. 2000;12:665–676. doi: 10.1016/S1074-7613(00)80217-4. [DOI] [PubMed] [Google Scholar]
- 87.Groves RW, et al. Effect of in vivo interleukin-1 on adhesion molecule expression in normal human skin. J. Invest. Dermatol. 1992;98:384–387. doi: 10.1111/1523-1747.ep12499816. [DOI] [PubMed] [Google Scholar]
- 88.Langeveld-Wildschut EG, et al. Clinical and immunologic variables in skin of patients with atopic eczema and either positive or negative atopy patch test reactions. J. Allergy Clin. Immunol. 2000;105:1008–1016. doi: 10.1067/mai.2000.106544. [DOI] [PubMed] [Google Scholar]
- 89.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 90.Campbell JJ, et al. CCR7 expression and memory T cell diversity in humans. J. Immunol. 2001;166:877–884. doi: 10.4049/jimmunol.166.2.877. [DOI] [PubMed] [Google Scholar]
- 91.Girolomoni G, Sebastiani S, Albanesi C, Cavani A. T-cell subpopulations in the development of atopic and contact allergy. Curr. Opin. Immunol. 2001;13:733–737. doi: 10.1016/S0952-7915(01)00287-4. [DOI] [PubMed] [Google Scholar]
- 92.Cavani A, et al. Human CD25+ regulatory T cells maintain immune tolerance to nickel in healthy, nonallergic individuals. J. Immunol. 2003;171:5760–5768. doi: 10.4049/jimmunol.171.11.5760. [DOI] [PubMed] [Google Scholar]
- 93.Mahnke K, Qian Y, Knop J, Enk AH. Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood. 2003;101:4862–4869. doi: 10.1182/blood-2002-10-3229. [DOI] [PubMed] [Google Scholar]
- 94.Wrone-Smith T, Nickoloff BJ. Dermal injection of immunocytes induces psoriasis. J. Clin. Invest. 1996;98:1878–1887. doi: 10.1172/JCI118989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kupper TS. Immunologic targets in psoriasis. N. Engl. J. Med. 2003;349:1987–1990. doi: 10.1056/NEJMp038164. [DOI] [PubMed] [Google Scholar]
- 96.Cather J, Menter A. Novel therapies for psoriasis. Am. J. Clin. Dermatol. 2002;3:159–173. doi: 10.2165/00128071-200203030-00003. [DOI] [PubMed] [Google Scholar]
- 97.Asadullah K, et al. IL-10 is a key cytokine in psoriasis. Proof of principle by IL-10 therapy: a new therapeutic approach. J. Clin. Invest. 1998;101:783–794. doi: 10.1172/JCI1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trepicchio WL, et al. Interleukin-11 therapy selectively downregulates type I cytokine proinflammatory pathways in psoriasis lesions. J. Clin. Invest. 1999;104:1527–1537. doi: 10.1172/JCI6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ghoreschi K, et al. Interleukin-4 therapy of psoriasis induces TH2 responses and improves human autoimmune disease. Nature Med. 2003;9:40–46. doi: 10.1038/nm804. [DOI] [PubMed] [Google Scholar]
- 100.Cather JC, Abramovits W. Investigational therapies for psoriasis. J. Am. Acad. Dermatol. 2003;49:S133–S138. doi: 10.1016/S0190-9622(03)01147-2. [DOI] [PubMed] [Google Scholar]
- 101.Williams JD, Griffiths CE. Cytokine blocking agents in dermatology. Clin. Exp. Dermatol. 2002;27:585–590. doi: 10.1046/j.1365-2230.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- 102.Bhushan M, et al. Anti-E-selectin is ineffective in the treatment of psoriasis: a randomized trial. Br. J. Dermatol. 2002;146:824–831. doi: 10.1046/j.1365-2133.2002.04743.x. [DOI] [PubMed] [Google Scholar]
- 103.Schon MP, et al. Efomycine M, a new specific inhibitor of selectin, impairs leukocyte adhesion and alleviates cutaneous inflammation. Nature Med. 2002;8:366–372. doi: 10.1038/nm0402-366. [DOI] [PubMed] [Google Scholar]
- 104.Tofte SJ, Hanifin JM. Current management and therapy of atopic dermatitis. J. Am. Acad. Dermatol. 2001;44:S13–S16. doi: 10.1067/mjd.2001.109811. [DOI] [PubMed] [Google Scholar]
- 105.Williams HC. Epidemiology of atopic dermatitis. Clin. Exp. Dermatol. 2000;25:522–529. doi: 10.1046/j.1365-2230.2000.00698.x. [DOI] [PubMed] [Google Scholar]
- 106.Hanifin JM. Epidemiology of atopic dermatitis. Monogr. Allergy. 1987;21:116–131. [PubMed] [Google Scholar]
- 107.Engler RJ, Kenner J, Leung DY. Smallpox vaccination: risk considerations for patients with atopic dermatitis. J. Allergy Clin. Immunol. 2002;110:357–365. doi: 10.1067/mai.2002.128052. [DOI] [PubMed] [Google Scholar]
- 108.Sidbury R, Hanifin JM. Old, new, and emerging therapies for atopic dermatitis. Dermatol. Clin. 2000;18:1–11. doi: 10.1016/S0733-8635(05)70141-8. [DOI] [PubMed] [Google Scholar]
- 109.Shum KW, Lawton S, Williams HC, Docherty G, Jones J. The British Association of Dermatologists audit of atopic eczema management in secondary care. Phase 2: audit of service process. Br. J. Dermatol. 2000;142:274–278. doi: 10.1046/j.1365-2133.2000.03297.x. [DOI] [PubMed] [Google Scholar]
- 110.Wuthrich B, Schmid-Grendelmeier P. The atopic eczema/dermatitis syndrome. Epidemiology, natural course, and immunology of the IgE-associated ('extrinsic') and the nonallergic ('intrinsic') AEDS. J. Investig. Allergol. Clin. Immunol. 2003;13:1–5. [PubMed] [Google Scholar]
- 111.Akdis C, Akdis M. Immunological differences between intrinsic and extrinsic types of atopic dermatitis. Clin. Exp. Allergy. 2003;33:1618–1621. doi: 10.1111/j.1365-2222.2003.01803.x. [DOI] [PubMed] [Google Scholar]
- 112.Bohle B, et al. Long-lived TH2 clones specific for seasonal and perennial allergens can be detected in blood and skin by their TCR-hypervariable regions. J. Immunol. 1998;160:2022–2027. [PubMed] [Google Scholar]
- 113.Grewe M, et al. A role for TH1 and TH2 cells in the immunopathogenesis of atopic dermatitis. Immunol. Today. 1998;19:359–361. doi: 10.1016/S0167-5699(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 114.Ong PY, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 115.Nomura I, et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J. Immunol. 2003;171:3262–3269. doi: 10.4049/jimmunol.171.6.3262. [DOI] [PubMed] [Google Scholar]
- 116.Grabbe S, Schwarz T. Immunoregulatory mechanisms involved in elicitation of allergic contact hypersensitivity. Am. J. Contact Dermat. 1996;7:238–246. doi: 10.1016/S1046-199X(96)90057-3. [DOI] [PubMed] [Google Scholar]
- 117.Kimber I, Dearman RJ. Allergic contact dermatitis: the cellular effectors. Contact Dermatitis. 2002;46:1–5. doi: 10.1034/j.1600-0536.2002.460101.x. [DOI] [PubMed] [Google Scholar]
- 118.Leung DY. Molecular basis of allergic diseases. Mol. Genet. Metab. 1998;63:157–167. doi: 10.1006/mgme.1998.2682. [DOI] [PubMed] [Google Scholar]
- 119.Cavani A, et al. Human CD4+ T lymphocytes with remarkable regulatory functions on dendritic cells and nickel-specific TH1 immune responses. J. Invest. Dermatol. 2000;114:295–302. doi: 10.1046/j.1523-1747.2000.00881.x. [DOI] [PubMed] [Google Scholar]
- 120.Aractingi S, Chosidow O. Cutaneous graft-versus-host disease. Arch. Dermatol. 1998;134:602–612. doi: 10.1001/archderm.134.5.602. [DOI] [PubMed] [Google Scholar]
- 121.Edelson RL. Cutaneous T cell lymphoma: the helping hand of dendritic cells. Ann. NY Acad. Sci. 2001;941:1–11. doi: 10.1111/j.1749-6632.2001.tb03705.x. [DOI] [PubMed] [Google Scholar]
- 122.Rook AH, et al. Pathogenesis of cutaneous T-cell lymphoma: implications for the use of recombinant cytokines and photopheresis. Clin. Exp. Immunol. 1997;107(Suppl. 1):16–20. [PubMed] [Google Scholar]
- 123.Yawalkar N, et al. Profound loss of T cell receptor repertoire complexity in cutaneous T cell lymphoma. Blood. 2003;102:4059–4066. doi: 10.1182/blood-2003-04-1044. [DOI] [PubMed] [Google Scholar]
- 124.Kemp EH, Waterman EA, Weetman AP. Immunological pathomechanisms in vitiligo. Expert Rev. Mol. Med. 2001;3:1–22. doi: 10.1017/S1462399401003362. [DOI] [PubMed] [Google Scholar]
- 125.Ogg GS, Dunbar R, Romero P, Chen JL, Cerundolo V. High frequency of skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmune vitiligo. J. Exp. Med. 1998;188:1203–1208. doi: 10.1084/jem.188.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clark WH, Jr, From L, Bernardino EA, Mihm MC. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res. 1969;29:705–727. [PubMed] [Google Scholar]
- 127.Ruiter DJ, Bhan AK, Harrist TJ, Sober AJ, Mihm MC., Jr. Major histocompatibility antigens and mononuclear inflammatory infiltrate in benign nevomelanocytic proliferations and malignant melanoma. J. Immunol. 1982;129:2808–2815. [PubMed] [Google Scholar]
- 128.Clark WH, Jr., et al. A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum. Pathol. 1984;15:1147–1165. doi: 10.1016/S0046-8177(84)80310-X. [DOI] [PubMed] [Google Scholar]
- 129.Ferradini L, et al. Analysis of T cell receptor variability in tumor-infiltrating lymphocytes from a human regressive melanoma. Evidence for in situ T cell clonal expansion. J. Clin. Invest. 1993;91:1183–1190. doi: 10.1172/JCI116278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mackensen A, et al. Evidence for in situ amplification of cytotoxic T-lymphocytes with antitumor activity in a human regressive melanoma. Cancer Res. 1993;53:3569–3573. [PubMed] [Google Scholar]
- 131.Mackensen A, et al. Direct evidence to support the immunosurveillance concept in a human regressive melanoma. J. Clin. Invest. 1994;93:1397–1402. doi: 10.1172/JCI117116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Salvi S, et al. Overexpression of the T-cell receptor β-chain variable region TCRBV14 in HLA-A2-matched primary human melanomas. Cancer Res. 1995;55:3374–3379. [PubMed] [Google Scholar]
- 133.Clark WH, Jr., et al. Model predicting survival in stage I melanoma based on tumor progression. J. Natl Cancer Inst. 1989;81:1893–1904. doi: 10.1093/jnci/81.24.1893. [DOI] [PubMed] [Google Scholar]
- 134.Pastorfide GC, et al. Image analysis of stage 1 melanoma (1.00–2.50 mm): lymphocytic infiltrates related to metastasis and survival. J. Cutan. Pathol. 1992;19:390–397. doi: 10.1111/j.1600-0560.1992.tb00611.x. [DOI] [PubMed] [Google Scholar]
- 135.Clemente CG, et al. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer. 1996;77:1303–1310. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 136.Weber LW, et al. Tumor immunity and autoimmunity induced by immunization with homologous DNA. J. Clin. Invest. 1998;102:1258–1264. doi: 10.1172/JCI4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bendandi M, et al. Complete molecular remissions induced by patient-specific vaccination plus granulocyte–monocyte colony-stimulating factor against lymphoma. Nature Med. 1999;5:1171–1177. doi: 10.1038/13928. [DOI] [PubMed] [Google Scholar]
- 138.Restifo NP, Rosenberg SA. Developing recombinant and synthetic vaccines for the treatment of melanoma. Curr. Opin. Oncol. 1999;11:50–57. doi: 10.1097/00001622-199901000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nair SK, et al. Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nature Med. 2000;6:1011–1017. doi: 10.1038/79519. [DOI] [PubMed] [Google Scholar]
- 140.Nestle FO, Banchereau J, Hart D. Dendritic cells: on the move from bench to bedside. Nature Med. 2001;7:761–765. doi: 10.1038/89863. [DOI] [PubMed] [Google Scholar]
- 141.Jager E, Jager D, Knuth A. Clinical cancer vaccine trials. Curr. Opin. Immunol. 2002;14:178–182. doi: 10.1016/S0952-7915(02)00318-7. [DOI] [PubMed] [Google Scholar]
- 142.Nestle FO. Vaccines and melanoma. Clin. Exp. Dermatol. 2002;27:597–601. doi: 10.1046/j.1365-2230.2002.01152.x. [DOI] [PubMed] [Google Scholar]
- 143.Dranoff G. GM-CSF-secreting melanoma vaccines. Oncogene. 2003;22:3188–3192. doi: 10.1038/sj.onc.1206459. [DOI] [PubMed] [Google Scholar]
- 144.Engleman EG. Dendritic cell-based cancer immunotherapy. Semin. Oncol. 2003;30:23–29. doi: 10.1016/S0093-7754(03)00229-X. [DOI] [PubMed] [Google Scholar]
- 145.Mackensen A, et al. Homing of intravenously and intralymphatically injected human dendritic cells generated in vitro from CD34+ hematopoietic progenitor cells. Cancer Immunol. Immunother. 1999;48:118–122. doi: 10.1007/s002620050555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Onaitis M, Kalady MF, Pruitt S, Tyler DS. Dendritic cell gene therapy. Surg. Oncol. Clin. N. Am. 2002;11:645–660. doi: 10.1016/S1055-3207(02)00027-3. [DOI] [PubMed] [Google Scholar]
- 147.Gilliet M, et al. Intranodal injection of semimature monocyte-derived dendritic cells induces T helper type 1 responses to protein neoantigen. Blood. 2003;102:36–42. doi: 10.1182/blood-2002-07-2274. [DOI] [PubMed] [Google Scholar]
- 148.Touloukian CE, Rosenberg SA. A survey of treatments used in patients with metastatic melanoma: analysis of 189 patients referred to the National Cancer Institute. Cancer J. Sci. Am. 1999;5:269–274. [PubMed] [Google Scholar]
- 149.Robert C, et al. Gene therapy to target dendritic cells from blood to lymph nodes. Gene Ther. 2003;10:1479–1486. doi: 10.1038/sj.gt.3302008. [DOI] [PubMed] [Google Scholar]
- 150.Dhodapkar MV, Steinman RM. Antigen-bearing immature dendritic cells induce peptide-specific CD8+ regulatory T cells in vivo in humans. Blood. 2002;100:174–177. doi: 10.1182/blood.V100.1.174. [DOI] [PubMed] [Google Scholar]
- 151.Henderson DA. Smallpox: clinical and epidemiologic features. Med. Health R. I. 2002;85:107–108. [PubMed] [Google Scholar]
- 152.McClain DJ, et al. Immunologic responses to vaccinia vaccines administered by different parenteral routes. J. Infect. Dis. 1997;175:756–763. doi: 10.1086/513968. [DOI] [PubMed] [Google Scholar]
- 153.Goldstein JA, Neff JM, Lane JM, Koplan JP. Smallpox vaccination reactions, prophylaxis, and therapy of complications. Pediatrics. 1975;55:342–347. [PubMed] [Google Scholar]
- 154.Leung DY, Soter NA. Cellular and immunologic mechanisms in atopic dermatitis. J. Am. Acad. Dermatol. 2001;44:S1–S12. doi: 10.1067/mjd.2001.109815. [DOI] [PubMed] [Google Scholar]
- 155.Jackson RJ, et al. Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J. Virol. 2001;75:1205–1210. doi: 10.1128/JVI.75.3.1205-1210.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ambach A, Bonnekoh B, Gollnick H. Perforin hyperreleasability and depletion in cytotoxic T cells from patients with exacerbated atopic dermatitis and asymptomatic rhinoconjunctivitis allergica. J. Allergy Clin. Immunol. 2001;107:878–886. doi: 10.1067/mai.2001.114240. [DOI] [PubMed] [Google Scholar]
- 157.Hirata T, et al. P-selectin glycoprotein ligand 1 (PSGL-1) is a physiological ligand for E-selectin in mediating T helper 1 lymphocyte migration. J. Exp. Med. 2000;192:1669–1676. doi: 10.1084/jem.192.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Abernathy-Carver K, Sampson H, Picker L, Leung D. Milk-induced eczema is associated with the expansion of T cells expressing cutaneous lymphocyte antigen. J. Clin. Invest. 1995;95:913–918. doi: 10.1172/JCI117743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tanaka T, et al. Interleukin-18 is elevated in the sera from patients with atopic dermatitis and from atopic dermatitis model mice, NC/Nga. Int. Arch. Allergy Immunol. 2001;125:236–240. doi: 10.1159/000053821. [DOI] [PubMed] [Google Scholar]
- 160.Girolomoni G, Pastore S. The role of keratinocytes in the pathogenesis of atopic dermatitis. J. Am. Acad. Dermatol. 2001;45:S25–S28. doi: 10.1067/mjd.2001.117021. [DOI] [PubMed] [Google Scholar]
- 161.Giustizieri ML, et al. Keratinocytes from patients with atopic dermatitis and psoriasis show a distinct chemokine production profile in response to T cell-derived cytokines. J. Allergy Clin. Immunol. 2001;107:871–877. doi: 10.1067/mai.2001.114707. [DOI] [PubMed] [Google Scholar]
- 162.Pastore S, Corinti S, La Placa M, Didona B, Girolomoni G. Interferon-γ promotes exaggerated cytokine production in keratinocytes cultured from patients with atopic dermatitis. J. Allergy Clin. Immunol. 1998;101:538–544. doi: 10.1016/S0091-6749(98)70361-6. [DOI] [PubMed] [Google Scholar]