

Key Points
Diabetic complications — the long-term damage to various organ systems — are a great cause of mortality and morbidity in both type 1 and type 2 diabetes. There are currently few therapeutic options to prevent or ameliorate these complications.
High blood glucose levels and the subsequent metabolic consequences of hyperglycaemia are widely considered the primary event that initiates diabetic complications, although there is accumulating evidence that impaired insulin signalling arising from insulin deficiency and insulin resistance may also have a pathogenic role.
Vascular dysfunction is a prominent complication of diabetes that is widely held to underlie damage to organ systems such as the macrovasculature, kidneys, eyes and nerves. Other consequences of diabetes, such as dyslipidaemia and hypertension, are key modifiers of vascular injury and act as accelerators of diabetic complications.
Numerous pathogenic mechanisms, including increased polyol pathway flux and mitochondrial activity, activation of protein kinase C and NADPH oxidase and signalling through the receptor for advanced glycation end products (RAGE) pathway, seem to form a central pathogenic axis that is common to most, if not all, of the complications of diabetes. These disorders all promote excess production of pro-oxidative molecules. Organ-specific mechanisms, such as diminished growth factor support and repair pathway activation, must also be considered.
Few animal models of diabetic complications faithfully reflect the advanced stages of organ pathology seen in humans. Current models can be viewed as potentially illustrating early biochemical and functional disorders of diabetes that ultimately lead to advanced pathology. New animal models are being developed using both a reductionist approach for examining specific gene products of interest and also by combining diverse molecular and physiological risk factors.
Control of blood glucose levels and lipids remains the most meaningful approach for preventing diabetic complications. This strategy is likely to be complemented by a diverse range of more focused therapeutics that have emerged from mechanistic studies in animal models and which are currently in clinical development. Some of these, such as those targeting cardiovascular disease, have the potential to affect several diabetic complications, whereas others focus on intervening in organ-specific pathogenic mechanisms. It is probable that combination therapies aimed at the hyperglycaemia-driven pathogenic axis and also at organ-specific disorders will provide the most effective approach to treating the diverse complications of diabetes.
Long-term diabetes increases the likelihood of developing complications such as macrovascular disease, nephropathy, retinopathy and neuropathy. This Review highlights the range of pathologies that are precipitated by hyperglycaemia and discusses recent developments in preclinical and clinical research for each of these complications.
Abstract
Long-term diabetes increases the likelihood of developing secondary damage to numerous systems, and these complications represent a substantial cause of morbidity and mortality. Establishing the causes of diabetes remains the key step towards eradicating the disease, but the prevention and amelioration of diabetic complications is equally important for the millions of individuals who already have the disease or are likely to develop it before prophylaxis or a cure become routinely available. In this Review, we focus on four common complications of diabetes, discuss the range of pathologies that are precipitated by hyperglycaemia and highlight emerging targets for therapeutic intervention.
Main
The increasing prevalence of diabetes, particularly among teenagers1, reinforces concerns over the appearance of the complications of long-term diabetes during what should be the most active and productive years of life. Patients with type 1 (insulin-dependent) or type 2 (insulin-independent) diabetes develop secondary complications, the risk of which is related to the duration of diabetes and the degree of glycaemic control2. The organs that are susceptible to diabetic complications exhibit insulin-independent glucose uptake and possess the glucose-metabolizing enzyme aldose reductase. Therefore, initial interest focused on hyperglycaemia and the subsequent increased glucose flux that is mediated by aldose reductase and the rest of the polyol pathway as a primary pathogenic insult that initiates diabetic complications3.
Unfortunately, almost 40 years of clinical investigation have been unable to show the efficacy of aldose reductase inhibitors to the satisfaction of European and North American regulatory bodies. The search for sites of therapeutic intervention has therefore extended to the downstream effects of aldose reductase activity and also to aldose reductase-independent mechanisms of glucose toxicity. Glucose-independent mechanisms of organ damage that arise from other physiological consequences of diabetes, such as impaired insulin and growth factor signalling, hyperlipidaemia and hypertension, could also be instigators of, or contributors to, the aetiology of specific diabetic complications. This mixture of universal and organ-specific mechanisms is reflected in the diverse range of therapeutic approaches that are currently being investigated (Fig. 1).
Figure 1. Selected therapeutic approaches showing either pan-complication or organ-specific efficacy in animal models of diabetes.
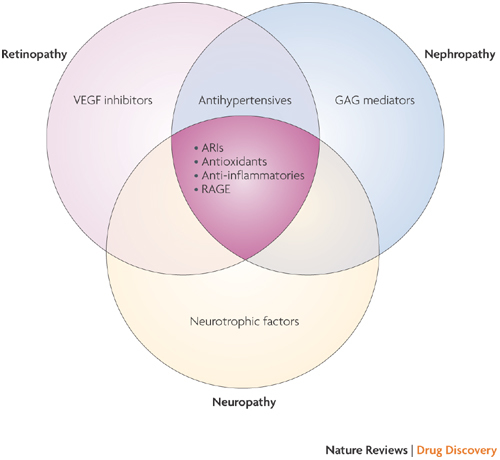
Vascular dysfunction contributes to all diabetic complications, and therapeutic approaches that target the vasculature (shown centre in dark pink, where the three areas overlap) show some efficacy against all complications. The kidneys, eyes and nerves could also be targeted by organ-specific approaches. ARI, aldose reductase inhibitor; GAG, glycosaminoglycan; RAGE, receptor for advanced glycation end products; VEGF, vascular endothelial growth factor.
The four most common complications of diabetes — macrovascular disease, nephropathy, retinopathy and neuropathy — share numerous mechanisms by which hyperglycaemia can disrupt cell and organ function (Fig. 2), with vascular dysfunction also affecting the kidneys, eyes and nervous system. In this Review, we discuss recent developments in preclinical and clinical research for each of these complications to illustrate therapeutic approaches that target either the full range of diabetic complications or the damage to individual organs.
Figure 2. Important glucotoxicity pathways contributing to diabetic complications.
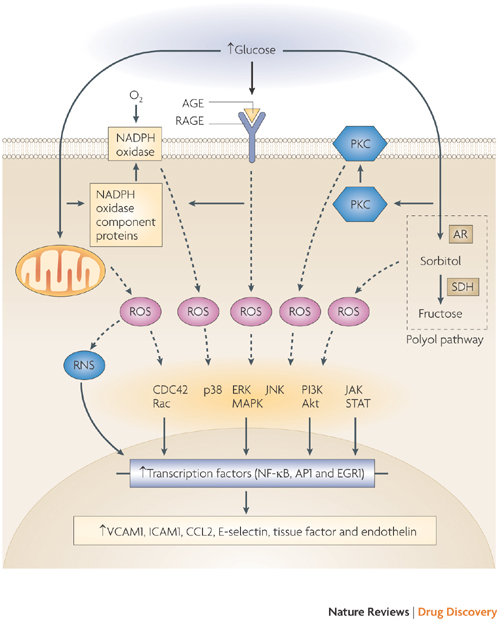
Organ damage can be triggered by both extracellular and intracellular hyperglycaemia. Increased extracellular glucose leads to non-enzymatic glycosylation of proteins and subsequent formation of advanced glycation end products (AGE) that interact with the receptor for AGE (RAGE) on the plasma membrane and promote the production of reactive oxygen species (ROS). Increased intracellular glucose drives mitochondrial activity, increases the activity of protein kinase C (PKC) and NADPH oxidase and promotes increased flux through the polyol pathway, all of which have many effects on cellular metabolism and phenotype. This figure highlights the consequences of excessive ROS production in the vasculature, where ROS-driven changes in cell phenotype are mediated by a range of signalling pathways and transcription factors. Cells of the kidneys, eyes and nervous system also undergo cell- and organ-specific phenotypic changes as a result of hyperglycaemia-mediated ROS production. ROS production, ROS-unrelated pathogenic consequences of hyperglycaemia and hyperglycaemia-independent mechanisms, such as impaired insulin signalling, are likely to collectively mediate the organ-specific pathologies of diabetic complications. Other pathways that are relevant to diabetic complications but are unrelated to hyperglycaemia per se, such as disruption of the renin–angiotensin system (Fig. 3), are described in the main text. AP1, activator protein 1; AR, aldose reductase; CCL2, CC-chemokine ligand 2 (also known as MCP1); CDC42, cell division cycle 42; EGR1, early growth response protein 1; ERK, extracellular signal-regulated kinase; ICAM1, intercellular adhesion molecule 1; JAK, Janus-activated kinase; JNK, Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; PI3K, phosphoinositide 3-kinase; RNS, reactive nitrogen species; SDH, sorbitol dehydrogenase; STAT, signal transducer and activator of transcription; VCAM1, vascular cell adhesion molecule 1.
Diabetes and macrovascular complications
Both type 1 (Refs 4, 5) and type 2 (Refs 6–8 diabetes have a largely irreversible and devastating effect on small and large blood vessels, and the consequences of vascular injury, such as hypertension, altered vascular permeability and ischaemia, can also contribute to the other complications of diabetes. Heightened oxidative stress and inflammation are increasingly recognized as being central pathogenic mechanisms and markedly alter patterns of gene expression in the vasculature9,10 shifting the balance from anti-inflammatory and anti-thrombotic homeostatic set points towards an increased pro-inflammatory and thrombogenic potential. As diabetes is further burdened by failure of vascular repair11,12, complications in many organs cause substantial morbidity and early mortality, with heart attack or stroke being the leading causes of mortality in both type 1 (Ref. 4) and type 2 (Ref. 13) diabetes.
Animal models and cardiovascular disease. Assorted animal models have been used to dissect the mechanisms underlying cardiovascular complications of diabetes and to identify possible therapeutic targets. Macrovascular complications have been studied using both rodent and larger animal models, such as rabbits, dogs, pigs and non-human primates; each species has its advantages and pitfalls. For example, although diabetes enhances vascular permeability in rodents14, advanced atherosclerosis is absent in this model, largely owing to the highly effective lipid-clearance mechanisms that are found in these species15. Rodent models that combine hyperglycaemia with dietary modulation or genetic modifications to induce hyperlipidaemia and promote atherosclerosis have therefore been developed. However, a consequence of combined hyperglycaemia and hyperlipidaemia in some of the well-studied mouse models, such as apolipoprotein E-deficient mice with concurrent type 1 diabetes, is that cholesterol levels are higher in diabetic animals compared with non-diabetic animals16. This makes it difficult to assess the relative contributions of glucose and/or lipids in driving the acceleration of atherosclerosis.
Two particularly creative models have been developed to address this issue. First, to assess the relative roles of hyperglycaemia and dyslipidaemia, mice lacking the low density lipoprotein receptor (LDLR) were crossed with transgenic mice expressing a viral protein under the control of the insulin promoter. When infected with the virus, the mice developed type 1 diabetes. In diabetic animals that were kept on a cholesterol-free diet, atherosclerosis was accelerated compared with non-diabetic animals, suggesting that hyperglycaemia drove accelerated atherosclerosis. However, when the mice were fed diets that were high in cholesterol (which rendered them diabetic), they developed severe hypertriglyceridaemia and advanced atherosclerosis17. These studies suggest that diabetes-associated dyslipidaemia accelerated lesion progression. Recent studies using this model have also shown that induction of type 1 diabetes in LDLR-deficient mice promoted plaque disruption, as measured by intraplaque haemorrhage18. Interestingly, plaque disruption was associated with the accumulation of monocytes that expressed S100 calcium-binding protein A9, a marker of inflammation.
The second model explores the role of the polyol pathway in the vascular complications of diabetes in mice, a species which normally has low levels of aldose reductase, by amplifying aldose reductase expression to physiologically relevant human levels. These aldose reductase-transgenic mice showed enhanced vulnerability to hyperglycaemia-induced atherosclerosis and ischaemia–reperfusion injury19,20. Importantly, hyperglycaemia seemed to be the primary factor driving accelerated atherosclerosis in aldose reductase-transgenic mice that were also deficient in LDLR, as lipid levels were the same in diabetic transgenic and wild-type animals. These and other animal models are helping to dissect the contribution of different pathogenic mechanisms to diabetic vascular damage.
The paucity of models for diabetic macrovascular complications has prompted the formation of the Animal Models of Diabetes Complications Consortium (AMDCC) to develop and evaluate new models21. However, although mouse models are suitable for dissecting and validating the specific role of hyperglycaemia in the pathogenesis of accelerated atherosclerosis in diabetes, they have definite shortcomings. For example, despite developing extensive and highly advanced atherosclerosis and intraplaque haemorrhage, current diabetic mouse models fail to reliably display evidence of thrombosis and overt myocardial infarction. By contrast, hyperglycaemic and hyperlipidaemic pigs and non-human primates with long-standing diabetes can have highly advanced lesions that are more similar to those observed in human subjects and, importantly, that might be vulnerable to thrombosis22,23.
One key component for accurate modelling may be the duration of diabetes, which may require many months to years to generate overt pathology. This has prompted extensive efforts to identify biomarkers of vascular injury in diabetes. Endothelial cell dysfunction, manifested as impaired vasodilatory responses to acetylcholine or reduced blood flow in human subjects, could prove useful in assessing the state of disease and, possibly, the effect of therapeutic intervention on vascular stress24. Studies that illustrate the potential diagnostic and predictive value of endothelial cell biopsy techniques in diabetes may also hold promise as an easily repeatable means to sample the vasculature and assess its inflammatory and thrombotic potential at any time point9,25.
Despite the limitations of the various animal models, these studies have shed light on some of the fundamental mechanisms that contribute to accelerated atherosclerosis in diabetes. When testing potential therapeutic targets, smaller animals could be used to probe mechanisms and to carry out early compound screening with a higher throughput, and larger animals could be subsequently used for the final testing of lead compounds and the validation of the mechanisms that were delineated in the smaller rodent species.
Mechanisms and interventions derived from animal models. The key to discovering treatments that target cardiovascular disease in individuals with diabetes lies in identifying the molecular species that promote perturbation of the vessel wall. Extensive epidemiological evidence suggests that glucose is one of the key players in this process26,27. Both the direct and indirect consequences of increased blood glucose levels contribute to the pathogenesis of accelerated cardiovascular disease in diabetes. In the macrovasculature, high levels of glucose probably synergize with superimposed stresses that are common to both diabetes and non-diabetic vascular disease, such as raised levels of serum lipids, hypertension and the sequelae of innate ageing processes, to continuously stress vascular cells. Such mechanisms lead not only to primary vascular dysfunction, but also to chronic cycles of stress that injure surrounding cells, which are dependent on intact vascular function. As summarized in Fig. 2, excess glucose directly stimulates activation of the polyol pathway3 and also the activity of mitochondria28, protein kinase C (PKC)29 and NADPH oxidase30,31, which results in the production of reactive oxygen species (ROS). Hyperglycaemia also leads to the formation of advanced glycation end products (AGE), which irrevocably alter the diabetic vasculature, leading to vascular stiffening owing to extensive protein cross-linking32. Moreover, extracellular AGE also bind and activate the signal transduction receptor RAGE (receptor of AGE). RAGE is a multi-ligand receptor, and its interactions with AGE and non-AGE pro-inflammatory ligands, such as S100–calgranulins and high-mobility group box 1 protein (HMGB1), are potent generators of accelerated vascular inflammation31,33. The importance of these pathways has been further suggested by recent studies involving the generation of unique mouse models. For example, the role of RAGE has been examined in both non-diabetic34 and diabetic35 mice deficient for the genes that encode apolipoprotein E and RAGE, and these studies have shown that RAGE has a pivotal role in atherosclerosis.
Clinical development of therapeutics. The insights gained from experimental models such as those described above have led to clinical trials investigating antagonism of the polyol pathway36, PKC isoforms (particularly PKCβ37,38), AGE39,40 and agents that counter the effect of the enhanced generation of ROS caused by high glucose levels, such as benfotiamine41. New small molecule ligands that antagonize RAGE and humanized chimeric soluble RAGE are also being tested in early clinical trials (see the 6-Month safety and efficacy study of TTP488 in patients with type 2 diabetes and persistent albuminuria). Long-term success in treating the blood vessels of patients with diabetes will probably lie in identifying combinations of these therapeutic targets, as it is likely that at distinct times and sites in the vessel wall, many mechanisms synergize to cause diabetic vascular stress. Whether suppression of these maladaptive messengers in the vessel wall will also facilitate the recruitment of progenitors and mediate vessel repair is a largely unexplored question. It is plausible that mechanisms which directly injure the vessel wall also disrupt the environmental cues and the flow of repair species and cells to the injured vessel. The long-term safety and tolerability of RAGE antagonism should be more fully investigated, particularly as studies in acute peripheral nerve injury suggest reparative roles for RAGE in inflammatory and axonal element signalling42,43.
As new targets and concepts are investigated (Table 1), it is important to note that inflammation and adaptive immune mechanisms that contribute to the pathogenesis of type 1 and type 2 diabetes also play a part in the subsequent macrovascular complications. In this context, strategies that broadly suppress inflammation, such as HMGH–CoA reductase inhibitors (statins)44 and peroxisome proliferator-activated receptor (PPAR) agonists45, have been considered relevant to the treatment of diabetic vascular injury. Statins are now considered almost routine in the management of both type 1 and type 2 diabetes, as various studies testing a range of these agents have shown a reduction in cardiovascular morbidity and mortality46,47.
Table 1.
Selected therapies in development for treating diabetic complications
| Drug name | Drug type | Presumed mechanism of action | Clinical trial phase |
|---|---|---|---|
| Omacor* | n-3 fatty acids | Enhances endothelial cell function and reduces vascular inflammation | IV |
| POMx* | Antioxidant derived from pomegranate juice | Reduces oxidative stress | IV |
| Rosiglitazone* | PPARγ agonist | Reduces insulin resistance and inhibits inflammatory cytokines | III and IV |
| Aliskiren*‡ | Renin inhibitor | Reduces hypertension | III (for angiopathy); III and IV (for nephropathy) |
| Alagebrium‡ | AGE cross-link breaker | Reduces AGE accumulation | II |
| TTP488‡ | RAGE antagonist | Reduced RAGE signalling | II |
| Pirfenidone‡ | Anti-fibrotic | Reduces interstitial fibrosis and glomerulosclerosis | II |
| Bevacizumab, ranibizumab and macugen§ | Anti-VEGF agents | Inhibits VEGF signalling, angiogenesis and vascular permeability | I–III |
| Candesartan§ | Angiotensin II inhibitor and antihypertensive agent | Inhibits angiogenesis and vascular permeability | III–IV |
| Infliximab§ | Monoclonal antibody against TNF | Inhibits TNF signalling | III |
| Doxycycline§ | Antibiotic | Inhibits inflammation | II |
| Triamcinolone§ | Corticosteroid | Inhibits inflammation and vascular permeability | I and II |
| Allopurinal, α-lipoic acid and nicotinamide‖ | Antioxidant cocktail | Reduces oxidative stress, inhibits PARP and increases blood flow | III |
| C-peptide‖ | Fragment of pro-insulin | Enhances insulin signalling or neurotrophic support | II |
| SB-509‖ | Zinc finger protein activator | Induces VEGF expression and has angiogenic and neurotrophic actions | II |
| MCC-257‖ | Sialic acid derivative | Enhances endogenous neurotrophic support | II |
| *Drug for which the indication is angiopathy‡Drug for which the indication is nephropathy.§Drug for which the indication is retinopathy.‖Drug for which the indication is neuropathy. Information was obtained from the database maintained at the ClinicalTrials.gov website. AGE, advanced glycation end products; PARP, poly(ADP-ribose) polymerase; PPAR, peroxisome proliferator-activated receptor; RAGE, receptor of AGE; TNF, tumour necrosis factor; VEGF, vascular endothelial growth factor. | |||
The case for PPAR agonists is less clear, despite good efficacy in experimental models of diabetes-associated atherosclerosis48. A recent study that evaluated the PPARγ agonist rosiglitazone using intravascular ultrasound suggests a trend towards reduced progression of atherosclerotic events in a cohort of subjects with type 2 diabetes49, although this drug has also been associated with increased cardiovascular events50. By contrast, another PPARγ agonist, pioglitazone, showed modest clinical benefits in a study on subjects with type 2 diabetes51. Dual PPARα–PPARγ agonists have been developed as a therapeutic approach to improved glucose and lipid homeostasis in patients with diabetes. However, fenofibrate did not clearly show benefits on cardiovascular outcomes52 despite a reduction in diabetic microvascular disease53, and muraglitazaar was withdrawn because of possible increased cardiovascular events54.
Although vascular inflammation may be the final common mediator and manifestation of diabetic vascular stress, there is little doubt that inhibiting these harmful pathways at earlier stages of hyperglycaemia is fundamental to preventing the devastating effects of diabetes on blood vessels. The importance of ambient hyperglycaemia per se in macrovascular disease may have to be reconsidered in the context of findings from two recent clinical trials, which explored the effects of tight glycaemic control in subjects with type 2 diabetes and established cardiovascular disease55,56. However, there are potential caveats to these studies. The lack of a positive effect on cardiovascular mortality in these studies could be related to the short duration of the trials (<5 years) or poor tolerance of the demanding and extensive therapeutic regimens by older subjects with type 2 diabetes and its many complications. It is also possible that irreversible vascular changes, such as advanced glycation57 or 'hyperglycaemic memory' resulting from previous periods of hyperglycaemia58, could also contribute, possibly through epigenetic mechanisms such as glucose-induced histone modifications that alter vascular gene expression59,60,61. As the effect of diabetes on plasma lipids and blood pressure could also have a substantial role in atherosclerosis, a therapeutic regimen that incorporates antihypertensive and lipid-lowering agents along with drugs that target the consequences of hyperglycaemia may ultimately prove to be the most effective approach, as recently highlighted in the follow-up of a study trialling this approach62.
Diabetic nephropathy
Diabetic nephropathy is now the most common cause of end-stage renal failure in the Western world63. From a clinical perspective, it is characterized by the onset of proteinuria and a subsequent decline in glomerular filtration rate and ultimate progression to uraemia, which is fatal if left untreated64. The main clinical associations that frequently precede overt diabetic nephropathy are hypertension and poor glycaemic control65. Once nephropathy is established, blood pressure often rises further, but glycaemic control can paradoxically improve as a result of reduced renal insulin clearance66. Both glucose-dependent pathways that are common to vasculopathy and other complications of diabetes (Fig. 2), and more organ-specific mechanisms that are linked to systemic and intraglomerular hypertension (Fig. 3), seem to play important parts in the development and progression of this disease67.
Figure 3. The proposed interactions between haemodynamic and metabolic disorders in diabetes that together mediate diabetic nephropathy.
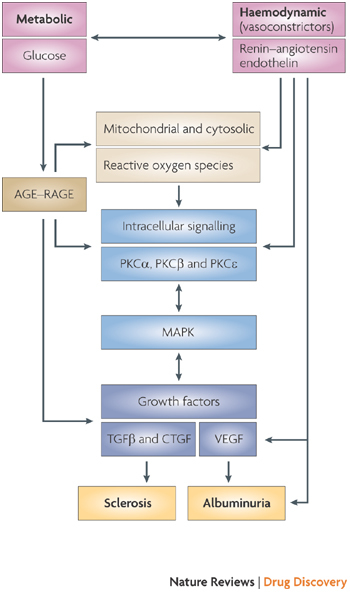
It is notable that many of the metabolic disorders of the kidney reflect those occurring in other organs that are compromised by diabetes (Fig. 2), whereas aspects of the haemodynamic component may be more specific to nephropathy. Therapeutic approaches that intervene in the core pathogenic mechanisms that are common to all diabetic complications may therefore require combination with organ-specific therapies for maximal benefit. AGE, advanced glycation end product; CTGF, connective tissue growth factor; MAPK, mitogen-activated protein kinase; PKC, protein kinase C; RAGE, receptor of AGE, TGFβ, transforming growth factor-β; VEGF, vascular endothelial growth factor.
The drugs currently used to treat diabetic renal disease largely target the hypertensive component. In particular, drugs that interrupt the renin–angiotensin system (RAS), such as angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor (AT2) antagonists, are currently considered first-line treatments for diabetic nephropathy, and this therapeutic strategy is incorporated into most national and international treatment guidelines68. This pharmacological approach has been extended by the recent production of the renin inhibitor aliskerin, which also seems to reduce proteinuria, particularly as part of dual therapy with an AT2 antagonist69. The most effective approach to blocking the RAS remains controversial, and various strategies are currently being investigated. With greater understanding of the complexity of the RAS and identification of new components of this pathway, such as ACE2, it has become increasingly evident that there is a complex interaction between the vasoconstrictor and vasodilator arms of the RAS70. Indeed, it seems that an alteration in one component, such as ACE2, can influence the renal response to agents such as ACE inhibitors70. Furthermore, there is often upregulation of upstream components such as renin with distal interruption of the RAS, which can be suppressed by vitamin D administration2,71.
Animal models of diabetic nephropathy. The classical model of streptozotocin-induced type 1 diabetes in rodents is still widely used and has a range of functional and structural changes that are comparable to human diabetic nephropathy72. These include early development of renal hypertrophy, progressive increases in albuminuria and changes to the renal ultrastructure, such as mesangial expansion and glomerular basement membrane thickening. Unfortunately, more advanced renal disease, specifically overt proteinuria, renal impairment and advanced structural lesions, are not prominent in this model72. New models, such as the Akita73 and OVE26 mice74, have been developed to address this concern and show more advanced lesions. Whether these new mouse strains will ultimately prove to be more useful preclinical models for testing new treatments remains to be determined, and much emphasis is currently being placed on the genetic background of the mice. For example, it has been suggested that manifestations of nephropathy in the Akita mouse model can differ depending on the genetic background of the mice73.
Mechanisms and interventions derived from animal models. Much of the initial research into diabetic nephropathy relied on detailed characterization of the progression of the disease at the functional and ultrastructural levels. These studies identified the phase of microalbuminuria or incipient diabetic nephropathy, characterized by modest increases in urinary excretion of albumin. It was then shown that this phase predicts the subsequent development of overt renal disease and is associated with key structural changes to the renal glomerulus75.
Recent studies have explored potential molecular and biochemical mechanisms that could be responsible for the progression of renal lesions in diabetes. Initial research focused on the mesangial cells within the glomerulus, but changes in other glomerular cells, including podocytes76, and progressive injury to the tubulointerstitium have subsequently been described77. Glucose-dependent pathways (Fig. 2), such as advanced glycation78, are clearly important components in the pathogenesis of diabetic renal disease and accompany organ-specific mechanisms involving disruption of vasoactive hormone pathways, such as the RAS79. Glucose and AGE-mediated activation of RAGE78,80, key intracellular signalling molecules such as PKC and mitogen-activated protein kinase, and angiotensin II also have a central role in this disease79. Furthermore, certain pro-sclerotic growth factors, such as transforming growth factor-β77,81 and connective tissue growth factor82, seem to promote extracellular matrix accumulation, a cardinal structural feature of the kidney in patients with diabetes. It is also becoming increasingly apparent that metabolic and haemodynamic pathways not only interact through common mediators, such as intracellular signalling molecules and growth factors, but can directly interact with each other. For example, angiotensin II can enhance AGE accumulation in the kidney and AGE can directly modulate expression of key components of the RAS83. Thus, it seems that metabolic and haemodynamic stimuli, triggered by the diabetic milieu, interact to amplify injury and perpetuate the progression of renal damage in diabetes (Fig. 3).
Clinical development of therapeutics. Over the past decade, there have been important advances in understanding the pathogenesis of diabetic nephropathy that have prompted ongoing clinical trials of new agents that are designed to delay and reverse diabetic renal disease. Efficacy of agents that influence advanced glycation pathways (such as soluble RAGE)78, inhibitors of renal AGE accumulation (such as the putative cross-link-breaker alagebrium)84, inhibitors of PKC activation (such as ruboxistaurin, an inhibitor of PKCβ)29 or inhibitors of vasoactive hormone pathways (such as endothelin antagonists) in animal models have all stimulated new clinical trials. Some of these clinical trials have now been completed85, whereas others are still in progress86 (Table 1). These trials are focusing primarily on albuminuria as an index of nephropathy, as well as assessing the decline in renal function. Because diabetic nephropathy takes many years to evolve and only occurs in a minority of subjects, these clinical trials have often been difficult to complete. Moreover, the US Food and Drug Administration currently considers renal impairment, and not proteinuria, as the appropriate renal end point of diabetic nephropathy. This requires studies that often involve more than 1,000 patients and a follow-up of up to 5 years87,88. Such studies are difficult to carry out in terms of recruitment and are expensive.
It is hoped that surrogate markers such as albuminuria may be accepted for approval by regulatory agencies to register drugs as renoprotective in diabetes89, although the finding that some subjects with diabetes develop renal impairment in the absence of albuminuria90 suggests that serial assessment of urinary albumin excretion may also not be ideal. Many research groups are trying to identify alternative biomarkers of renoprotection, and much of this work focuses on urine proteomics91. This research is at an early phase, and a biomarker has not yet been established. Ultimately, the goal is to develop more robust strategies for monitoring diabetic nephropathy and in particular to generate a more reliable approach to identifying truly renoprotective regimens that not only decrease albuminuria but also reduce renal morphological injury and preserve renal function.
Diabetic retinopathy
Diabetes is the leading cause of new cases of blindness among adults aged 20–74 years92. It is characterized by a range of retinal lesions and abnormalities that indicate vascular damage (capillary microaneurysms, capillary degeneration, increased vascular permeability and new vessel formation) and death or dysfunction of the neural retina ('cotton wool spots', alterations in retinal electrophysiology, loss of colour or hue discrimination). Clinically, diabetic retinopathy has been separated into non-proliferative and proliferative disease stages. Only the late stages of the retinopathy, especially neovascularization and retinal oedema, have adverse effects on vision93, but these disorders seem to be dependent on changes that develop in the earlier stages of the disease. New vessels growing out of the retina into the normally avascular vitreous fluid are particularly threatening for vision owing to excessive leakage from these new vessels (pre-retinal haemorrhage) or to the development of a fibrovascular membrane93. Available evidence suggests that occlusion or degeneration of retinal capillaries is strongly associated with the development and progression of diabetic retinopathy94, presumably by contributing to the development of ischaemia and the subsequent release of hypoxia-inducible vasoproliferative factors. Accumulation of fluid within the retina also contributes to the visual impairment in diabetes, in part by distorting the retinal architecture93.
Diabetic retinopathy takes many years to develop, and almost all patients with type 1 (Ref. 95) and type 2 (Ref. 96) diabetes exhibit some lesions after 20 years of disease. Nevertheless, only a fraction of these patients will progress to having visual impairments. There does not seem to be a difference in the clinical picture of diabetic retinopathy between patients with type 1 and type 2 diabetes.
Animal models of retinopathy. Numerous species, including monkeys, dogs, cats, pigs, hamsters, rats and mice, have been used as models for the study of diabetic retinopathy97. All mammalian species studied to date develop at least the early stages of retinopathy, including degeneration of retinal capillaries. Development of retinopathy in many of these models occurs more rapidly than in humans, with early lesions developing within months to years of the onset of diabetes. The severity of retinal disease increases with duration of diabetes, but remains mild compared with that seen in many patients with diabetes. This is due in part to the limited lifespan of laboratory animals. With the exception of occasional contrary claims, diabetes alone has not been found to cause pre-retinal (intravitreal) neovascularization in any animal model98. Again, this is probably due in part to insufficient severity of capillary degeneration and other lesions during the limited time over which these models have been studied. No data have yet been generated to suggest that the current animal models are inappropriate reflections of human diabetic retinopathy because they are mechanistically inadequate. Instead, the development and appearance of early retinopathy is similar in many diverse animal models of diabetes, in which the differences in the severity of retinopathy are related to the differences in the severity of hyperglycaemia.
Investigators who are interested in studying or inhibiting retinal neovascularization in vivo have turned to non-diabetic models in which retinal neovascularization occurs after branch vein occlusion, oxygen-induced retinopathy99 or overexpression of growth factors such as vascular endothelial growth factor (VEGF)100,101 or insulin-like growth factor102 in the eye. Because degeneration or occlusion of retinal capillaries in diabetes has been closely linked to eventual retinal neovascularization in humans, inhibition of diabetes-induced capillary degeneration is also used to evaluate therapeutic interventions103.
Retinal neurons, particularly the retinal ganglion cells that connect the eye to the brain, have also been reported to degenerate in diabetic rats104,105 and some strains of diabetic mice106. Diabetes-induced retinal neuron loss seems to begin before death of retinal vascular cells occurs but is not dramatic in these models, again perhaps owing to the short duration of the study. Despite this progressive neuronal loss, animal models have not yet been found to show evidence of visual impairment or blindness because of diabetes.
There remain important gaps in our understanding of the pathogenesis of retinal neovascularization in diabetes, the role of neural dysfunction and neurodegeneration in loss of vision and in the translation of efforts to inhibit retinal capillary degeneration in animal studies into the prevention of vision loss in patients with diabetes. Many cell types, both within and outside the retina, are likely to contribute to the development of diabetic retinopathy, and it is becoming apparent that the interrelationship between the vascular, neural, myeloid and glial cells needs further study in both health and disease. Ongoing efforts to develop genetically modified animals may offer new ways to investigate the pathogenesis of diabetic retinopathy and identify new therapeutic targets.
Mechanisms and interventions derived from animal models. Although it is widely accepted that diabetic retinopathy is caused by poor glycaemic control, the progression of retinopathy does not reverse, or even immediately halt, when better glycaemic control is achieved26,107. The observation that diabetic-like retinopathy develops following a long-term increase in galactose levels in the blood, even in the absence of diabetes, provides strong evidence that hyperglycaemia per se, as opposed to insulin deficiency or alterations in lipid profile that are characteristic of poor diabetic control, is sufficient to initiate the development of aspects of retinopathy in mice108 and rats109,110. However, disorders that commonly parallel hyperglycaemia in patients with diabetes, such as altered blood pressure and dyslipidaemia, have been found to influence the rate of progression of retinopathy27,111. In recent years, there has been a great increase in the number of potential therapies that are reported to inhibit the diabetes-induced degeneration of retinal capillaries and neurons as well as the increase in retinal vascular permeability in animals. These range from pan-complication therapies, such as aldose reductase inhibitors, to tissue-specific approaches, such as anti-VEGF agents103. Likewise, the range of therapies that have been found to inhibit retinal neovascularization in non-diabetes animal models is expanding and includes both those with potential efficacy against many diabetic complications, such as PKCβ inhibitors, and those that specifically inhibit mechanisms of retinopathy, such as blocking VEGF signalling112.
A large group of diabetes-induced biochemical and physiological abnormalities in the retina, which were previously regarded as being independent of each other, are now known to associate in the context of inflammation103,113. The non-proliferative stages of diabetic retinopathy include altered vascular permeability and function, vascular degeneration, and neural dysfunction and degeneration. The interplay between these abnormalities is an ongoing area of research, and it is conceivable that individual therapies might not have comparable effects on each of these different areas.
Clinical development of therapeutics. Numerous clinical studies over the past several decades have provided strong evidence that surrogate markers that quantify the severity of retinopathy, such as the Early Treatment of Diabetic Retinopathy Study (ETDRS) retinopathy grading scale114,115, can predict progression of the retinopathy and can indicate inhibition of disease progression. Randomized, multi-centre clinical trials have similarly provided valuable data on the ability of treatments, such as those providing improved glycaemic control, to inhibit the development and/or progression of retinopathy. Intensive treatment to normalize blood glucose levels resulted in a 63% reduction in retinopathy progression and a significant inhibition of other microvascular complications26,116 in patients with diabetes. Furthermore, laser photocoagulation reduced the risk of severe visual loss by more than 50% in the eyes of patients with high-risk characteristics117.
On the basis of positive preclinical studies118, a clinical study recently assessed the role of the angiotensin II antagonist candesartan in both type 1 and type 2 diabetes with or without early diabetic retinopathy119,120. Overall, this agent had a positive effect, with reduced progression and in some groups regression of retinopathy as measured by the ETDRS scale. There is also increasing evidence of benefits from intravitreal injections of steroids such as triamcinolone, often in conjunction with laser photocoagulation on vision-threatening retinopathy, including diffuse macular oedema121,122. However, there are risks associated with intraocular steroid therapy, including increased intraocular pressure and enhanced cataract formation. Several other clinical trials have not produced the outcomes that were hoped for, probably owing in part to insufficient study duration, slow development or progression of retinopathy, insufficient inhibition of the therapeutic target or a lack of involvement of the therapeutic target in the pathogenesis of the retinopathy123,124,125,126.
The development of non-invasive methods to assess the vascular and neural components of the eye is offering new opportunities to investigate the pathogenesis of retinopathy and to document responses to therapeutic intervention. Directed therapies against abnormalities that are believed to play key parts in the later stages of diabetic retinopathy, including inhibitors of PKC127,128 or VEGF129, are providing therapeutic tools to inhibit clinically important aspects of retinopathy in patients with diabetes (Table 1). However, it is important to recognize that there is a disconnection between what is studied in animal models using high resolution microscopy and isolated tissue preparations, and what can currently be measured in patients with diabetes using colour fundus photographs, fluorescein angiograms and retinal function tests. New techniques, such as optical coherence tomography130 and magnetic resonance imaging131,132, may be useful for both animal and clinical investigations and will hopefully lead to more interaction between preclinical and clinical scientists.
Diabetic neuropathy
Diabetic neuropathy affects the somatic and autonomic divisions of the peripheral nervous system, and there is a growing appreciation that the spinal cord133 and higher central nervous system (CNS)134 may also be damaged. Over half of all patients with diabetes develop some form of neuropathy135, resulting in sensory loss, pain and autonomic dysfunction. These manifestations of neuropathy, along with their contribution to impaired wound healing and cardiovascular and erectile dysfunction, can severely reduce a patient's quality of life. As with the other complications discussed above, duration of diabetes and lack of adequate glycaemic control are important risk factors for neuropathy in both type 1 and type 2 diabetes26,116. No therapy for diabetic degenerative neuropathy, other than maintenance of normoglycaemia, is approved by regulatory bodies in Europe and the United States, and current pain management strategies are not consistently effective and do not target the causes of diabetes-induced pain. The development of new therapies has been hindered by an incomplete understanding of the aetiological mechanisms involved, which is largely a reflection of the lack of a suitable animal model.
Animal models of neuropathy. Most studies have been carried out in rodent models of type 1 or type 2 diabetes, in which hyperglycaemia is induced by genetic, pharmacological or dietary manipulations. Short-term rodent models of diabetes quickly develop a slowing of nerve conduction but generally lack overt demyelination and fibre loss in nerve trunks, which are prominent features of clinical diabetic neuropathy136. Longer durations of diabetes may produce discernable pathology in nerve trunks of some rodent models137,138, and the retraction of small sensory fibre terminals in the skin of short-term rodent models of diabetes139,140 offers a measurement of nerve pathology that can be used in drug screening assays. The current rodent models of diabetic neuropathy may best reflect initial biochemical and functional disorders that precede degenerative neuropathy and could be useful for understanding early pathogenic events. However, there remains a prominent knowledge gap regarding the mechanisms of demyelination and neurodegeneration, such that a leap of faith has been required when translating any therapy that targets degenerative neuropathy from preclinical studies to clinical trials.
The lack of overt degenerative neuropathy in most rodent models of diabetes may be due to their short lifespan and physically shorter axons. Larger, long-lived diabetic animals have therefore also been used as alternative models. Diabetic dogs develop a slowing of nerve conduction and corneal hyposensitivity after years of hyperglycaemia141,142, but degenerative neuropathy is minimal143. Diabetic monkeys were recently shown to have epidermal nerve fibre loss144, and studies of diabetic domestic cats have identified a degenerative neuropathy (Fig. 4) that replicates many features of the human condition145,146. The extent to which the growing population of domestic cats with diabetes can be used to test potential therapies for degenerative neuropathy and act as a bridge between traditional preclinical models and clinical trials is currently being evaluated.
Figure 4. Peroneal nerve biopsy from a diabetic cat.
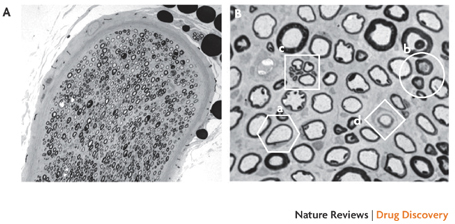
This image shows nerve fibre loss (A) and the presence of pathological features (B), including thin myelin (a), dystrophic axons (b), a cluster of small regenerating axons (c) and a putative supernumenary Schwann cell (d) that are rare in the common rodent models of diabetic neuropathy. The pathologic changes found in the nerves of diabetic domestic cats closely resemble those seen in patients with diabetes146. The growing number of domestic cats with diabetes may offer an experimental bridge between preclinical studies in rodents and clinical trials that allows direct measurement of efficacy against degenerative diabetic neuropathy and may also provide an additional market for such drugs. Images courtesy of A. Mizisin, University of California, San Diego, USA.
Mechanisms and interventions derived from animal models. Prevention, or preferably reversal, of the slowing of large-fibre conduction in diabetic rodents has historically been the preclinical 'gold standard' for establishing the therapeutic potential of drugs targeting degenerative diabetic neuropathy. Over 100 compounds have been reported to prevent or reverse the slowing of large-fibre conduction, and the successful agents seem to target diverse pathogenic mechanisms. However, these have yet to translate to a successful therapy for diabetic neuropathy. Whether this reflects a complex and interconnected pathogenesis of diabetic neuropathy or the unsuitability of conduction defects in diabetic rodents for predicting efficacy in humans remains controversial. A more cautious approach that involves showing efficacy against both functional and structural indices of neuropathy may be warranted.
Many of the potential therapies that have been investigated to treat diabetic peripheral neuropathy have targeted mechanisms such as vascular dysfunction147, oxidative stress148 and RAGE signalling149 all of which are common to all the diabetic complications (Figs 1,2). Evidence that impaired neurotrophic support contributes to diabetic peripheral and autonomic neuropathy150,151 suggests a more organ-specific pathogenic mechanism (Fig. 1), although some aspects of neurotrophic insufficiency are clearly downstream consequences of hyperglycaemia152,153. Several neuroactive proteins and polypeptides have been investigated as replacement therapies to counteract impaired neurotrophic support150 but difficulties with delivery routes and systemic effects have impeded clinical development. Small molecules that promote endogenous production of neurotrophic factors have also shown some success in diabetic rodents154,155,156. Most recently, gene therapy to facilitate endogenous production of nerve growth factor157,158, hepatocye growth factor159, neurotrophin 3 (Ref. 160) or VEGF161,162 has been explored. Advancement of this approach must clearly incorporate the goal of delivering localized and regulated production of such factors.
Impaired signalling through insulin receptors, whether driven by insulin deficiency or resistance, is also emerging as a possible primary pathogenic mechanism that contributes to diabetes-induced damage to the nervous system. Peripheral nerves have insulin receptors163 and insulin treatment regimes that do not modulate circulating glucose levels can protect against neurochemical164, behavioural165 and functional166,167 indices of neuropathy in diabetic rodents. This suggests that insulin acts as a support factor for peripheral nerves.
As well as impeding neurotrophic support, reduced insulin signalling can also activate neurotoxic mechanisms. This is becoming increasingly apparent in the CNS, where deficient insulin signalling has been linked to accumulation of amyloid-β and hyperphosphorylation of the neuronal protein tau in models of Alzheimer's disease (Ref. 168). Amyloid-β and hyperphosphorylated tau also accumulate in the brain of diabetic rats169 and mice170,171, in which there is evidence of synaptic and neuronal loss. There is continuing debate regarding the extent to which patients with type 1 and type 2 diabetes are prone to developing cognitive impairments and manifestations of dementia172,173. Nevertheless, the apparent convergence of pathogenic mechanisms for these two common diseases of ageing on defective insulin signalling is attracting considerable interest174 and may reveal new targets for therapeutic intervention.
Therapeutic strategies to treat the pain associated with diabetes have traditionally drawn on experience from other neuropathic pain states rather than targeting diabetes-specific mechanisms; an example of this is the widespread use of tricyclic antidepressants175. The anticonvulsants gabapentin and pregabalin176,177 and the antidepressant duloxetine178 have been used for alleviating diabetes-induced neuropathic pain, albeit with caveats concerning their efficacy, side effects, cost effectiveness179 and a lack of effect on the underlying degenerative neuropathy. Although these treatments have emerged from broad anti-neuropathic pain programmes, preclinical studies suggest that a wider therapeutic window may exist in diabetes because of the increased expression of the presumed targets180,181. Reports that diabetes increases the expression of other potential targets, such as peripheral κ-opioid receptors182 and spinal cyclooxygenase 2 (Ref. 183), may provide additional avenues for drug development.
Clinical development of therapeutics. Many agents have entered clinical trials to treat diabetic neuropathy. Twenty five years of failure to translate the mechanistic and therapeutic findings in animal models to a clinically effective treatment for diabetic degenerative neuropathy has prompted sharp divides in the research community. Specifically, some researchers see the animal models as being inadequate tools for discovering effective therapeutics and others think that the clinical trials have been poorly designed and executed or have relied on inappropriate or unreliable end points; both views probably have some merit.
Approaches currently under investigation range from refinement of aldose reductase inhibition and other interventions that may have pan-organ efficacy to the neuron-specific targeting of neurotrophic support mechanisms (Table 1). Previous clinical trials have largely relied on the slowing of nerve conduction and on sensory testing as quantifiable predictors of progressive degenerative neuropathy184,185. Sural nerve biopsies have also been used as a direct measure of nerve pathology186 but the technique is invasive, difficult to quantify and does not allow multiple measurements. The continued failure to show an acceptable clinical efficacy of therapies that were developed from preclinical screening against slowing of nerve conduction in diabetic rodents could be due to mechanistic differences between species or the lack of reliable measures of early neuropathy.
One advance that may assist drug development studies is the emerging use of skin biopsies as a minimally invasive measure of small fibre distal degenerative neuropathy187,188. Epidermal fibre loss can be detected even before the onset of clinically overt diabetes189 and is associated with functional indices of sensory loss190. Although the technique has currently been restricted to the evaluation of small sensory fibres and not of myelinated sensory, motor or autonomic fibres that are also affected by diabetes, it offers the advantage of providing a direct measurement of fibre loss that may ultimately result in loss of thermal sensation. Repeated biopsies can be carried out and allow monitoring of the progression of neuropathy and the effects of drug treatment.
The use of corneal confocal microscopy to view changes in sensory nerve fibres in the eyes of patients with diabetes is also currently being evaluated against other measures of early neuropathy191. Corneal confocal microscopy offers the additional advantage of being entirely non-invasive, which allows many sequential measurements. It also facilitates local and topical drug delivery and can be used to detect nerve regeneration192. Skin biopsy and corneal microscopy may therefore prove useful additions to future clinical trials of drugs that target diabetic neuropathy.
Current and future challenges
It is likely that refinement of pancreatic transplantation and other approaches to maintaining long-term regulation of insulin and glucose levels, such as the artificial pancreas, will ultimately reduce the incidence and severity of diabetic complications. However, initial periods of poorly controlled diabetes can have a protracted negative effect on the subsequent protection that is afforded by improved glycaemic control193, and numerous patients have, or are in the process of developing, diabetic complications. These caveats provide sufficient concern to support efforts directed at understanding the pathogenic mechanisms that cause diabetic complications and at developing therapeutic interventions.
The effects of diabetes on the organ systems described above emphasize that the main complications share numerous glucose-driven pathologic mechanisms (Figs 1,2). Targeting glucose-mediated vascular dysfunction with approaches such as inhibition of RAGE signalling, aldose reductase activity and oxidative stress is a particularly appealing therapeutic approach as it offers potential efficacy against multiple complications. Moreover, some of these targets may also have additional organ-specific effects that are independent of their impact on vascular function. For example, aldose reductase also localizes in Schwann cells of peripheral nerves and in cells of the kidneys and eyes194,195, whereas RAGE expression is induced or increased by diabetes in each of these organs independently of its expression in blood vessels78,196,197. Some treatments therefore have the potential to disrupt both universal and organ-specific pathogenic mechanisms.
The limitations of current rodent models of diabetic complications, which tend to show early metabolic and functional disorders but lack marked structural pathology, are a cause for concern. The use of these models is driven in part by the desire of researchers to accelerate the onset of complications in inexpensive species that are amenable to genetic manipulation. Although current rodent models are useful for identifying the initial pathogenesis of diabetic complications and for studying the effect of potential prophylactic and early interventional therapies, there is persistent uncertainly as to whether investigators are studying mechanisms and drugs that are pertinent to overt pathological damage in humans. Another recurring theme is the difficulty in designing viable clinical trials and identifying which end points should be used as indicators of therapeutic efficacy. It is clear that the regulatory bodies are challenged by conflicting requirements to establish consistent end points which can be used in trials that may take many years to complete, while needing to have the flexibility to adopt new surrogate biomarkers as they are identified and validated by preclinical and clinical studies.
The fact that diabetes disrupts such a diverse range of highly specialized organs also presents substantial challenges for drug development. The example of VEGF, for which inhibitors are being developed to treat diabetic retinopathy129 and increased expression provoked to treat neuropathy162 illustrates the complexities that have to be considered when addressing individual complications within the context of a disease that impairs so many systems. Nevertheless, new animal models, creative drug delivery systems and improvements in clinical trial design and biomarkers will hopefully combine to accelerate the development of therapies for diabetic complications and improve approaches that are already being investigated. A single drug that is effective against all diabetic complications may not be a realistic goal. Therapeutic strategies that incorporate interventions targeting the glucose-mediated pathogenic axis outlined in Fig. 2, in combination with those that address organ-specific and glucose-independent mechanisms, may provide the most successful approach to drug development in this area.
Acknowledgements
The authors are supported by the Juvenile Diabetes Research Foundation (M.E.C., N.A.C., T.S.K., A.M.S.), the National Institutes of Health (M.E.C., N.A.C., T.S.K., A.M.S.), the National Health and Medical Research Council of Australia (M.E.C.) and the Medical Research Service of the Department of Veterans Affairs (T.S.K.).
Glossary
- Hyperglycaemia
A protracted increase in blood glucose levels beyond the usual fasting and/or postprandial ranges. Frequently used as a synonym for diabetes.
- Polyol pathway
A glucose-metabolizing pathway comprising the enzymes aldose reductase and sorbitol dehydrogenase that is found in organs that develop diabetic complications.
- Aldose reductase inhibitors
The class of drugs that block the activity of the enzyme aldose reductase, which have been widely studied as a potential means of preventing or ameliorating diabetic complications.
- Hypertension
A chronic increase in blood pressure (usually measured as arterial blood pressure) that is associated with many diseases, including diabetes.
- Nephropathy
Damage to the kidneys.
- Retinopathy
Damage to the retina of the eye.
- Neuropathy
Damage to the nervous system (usually applied to the peripheral nerves).
- Atherosclerosis
A chronic inflammatory response in the wall of large blood vessels during which the vessel wall hardens owing to accumulation of plaques that are formed following oxidation of low density lipoproteins in the blood by reactive oxygen species.
- Endothelial cell
Simple squamous epithelial cell that lines the lumen of all blood vessels and is considered an important site of diabetes-induced damage to blood vessels and subsequently other organs.
- Protein kinase C
A family of enzymes that, when activated, translocate to the plasma membrane and facilitate phosphorylation of other proteins, thereby activating or deactivating them. Aberrant activation of PKC has been implicated in the pathogenesis of many diabetic complications.
- NADPH oxidase
A membrane-bound enzyme complex that uses NADPH to catalyse the conversion of oxygen to superoxide and thereby generates reactive oxygen species. Widely studied as a mechanism of cell-mediated bacterial killing, excessive activity of NADPH oxidase isoforms has also been implicated in mechanisms of diabetic complications.
- Advanced glycation end product
A product of the irreversible addition of glucose to proteins or fats. It is produced following a sequence of Amadori, Schiff and Maillard reactions, which can change the function of the recipient molecule.
- Proteinuria
The appearance of serum proteins (for example, albumin) in the urine that is associated with early stages of diabetes-induced damage to the filtration system of the kidney.
- Vasculopathy
Damage to blood vessels. Usually divided into damage to large (macrovascular disease) and small (microvascular disease) blood vessels.
- Streptozotocin
(STZ). A glucosamine-nitrosurea compound, originally isolated from Streptomyces achromogenes, that enters the pancreatic β-cell by a glucose transporter system and damages DNA by alkylation, leading to cell death and subsequent insulin deficiency. STZ is used to induce type 1 diabetes in animals and provides the most widely studied models of diabetic complications.
- Renal glomerulus
Capillary bed of the renal corpuscle from which plasma is extruded and filtered before entering the tubular system of the kidney. The filtering system includes the endothelial cells of the capillary, the glomerular basement membrane, podocytes and associated mesangial cells. Disruption of one or more of these cells and structures can impair blood filtration and contribute to diabetic nephropathy.
- Microaneurysm
Small, focal points of damage to a blood vessel (usually capillary) wall, leading to pressure-induced swelling that may cause the vessel to rupture and allow leakage of blood (haemorrhage) into the surrounding environment.
- Neovascularization
Growth of new blood vessels. Can be beneficial when blood vessels have been damaged, but can cause diabetic complications such as proliferative retinopathy if uncontrolled or inappropriate.
- Oedema
Swelling of the extracelluar tissue space by influx of fluid in response to the accumulation of osmotically active molecules. Oedema has a protective role following tissue injury but can also cause damage if it is prolonged or develops inappropriately.
- Glial cell
A non-neuronal cell of the nervous system, including Schwann cells (in peripheral nerve), astrocytes and oligodendrocytes (in brain and spinal cord) that regulates the environment surrounding neurons. Smaller microglial cells are scavengers of cellular debris that can accumulate in the nervous system, particularly following damage.
- Demyelination
Retraction of the myelin sheath from around a neuron that is caused by damage to, or death of, myelin-forming Schwann cells (in the peripheral nervous system) or oligodendrocytes (in the central nervous system). Demyelination is a prominent feature of diabetic neuropathy but has been difficult to model in rodent models of diabetes.
- Epidermal nerve fibre
A small unmyelinated sensory neuron of the peripheral nervous system, the peripheral terminal of which projects above the dermis of the skin into the epidermis and allows sensation of heat pain and other stimuli. Also referred to as an intra-epidermal nerve fibre.
- Oxidative stress
Inappropriate oxidation of proteins, lipids and DNA that may change the function of these molecules, caused by excessive production of reactive oxygen species beyond the local capacity to remove them.
- Neurotrophic factor
A term generally used to encompass any molecule that supports neuronal growth and repair or prevents neuronal death.
- Sural nerve
A distal sensory branch of the sciatic nerve trunk that passes close to the dermis at the ankle and can therefore be biopsied.
- Corneal confocal microscopy
An imaging technique that allows sensory neurons in the cornea of the eye to be viewed without tissue incision or invasion.
Biographies
Nigel A. Calcutt obtained his B.Sc. in zoology and Ph.D. in physiology and pharmacology from Nottingham University, UK. After postdoctoral work in pharmacology at St Bartholomew's Hospital, London, UK, he moved to the University of California, San Diego, USA, where he is now Professor in the Department of Pathology. His research interests include mechanisms of neuropathies that are associated with diabetes and other diseases, and the development of animal models and therapeutics for these disorders.
Mark E. Cooper studied medicine at the University of Melbourne, Australia, followed by training in endocrinology and a Ph.D. in the Department of Medicine at the same institute. He is currently Associate Director of the Baker IDI Heart and Diabetes Institute in Melbourne, where he heads the Diabetes and Metabolism Division. He studies diabetic nephropathy and vascular disease, with a particular interest in preclinical studies exploring new therapeutics.
Timothy S. Kern obtained his Ph.D. in pathology from the University of Wisconsin–Madison, USA. After postdoctoral training in ophthalmology at this university, he joined the faculty until moving to the Department of Medicine at Case Western Reserve University, Cleveland, Ohio, USA. His current research interest is on the role of inflammation in the development of diabetic retinopathy and other complications of diabetes, and the development of therapeutic approaches to inhibit retinopathy.
Ann Marie Schmidt is the Gerald and Janet Carrus Professor of Surgical Science in the Department of Surgery at Columbia University, New York, USA. Her current research is focused on the biology of the receptor for advanced glycation end products and its influence in diabetic complications, atherosclerosis inflammation and neurodegeneration.
Related links
FURTHER INFORMATION
Competing interests
M.E.C. has received research grants from Synvista, Speedel and Astra Zeneca, which are the manufacturers of algebrium, avosentan and candesartan, respectively. He has also received honoraria from GlaxoSmithKline, Servier and Astra Zeneca, which are the manufacturers of rosiglitazone, perindopril and candesartan, respectively.
References
- 1.Botero D, Wolfsdorf JI. Diabetes mellitus in children and adolescents. Arch. Med. Res. 2005;36:281–290. doi: 10.1016/j.arcmed.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Turner RC, Holman RR. Lessons from UK prospective diabetes study. Diabetes Res. Clin. Pract. 1995;28:S151–S157. doi: 10.1016/0168-8227(95)01105-m. [DOI] [PubMed] [Google Scholar]
- 3.Gabbay KH. The sorbitol pathway and the complications of diabetes. N. Engl. J. Med. 1973;288:831–836. doi: 10.1056/NEJM197304192881609. [DOI] [PubMed] [Google Scholar]
- 4.Nathan DM, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nathan DM, et al. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N. Engl. J. Med. 2003;348:2294–2303. doi: 10.1056/NEJMoa022314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner R, Cull C, Holman R. United Kingdom Prospective Diabetes Study 17: a 9-year update of a randomized, controlled trial on the effect of improved metabolic control on complications in non-insulin-dependent diabetes mellitus. Ann. Intern. Med. 1996;124:136–145. doi: 10.7326/0003-4819-124-1_part_2-199601011-00011. [DOI] [PubMed] [Google Scholar]
- 7.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HAW. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008;359:1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- 8.Holman RR, Paul SK, Bethel MA, Neil HA, Matthews DR. Long-term follow-up after tight control of blood pressure in type 2 diabetes. N. Engl. J. Med. 2008;359:1565–1576. doi: 10.1056/NEJMoa0806359. [DOI] [PubMed] [Google Scholar]
- 9.Feng L, et al. Chronic vascular inflammation in patients with type 2 diabetes: endothelial biopsy and RT-PCR analysis. Diabetes Care. 2005;28:379–384. doi: 10.2337/diacare.28.2.379. [DOI] [PubMed] [Google Scholar]
- 10.Crimi E, Ignarro LJ, Napoli C. Microcirculation and oxidative stress. Free Radic. Res. 2007;41:1364–1375. doi: 10.1080/10715760701732830. [DOI] [PubMed] [Google Scholar]
- 11.Fadini GP, et al. Number and function of endothelial progenitor cells as a marker of severity for diabetic vasculopathy. Arterioscler. Thromb. Vasc. Biol. 2006;26:2140–2146. doi: 10.1161/01.ATV.0000237750.44469.88. [DOI] [PubMed] [Google Scholar]
- 12.Tepper OM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–2786. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 13.Dale AC, Vatten LJ, Nilsen TI, Midthjell K, Wiseth R. Secular decline in mortality from coronary heart disease in adults with diabetes mellitus: cohort study. BMJ. 2008;337:99–102. doi: 10.1136/bmj.39582.447998.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waltman S, Krupin T, Hanish S, Oestrich C, Becker B. Alteration of the blood–retinal barrier in experimental diabetes mellitus. Arch. Ophthalmol. 1978;96:878–879. doi: 10.1001/archopht.1978.03910050480018. [DOI] [PubMed] [Google Scholar]
- 15.Galman C, et al. Age-induced hypercholesterolemia in the rat relates to reduced elimination but not increased intestinal absorption of cholesterol. Am. J. Physiol. Endocrinol. Metab. 2007;293:E737–E742. doi: 10.1152/ajpendo.00166.2007. [DOI] [PubMed] [Google Scholar]
- 16.Park L, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nature Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 17.Renard CB, et al. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J. Clin. Invest. 2004;114:659–668. doi: 10.1172/JCI17867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johansson F, et al. Type 1 diabetes promotes disruption of advanced atherosclerotic lesions in LDL receptor-deficient mice. Proc. Natl Acad. Sci. USA. 2008;105:2082–2087. doi: 10.1073/pnas.0709958105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hwang YC, et al. Central role for aldose reductase pathway in myocardial ischemic injury. FASEB J. 2004;18:1192–1199. doi: 10.1096/fj.03-1400com. [DOI] [PubMed] [Google Scholar]
- 20.Vikramadithyan RK, et al. Human aldose reductase expression accelerates diabetic atherosclerosis in transgenic mice. J. Clin. Invest. 2005;115:2434–2443. doi: 10.1172/JCI24819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsueh W, et al. Recipes for creating animal models of diabetic cardiovascular disease. Circ. Res. 2007;100:1415–1427. doi: 10.1161/01.RES.0000266449.37396.1f. [DOI] [PubMed] [Google Scholar]
- 22.Clarkson TB, Koritnik DR, Weingand KW, Miller LC. Nonhuman primate models of atherosclerosis: potential for the study of diabetes mellitus and hyperinsulinemia. Metabolism. 1985;34:51–59. doi: 10.1016/s0026-0495(85)80010-x. [DOI] [PubMed] [Google Scholar]
- 23.Gerrity RG, Natarajan R, Nadler JL, Kimsey T. Diabetes-induced accelerated atherosclerosis in swine. Diabetes. 2001;50:1654–1665. doi: 10.2337/diabetes.50.7.1654. [DOI] [PubMed] [Google Scholar]
- 24.Johnstone MT, et al. Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. Circulation. 1993;88:2510–2516. doi: 10.1161/01.cir.88.6.2510. [DOI] [PubMed] [Google Scholar]
- 25.Colombo PC, et al. Endothelial cell activation in patients with decompensated heart failure. Circulation. 2005;111:58–62. doi: 10.1161/01.CIR.0000151611.89232.3B. [DOI] [PubMed] [Google Scholar]
- 26.[No authors listed.] The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N. Engl. J. Med.329, 977–986 (1993). A landmark paper that clearly shows the role of insulin deficiency and its consequences (including hyperglycaemia) in the development of retinopathy, nephropathy and neuropathy during type 1 diabetes. [DOI] [PubMed]
- 27.UK Prospective Diabetes Study Group Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ. 1998;317(7160):703–713. [PMC free article] [PubMed] [Google Scholar]
- 28.Nishikawa T, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 29.Ishii H, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC β inhibitor. Science. 1996;272:728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 30.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wautier MP, et al. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001;280:E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 32.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N. Engl. J. Med. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 33.Herold K, et al. Receptor for advanced glycation end products (RAGE) in a dash to the rescue: inflammatory signals gone awry in the primal response to stress. J. Leukoc. Biol. 2007;82:204–212. doi: 10.1189/jlb.1206751. [DOI] [PubMed] [Google Scholar]
- 34.Harja E, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J. Clin. Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soro-Paavonen A, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzen S, Buyukbingol E. Recent studies of aldose reductase enzyme inhibition for diabetic complications. Curr. Med. Chem. 2003;10:1329–1352. doi: 10.2174/0929867033457377. [DOI] [PubMed] [Google Scholar]
- 37.Joy SV, et al. Ruboxistaurin, a protein kinase C β inhibitor, as an emerging treatment for diabetes microvascular complications. Ann. Pharmacother. 2005;39:1693–1699. doi: 10.1345/aph.1E572. [DOI] [PubMed] [Google Scholar]
- 38.McGill JB, et al. Clinical safety of the selective PKC-β inhibitor, ruboxistaurin. Expert Opin. Drug Saf. 2006;5:835–845. doi: 10.1517/14740338.5.6.835. [DOI] [PubMed] [Google Scholar]
- 39.Kass DA, et al. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation. 2001;104:1464–1470. doi: 10.1161/hc3801.097806. [DOI] [PubMed] [Google Scholar]
- 40.Zieman SJ, et al. Advanced glycation endproduct crosslink breaker (alagebrium) improves endothelial function in patients with isolated systolic hypertension. J. Hypertens. 2007;25:577–583. doi: 10.1097/HJH.0b013e328013e7dd. [DOI] [PubMed] [Google Scholar]
- 41.Stirban A, et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care. 2006;29:2064–2071. doi: 10.2337/dc06-0531. [DOI] [PubMed] [Google Scholar]
- 42.Rong LL, et al. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J. 2004;18:1812–1817. doi: 10.1096/fj.04-1899com. [DOI] [PubMed] [Google Scholar]
- 43.Rong LL, et al. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004;18:1818–1825. doi: 10.1096/fj.04-1900com. [DOI] [PubMed] [Google Scholar]
- 44.Ludwig S, Shen GX. Statins for diabetic cardiovascular complications. Curr. Vasc. Pharmacol. 2006;4:245–251. doi: 10.2174/157016106777698388. [DOI] [PubMed] [Google Scholar]
- 45.Panunti B, Fonseca V. Effects of PPAR gamma agonists on cardiovascular function in obese, non-diabetic patients. Vascul. Pharmacol. 2006;45:29–35. doi: 10.1016/j.vph.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 46.Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361:2005–2016. doi: 10.1016/s0140-6736(03)13636-7. [DOI] [PubMed] [Google Scholar]
- 47.Kearney PM, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117–125. doi: 10.1016/S0140-6736(08)60104-X. [DOI] [PubMed] [Google Scholar]
- 48.Calkin AC, et al. Rosiglitazone attenuates atherosclerosis in a model of insulin insufficiency independent of its metabolic effects. Arterioscler. Thromb. Vasc. Biol. 2005;25:1903–1909. doi: 10.1161/01.ATV.0000177813.99577.6b. [DOI] [PubMed] [Google Scholar]
- 49.Ratner RE, et al. Assessment on the Prevention of Progression by Rosiglitazone on Atherosclerosis in diabetes patients with Cardiovascular History (APPROACH): study design and baseline characteristics. Am. Heart J. 2008;156:1074–1079. doi: 10.1016/j.ahj.2008.07.025. [DOI] [PubMed] [Google Scholar]
- 50.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 51.Dormandy JA, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 52.Keech A, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 53.Keech AC, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370:1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- 54.Nissen SE, Wolski K, Topol EJ. Effect of muraglitazar on death and major adverse cardiovascular events in patients with type 2 diabetes mellitus. JAMA. 2005;294:2581–2586. doi: 10.1001/jama.294.20.joc50147. [DOI] [PubMed] [Google Scholar]
- 55.Gerstein HC, et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel A, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008;358:2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 57.Goh S-Y, Cooper ME. Clinical review: the role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metabolism. 2008;93:1143–1152. doi: 10.1210/jc.2007-1817. [DOI] [PubMed] [Google Scholar]
- 58.Chalmers J, Cooper ME. UKPDS and the legacy effect. N. Engl. J. Med. 2008;359:1618–1620. doi: 10.1056/NEJMe0807625. [DOI] [PubMed] [Google Scholar]
- 59.El-Osta A, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008;205:2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mack CP. An epigenetic clue to diabetic vascular disease. Circ. Res. 2008;103:568–570. doi: 10.1161/CIRCRESAHA.108.184358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Villeneuve LM, et al. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc. Natl Acad. Sci. USA. 2008;105:9047–9052. doi: 10.1073/pnas.0803623105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaede P, Lund-Andersen H, Parving H-H, Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N. Engl. J. Med. 2008;358:580–591. doi: 10.1056/NEJMoa0706245. [DOI] [PubMed] [Google Scholar]
- 63.Gilbertson DT, et al. Projecting the number of patients with end-stage renal disease in the United States to the year 2015. J. Am. Soc. Nephrol. 2005;16:3736–3741. doi: 10.1681/ASN.2005010112. [DOI] [PubMed] [Google Scholar]
- 64.Mogensen CE, Christensen CK, Vittinghus E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes. 1983;32(Suppl. 2):64–78. doi: 10.2337/diab.32.2.s64. [DOI] [PubMed] [Google Scholar]
- 65.[No authors listed.]UK Prospective Diabetes Study (UKPDS). X. Urinary albumin excretion over 3 years in diet-treated type 2, (non-insulin-dependent) diabetic patients, and association with hypertension, hyperglycaemia and hypertriglyceridaemia. Diabetologia36, 1021–1029 (1993). [PubMed]
- 66.Amico JA, Klein I. Diabetic management in patients with renal failure. Diabetes Care. 1981;4:430–434. doi: 10.2337/diacare.4.3.430. [DOI] [PubMed] [Google Scholar]
- 67.Cooper ME. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet. 1998;352:213–219. doi: 10.1016/S0140-6736(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 68.Molitch ME, et al. Nephropathy in diabetes. Diabetes Care. 2004;27:S79–S83. doi: 10.2337/diacare.27.2007.s79. [DOI] [PubMed] [Google Scholar]
- 69.Parving H-H, Persson F, Lewis JB, Lewis EJ, Hollenberg NK. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N. Engl. J. Med. 2008;358:2433–2446. doi: 10.1056/NEJMoa0708379. [DOI] [PubMed] [Google Scholar]
- 70.Tikellis C, et al. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes. 2008;57:1018–1025. doi: 10.2337/db07-1212. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Z, et al. Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: blockade of compensatory renin increase. Proc. Natl Acad. Sci. USA. 2008;105:15896–15901. doi: 10.1073/pnas.0803751105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Breyer MD, et al. Mouse models of diabetic nephropathy. J. Am. Soc. Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 73.Gurley SB, et al. Impact of genetic background on nephropathy in diabetic mice. Am. J. Physiol. Renal Physiol. 2006;290:F214–F222. doi: 10.1152/ajprenal.00204.2005. [DOI] [PubMed] [Google Scholar]
- 74.Zheng S, et al. Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes. 2004;53:3248–3257. doi: 10.2337/diabetes.53.12.3248. [DOI] [PubMed] [Google Scholar]
- 75.Mauer SM, et al. Structural–functional relationships in diabetic nephropathy. J. Clin. Invest. 1984;74:1143–1155. doi: 10.1172/JCI111523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 77.Oldfield MD, et al. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE) J. Clin. Invest. 2001;108:1853–1863. doi: 10.1172/JCI11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wendt TM, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 2003;162:1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cooper ME. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia. 2001;44:1957–1972. doi: 10.1007/s001250100000. [DOI] [PubMed] [Google Scholar]
- 80.Flyvbjerg A, et al. Long-term renal effects of a neutralizing RAGE antibody in obese type 2 diabetic mice. Diabetes. 2004;53:166–172. doi: 10.2337/diabetes.53.1.166. [DOI] [PubMed] [Google Scholar]
- 81.Ziyadeh FN, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-β antibody in db/db diabetic mice. Proc. Natl Acad. Sci. USA. 2000;97:8015–8020. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burns WC, et al. Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J. Am. Soc. Nephrol. 2006;17:2484–2494. doi: 10.1681/ASN.2006050525. [DOI] [PubMed] [Google Scholar]
- 83.Thomas MC, et al. Interactions between renin angiotensin system and advanced glycation in the kidney. J. Am. Soc. Nephrol. 2005;16:2976–2984. doi: 10.1681/ASN.2005010013. [DOI] [PubMed] [Google Scholar]
- 84.Forbes JM, Cooper ME, Oldfield MD, Thomas MC. Role of advanced glycation end products in diabetic nephropathy. J. Am. Soc. Nephrol. 2003;14:S254–S258. doi: 10.1097/01.asn.0000077413.41276.17. [DOI] [PubMed] [Google Scholar]
- 85.Tuttle KR, et al. The effect of ruboxistaurin on nephropathy in type 2 diabetes. Diabetes Care. 2005;28:2686–2690. doi: 10.2337/diacare.28.11.2686. [DOI] [PubMed] [Google Scholar]
- 86.Deelman L, Sharma K. Mechanisms of kidney fibrosis and the role of antifibrotic therapies. Curr. Opin. Nephrol. Hypertens. 2009;18:85–90. doi: 10.1097/MNH.0b013e32831c50a1. [DOI] [PubMed] [Google Scholar]
- 87.Brenner BM, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 88.Lewis EJ, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001;345:851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 89.Bakris GL, et al. The future of clinical trials in chronic renal disease: outcome of an NIH/FDA/Physician Specialist Conference. Evaluation of Clinical Trial Endpoints in Chronic Renal Disease Study Group. J. Clin. Pharmacol. 2000;40:815–825. doi: 10.1177/00912700022009549. [DOI] [PubMed] [Google Scholar]
- 90.MacIsaac RJ, et al. Nonalbuminuric renal insufficiency in type 2 diabetes. Diabetes Care. 2004;27:195–200. doi: 10.2337/diacare.27.1.195. [DOI] [PubMed] [Google Scholar]
- 91.Otu HH, et al. Prediction of diabetic nephropathy using urine proteomic profiling 10 years prior to development of nephropathy. Diabetes Care. 2007;30:638–643. doi: 10.2337/dc06-1656. [DOI] [PubMed] [Google Scholar]
- 92.Centers for Disease Control and Prevention. National diabetes fact sheet: general information and national estimates on diabetes in the United States (Department of Health and Human Services,Centers for Disease Control and Prevention, Atlanta, Georgia, USA, 2005).
- 93.Frank RN. Diabetic retinopathy. N. Engl. J. Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 94.Bresnick GH, Engerman R, Davis MD, de Venecia G, Myers FL. Patterns of ischemia in diabetic retinopathy. Trans. Sect. Ophthalmol. Am. Acad. Ophthalmol. Otolaryngol. 1976;81:OP694–OP709. [PubMed] [Google Scholar]
- 95.Roy MS, et al. The prevalence of diabetic retinopathy among adult type 1 diabetic persons in the United States. Arch. Ophthalmol. 2004;122:546–551. doi: 10.1001/archopht.122.4.546. [DOI] [PubMed] [Google Scholar]
- 96.Kempen JH, et al. The prevalence of diabetic retinopathy among adults in the United States. Arch. Ophthalmol. 2004;122:552–563. doi: 10.1001/archopht.122.4.552. [DOI] [PubMed] [Google Scholar]
- 97.Engerman RL, Kern TS. Retinopathy in animal models of diabetes. Diabetes Metab. Rev. 1995;11:109–120. doi: 10.1002/dmr.5610110203. [DOI] [PubMed] [Google Scholar]
- 98.Kern TS. Retinal and Choroidal Angiogenesis. 2008. pp. 81–102. [Google Scholar]
- 99.Madan A, Penn JS. Animal models of oxygen-induced retinopathy. Front. Biosci. 2003;8:d1030–d1043. doi: 10.2741/1056. [DOI] [PubMed] [Google Scholar]
- 100.Ohno-Matsui K, et al. Inducible expression of vascular endothelial growth factor in adult mice causes severe proliferative retinopathy and retinal detachment. Am. J. Pathol. 2002;160:711–719. doi: 10.1016/S0002-9440(10)64891-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van Eeden PE, et al. Characterisation of a model for retinal neovascularisation. VEGF model characterisation. Adv. Exp. Med. Biol. 2006;572:163–168. doi: 10.1007/0-387-32442-9_24. [DOI] [PubMed] [Google Scholar]
- 102.Ruberte J, et al. Increased ocular levels of IGF-1 in transgenic mice lead to diabetes-like eye disease. J. Clin. Invest. 2004;113:1149–1157. doi: 10.1172/JCI19478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kern TS. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp. Diabetes Res. 2007;2007:95–103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barber AJ, et al. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Scott TM, Foote J, Peat B, Galway G. Vascular and neural changes in the rat optic nerve following induction of diabetes with streptozotocin. J. Anat. 1986;144:145–152. [PMC free article] [PubMed] [Google Scholar]
- 106.Barber AJ, et al. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest. Ophthalmol. Vis. Sci. 2005;46:2210–2218. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 107.Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36:808–812. doi: 10.2337/diab.36.7.808. [DOI] [PubMed] [Google Scholar]
- 108.Kern TS, Engerman RL. A mouse model of diabetic retinopathy. Arch. Ophthalmol. 1996;114:986–990. doi: 10.1001/archopht.1996.01100140194013. [DOI] [PubMed] [Google Scholar]
- 109.Engerman RL, Kern TS. Experimental galactosemia produces diabetic-like retinopathy. Diabetes. 1984;33:97–100. doi: 10.2337/diab.33.1.97. [DOI] [PubMed] [Google Scholar]
- 110.Robison WG, Jr, Nagata M, Laver N, Hohman TC, Kinoshita JH. Diabetic-like retinopathy in rats prevented with an aldose reductase inhibitor. Invest. Ophthalmol. Vis. Sci. 1989;30:2285–2292. [PubMed] [Google Scholar]
- 111.Barile GR, et al. The RAGE axis in early diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2005;46:2916–2924. doi: 10.1167/iovs.04-1409. [DOI] [PubMed] [Google Scholar]
- 112.Dorrell M, Uusitalo-Jarvinen H, Aguilar E, Friedlander M. Ocular neovascularization: basic mechanisms and therapeutic advances. Surv. Ophthalmol. 2007;52:S3–S19. doi: 10.1016/j.survophthal.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 113.Adamis AP, Berman AJ. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin. Immunopathol. 2008;30:65–84. doi: 10.1007/s00281-008-0111-x. [DOI] [PubMed] [Google Scholar]
- 114.[No authors listed.] Fundus photographic risk factors for progression of diabetic retinopathy. ETDRS report number 12. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology98, 823–833 (1991). [PubMed]
- 115.[No authors listed.] Grading diabetic retinopathy from stereoscopic color fundus photographs — an extension of the modified Airlie House classification. ETDRS report number 10. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology98, 786–806 (1991). [PubMed]
- 116.[No authors listed.] Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet352, 837–853 (1998). [PubMed]
- 117.Neubauer AS, Ulbig MW. Laser treatment in diabetic retinopathy. Ophthalmologica. 2007;221:95–102. doi: 10.1159/000098254. [DOI] [PubMed] [Google Scholar]
- 118.Moravski CJ, et al. The renin–angiotensin system influences ocular endothelial cell proliferation in diabetes: transgenic and interventional studies. Am. J. Pathol. 2003;162:151–160. doi: 10.1016/S0002-9440(10)63806-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chaturvedi N, et al. Effect of candesartan on prevention (DIRECT-Prevent 1) and progression (DIRECT-Protect 1) of retinopathy in type 1 diabetes: randomised, placebo-controlled trials. Lancet. 2008;372:1394–1402. doi: 10.1016/S0140-6736(08)61412-9. [DOI] [PubMed] [Google Scholar]
- 120.Sjolie AK, et al. Effect of candesartan on progression and regression of retinopathy in type 2 diabetes (DIRECT-Protect 2): a randomised placebo-controlled trial. Lancet. 2008;372:1385–1393. doi: 10.1016/S0140-6736(08)61411-7. [DOI] [PubMed] [Google Scholar]
- 121.Gillies MC, Islam FM, Zhu M, Larsson J, Wong TY. Efficacy and safety of multiple intravitreal triamcinolone injections for refractory diabetic macular oedema. Br. J. Ophthalmol. 2007;91:1323–1326. doi: 10.1136/bjo.2006.113167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kang SW, Park SC, Cho HY, Kang JH. Triple therapy of vitrectomy, intravitreal triamcinolone, and macular laser photocoagulation for intractable diabetic macular edema. Am. J. Opthalmol. 2007;144:878–885. doi: 10.1016/j.ajo.2007.07.044. [DOI] [PubMed] [Google Scholar]
- 123.[No authors listed.] A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Sorbinil Retinopathy Trial Research Group. Arch. Ophthalmol.108, 1234–1244 (1990). [DOI] [PubMed]
- 124.[No authors listed.] Effects of aspirin treatment on diabetic retinopathy. ETDRS report number 8. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology98, 757–765 (1991). [PubMed]
- 125.The PKC-DRS Study Group. The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the Protein Kinase C β Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomized clinical trial. Diabetes54, 2188–2197 (2005). [DOI] [PubMed]
- 126.The PKC-DMES Study Group. Effect of ruboxistaurin in patients with diabetic macular edema: thirty-month results of the randomized PKC-DMES clinical trial. Arch. Ophthalmol.125, 318–324 (2007). [DOI] [PubMed]
- 127.Aiello LP, et al. Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology. 2006;113:2221–2230. doi: 10.1016/j.ophtha.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 128.Davis MD, et al. Effect of ruboxistaurin on the visual acuity decline associated with long-standing diabetic macular edema. Invest. Ophthalmol. Vis. Sci. 2009;50:1–4. doi: 10.1167/iovs.08-2473. [DOI] [PubMed] [Google Scholar]
- 129.Starita C, Patel M, Katz B, Adamis AP. Vascular endothelial growth factor and the potential therapeutic use of pegaptanib (macugen) in diabetic retinopathy. Dev. Ophthalmol. 2007;39:122–148. doi: 10.1159/000098504. [DOI] [PubMed] [Google Scholar]
- 130.Iwasaki T, et al. Three-dimensional optical coherence tomography of proliferative diabetic retinopathy. Br. J. Ophthalmol. 2008;92:713. doi: 10.1136/bjo.2007.135319. [DOI] [PubMed] [Google Scholar]
- 131.Berkowitz BA, Roberts R, Stemmler A, Luan H, Gradianu M. Impaired apparent ion demand in experimental diabetic retinopathy: correction by lipoic acid. Invest. Ophthalmol. Vis. Sci. 2007;48:4753–4758. doi: 10.1167/iovs.07-0433. [DOI] [PubMed] [Google Scholar]
- 132.Trick GL, Edwards P, Desai U, Berkowitz BA. Early supernormal retinal oxygenation response in patients with diabetes. Invest. Ophthalmol. Vis. Sci. 2006;47:1612–1619. doi: 10.1167/iovs.05-0833. [DOI] [PubMed] [Google Scholar]
- 133.Selvarajah D, et al. Early involvement of the spinal cord in diabetic peripheral neuropathy. Diabetes Care. 2006;29:2664–2669. doi: 10.2337/dc06-0650. [DOI] [PubMed] [Google Scholar]
- 134.Wessels AM, et al. Microvascular disease in type 1 diabetes alters brain activation: a functional magnetic resonance imaging study. Diabetes. 2006;55:334–340. doi: 10.2337/diabetes.55.02.06.db05-0680. [DOI] [PubMed] [Google Scholar]
- 135.Pirart J. [Diabetes mellitus and its degenerative complications: a prospective study of 4,400 patients observed between 1947 and 1973 (author's transl.)] Diabete Metab. 1977;3:97–107. [PubMed] [Google Scholar]
- 136.Sharma AK, Thomas PK. Peripheral nerve structure and function in experimental diabetes. J. Neurol. Sci. 1974;23:1–15. doi: 10.1016/0022-510x(74)90136-1. [DOI] [PubMed] [Google Scholar]
- 137.Brussee V, et al. Distal degenerative sensory neuropathy in a long term type 2 diabetes rat model. Diabetes. 2008;57:1664–1673. doi: 10.2337/db07-1737. [DOI] [PubMed] [Google Scholar]
- 138.Kamiya H, Zhang W, Sima AA. Degeneration of the Golgi and neuronal loss in dorsal root ganglia in diabetic BioBreeding/Worcester rats. Diabetologia. 2006;49:2763–2774. doi: 10.1007/s00125-006-0379-0. [DOI] [PubMed] [Google Scholar]
- 139.Bianchi R, et al. Erythropoietin both protects from and reverses experimental diabetic neuropathy. Proc. Natl Acad. Sci. USA. 2004;101:823–828. doi: 10.1073/pnas.0307823100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen YS, Chung SS, Chung SK. Noninvasive monitoring of diabetes-induced cutaneous nerve fiber loss and hypoalgesia in Thy1–YFP transgenic mice. Diabetes. 2005;54:3112–3118. doi: 10.2337/diabetes.54.11.3112. [DOI] [PubMed] [Google Scholar]
- 141.Engerman RL, Kern TS, Larson ME. Nerve conduction and aldose reductase inhibition during 5 years of diabetes or galactosaemia in dogs. Diabetologia. 1994;37:141–144. doi: 10.1007/s001250050084. [DOI] [PubMed] [Google Scholar]
- 142.Good KL, Maggs DJ, Hollingsworth SR, Scagliotti RH, Nelson RW. Corneal sensitivity in dogs with diabetes mellitus. Am. J. Vet. Res. 2003;64:7–11. doi: 10.2460/ajvr.2003.64.7. [DOI] [PubMed] [Google Scholar]
- 143.Walker D, et al. Nerve pathology in the type 1 diabetic dog: effects of treatment with sulindac. J. Peripher. Nerv. Syst. 2001;6:219–226. doi: 10.1046/j.1529-8027.2001.01023.x. [DOI] [PubMed] [Google Scholar]
- 144.Pare M, et al. Differential hypertrophy and atrophy among all types of cutaneous innervation in the glabrous skin of the monkey hand during aging and naturally occurring type 2 diabetes. J. Comp. Neurol. 2007;501:543–567. doi: 10.1002/cne.21262. [DOI] [PubMed] [Google Scholar]
- 145.Estrella JS, et al. Endoneurial microvascular pathology in feline diabetic neuropathy. Microvasc. Res. 2008;75:403–410. doi: 10.1016/j.mvr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mizisin AP, et al. Comparable myelinated nerve pathology in feline and human diabetes mellitus. Acta Neuropathol. 2007;113:431–442. doi: 10.1007/s00401-006-0163-8. [DOI] [PubMed] [Google Scholar]
- 147.Cameron NE, Eaton SE, Cotter MA, Tesfaye S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia. 2001;44:1973–1988. doi: 10.1007/s001250100001. [DOI] [PubMed] [Google Scholar]
- 148.Pop-Busui R, Sima A, Stevens M. Diabetic neuropathy and oxidative stress. Diabetes Metab. Res. Rev. 2006;22:257–273. doi: 10.1002/dmrr.625. [DOI] [PubMed] [Google Scholar]
- 149.Toth C, Martinez J, Zochodne DW. RAGE, diabetes, and the nervous system. Curr. Mol. Med. 2007;7:766–776. doi: 10.2174/156652407783220705. [DOI] [PubMed] [Google Scholar]
- 150.Calcutt NA, Jolivalt CG, Fernyhough P. Growth factors as therapeutics for diabetic neuropathy. Curr. Drug Targets. 2008;9:47–59. doi: 10.2174/138945008783431727. [DOI] [PubMed] [Google Scholar]
- 151.Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nature Rev. Neurosci. 2008;9:36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- 152.Mizisin AP, Calcutt NA, DiStefano PS, Acheson A, Longo FM. Aldose reductase inhibition increases CNTF-like bioactivity and protein in sciatic nerves from galactose-fed and normal rats. Diabetes. 1997;46:647–652. doi: 10.2337/diab.46.4.647. [DOI] [PubMed] [Google Scholar]
- 153.Suzuki T, Sekido H, Kato N, Nakayama Y, Yabe-Nishimura C. Neurotrophin-3-induced production of nerve growth factor is suppressed in Schwann cells exposed to high glucose: involvement of the polyol pathway. J. Neurochem. 2004;91:1430–1438. doi: 10.1111/j.1471-4159.2004.02824.x. [DOI] [PubMed] [Google Scholar]
- 154.Calcutt NA, Freshwater JD, Hauptmann N, Taylor EM, Mizisin AP. Protection of sensory function in diabetic rats by neotrofin. Eur. J. Pharmacol. 2006;534:187–193. doi: 10.1016/j.ejphar.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 155.Riaz S, Malcangio M, Miller M, Tomlinson DR. A vitamin D(3) derivative (CB1093) induces nerve growth factor and prevents neurotrophic deficits in streptozotocin-diabetic rats. Diabetologia. 1999;42:1308–1313. doi: 10.1007/s001250051443. [DOI] [PubMed] [Google Scholar]
- 156.Kakinoki B, et al. Orally active neurotrophin-enhancing agent protects against dysfunctions of the peripheral nerves in hyperglycemic animals. Diabetes. 2006;55:616–621. doi: 10.2337/diabetes.55.03.06.db05-1091. [DOI] [PubMed] [Google Scholar]
- 157.Goss JR, et al. Herpes simplex-mediated gene transfer of nerve growth factor protects against peripheral neuropathy in streptozotocin-induced diabetes in the mouse. Diabetes. 2002;51:2227–2232. doi: 10.2337/diabetes.51.7.2227. [DOI] [PubMed] [Google Scholar]
- 158.Walwyn WM, et al. HSV-1-mediated NGF delivery delays nociceptive deficits in a genetic model of diabetic neuropathy. Exp. Neurol. 2006;198:260–270. doi: 10.1016/j.expneurol.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 159.Kato N, et al. Nonviral gene transfer of human hepatocyte growth factor improves streptozotocin-induced diabetic neuropathy in rats. Diabetes. 2005;54:846–854. doi: 10.2337/diabetes.54.3.846. [DOI] [PubMed] [Google Scholar]
- 160.Pradat PF, et al. Continuous delivery of neurotrophin 3 by gene therapy has a neuroprotective effect in experimental models of diabetic and acrylamide neuropathies. Hum. Gene Ther. 2001;12:2237–2249. doi: 10.1089/10430340152710577. [DOI] [PubMed] [Google Scholar]
- 161.Chattopadhyay M, et al. HSV-mediated gene transfer of vascular endothelial growth factor to dorsal root ganglia prevents diabetic neuropathy. Gene Ther. 2005;12:1377–1384. doi: 10.1038/sj.gt.3302533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Price SA, et al. Gene transfer of an engineered transcription factor promoting expression of VEGF-A protects against experimental diabetic neuropathy. Diabetes. 2006;55:1847–1854. doi: 10.2337/db05-1060. [DOI] [PubMed] [Google Scholar]
- 163.Sugimoto K, Murakawa Y, Zhang W, Xu G, Sima AA. Insulin receptor in rat peripheral nerve: its localization and alternatively spliced isoforms. Diabetes Metab. Res. Rev. 2000;16:354–363. doi: 10.1002/1520-7560(200009/10)16:5<354::aid-dmrr149>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 164.Huang TJ, et al. Insulin prevents depolarization of the mitochondrial inner membrane in sensory neurons of type 1 diabetic rats in the presence of sustained hyperglycemia. Diabetes. 2003;52:2129–2136. doi: 10.2337/diabetes.52.8.2129. [DOI] [PubMed] [Google Scholar]
- 165.Hoybergs YM, Meert TF. The effect of low-dose insulin on mechanical sensitivity and allodynia in type I diabetes neuropathy. Neurosci. Lett. 2007;417:149–154. doi: 10.1016/j.neulet.2007.02.087. [DOI] [PubMed] [Google Scholar]
- 166.Brussee V, Cunningham FA, Zochodne DW. Direct insulin signaling of neurons reverses diabetic neuropathy. Diabetes. 2004;53:1824–1830. doi: 10.2337/diabetes.53.7.1824. [DOI] [PubMed] [Google Scholar]
- 167.Pierson CR, Zhang W, Murakawa Y, Sima AA. Insulin deficiency rather than hyperglycemia accounts for impaired neurotrophic responses and nerve fiber regeneration in type 1 diabetic neuropathy. J. Neuropathol. Exp. Neurol. 2003;62:260–271. doi: 10.1093/jnen/62.3.260. [DOI] [PubMed] [Google Scholar]
- 168.Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer's disease. Exp. Gerontol. 2007;42:10–21. doi: 10.1016/j.exger.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 169.Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–1824. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- 170.Burdo Joseph R., Chen Qi, Calcutt Nigel A., Schubert David. The pathological interaction between diabetes and presymptomatic Alzheimer's disease. Neurobiology of Aging. 2009;30(12):1910–1917. doi: 10.1016/j.neurobiolaging.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 171.Jolivalt CG, et al. Defective insulin signaling pathway and increased GSK-3 activity in the brain of diabetic mice: parallels with Alzheimer's disease and correction by insulin. J. Neurosci. Res. 2008;86:3265–3274. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jacobson AM, et al. Long-term effect of diabetes and its treatment on cognitive function. N. Engl. J. Med. 2007;356:1842–1852. doi: 10.1056/NEJMoa066397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kloppenborg RP, van den Berg E, Kappelle LJ, Biessels GJ. Diabetes and other vascular risk factors for dementia: which factor matters most? A systematic review. Eur. J. Pharmacol. 2008;585:97–108. doi: 10.1016/j.ejphar.2008.02.049. [DOI] [PubMed] [Google Scholar]
- 174.Li L, Holscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res. Rev. 2007;56:384–402. doi: 10.1016/j.brainresrev.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 175.Attal N, et al. EFNS guidelines on pharmacological treatment of neuropathic pain. Eur. J. Neurol. 2006;13:1153–1169. doi: 10.1111/j.1468-1331.2006.01511.x. [DOI] [PubMed] [Google Scholar]
- 176.Backonja M, Glanzman RL. Gabapentin dosing for neuropathic pain: evidence from randomized, placebo-controlled clinical trials. Clin. Ther. 2003;25:81–104. doi: 10.1016/s0149-2918(03)90011-7. [DOI] [PubMed] [Google Scholar]
- 177.Freynhagen R, Strojek K, Griesing T, Whalen E, Balkenohl M. Efficacy of pregabalin in neuropathic pain evaluated in a 12-week, randomised, double-blind, multicentre, placebo-controlled trial of flexible- and fixed-dose regimens. Pain. 2005;115:254–263. doi: 10.1016/j.pain.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 178.Raskin J, et al. A double-blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain Med. 2005;6:346–356. doi: 10.1111/j.1526-4637.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- 179.Ruessmann Heinz–Jürgen. Switching from pathogenetic treatment with α-lipoic acid to gabapentin and other analgesics in painful diabetic neuropathy: a real-world study in outpatients. Journal of Diabetes and its Complications. 2009;23(3):174–177. doi: 10.1016/j.jdiacomp.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 180.Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann. Neurol. 2002;52:786–792. doi: 10.1002/ana.10364. [DOI] [PubMed] [Google Scholar]
- 181.Luo ZD, et al. Injury type-specific calcium channel α2δ1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J. Pharmacol. Exp. Ther. 2002;303:1199–1205. doi: 10.1124/jpet.102.041574. [DOI] [PubMed] [Google Scholar]
- 182.Jolivalt CG, Jiang Y, Freshwater JD, Bartoszyk GD, Calcutt NA. Dynorphin A, kappa opioid receptors and the antinociceptive efficacy of asimadoline in streptozotocin-induced diabetic rats. Diabetologia. 2006;49:2775–2785. doi: 10.1007/s00125-006-0397-y. [DOI] [PubMed] [Google Scholar]
- 183.Ramos KM, Jiang Y, Svensson CI, Calcutt NA. Pathogenesis of spinally mediated hyperalgesia in diabetes. Diabetes. 2007;56:1569–1576. doi: 10.2337/db06-1269. [DOI] [PubMed] [Google Scholar]
- 184.Bird SJ, Brown MJ, Spino C, Watling S, Foyt HL. Value of repeated measures of nerve conduction and quantitative sensory testing in a diabetic neuropathy trial. Muscle Nerve. 2006;34:214–224. doi: 10.1002/mus.20577. [DOI] [PubMed] [Google Scholar]
- 185.Bril V, Buchanan RA. Long-term effects of ranirestat (AS-3201) on peripheral nerve function in patients with diabetic sensorimotor polyneuropathy. Diabetes Care. 2006;29:68–72. doi: 10.2337/diacare.29.01.06.dc05-1447. [DOI] [PubMed] [Google Scholar]
- 186.Greene DA, Arezzo JC, Brown MB. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Zenarestat Study Group. Neurology. 1999;53:580–591. doi: 10.1212/wnl.53.3.580. [DOI] [PubMed] [Google Scholar]
- 187.Lauria G, et al. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur. J. Neurol. 2005;12:747–758. doi: 10.1111/j.1468-1331.2005.01260.x. [DOI] [PubMed] [Google Scholar]
- 188.Lauria G, Devigili G. Skin biopsy as a diagnostic tool in peripheral neuropathy. Nature Clin. Pract. Neurol. 2007;3:546–557. doi: 10.1038/ncpneuro0630. [DOI] [PubMed] [Google Scholar]
- 189.Smith AG, Ramachandran P, Tripp S, Singleton JR. Epidermal nerve innervation in impaired glucose tolerance and diabetes-associated neuropathy. Neurology. 2001;57:1701–1704. doi: 10.1212/wnl.57.9.1701. [DOI] [PubMed] [Google Scholar]
- 190.Pittenger GL, et al. Intraepidermal nerve fibers are indicators of small-fiber neuropathy in both diabetic and nondiabetic patients. Diabetes Care. 2004;27:1974–1979. doi: 10.2337/diacare.27.8.1974. [DOI] [PubMed] [Google Scholar]
- 191.Quattrini C, et al. Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes. 2007;56:2148–2154. doi: 10.2337/db07-0285. [DOI] [PubMed] [Google Scholar]
- 192.Mehra S, et al. Corneal confocal microscopy detects early nerve regeneration after pancreas transplantation in patients with type 1 diabetes. Diabetes Care. 2007;30:2608–2612. doi: 10.2337/dc07-0870. [DOI] [PubMed] [Google Scholar]
- 193.Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA287, 2563–2569 (2002). [DOI] [PMC free article] [PubMed]
- 194.Ludvigson MA, Sorenson RL. Immunohistochemical localization of aldose reductase. II. Rat eye and kidney. Diabetes. 1980;29:450–459. doi: 10.2337/diab.29.6.450. [DOI] [PubMed] [Google Scholar]
- 195.Ludvigson MA, Sorenson RL. Immunohistochemical localization of aldose reductase. I. Enzyme purification and antibody preparation — localization in peripheral nerve, artery, and testis. Diabetes. 1980;29:438–449. doi: 10.2337/diab.29.6.438. [DOI] [PubMed] [Google Scholar]
- 196.Pachydaki SI, et al. Upregulation of RAGE and its ligands in proliferative retinal disease. Exp. Eye Res. 2006;82:807–815. doi: 10.1016/j.exer.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 197.Toth C, et al. Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes. 2008;57:1002–1017. doi: 10.2337/db07-0339. [DOI] [PubMed] [Google Scholar]