

Translating basic research into viable therapies is challenged by many obstacles. In this Opinion article, Ian Sabroe and colleagues identify some of these obstacles and suggest a series of strategies to maximize the potential of translational research.
Abstract
Although there is overwhelming pressure from funding agencies and the general public for scientists to bridge basic and translational studies, the fact remains that there are significant hurdles to overcome in order to achieve this goal. The purpose of this Opinion article is to examine the nature of these hurdles and to provide food for thought on the main obstacles that impede this process.
Main
Molecular biology is barely out of its infancy. It has achieved much, including the sequencing of the entire human genome, but it is already under serious attack for its failure to deliver effective therapies in many areas. This Opinion article will provide a subjective view of our understanding of translational research, identify obstacles to its successful development, and propose a series of initiatives to improve the effectiveness of translational research strategies.
What constitutes translational biology?
Originating from Latin, translation means to 'carry across'. In biomedical research, the goal of translational science is to develop a thorough operational understanding of the human organism in health and disease, with the goal of 'carrying across' this knowledge to alleviate disease and suffering and to improve the quality of human existence. To be translational in medicine we must acquire knowledge from the broad arena of molecular and cellular biology and then apply this knowledge to human disease.
The quality of our scientific output (perceived as a change in disease incidence and/or the development of a therapy) is largely dependent on the quality of the input data and the methods for their processing and interpretation, although the process of generating effective translational science is not as linear (that is, from molecules to models to humans) as is often thought. Failure to ask the appropriate questions of optimized systems leads to the acquisition of knowledge that might be less relevant than anticipated.
Further corruption of the process comes as a result of limitations in our models, which are often not fully appreciated (or are simply ignored) at the time of the study. Additionally, grant agencies must be sufficiently flexible to allow researchers to follow up on novel observations because many of the most exciting developments arise from unexpected findings. Determining what research is intrinsically translational, or has been translated effectively, is therefore surprisingly difficult, and in a healthy global biomedical research environment, translation will continue to mean very different things to different groups.
Few scientists will see a process through from the conception of a hypothesis to the development of a specific medical therapy. As scientists, we are nonetheless required to measure our performance in terms of our 'translational potential', particularly when it comes to justifying and generating funding and publications. Therefore, in the absence of being able to measure contributions to health directly, we often quantify individual success using surrogate markers (such as publications and their impact, prestige, and funding), which depend on the prevailing concepts of what constitutes importance. However, these markers may be flawed for this purpose.
A more global assessment of the output of translational science might consider whether the scientific community has improved specific disease outcomes, quality of life or life expectancy. Specific examples of global success might not be as common as we would wish, but increased successes in the areas of organ transplantation or the ability to eradicate diseases such as measles virus highlight our ability to be successful. The improved treatment of many cancers through the combination of good science and high-quality clinical trials of new therapies or combinations of therapies has been strikingly impressive and provides many examples of highly effective translational science1,2. There remains, however, a lack of available mechanisms by which to relate our individual contributions to the global progress of translational science, and many factors conspire to impede our progress.
What inhibits translational biology?
The translation of basic scientific research faces a myriad of hurdles, both obvious and occult. These revolve around our understanding of the nature of the translational process, the integration of the outputs of different technological approaches to disease, the use of models, access to tissues and appropriate materials, and the need for support in increasingly complex areas such as ethics and bioinformatics. In addition, owing to technological advances, well-meaning safeguards of personal privacy have been imposed in relation to carrying out research in humans, and these have, in fact, greatly impeded progress in translational studies.
Problems with the models. Entirely appropriate restrictions on what research can be done in humans have contributed to the status quo in which the mouse has become an indispensable model for translational biology; however, it is often not possible to predict biological responses in humans accurately based solely on results obtained from animal models3,4,5,6,7,8,9,10. Within immunity, exposure of mice to their own species-specific commensals and pathogenic organisms might contribute to a species-specific immunological phenotype4,9 that affects the translational relevance. For example, studies of the effects of IRAK4 (interleukin-1 receptor-associated kinase 4) deficiency have revealed increased susceptibility to a greater range of pathogens in mice than is observed in humans who are deficient for IRAK4 (Refs 11–13).
Where good potential translational concepts are generated from models, determining how to move to the human can be challenging, as highlighted by difficulties in calculating tolerogenic doses of insulin in the prevention of diabetes10. In addition, technical limitations, or the dominance of prevailing models, can sometimes limit the scope of in vivo work, as illustrated by studies of airway disease, where studies in mouse models are focused predominantly on the important T helper 2 (TH2) component of asthma but are poor models for the contribution of other mediators. Mouse models are also complicated by our limited understanding of the phenotypes of human diseases14,15. Designing perfect models of diseases that we do not fully understand is a tall order, and chronic diseases that involve life-long interactions between the host and the environment present a particularly difficult challenge5,6,7,8. Developing truly chronic models of disease is, however, heavily militated against owing to ethical concerns about prolonged suffering. Moreover, the typically short-term nature of research funding, where outputs must be deliverable at a reasonable cost and within the time frame of a project grant, hinders the development of chronic models of disease.
It is equally clear that work on a single cell line is often poorly predictive of the behaviour of the whole organism. Although the use of primary cells from normal tissues can overcome some of these problems, the phenotype of these cells might be altered by their removal from the microenvironmental influences in vivo, and they often show markedly different responses to those of primary cells that are obtained from diseased tissue. Therefore, rodent models remain essential and cannot be replaced by in vitro approaches at present, but are an imperfect translational conduit for both biological and practical reasons (such as the difficulty in establishing chronic disease models).
Other models such as flies and zebrafish (Danio rerio) have many advantages for forward genetics and other related studies, but have substantially greater differences to humans than mammalian models. Primates are the most compatible mammalian models for immunological research, and have provided invaluable insights into diseases such as HIV, but their broader use is not feasible because of major ethical concerns, as well as other practical and cost-related issues. Even these models can have limitations. Recently, differential expression of SIGLECs (sialic-acid-binding immunoglobulin-like lectins), which are inhibitory receptors thought to downregulate immune-cell activation, has been noted between apes and humans16, and such differences in immune responses between species require careful consideration, as highlighted by the development of a 'cytokine storm' when individuals received an agonistic CD28-specific antibody that did not show the same effects in monkeys17,18.
Complex ethical dimensions and policy issues. Research is being increasingly hindered by bureaucracy. Evident at many levels in research management, nowhere is this more of a problem than in the areas of access to human samples and tissues, and in the establishment of clinical collaborations. Liaison with industry also provides additional challenges10. Furthermore, some of the most exciting areas of current research, such as work with embryonic stem cells, are subject to special ethical concerns. Even the simplest research involving humans, or archived specimens from humans, is often encumbered by a multistaged, intimidating application process that might dissuade individuals from carrying out the proposed study. Overcautious interpretation by ethics committees and regulatory authorities of what is ethical results in restrictive practices that can, for example, prevent re-screening of DNA banks for relevant markers and mutations, or apply over-elaborate protection of clinical phenotype data.
Although it is absolutely clear that robust systems are needed to prevent exploitation of patients or clinical data, it is often unclear how the ethical standards that are applied to the review of proposed studies are developed. Apparently arbitrary standards that might be perceived to be more ethical simply because they are more restrictive seem to be ever more on the increase. It is also unethical to excessively hinder research, but this imperative sometimes seems to come second to apparently minor scruples over details such as the methods of recruitment. Although the introduction of national standards to ensure timely review of proposals has, in the United Kingdom, begun to address these issues, increasingly complex review processes are frustrating and sometimes of questionable ethical value.
The area of personal privacy is particularly complex. The majority public view is supportive of the use of some personal data in confidential research19. However, although many individuals are less concerned that well-regulated data usage is an invasion of privacy than some ethics committees seem to be, due consideration is needed of the views of the minorities who are more concerned with their privacy. The future potential for apparently confidential genetic research resulting in the identification of individuals through the cross-referencing of DNA databases is clearly of concern20,21. In addition, governments tend to respond to highly public episodes of research offences or clinical misconduct with legislation. This runs the risk of creating additional administrative burdens for the majority of individual scientists who practise ethically and well, without ensuring the prevention of future wilful misdeeds by a rare minority. Increasingly, clinical practice is becoming more defensive and less willing to engage in research as a response to a society that is becoming more risk-adverse, litigious and blame-centred when adverse events occur. This generates further barriers to research in humans and requires a frank reappraisal of what experimentation is acceptable in humans or on samples that are derived from humans.
Additional problems arise in setting research priorities. Without effective public input, we run the risk that research priorities become the whim of changing government focuses, which lack the stable, long-term input necessary for meaningful scientific advancement. Although charities and private foundations that are centred on specific diseases provide important contributions, their focus is often too narrow to tackle the wider research agenda. This has led to a culture of increasing financial pressures that are governed by short-term policies, which require rapid outputs, and piecemeal funding, stifling longer term, more open-ended research that might produce real translational progress10. For example, political pressures to study agents of bioterrorism or emerging infectious diseases such as SARS (severe acute respiratory syndrome) can be welcomed and are clearly important; however, more long-term commitment to studying infections of historical global importance is also essential.
With respect to developing long-term goals, the National Institutes of Health (NIH) Method to Extend Research in Time (MERIT) Award programme in the United States provides an excellent example of how this might be accomplished. Experienced researchers who have superior competing renewal scores (scoring between the 0.1 and 5th percentiles), and who have shown a long-term commitment to and success in research may be selected for financial support for up to 10 years.
Therefore, MERIT Award recipients have a much longer window of opportunity to move studies along from basic to translational research while being provided with the necessary freedom from the administrative burdens associated with more frequent grant submissions. Unfortunately, these awards are extremely limited in number and do not apply to the majority of outstanding NIH grantees. Examples of what can be accomplished by long-term funding include the substantial contribution to the development and subsequent humanization of monoclonal antibodies at the UK Medical Research Council (MRC) Laboratory for Molecular Biology in Cambridge.
Solutions
A positive definition of translational biology. Translational biomedicine must be seen by scientists and the wider public in the context of a broad-based body of science that has the potential to contribute, in the near or long term, to the advancement of human health. Increased financial and political pressures on scientists to maximize directly measurable patient benefit from research carried out over short time frames runs the risk of destroying a healthy science base and should be actively resisted. A continued and robust engagement of the public is mandatory for scientists, because unless the public values the broad sweep of translational biomedical science, funding will inevitably be diverted away from science or applied only to those areas of research that immediately influence patient care.
As scientists, we must learn to articulate not only the promise of science, but also the difficulties that are associated with moving an idea along to the product stage, so that unrealistic expectations of perfect medications for every disease in short time frames are not raised. We must also wrestle the debate on the usefulness of in vivo models away from the more extreme ends of the anti-vivisection movement so that a constructive, rather than confrontational, discussion can evolve.
Equally, we must foster a culture of research in the curricula of medical students, and promote the goals of translational research to basic science undergraduate and graduate students. We must seek to marry the skills of basic scientists with those of clinicians to convey the idea that strong translational research underlies improved health and is inseparable from the provision of good standards of clinical care.
Improving our use of the models? In many biomedical fields, a series of debates have highlighted deficiencies in our current in vivo models. Strain differences22 and variance in experimental conditions between research groups pose major challenges in comparing the results derived from different studies. A collaborative investment in phenotyping human disease by clinicians and basic scientists is required for developing robust models. There is clearly an opportunity to define the characteristics, behaviour and relevance of model systems, particularly with a view to standardizing optimal strains and protocols. For example, multiple models exist for common diseases such as asthma23 and rheumatoid arthritis24, although harmonizing protocols and strains for particular features of a disease must avoid the loss of sufficient diversity in order to allow the finding of the unexpected.
This challenge could be met by interest groups or individuals working in a particular area, by large national funding agencies and/or international funding initiatives, or alternatively by specialist societies. Such debates might facilitate the comparison of data between laboratories and between species, and might highlight the components of specific diseases that are ripe for the development of new in vivo models and protocols (for example, there remains a great need to more effectively model the role of the innate immune system in acute and chronic asthma), broadening the number of disease processes or phenotypes that are modelled in pathology.
Although much work will continue to focus on the mouse, for some diseases comparative or independent studies in other species will continue to be important. Beyond the scope of this article, there are issues ahead with respect to subject selection for early clinical trials and the development of individualized treatment regimes that are based on pharmacogenetic and individual disease phenotypes25,26.
The tendency to view translational research as a linear process in which mice are the gateway between basic science and humans does rodent models a great disservice. In particular, the classical route of identifying genes in vitro, followed by generating knockout animals in vivo, has, in general, been poorly predictive of the consequences of targeting specific molecules in humans. Advances in medical sciences that have emerged in this manner are vastly outnumbered by those that arise by serendipity or through less predictable routes. A more holistic integration of in vivo models with in vitro science and studies in the human is needed when summarizing the translational relevance of a system, in which in vivo models contribute strongly to an iterative strategy, but are not themselves the final arbiters (Fig. 1).
Figure 1. Developing interactive models.
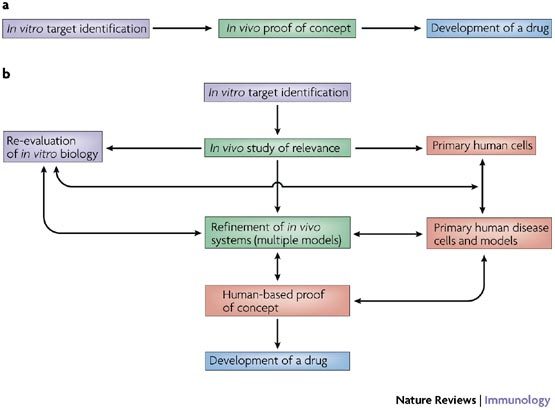
a | In vivo experimentation, perhaps in particular the generation and characterization of knockout mouse strains in experimental models of disease, is often viewed as the gatekeeper between in vitro science and the generation of drug targets that are appropriate for human disease. Although central to an effective understanding of immune biology and the role of new candidate drug targets, the predictive value of in vivo experimentation is less than desired, particularly in the context of studies of single knockouts in specific disease models and mouse strains. b | An example of a more holistic network in which multiple lines of evidence allow the refinement of objectives and target relevance in order to increase the chance of successful drug discovery. Such approaches reflect the approach of many researchers, but (acknowledging that no branch of science is 'easy') the main difficulties associated with undertaking human-based research run the risk of degrading the quality of data that arise from an integrated scientific approach.
The potential to identify medicines for use in human disease by screening less complex biological systems has long been recognized in the pharmaceutical industry, and there is now considerable interest in the use of model organisms such as Caenorhabditis elegans and zebrafish in high-throughput screens for new drugs27,28. It is becoming increasingly evident that these systems can be used to identify therapeutic targets in the immune system29. In this way, understanding the biology of pathways and gene products is deferred, and the ability of a compound to intervene in complex biological processes is directly tested, as highlighted in recent studies of calcium-channel antagonists in C. elegans27.
Combining the use of mice and humans in effective strategies. It seems self-evident that research which aspires to influence pathogenesis in humans needs to be carried out on the human system. At the same time, we must also anticipate the potential for doing harm that might accompany any new approaches to treating diseases, which militates against a rapid progression to phase I human trials. Nonetheless, the extraordinary difficulties that are associated with carrying out human studies in parallel with animal models has facilitated a culture in which such studies rarely occur, and in which prioritizing research is not driven by the inclusion of such processes. Indeed, almost quixotically, work in human tissues and cell lines is sometimes not deemed to be of importance until verified in a mouse with a targeted mutation. Bridging this divide requires the scientific community to value more highly the studies that seek to bring mouse and human studies together, and to appreciate that in human studies a lack of phenotype or subtle modifications of processes might be as important as models that generate dramatic phenotypes.
Increasing use of complex tissue models. From simple co-culture models of normal human tissues and ultimately to the generation of whole organs or representations of whole organs30 in the laboratory, we are now beginning to produce in vitro human systems that can complement essential work that is currently only achievable in vivo. Increasing success with new gene-delivery systems, combined with new technologies such as gene knockdown by RNA interference, might allow us to overcome the inability to study humans with targeted gene deficiencies. Such models are in their infancy, and cell-culture-based approaches are, in many scientists' views, farther from human biology than techniques that investigate biology in mice. It would seem that both are required, and therefore, a major thrust to develop standardized co-culture models that are based on primary human cells from healthy and diseased subjects would provide a complementary scientific base to our strong expertise in the mouse. Our commitment to this development is essential, not only to boost the translation of science to the human, but also to ensure that we honour the principles of reduction, refinement and replacement (minimizing the number of animals that are used and finding alternatives wherever feasible) that are central to all animal experimentation.
We are faced with many difficulties in generating appropriate human tissue models, including defining differences in similar cell types that are pathologically relevant (for example, comparing the biology of endothelial cells isolated from the umbilical cord with those isolated from different microvascular beds). We also need to determine the optimal representative culture conditions for primary cells. For example, when should epithelial cells be studied at an air–liquid interface, or when should leukocyte–endothelial interactions be studied under flow? Importantly, diseased tissues are often modified by inflammation, genetic traits and epigenetic processes that require further consideration when translating from the biology of health to the biology of disease. Nevertheless, we have recently shown that simple co-cultures of primary human cells can in some circumstances replicate inflammatory systems that are observed in vivo in mice31,32; such co-culture models are becoming increasingly common. The future ability to study systems that resemble organs or complete tissues, and verifying the work of simple co-culture experiments in such systems, offers a future we should not only embrace, but also be actively working towards at every opportunity.
Increasing use of complex therapeutic strategies. It is routine in clinical practice to combine drugs with similar or differing modes of action when treating disease. There is also increasing appreciation of the roles of cellular and molecular networks in the aetiology of disease33, a fact that is implicitly recognized by the need for in vivo models in which complex interactions between tissues and cells can be studied without a need to identify the full panoply of the systems involved (Box 1). In the context of an inflammatory disease, we have recently proposed that these networks are best described by the terminology of 'contiguous immunity', whereby, in disease, temporally and spatially contiguous networks comprising multiple processes of innate and adaptive immunity are in continual dialogue and evolution33. It is curious, then, that so many studies aim to land a single 'killer blow' on pathological processes, rather than considering the impact of therapeutic combinations. Our increasingly deeper understanding of the complexity of the inflammatory process often results in the targeting of downstream components for which there might be redundancy or very specific functionality. The targeting of such systems in combination, although complicated to achieve and less satisfying with respect to the identification of single clear targets, is nonetheless likely to have relevance for successful translation.
Allowing access to tissues and overcoming ethical anxieties. The diversion of research funding into bureaucracy and the delays in productivity that result from meeting statutory requirements for ethical practice contribute to the erosion of research capacity. Research cannot do without governance, but it is disappointing that increasing regulation has not been met by increased support for ethical human-based research, and although many processes conspire to make research harder, few exist to make it easier. A simplification of the regulations and national-level funding that provides templates for ethical applications, together with training and support for investigators, would do much to make this easier. Because many projects involve international collaborations, simplified standard international regulations would also greatly facilitate progress.
Many research projects have conceptually similar goals (such as the tackling of inflammation in a tissue), and generic pre-approved protocols requiring modest local adaptation would obviate many problems. In the United Kingdom, the proposed establishment of a single health research board to coordinate government-funded health research provides an opportunity to develop programmes that examine the effects of government regulation on research and mechanisms for overcoming the issues discussed earlier. Research on human tissues could be enhanced by improved access to normal and diseased specimens. The establishment of central and devolved services whose function would be to source human tissues in an ethical manner, characterize the donors and tissues anonymously, and make these resources freely available to investigators in research-active countries would transform biological research (indeed, some progress is being made here, but there is still much to be done).
An emphasis on dialogue with scientists and flexibility with respect to supporting science would be required to underpin access to these resources by researchers. Where materials are scarce, such systems could be supported by expertise in cell-line immortalization. The development of the Lung Tissue Research Consortium by the National Heart, Lung and Blood Institute is an exemplar of a clinical data and tissue bank that offers enormous potential for research in lung disease; in addition, the development of the UK Biobank shows it is feasible for human resources to be sourced ethically in ways that allow relatively broad use in medical research according to need. The proposed tissue banks would promote a positive view of human science working in parallel with mouse biology, which also requires commitment and support from the public to make it work.
'Omics'. One area in which large-scale biology is excelling is the use of public-access databases (such as those pertaining to gene expression (genomics) or the production of metabolites (metabolomics))34, but we are missing opportunities to further expand these databases. Local and national guidelines define the optimal management of many diseases, but it can be argued that the providers of health care are not given appropriate opportunities to engage with the scientists whose work is developing the future treatments for these diseases. Scientific initiatives that are associated with national care plans could drive disease phenotyping and tissue collection for tissue, DNA, RNA, protein and metabolite databanks with good public access. Good clinical practice not only should focus on ensuring that existing best practice is reliably reproduced, but also should put equal weight to research and development of practice through translational research. It is not just the priority of scientists to engage with clinicians: the imperative is equally strong that clinicians should engage more with basic scientists. In both cases it is important that such engagement is facilitated and supported by national policy.
Another major challenge is to take the input of a large amount of descriptive data generated by an 'omics' approach and both interpret and reapply it to a translational problem in a hypothesis-driven manner. As we learn to integrate complex data sets with in vitro cell biology and in vivo models, we might begin to generate virtual phenotypes, allowing conclusions to be drawn on the basis of the relatedness of a series of data sets to the questions asked. In essence, all researchers do this when they read published data, but the process is inherently subjective and formalizing such integrated biology could be more objective and so better inform future experiments.
Conclusion
We are now experiencing an unprecedented blossoming in available technologies, unparalleled through history, which makes biomedical science extraordinarily exciting. With this comes an ever greater financial burden and increasingly complicated ethical issues. An increasing focus on the need for effective translation from the use of these resources highlights the many obstacles that impede such progress. We have highlighted these difficulties, and have suggested strategies to rejuvenate and maximize our translational potential.
Box 1 | Examples of networked communication in immunology.
The difficulty in designing in vitro and in vivo experiments to model human disease has inevitably required and generated simplifications of pathology, often leading to relatively linear models of disease (for example, tissue damage, followed by antigen presentation, the generation of immunological memory and autoantibodies, and a resulting autoimmune disease). In reality, pathology is generated by networks that can exhibit substantial plasticity over the course of a disease. These principles are illustrated in the figure. A disease process — pathology — is represented at the centre of a series of simple conceptual networks, components of which are left intentionally blank to avoid attempting to define specific diseases. Each component (or node) within the network might have different roles at different times in a disease, or if active at more than one tissue site, might even have different roles simultaneously in a disease process. Therefore, the depicted connections are plastic over time and in individual microenvironments.
In a cellular network. Pathology can be considered in the context of networks of cells that are recruited or resident at inflammatory sites, whose communication through cytokines and other molecules regulates inflammation.
In a cytokine network. Pathology is driven by the interrelated actions of cytokines, again forming a dynamic plastic network.
In a process network. Process behaviour (for example, angiogenesis, scarring and leukocyte recruitment) will contribute differently to pathology at specific tissue locations and different times in the disease. As an illustration, wound-healing responses might contribute to the resolution of normal tissue architecture, the development of fibrosis, or the regulation of inflammatory cell recruitment and survival. Depending on the nature and duration of the stimulus driving the wound-healing process, and the location in which it is set, multiple resulting pathological phenotypes are feasible.
It is the nature of tissues to contain multiple cells and supporting structures that are physically associated, with many more cells that can transit through or become resident. We have proposed that the immunity seen in pathology rarely falls clearly into categories of innate and adaptive immune, or T helper 1 (TH1) and TH2 responses. Rather, pathology is generated by a networked interaction that changes over time. The networked relationships of immune pathology might be better described as 'contiguous immunity', in which multiple processes or networks can be operational and in dialogue in the same space (physically contiguous); equally, processes might be linked together in evolving patterns (temporally contiguous). Understanding these networks, and, where necessary, developing new models to elucidate and target them, is essential to effective translational biology. IFN, interferon; IL, interleukin.
Acknowledgements
We thank the many scientists whose stimulating conversations have in some measure been represented in these pages. I.S. is supported by a Medical Research Council (MRC) Senior Clinical Fellowship, S.R. is supported by an MRC Clinician Scientist Fellowship, and D.D. is supported by a Wellcome Trust Senior Clinical Fellowship. The views expressed in this article are not necessarily those of the authors' host institutions or funding bodies.
Biographies
Ian Sabroe is a Medical Research Council Senior Clinical Fellow at the University of Sheffield, UK. He is also a respiratory physician with a special interest in the treatment of severe asthma. He undertook his Ph.D. and early postdoctoral studies at Imperial College London, UK, working principally on chemokine biology and the recruitment of neutrophils and eosinophils. He moved to Sheffield in 2000 to continue his work in innate immunity, where his laboratory focuses on the roles of Toll-like receptors in the regulation of neutrophil and tissue cell function, particularly as they pertain to lung disease.
David Dockrell is a graduate of Trinity College Dublin, Ireland, and undertook postgraduate training at the Mayo Clinic, Rochester, Minnesota, USA. He is a clinician scientist and studying aspects of the pathogenesis of infectious disease, focusing on the role of the macrophage in the innate immune response to infection, in particular Streptococcus pneumoniae and pulmonary infection. His group have focused on how the induction of apoptosis regulates both microbial killing and controls the inflammatory response during infectious diseases using both in vitro and in vivo models. He is a Wellcome Senior Clinical Fellow at the University of Sheffield and an infectious diseases clinician.
Stefanie Vogel is currently Professor of Microbiology and Immunology at the University of Maryland, Baltimore, USA, and a recipient of the prestigious US National Institutes of Health (NIH) MERIT Award. She obtained her Ph.D. in 1977 at the University of Maryland, College Park, USA, did her postdoctoral training at the NIH, and joined the faculty of Uniformed Services University, USA, in 1980. In 2002, Vogel and her laboratory moved to the University of Maryland, School of Medicine in Baltimore, where she teaches medical and graduate programmes and continues her long-standing work on the innate immune response to infection and malignancy.
Stephen Renshaw is a Medical Research Council Clinician Scientist Fellow, combining research at the University of Sheffield with clinical work in respiratory medicine. Having studied medicine in Cambridge and Oxford, UK, he obtained his Ph.D. from the University of Sheffield in 2001. His research has investigated the role of death receptor signalling in the regulation of neutrophil apoptosis, and he is now establishing a research group using the unique advantages of the zebrafish to elucidate the molecular controls of neutrophil apoptosis and inflammation resolution in vivo.
Moira Whyte is Professor of Respiratory Medicine and heads the Section of Infection, Inflammation and Immunity in the School of Medicine and Biomedical Sciences at the University of Sheffield. She trained at the Royal Postgraduate Medical School, UK, undertaking a Ph.D. in the mechanisms of neutrophil apoptosis, and had a period of postdoctoral training at the Imperial Cancer Research Fund, London, before moving to Sheffield in 1996. Her laboratory focuses on the regulation of immune cell apoptosis in the context of host–pathogen interactions and her clinical interests are in airway inflammation and interstitial lung disease.
Steven Dower studied for a D.Phil. (1975–1978) on the analysis of antibody–antigen interactions. From 1979 to 1982 he was a Fogarty Fellow at the US National Cancer Institute, studying the activation of Fc receptors by immune complexes. Subsequently he was Head of Biochemistry at Immunex (now part of Amgen) and worked on the structure and function of cytokine receptors. This led to the development of Enbrel, the most widely used 'new-generation' anti-inflammatory drug. Since 1995 he has been Chair of Molecular Immunology at the University of Sheffield where he has studied the role of cytokine receptors and Toll-like receptors in innate immunity.
Related links
FURTHER INFORMATION
Competing interests
The authors declare no competing financial interests.
References
- 1.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 2.Levine MN, Whelan T. Adjuvant chemotherapy for breast cancer — 30 years later. N. Engl. J. Med. 2006;355:1920–1922. doi: 10.1056/NEJMe068204. [DOI] [PubMed] [Google Scholar]
- 3.Gelfand EW. Pro: mice are a good model of human airway disease. Am. J. Respir. Crit. Care Med. 2002;166:5–6. doi: 10.1164/rccm.2204023. [DOI] [PubMed] [Google Scholar]
- 4.Gordon CJ, Grafton G, Wood PM, Larche M, Armitage RJ. Modelling the human immune response: can mice be trusted? Commentary. Curr. Opin. Pharmacol. 2001;1:431–435. doi: 10.1016/S1471-4892(01)00074-1. [DOI] [PubMed] [Google Scholar]
- 5.Kumar RK, Foster PS. Modeling allergic asthma in mice: pitfalls and opportunities. Am. J. Respir. Cell Mol. Biol. 2002;27:267–272. doi: 10.1165/rcmb.F248. [DOI] [PubMed] [Google Scholar]
- 6.Persson CG. Con: mice are not a good model of human airway disease. Am. J. Respir. Crit. Care Med. 2002;166:6–7. doi: 10.1164/rccm.2204001. [DOI] [PubMed] [Google Scholar]
- 7.Persson CG, Erjefalt JS, Korsgren M, Sundler F. The mouse trap. Trends Pharmacol. Sci. 1997;18:465–467. doi: 10.1016/S0165-6147(97)90684-2. [DOI] [PubMed] [Google Scholar]
- 8.Roep BO, Atkinson M, von Herrath M. Satisfaction (not) guaranteed: re-evaluating the use of animal models of type 1 diabetes. Nature Rev. Immunol. 2004;4:989–997. doi: 10.1038/nri1502. [DOI] [PubMed] [Google Scholar]
- 9.Wiles S, Hanage WP, Frankel G, Robertson B. Modelling infectious disease — time to think outside the box? Nature Rev. Microbiol. 2006;4:307–312. doi: 10.1038/nrmicro1386. [DOI] [PubMed] [Google Scholar]
- 10.von Herrath MG, Nepom GT. Lost in translation: barriers to implementing clinical immunotherapeutics for autoimmunity. J. Exp. Med. 2005;202:1159–1162. doi: 10.1084/jem.20051224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medvedev AE, et al. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J. Exp. Med. 2003;198:521–531. doi: 10.1084/jem.20030701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Picard C, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki N, et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416:750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 14.Wenzel SE. Phenotypes in asthma: useful guides for therapy, distinct biological processes, or both? Am. J. Respir. Crit. Care Med. 2004;170:579–580. doi: 10.1164/rccm.2407005. [DOI] [PubMed] [Google Scholar]
- 15.Wenzel SE, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am. J. Respir. Crit. Care Med. 1999;160:1001–1008. doi: 10.1164/ajrccm.160.3.9812110. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A. Loss of Siglec expression on T lymphocytes during human evolution. Proc. Natl Acad. Sci. USA. 2006;103:7765–7770. doi: 10.1073/pnas.0510484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen J. Immunology. Differences in immune cell 'brakes' may explain chimp–human split on AIDS. Science. 2006;312:672–673. doi: 10.1126/science.312.5774.672b. [DOI] [PubMed] [Google Scholar]
- 18.Suntharalingam G, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 19.Barrett G, Cassell JA, Peacock JL, Coleman MP. National survey of British public's views on use of identifiable medical data by the National Cancer Registry. BMJ. 2006;332:1068–1072. doi: 10.1136/bmj.38805.473738.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin Z, Altman RB, Owen AB. Confidentiality in genome research. Science. 2006;313:441–442. doi: 10.1126/science.313.5786.441b. [DOI] [PubMed] [Google Scholar]
- 21.McGuire AL, Gibbs RA. Genetics. No longer de-identified. Science. 2006;312:370–371. doi: 10.1126/science.1125339. [DOI] [PubMed] [Google Scholar]
- 22.Shinagawa K, Kojima M. Mouse model of airway remodeling: strain differences. Am. J. Respir. Crit. Care Med. 2003;168:959–967. doi: 10.1164/rccm.200210-1188OC. [DOI] [PubMed] [Google Scholar]
- 23.Kips JC, et al. Murine models of asthma. Eur. Respir. J. 2003;22:374–382. doi: 10.1183/09031936.03.00026403. [DOI] [PubMed] [Google Scholar]
- 24.Brand DD. Rodent models of rheumatoid arthritis. Comp. Med. 2005;55:114–122. [PubMed] [Google Scholar]
- 25.Fan C, et al. Concordance among gene-expression-based predictors for breast cancer. N. Engl. J. Med. 2006;355:560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]
- 26.Potti A, et al. A genomic strategy to refine prognosis in early-stage non-small-cell lung cancer. N. Engl. J. Med. 2006;355:570–580. doi: 10.1056/NEJMoa060467. [DOI] [PubMed] [Google Scholar]
- 27.Kwok TC, et al. A small-molecule screen in C. elegans yields a new calcium channel antagonist. Nature. 2006;441:91–95. doi: 10.1038/nature04657. [DOI] [PubMed] [Google Scholar]
- 28.Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nature Rev. Drug Discov. 2005;4:35–44. doi: 10.1038/nrd1606. [DOI] [PubMed] [Google Scholar]
- 29.Renshaw, S. A. et al. A transgenic zebrafish model of neutrophilic inflammation. Blood (in the press). [DOI] [PubMed]
- 30.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nature Rev. Mol. Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 31.Morris GE, et al. Cooperative molecular and cellular networks regulate Toll-like receptor-dependent inflammatory responses. FASEB J. 2006;20:2153–2155. doi: 10.1096/fj.06-5910fje. [DOI] [PubMed] [Google Scholar]
- 32.Morris GE, et al. Agonists of Toll-like receptors 2 and 4 activate airway smooth muscle via mononuclear leukocytes. Am. J. Respir. Crit. Care Med. 2005;171:814–822. doi: 10.1164/rccm.200403-406OC. [DOI] [PubMed] [Google Scholar]
- 33.Sabroe, I. et al. Pulmonary perspective: targeting the networks that underpin contiguous immunity in asthma and COPD. Am. J. Respir. Crit. Care Med. (in the press). [DOI] [PubMed]
- 34.Nobeli I, Thornton JM. A bioinformatician's view of the metabolome. BioEssays. 2006;28:534–545. doi: 10.1002/bies.20414. [DOI] [PubMed] [Google Scholar]