

Key Points
To date, although many viral infections can be successfully prevented via vaccination, we lack effective knowledge of vaccines for numerous important human pathogens, including hepatitis C virus (HCV) and human immunodeficiency virus (HIV). Accordingly, antiviral drugs will be needed to treat many viral diseases. Virally encoded enzymes and cellular enzymes adapted for use by viruses for replication might represent useful targets for antiviral drugs.
Drugs that target either a viral or cellular polypeptide hold different implications. Inhibitors of unique viral functions have a lower risk of toxicity, whereas inhibitors of cellular enzymes that are used by viruses have a narrower window for efficacy without creating toxicity.
All viruses seem to require a helicase function for replication. HCV encodes a viral RNA helicase, and recent findings have shown that HIV-1 adapts a cellular RNA helicase for its viral lifecycle. These observations raise the possibility of small-molecule helicase inhibitors as a general mode of antiviral therapy.
Helicases fall into three super-families (SF1, SF2 and SF3) with conserved motifs. The conserved motifs are associated with conserved helicase function. However, outside of the conserved motifs the primary sequences and tertiary structures between helicases are differ greatly. In this regard, differences in primary sequence and tertiary structure between the helicase of a viral pathogen and that of cellular helicases can be exploited to confer specificity to an antiviral inhibitor.
The conformation of an active helicase can be broadly divided into an 'open' and a 'closed' complex. Strategies for identifying small-molecule helicase inhibitors include: inhibiting NTPase activity by direct competition with NTP binding; competitively inhibit nucleic-acid binding; inhibiting NTP hydrolysis or NDP release by blocking the movement of domain 2; inhibiting the process that couples NTP hydrolysis to translocation and unwinding of nucleic acid; inhibiting unwinding by sterically blocking helicase translocation; and inhibiting unwinding. Other potential inhibitory mechanisms include those that change the physical conformation of the helicase, or those that disrupt helicase turnover, or those that inhibit helicase interaction with other crucial proteins.
Preclinical proof of concept for helicase inhibitors as antiviral agents has been obtained for HSV. This breakthrough finding provides the best evidence to date that it is possible to develop selective, potent inhibitors of a viral helicase as antiviral agents. Searches are ongoing for antihelicase molecules that have activity against HCV or HIV-1.
Abstract
Although there has been considerable progress in the development of antiviral agents in recent years, there is still a pressing need for new drugs both to improve on the properties of existing agents and to combat the problem of viral resistance. Helicases, both viral and human, have recently emerged as novel targets for the treatment of viral infections. Here, we discuss the role of these enzymes, factors affecting their potential as drug targets and progress in the development of agents that inhibit their activity using the hepatitis C virus-encoded helicase NS3 and the cellular helicase DDX3 adopted for use by HIV-1 as examples.
Main
Viruses are obligate cellular parasites. It is therefore not surprising that viruses share similar metabolic strategies with their hosts. For many important functions, viruses either encode proteins closely related to host proteins or have evolved the ability to directly co-opt the services of cellular factors. To date, although many viral infections can be successfully prevented via vaccination, we lack effective knowledge of vaccines for numerous important human pathogens, including hepatitis C virus (HCV) and human immunodeficiency virus (HIV). Accordingly, antiviral drugs are likely to remain a significant mainstay for treating many viral diseases. Regrettably, medically licensed antivirals currently number less than 501, which, nevertheless, represent substantial progress, as 15 years ago that were fewer than five antivirals in clinical use.
Target the host or the virus?
In considering how to design antiviral drugs, one envisions two broad strategies. Because viruses replicate by using self-encoded proteins or by seizing control of cellular factors, agents designed to interrupt viral replication could, in principle, target with equal effectiveness either a viral or cellular polypeptide2. Interestingly, the two strategies hold different implications. In the case of a unique viral function, such as capsid formation, there is a lower risk of creating inhibitors with toxic effects on the host, similar to the advantage antibacterial drugs have in inhibiting bacterial targets that do not exist in their host. In the case of a target such as a viral HELICASE, there is a smaller window for specificity because the viral and cellular enzymes catalyse a similar enzymatic reaction. However, as the viral and cellular enzymes are not identical, traditional medicinal chemistry and structure-based drug design can exploit the difference between host and viral enzymes to create drugs with high specificity for the virus.
The Achilles heel of antiviral therapy is RESISTANCE3. Unless a drug is incredibly potent (reducing the size of the replicating pool of virus rapidly), and therefore requires only a short duration of treatment (reducing the time for the resistant viruses to amplify), resistance to treatment will arise over time, as observed with HIV or HBV patients on therapy. Targeting a cellular factor that is required for viral replication should help overcome the problem of viral resistance. Theoretically, the drug could be pan-antiviral and inhibit all viruses that are dependent on the same host factor. Operationally, we define pan-antiviral to mean that the inhibitor targets more than one family of viruses. This latter intervention strategy limits the development of resistant viruses, but it has a major disadvantage in generally causing greater toxicity to the host. Empirically, how one focuses one's antiviral drug design can be influenced by whether the virus replicates largely autonomously of the host, using predominantly virally encoded genes (for example, herpes simplex virus (HSV)), or whether the virus is intimately associated with the host's metabolism (for example, integrated proviruses). In this review, we discuss in a non-exhaustive fashion the HCV-encoded helicase NS3, and the cellular helicase DDX3 adopted for use by HIV-1, as two illustrative examples of potential antiviral targets.
Helicase motifs, structure and function
At the most basic level, helicases are motor enzymes that use energy derived from NTP hydrolysis to unwind double-stranded nucleic acids4,5,6,7. Further classification depends on whether the helicase can bind single-stranded nucleic acid, unwind RNA or DNA or both, and the direction of unwinding (3′ to 5′ or 5′ to 3′), and whether certain signature motifs are present in the primary sequence. Helicases have been divided into three super-families (SF1, SF2 and SF3) and two small families based on sequence comparisons and conserved motifs8. Examples of members of SF1, SF2 and SF3 are shown in Table 1. Figure 1a shows a schematic illustration of the seven highly conserved motifs among the two largest families (SF1 and SF2). Although, as discussed later, the conserved motifs are associated with conserved helicase function, a comparison of the primary sequence of two SF2 helicases (Fig. 1b), HCV helicase and human DDX3 reveals a paucity of identical residues (highlighted in green) and a tremendous divergence in sequence outside of the conserved motifs (highlighted in red). The differences in primary sequence and tertiary structure between the helicase of a viral pathogen and that of cellular helicases can be exploited to confer specificity to an antiviral inhibitor.
Table 1.
Examples of viral and cellular helicases
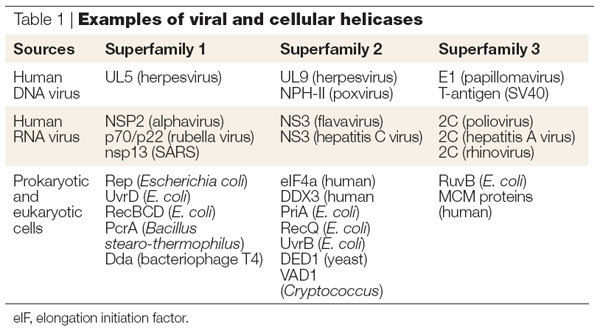
Figure 1. Illustrative structures and alignments of DEAD box helicases from superfamily 2 (SF II).
a | Schematic illustration of a two-domain SF2 helicase with the consensus motifs as indicated. b | Sequence alignment of the hepatitis C virus (HCV) NS3 helicase and the DDX3 cellular helicase used by HIV-1 as a Rev co-factor. Analogous motifs between the two SF2 helicases are highlighted. Note the paucity of sequence relatedness outside of the helicase motifs. c | The three-domain structure of HCV NS3 helicase with the bound poly(U) (PDB code: 1A1V). Domains 1, 2 and 3 are coloured in magenta, yellow and cyan, respectively. d | The poly(U), coloured orange, binds at the interface of domain 3 with the first two domains. Illustration of position of consensus motifs (highlighted in orange) on the inner faces of domains 1 and 2 using HCV helicase structure. The position of an oligonucleotide from a co-complex structure is shown; however, this is not conserved in other helicases.
The structure of a number of helicases has been solved and a recent review on helicase structure and function is available9. The total number of domains can vary from four (that is, PcrA/Rep), to three (that is, HCV helicase) to two (that is, elongation initiation factor 4A (eIF4A)). The different domains of a three-domain helicase (HCV helicase) are shown schematically in Fig. 1c, in which domain 1 (magenta), domain 2 (yellow) and domain 3 (cyan) are clearly delineated. Domains 1 and 2 form a core that is conserved in all helicases, and contain most of the conserved sequence motifs at the interface between the two domains, as seen in the example of HCV helicase (Fig. 1d). Surprisingly, despite widely disparate protein sequences, similar structural elements are conserved in all the structures solved to date. This is illustrated by an overlay of the structures for HCV RNA helicase10 (red; three domains) and PcrA DNA helicase11 (cyan; four domains) in Fig. 2a. The position and length of the α-helices and the number and orientation of β-strands in the core of both enzymes (top two domains) are very similar. By contrast, a divergence is clearly seen in domain 3 (bottom left) and HCV helicase has no counterpart to domain 4 of PcrA helicase (cyan, bottom right).
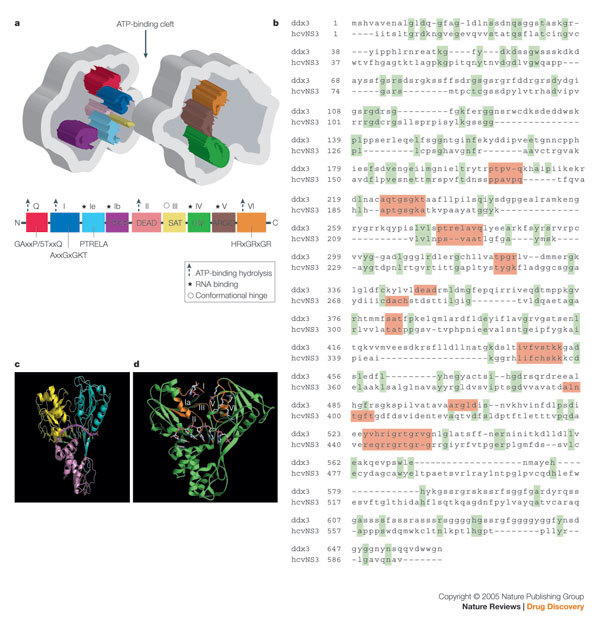
Figure 2. Example of a crucial structural element that is not conserved in the consensus sequence motifs.
a | The overlay of the crystal structures of hepatitis C virus (HCV) helicase (red; PDB code: 1HEI) and PcrA helicase (cyan; PDB code: 1QHH). The ATP is coloured magenta. The HCV helicase has three domains, whereas the PcrA helicase has four domains. The overlay shows the overall structural homology of three domains of the two structures, although they share very little sequence similarity. b | Identification of a conserved functional element in motif VI and IV by structure in HCV RNA Helicase and PcrA DNA helicase
All helicases bind NTP using two structural elements — a phosphate-binding P-loop, also known as motif I/Walker A motif, and motif II, a Mg2+ co-factor-binding loop also known as Walker B8,11,12. SF2 RNA helicases are also called DEA(D/H)-box helicases after the signature sequence of Walker B motif. As highlighted in orange in Fig. 1b, the residues in the conserved motifs required for binding and hydrolysing NTP lie at the interface between of domains 1 and 2 in HCV helicase. There is considerable flexibility in the distance between the two domains, although NTP hydrolysis can only occur when the domains are in a 'closed' configuration. Additional crystallographic and mutational analyses have identified motifs that contribute to oligonucleotide binding, such as Ia and IV8. The TxGx motif spans the bottom of the cleft between domains 1 and 2 and acts as a 'hinge' for the movement of domain 2. The exact changes in physical conformation and the path the RNA or DNA takes as one strand is destabilized and 'unwound' from the other are not well understood, but they do not seem to be conserved between different helicases. Complicating matters further, some helicases seem to function as monomers (for example, HCV helicase), others as dimers (for example, HSV UL5) and yet others as hexamers (for example, simian virus 40 (SV40) T antigen). Many putative helicases have so far only been identified by the presence of a DEAD/H box in the primary sequence and await biochemical verification of bona fide helicase function.
However, not all of the crucial elements for helicase function can be detected from the primary sequence. Structural analyses have identified crucial residues that are conserved only in space, but not in the primary sequence. An example is shown in Fig. 2b, in which the crystal structures of the interface between domains 1 (left) and 2 (right) of HCV helicase10 (in green) and PcrA helicase11 (in blue) are superimposed. Two pairs of arginine residues on the inner face of domain 2 are highlighted (circles), which have been shown by mutational analyses to be essential for NTP hydrolysis. The lower pair, Arg-610 of PcrA (yellow sticks) and Arg-464 of HCV helicase (white sticks) are both in motif VI and are structurally and functionally homologous to each other13. In the upper pair, Arg-467 of HCV helicase also shows close functional and structural homology with Arg-287 of PcrA, but they derive from two different sequence motifs. The HCV residue in the pair is encoded in motif VI, whereas the analogous PcrA residue comes from motif IV, a relationship that cannot be derived by homology searches and sequence gazing14.
Cellular functions of RNA helicases
DEAD box15 proteins are the most numerous members of helicase superfamily 2 (Table 1) and are ubiquitous in eukaryotic genomes. As shown in Fig. 3, although precise substrates remain undefined, DEAD helicases are involved pleiotropically in many aspects of RNA metabolism, including transcription, mRNA splicing, mRNA export, translation, RNA stability and mitochondrial gene expression16,17,18,19,20. Despite their large number, each RNA helicase seems to be individually essential, because loss of one DEAD box protein in yeast cannot be complemented by another overexpressed family member21.
Figure 3. Schematic representations of nuclear and cytoplasmic functions attributed to RNA helicases.
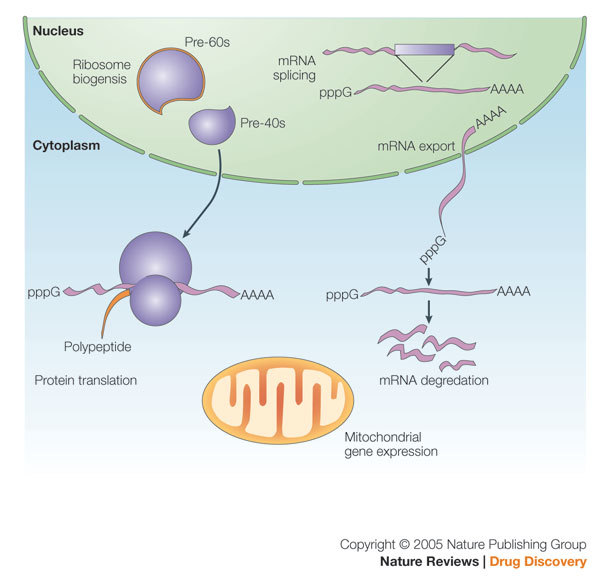
Various helicases have been implicated in transcription, mRNA splicing, mRNA transport, mRNA translation, ribosome biogenesis and mitochondrial gene expression.
Unwinding of highly structured RNAs might be reasoned to be important for eukaryotic transcription. However, direct evidence for such a role by of an RNA helicase has been elusive. Once transcribed, RNAs are rapidly packaged into ribonucleoprotein complexes (RNPs22) for further processing. In such a setting, RNA helicases can play roles in RNA–RNA and RNA–protein23 remodelling. There is evidence that helicases such as UAP56, Brr2, Prp16, Prp22, and Prp43 have roles in RNA splicing24, whereas others, such as Dbp525,26 and DDX327, serve to chaperone RNAs from the nucleus into the cytoplasm. Translation of mRNAs in the cytoplasm is facilitated by helicases such as eIF4a and Ded1, whereas Rh1B, Ski2, Dob1 and Dhh1 helicases modulate the stability of mRNAs24. DEAD box helicases also act in ribosome biogenesis through regulation of small nucleolar (snoRNAs) and ribosomal (rRNAs) RNA interactions28,29.
Human helicases as antiviral drug targets
A correlation exists between genome size and the encoding of a helicase by (+ strand) RNA viruses30. Although all viruses are thought to functionally need helicase(s), in some cases the larger entities have the luxury of self-encoding such an enzyme, whereas size-constrained, smaller viruses can adapt to use a cellular helicase. In general, RNA viruses that replicate in the cytoplasm mostly self-encode a helicase, whereas those that replicate in the nucleus often utilize a cellular helicase. Consistent with the latter notion, HIV-1 was shown in two recent studies to co-opt the activity of a cellular helicase27,31. Human helicase members of superfamily 2, DDX131 and DDX3 are required for the viral Rev protein to export unspliced/partially spliced HIV-1 mRNAs from the nucleus into the cytoplasm. DDX1 or DDX3 can therefore be added to the list of potential treatment targets of HIV-1-encoded enzymes (that is, reverse transcriptase, protease and integrase).
Viral helicases as antiviral drug targets
The potential of helicases as antiviral drug targets has recently been reviewed32,33,34,35,36. Unlike retroviruses, two other human viruses, HSV and human papillomavirus (HPV), physically encode their own helicases. The HPV E1 protein37 is a member of superfamily 3 (Table 1). A number of laboratories have developed high-throughput helicase or E1-dependent polymerase screens for inhibitors of HPV replication. To date, no successful clinical drug candidates derived from these efforts have been reported.
The HSV UL5 and UL9 genes encode helicases in superfamily 1 and 2, respectively38. UL5 forms a heterotrimeric helicase–primase complex with UL8 and UL5239 that is responsible for unwinding duplex viral DNA at replication forks and laying down Okazaki primers for elongation.
Preclinical proof of concept for helicase inhibitors as antiviral agents has been obtained for HSV32,33,35. Using high-throughput screening (HTS)34, thiazolylphenyl amino-thiazole40,41, (dichloroanilino)purines andpyrimidines42 and thiazolylsulphonamide inhibitors43 were identified by HTS of the inhibition of HSV UL5/8/52 primase–helicase complex. Optimization of the screening hits resulted in compounds that inhibited HSV growth in cell culture with little cytotoxicity and which were orally active in an animal model of HSV. Mechanistically, the thiazolylphenyl-containing drugs seem to stabilize helicase–primase binding to polynucleotide substrates and halt further catalytic cycles. The mechanism of antiviral action was confirmed when several groups independently selected resistant viruses with single point mutations in the UL5 DNA helicase gene for both classes of compounds, confirming the mechanism of action of the inhibitors. Although the UL5 mutants showed little to no reduction in replicative fitness compared with wild-type virus in cultured cells44, the fitness of such mutants in the clinical setting is the most important measure of fitness. Nonetheless, this is the most successful demonstration to date that it is possible to develop selective, potent inhibitors of a viral helicase as antiviral agents in a preclinical setting.
HCV helicase as an antiviral drug target
The successful results of the HSV helicase inhibitors were not made public until several years after many companies had started screening for HCV helicase inhibitors. However, at the time there was widespread optimism that the HCV helicase would turn out to be a good target for antiviral drug discovery. Bolstered by the successful examples of anti-HIV drug design, structure-based drug design efforts have focused on finding inhibitors of three essential viral enzymes: the NS3 serine protease45, the NS3 helicase10,46,47 and the NS5B RNA-dependent RNA polymerase48,49,50. In the NS3 bifunctional protein the first 181 amino acids form a serine proteinase, whereas the carboxy-terminal 181–631 amino acids form a helicase. From a biological standpoint, protease and helicase activities co-exist in vivo, and either could serve as a useful anti-HCV target. The helicase and polymerase form a complex with other host and viral proteins to create the viral replicase multi-protein complex51,52,53. In order for HCV to multiply, negative-stranded RNA replicative intermediates must be synthesized by the replicase complex using incoming positive-stranded RNA from the infecting virus as the template. The negative-stranded replicative intermediate is then used as an intermediate template to synthesize positive-stranded progeny RNA, which is packaged into viral capsids. Because the positive and negative RNA strands are complementary, HCV helicase is thought to be required for strand separation. Additional functions in HCV replication might include the melting of highly stable secondary structures, which are known to be present in HCV RNA, in order to increase translational efficiency of the polyprotein or to increase replication rate and fidelity54,55. Currently, very little is known about the crucial protein–protein interactions in HCV, except that the NS4A cofactor interacts with NS3. Microscopic studies indicate that replicase, as well as most of the HCV proteins, congregate on the ER to form a 'membraneous web'. It is thought that cellular factors are probably present in this membrane-associated complex or complexes. Future efforts at mapping protein–protein interactions, both between viral proteins and cellular factors, will be important for successful drug discovery.
A consideration in assay design is the form of the protein to use in the enzymatic screen, particularly in the case of multi-domain or multi-functional proteins. In the native state, the helicase domain is part of the NS3·4A complex, which also contains the NS3 serine protease domain and its NS4A cofactor56. Figure 4 shows a crystal structure of full-length NS3 in a covalent complex with an NS4A peptide57. Comparison of the three helicase domains in the full-length protein with the structure of the helicase domain alone shown in Fig. 1c reveals that the structures of the two are very similar. The position of an ATP molecule (shown in red) at the top of the cleft between domains 1 and 2, and the position of the bound oligonucleotide from the crystal structure of the catalytic domain alone, reveals that the presence of the protease domain does not inhibit access to these sites. The protease domain shown in Fig. 4 is coloured blue and the 4A cofactor is in green; the location of the active site is indicated by the position of VX-950, a peptidomimetic NS3·4A protease inhibitor created using structure-based drug design, which shows that the presence of the helicase domain is unlikely to block access of protease substrates or inhibitors. These structural studies support the notion that in the absence of other replicase proteins, the helicase domain can largely function independently of the protease domain and that use of the helicase domain alone could be a reasonable choice for an assay format. However, biochemical studies comparing the helicase activity of the full-length NS3 protein with the truncated helicase domain suggest that the full-length domain is more efficient at unwinding, and subtle differences exist in the mechanism of unwinding for the full-length and truncated domain58,59,60,61,62 in the presence and absence of NS463. It is therefore possible that different inhibitors might score positively in a screen using different forms of the enzyme.
Figure 4. The structure of full-length NS3 of HCV (PDB code: 1CU1).

The three domains of the helicase in the front are coloured as in Fig. 1a. The amino-terminal protease domain is coloured blue. A protease inhibitor, VX-950 (coloured pink), is modelled into the active site of the protease at the interface of the carboxy-terminal third domain of the helicase. The poly(U) (orange) and ATP (red) were modelled into the structure by overlaying the full-length NS3 structure with the structures of helicase domain bound with poly(U) (PDB code: 1A1V) and the PcrA helicase bound with ATP (PDB code: 1QHH). It is clear from this picture that NS3 can function independently as a helicase and as a protease.
Strategies for discovering inhibitors
In considering drug designs against helicases, one can begin with several general conceptual strategies. Helicases have multiple enzymatic activities and functional domains that present multiple potential mechanisms of action for the design of an inhibitor. This review will present a general description of helicase activity, suitable for the general reader. An in-depth description can be found in detailed physical and kinetic analyses of periodic cycles of RNA unwinding and pausing by HCV NS3 helicase, which have been investigated by several laboratories59,64,65,66,67,68. As schematically shown in Fig. 5a, the conformation of an active helicase can be broadly divided into an 'open' and a 'closed' complex of domains 1 and 2, with a transition between the two. As shown in Fig. 5b, assays can be used to identify small-molecule helicase inhibitors that have the following effects: they inhibit NTPase activity by direct competition with NTP binding69; competitively inhibit nucleic-acid binding; inhibit NTP hydrolysis or NDP release by blocking the movement of domain 2; inhibit the process that couples NTP hydrolysis to translocation and unwinding of nucleic acid; inhibit unwinding by sterically blocking helicase translocation70; and inhibit unwinding (reviewed in Refs 14,71). For all these mechanisms, unwinding and binding assays can be deployed.
Figure 5. Helicase as a drug target: assays for multiple potential mechanisms of action.
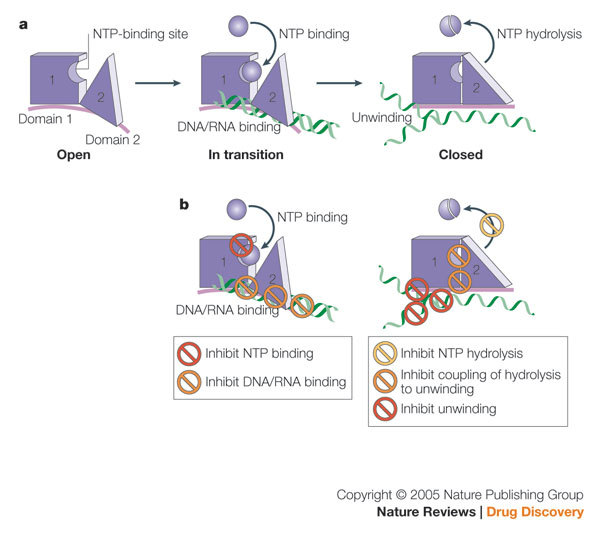
a | Schematic view of helicase reaction. b | Potential mechanisms of action for a small-molecule inhibitor. See discussion in text.
A separate category of potential inhibitors not depicted in Fig. 5 are those that change the physical conformation of the helicase, inhibiting the natural range of motion required for unwinding or altering the interface between domains 1 and 2. Other non-enzymatic mechanisms of action for new helicase inhibitors could include disruption of the turnover of NS3 protein or inhibiting crucial interactions with other proteins in the replicase complex.
The solution of the crystal structure of HCV helicase complexed with oligonucleotide, as well as mutagenesis studies, have identified key residues that are essential for enzyme activity or translocation of the RNA substrate13,72. However, there is no consensus on the mechanism of unwinding, and three different models have been proposed by three different groups who have published crystal structures of HCV helicase.
It is important to inhibit functions that are essential for helicase activity. As all helicases share some common enzyme reactions (for example, NTP hydrolysis and translocation) in their mechanisms of action, it might be difficult to avoid the possibility of hitting an unintended cellular helicase and generating unwanted side effects. Electrostatic analysis of the HCV helicase shows both active-site and non-active-site locations73 that could be exploited for drug design. Although it might be tempting to inhibit oligomerization, inhibition of protein–protein interactions with a small molecule could be difficult because such interactions usually have multiple points of contact over a broad surface area, and blocking one such contact with a small molecule is usually not sufficient to block binding of the two proteins. This kind of mechanism is more suitably targeted by antibodies and antibody mimics. Likewise, targeting additional domains that are unique to a helicase in an attempt to gain specificity — such as the two novel conserved motifs in HCV helicase74 — is not practical unless a specific assay is available for a function known to be associated with the domain or an approach is used to develop reagents that bind to the region of interest. Recombinant human antibodies75,76 and RNA aptamers77,78 have been developed that bind to HCV helicase and which inhibit HCV helicase activity.
Of all these potential assays, most laboratories have relied on a simple unwinding assay for primary HTS, such as a scintillation proximity assay (SPA)79,80 or a fluorometric assay81. Theoretically, an unwinding assay should increase the odds of finding an inhibitor because inhibition of any of the potential mechanisms of action listed above, except those requiring a cellular environment (for example, turnover and replicase-complex formation), should result in 'hits' (for example, low-potency chemical starting points for medicinal chemistry optimization). The choice of a more stable DNA substrate versus the more 'natural' RNA substrate is available for HCV helicase because it can unwind both RNA and DNA homo- and heteroduplexes. Biochemical assays for detecting inhibitors of nucleic-acid binding or ATPase activity can be run in HTS mode, but are primarily used to elucidate the mechanism of action of an inhibitor. There is no reason why an unwinding assay could not also find inhibitors that cause conformational changes to the helicase.
However, in most screenings the initial 'hits' or candidate inhibitors are usually not very potent (μM IC50), and are useful in the sense that one hopes to evolve them into more potent compounds. Empirically, in screens for HCV helicase, very few candidates were found; one explanation for this dearth of results could be that non-potent hits simply could not bind with sufficiently high affinity to helicase to inhibit the unwinding reaction. An alternative to an unwinding assay is to identify molecules that simply bind to the helicase and alter its conformation. Accordingly, the strategy is to evolve initial binders that change conformation into tighter binders that can also inhibit unwinding. In scoring for binding and conformational perturbation, one could possibly find more hits than scoring for unwinding.
Efforts to develop HCV inhibitors
Of the three major HCV enzymatic targets for drug discovery, NS3 protease inhibitors have been the most successful to date. Proof of concept for this class of inhibitors has been demonstrated by Boehringer Ingelheim and Vertex Pharmaceuticals using BILN-206182,83 and VX-95084, respectively. Both compounds decreased HCV viral load in patients by ∼2–3 logs in the first 3 days of dosing. In some patients treated with VX-950, the HCV viral load dropped from 106 infectious units (IU) per ml at the start of treatment to below the limit of detection (<10 IU per ml) during 14 days of dosing. Several other HCV protease inhibitors from different companies are currently in clinical trials. The speed with which HCV protease inhibitors have approached the clinic is remarkable given that the active site of the enzyme is notorious for being 'flat and featureless'. In addition, most high-throughput protease screens of large compound libraries have not produced good hits. However, it has been possible to use the HCV protease substrate and cleavage products as starting points for structure-based design to create the peptidomimetic inhibitors that are in clinical development today.
In contrast, HCV polymerase high-throughput assays for elongation have generally been fruitful and yielded a diverse group of active-site and non-active-site inhibitors with different scaffolds. The availability of multiple starting points for HCV polymerase inhibitors means that there is a wealth of companies with preclinical inhibitors, some of which have shown good potency and good bioavailability. In contrast to HCV protease inhibitors, a number of polymerase inhibitors have entered the clinic and failed to significantly decrease HCV viral load in patients. NM283 (Idenix) has provided proof of concept for active-site polymerase inhibitors with ∼1.2 log reduction in viral titre after treating patients for 15 days85.
Because there are multiple mechanisms of action that could inhibit HCV helicase, many have been surprised at the meagre array of hits from helicase screens or focused chemical libraries54, many of which were nucleic-acid binders or intercalators with low potency. The ATP-binding site has been successfully targeted by kinase and gyrase inhibitors in which the binding site is a distinct and well-defined 'pocket'. By contrast, only weak inhibitors have been found that inhibit the HCV binding site86. One explanation might be that the HCV helicase NTP-binding site at the interface of domains 1 and 2 has little specificity — it binds all NTPs primarily through the phosphate, not the base, of the nucleotide. In all helicases, the NTP-binding pocket forms transiently as domain 2 approaches domain 1, a movement that results in hydrolysis of ATP and concomitant opening or separation of domain 2 from domain 1. Most of the crucial residues for helicase activity and the conserved motifs are arrayed on the inside face between domains 1 and 2 in which the potential target-binding sites are only formed transiently when the two domains close. The constant movement of domains 1 and 2 between an 'open' and a 'closed' configuration and back again means that the enzyme is likely to often be in transition87. We have yet to understand the necessary principles required to inhibit such a dynamic protein system.
It is not hard to imagine that the development of HCV helicase inhibitors for clinical use has been slow because the enzyme is a moving target and undergoes significant transient conformational changes that require the coupling of NTP hydrolysis to unwinding67,72,74,88. The degree of movement is illustrated in Fig. 6A, which is a superimposition of eight independent HCV helicase crystal structures, and Fig. 6B, which is a superimposition of four of the above eight independent HCV helicase structures. Domains 1 and 3 are relatively rigid with respect to each other (the traces lie on top of each other), whereas domain 2 has a large freedom of motion. This is indicated in Fig. 6B by the silver ball identifying a single carbon atom in domain 2. Finally, small-molecule drug design requires a reasonable starting point, whether it is a substrate mimic or a hit from screening. It is not obvious from the HCV helicase crystal structures that the 'gate keeper' residues, which are crucial for binding the (unwinding) nucleic acid, form a binding pocket suitable for structure-based inhibitor design.
Figure 6. A moving target is hard to hit.
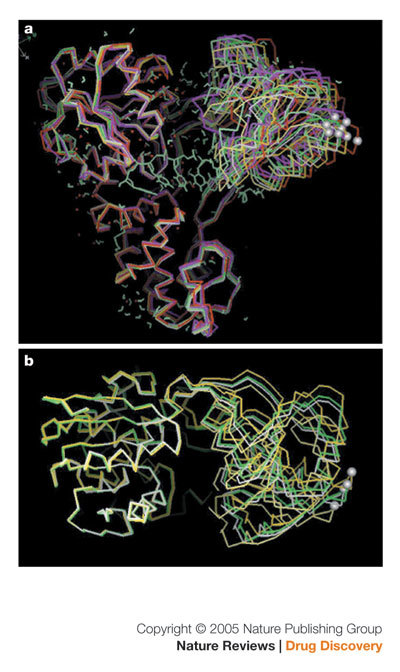
Superimposition of independent hepatitis C virus helicase structures. a | Superimposition of eight independently derived structures ('side view'). b | Superimposition of four independently derived structures ('top view').
Concluding remarks
Globally, HIV-1 infects more than 40 million individuals; HCV is estimated to have more than 170 million carriers. All cells and viruses require a helicase function, not necessarily their own, for nucleic-acid replication. The requirement for long-term treatment regimens for HIV allows for the generation of drug-resistant mutations during the course of therapy. To minimize the possibilities of generating resistance, targeting a cellular enzyme required by HIV-1 could be a viable option. Many non-anti-infective drug therapies target cellular enzymes, such as angiotensin-converting enzyme (ACE) to treat hypertension, congestive heart failure, myocardial infarction, endothelial dysfunction and renal disease89. The potential for off-target-induced cytotoxicity is significant, irrespective of whether one is targeting a cellular or viral helicase, because of similarities in structure and function. However, the recently characterized requirement for a helicase in Rev-dependent HIV-1 gene expression could be a new means to treat HIV-1 without generating drug-resistant viruses. In our laboratory (K.-T.J.), preliminary evidence has been found that ring-expanded nucleoside analogues, previously used successfully as NTPase/helicase inhibitors of West Nile virus90, have substantial anti-HIV-1 activity in cultured cells at non-cytotoxic doses. The HSV helicase–primase inhibitors offer proof of principle that a viral helicase complex can be specifically differentiated from and selectively inhibited among a pool of related cellular helicases in cell-based experiments and in an infectious animal model without significant cytotoxicities. Although the conserved helicase motifs within the helicases are very similar, individual helicases are widely divergent in their coding sequences, raising the possibility that each individual protein could be targeted with specificity in a knowledge-based manner. As highlighted in Table 1, an emerging picture is that viruses either encode their own helicase or use a cellular helicase for replication. For example, poxviruses, papovaviruses and pathogenic RNA viruses, such as West Nile virus, severe acute respiratory syndrome (SARS) coronavirus and Dengue fever virus, all encode viral helicases that could potentially be future antiviral targets. Regrettably, other than the rare drug candidates that we have mentioned in this review, there currently are no clinically useful candidate compounds for viral helicases. Cumulated successes against virus-encoded helicases are likely to precede effective drug targeting of cellular helicases used by viruses.
Acknowledgements
We would like to acknowledge M. Sintchak and J. Kim for their contribution to the HCV helicase structure figures and thank M. Briggs, C. Lin, M. Murcko, S. Lyons, J. Thomson, A. Elmo and V. Yedavalli for helpful discussions and editorial assistance.
Glossary
- HELICASE
An enzyme that unwinds double-stranded nucleic acids.
- RESISTANCE
Change in pathogens that render them insensitive to previously effective drugs.
- DEAD
Short acronym for the amino acids aspartate, glutamate, alanine and aspartate.
Biographies
Ann D. Kwong is a Senior Research Fellow and Head of Infectious Disease Biology at Vertex Pharmaceuticals, Inc., Cambridge, Massachusetts, USA. She received her Ph.D. in Virology from Niza Frenkel at the University of Chicago, Illinois, and performed her postdoctoral studies on SV40 in vitro DNA replication with Jerry Hurwitz at Memorial Sloan-Kettering Cancer Centre, New York. She worked on HSV and HCV antiviral drug discovery at Schering-Plough Research Institute in Kenilworth, New Jersey, before moving to Vertex. Her group has provided the biological support and understanding for teams dedicated to a structure-based drug design approach for the development of inhibitors of both viral and cellular targets for the treatment of HCV-infected patients.
B. Govinda Rao is a Research Fellow in the Applications Modeling group at Vertex Pharmaceuticals, Inc., Cambridge, Massachusetts, USA. He received his Ph.D. in Structural and Solid State Chemistry at the Indian Institute of Science, Bangalore, India, and carried out postdoctoral research on free energy simulations of enzyme–inhibitor complexes at the Scripps Research Institute, La Jolla, California, before joining Vertex in 1990. Rao is a co-inventor of the marketed AIDS drug agenerase and contributed to the design of VX-950, a HCV protease inhibitor undergoing clinical trials.
Kuan-Teh Jeang heads the Molecular Virology Section in the Laboratory of Molecular Microbiology, National Institute of Allergy and Infectious Disease (NIAID), National Institutes of Health. He received his M.D. and Ph.D. degrees from the Medical Scientist Training Program (MSTP) of the Johns Hopkins University School of Medicine and did a year of medical internship at the University of Iowa Hospital. In 1985, he joined the National Cancer Institute as a medical staff fellow, and moved to the NIAID in 1988. He has published more than 200 peer-reviewed articles and chapters. His research interests include studying the molecular mechanisms that regulate gene expression of HIV-1 and HTLV-1 in human cells.
Related links
DATABASES
Entrez Gene
Accession codes
Accessions
Protein Data Bank
Competing interests
The authors declare no competing financial interests.
References
- 1.De Clercq E. Antivirals and antiviral strategies. Nature Rev. Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Clercq E. Strategies in the design of antiviral drugs. Nature Rev. Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 3.Richman DD. The implications of drug resistance for strategies of combination antiviral chemotherapy. Antiviral Res. 1996;29:31–33. doi: 10.1016/0166-3542(95)00911-6. [DOI] [PubMed] [Google Scholar]
- 4.Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 5.Soultanas P, Wigley DB. Unwinding the 'Gordian knot' of helicase action. Trends Biochem. Sci. 2001;26:47–54. doi: 10.1016/s0968-0004(00)01734-5. [DOI] [PubMed] [Google Scholar]
- 6.Singleton MR, Wigley DB. Modularity and specialization in superfamily 1 and 2 helicases. J. Bacteriol. 2002;184:1819–1826. doi: 10.1128/JB.184.7.1819-1826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levin MK, Patel SS. Molecular Motors. 2002. pp. 179–198. [Google Scholar]
- 8.Gorbalenya AE, Koonin EV. Helicases: amino acid sequence comparisons and structure–function relationships. Curr. Opin. Struc. Biol. 1993;3:419–429. [Google Scholar]
- 9.Caruthers J, McKay D. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 10.Kim J, et al. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 1998;6:89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 11.Subramanya HS, Bird LE, Brannigan JA, Wigley DB. Crystal structure of a DExx box DNA helicase. Nature. 1996;384:379–383. doi: 10.1038/384379a0. [DOI] [PubMed] [Google Scholar]
- 12.Walker JE, Runswick MJ, Gay NJ. Distantly related sequences in the α- and β-subunits of ATP synthetase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim DW, Kim J, Gwack Y, Han JH, Choe J. Mutational analysis of the hepatitis C Virus RNA Helicase. J. Virol. 1997;71:9400–9409. doi: 10.1128/jvi.71.12.9400-9409.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwong AD, Kim JL, Lin C. Structure and function of hepatitis C virus NS3 helicase. Curr. Top. Microbiol. Immunol. 2000;242:171–196. doi: 10.1007/978-3-642-59605-6_9. [DOI] [PubMed] [Google Scholar]
- 15.Linder P, et al. Birth of the D-E-A-D box. Nature. 1989;337:121–122. doi: 10.1038/337121a0. [DOI] [PubMed] [Google Scholar]
- 16.Lorsch JR. RNA chaperones exist and DEAD box proteins get a life. Cell. 2002;109:797–800. doi: 10.1016/s0092-8674(02)00804-8. [DOI] [PubMed] [Google Scholar]
- 17.Tanner NK, Linder P. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell. 2001;8:251–262. doi: 10.1016/s1097-2765(01)00329-x. [DOI] [PubMed] [Google Scholar]
- 18.Luking A, Stahl U, Schmidt U. The protein family of RNA helicases. Crit. Rev. Biochem. Mol. Biol. 1998;33:259–296. doi: 10.1080/10409239891204233. [DOI] [PubMed] [Google Scholar]
- 19.Linder P, Stutz F. mRNA export: travelling with DEAD box proteins. Curr. Biol. 2001;11:R961–R963. doi: 10.1016/s0960-9822(01)00574-7. [DOI] [PubMed] [Google Scholar]
- 20.de la Cruz J, Kressler D, Linder P. Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci. 1999;24:192–198. doi: 10.1016/s0968-0004(99)01376-6. [DOI] [PubMed] [Google Scholar]
- 21.Kessler MM, et al. Hrp1, a sequence-specific RNA-binding protein that shuttles between the nucleus and the cytoplasm, is required for mRNA 3′-end formation in yeast. Genes Dev. 1997;11:2545–2556. doi: 10.1101/gad.11.19.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nature Rev. Mol. Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 23.Fairman ME, et al. Protein displacement by DExH/D “RNA helicases” without duplex unwinding. Science. 2004;304:730–734. doi: 10.1126/science.1095596. [DOI] [PubMed] [Google Scholar]
- 24.Rocak S, Linder P. DEAD-box proteins: the driving forces behind RNA metabolism. Nature Rev. Mol. Cell. Biol. 2004;5:232–241. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- 25.Tseng SS, et al. Dbp5p, a cytosolic RNA helicase, is required for poly(A)+ RNA export. EMBO J. 1998;17:2651–2662. doi: 10.1093/emboj/17.9.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitt C, et al. Dbp5, a DEAD-box protein required for mRNA export, is recruited to the cytoplasmic fibrils of nuclear pore complex via a conserved interaction with CAN/Nup159p. EMBO J. 1999;18:4332–4347. doi: 10.1093/emboj/18.15.4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell. 2004;119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 28.Daugeron MC, Linder P. Dbp7p, a putative ATP-dependent RNA helicase from Saccharomyces cerevisiae, is required for 60S ribosomal subunit assembly. RNA. 1998;4:566–581. doi: 10.1017/s1355838298980190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang WQ, Clark JA, Fournier MJ. The rRNA-processing function of the yeast U14 small nucleolar RNA can be rescued by a conserved RNA helicase-like protein. Mol. Cell Biol. 1997;17:4124–4132. doi: 10.1128/mcb.17.7.4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koonin EV, Dolja VV. Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993;28:375–430. doi: 10.3109/10409239309078440. [DOI] [PubMed] [Google Scholar]
- 31.Fang J, et al. A DEAD box protein facilitates HIV-1 replication as a cellular co-factor of Rev. Virology. 2004;330:471–480. doi: 10.1016/j.virol.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 32.Kleymann G, et al. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nature Med. 2002;8:392–398. doi: 10.1038/nm0402-392. [DOI] [PubMed] [Google Scholar]
- 33.Crumpacker CS, Schaffer PA. New anti-HSV therapeutics target the helicase-primase complex. Nature Med. 2002;8:327–328. doi: 10.1038/nm0402-327. [DOI] [PubMed] [Google Scholar]
- 34.Jones PS. Strategies for antiviral drug discovery. Antivir. Chem. Chemother. 1998;9:283–302. [PubMed] [Google Scholar]
- 35.Frick DN. Helicases as antiviral drug targets. Drug News Perspect. 2003;16:355–362. doi: 10.1358/dnp.2003.16.6.829307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadaré G, Haenni A-L. Minireview: virus-encoded RNA helicases. J. Virol. 1997;71:2583–2590. doi: 10.1128/jvi.71.4.2583-2590.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson VG, West M, Woytek K, Rangasamy D. Papillomavirus E1 proteins: form, function, and features. Virus Genes. 2002;24:275–290. doi: 10.1023/a:1015336817836. [DOI] [PubMed] [Google Scholar]
- 38.Marintcheva B, Weller SK. A tale of two HSV-1 helicases: roles of phage and animal virus helicases in DNA replication and recombination. Prog. Nucleic Acid Res. Mol. Biol. 2001;70:77–118. doi: 10.1016/s0079-6603(01)70014-1. [DOI] [PubMed] [Google Scholar]
- 39.Crute JJ, et al. Herpes simplex virus 1 helicase-primase: a complex of three herpes-encoded gene products. Proc. Natl Acad. Sci. USA. 1989;86:2186–2189. doi: 10.1073/pnas.86.7.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spector FC, Liang L, Giordano H, Sivaraja M, Peerson MG. T157602, a 2-amino-thiazole inhibits HSV replication by interacting with the UL5 component of the UL5/8/52 helicase primase complex. Antiviral Res. 1998;37:A43. doi: 10.1128/jvi.72.9.6979-6987.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spector FC, Liang L, Giordano H, Sivaraja M, Peterson MG. Inhibition of herpes simplex virus replication by a 2-amino thiazole via interactions with the helicase component of the UL5–UL8–UL52 complex. J. Virol. 1998;72:6979–6987. doi: 10.1128/jvi.72.9.6979-6987.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crute JJ, et al. Inhibition of herpes simplex virus type 1 helicase–primase by (dichloroanilino)purines and-pyrimidines. J. Med. Chem. 1995;38:1820–1825. doi: 10.1021/jm00010a027. [DOI] [PubMed] [Google Scholar]
- 43.Betz UA, Fischer R, Kleymann G, Hendrix M, Rubsamen-Waigmann H. Potent in vivo antiviral activity of the herpes simplex virus primase–helicase inhibitor BAY 57–1293. Antimicrob. Agents Chemother. 2002;46:1766–1772. doi: 10.1128/AAC.46.6.1766-1772.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liuzzi M, et al. Aminothiazolyl-phenyl-based inhibitors of HSV helicase-primase: a novel class of orally active antiherpetic agents. Antiviral Res. 1998;37:A42. [Google Scholar]
- 45.Kim JL, et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996;87:343–355. doi: 10.1016/s0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 46.Yao N, et al. Structure of the hepatitis C virus RNA helicase domain. Nature Struct. Biol. 1997;4:463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 47.Cho H-S, et al. Crystal structure of RNA helicase from genotype 1b hepatitis C virus. J. Biol. Chem. 1998;273:15045–15052. doi: 10.1074/jbc.273.24.15045. [DOI] [PubMed] [Google Scholar]
- 48.Lesburg CA, et al. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nature Struct. Biol. 1999;6:937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 49.Bressanelli S, Tomei L, Rey FA, De Francesco R. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 2002;76:3482–3492. doi: 10.1128/JVI.76.7.3482-3492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ago H, et al. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure Fold. Des. 1999;7:1417–1426. doi: 10.1016/s0969-2126(00)80031-3. [DOI] [PubMed] [Google Scholar]
- 51.Salonen A, Ahola T, Kaariainen L. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 2005;285:139–173. doi: 10.1007/3-540-26764-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma H, et al. Inhibition of native hepatitis C virus replicase by nucleotide and non-nucleoside inhibitors. Virology. 2005;332:8–15. doi: 10.1016/j.virol.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 53.Moradpour D, et al. Membrane association of the RNA-dependent RNA polymerase is essential for hepatitis C virus RNA replication. J. Virol. 2004;78:13278–13284. doi: 10.1128/JVI.78.23.13278-13284.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phoon CW, Ng PY, Ting AE, Yeo SL, Sim MM. Biological evaluation of hepatitis C virus helicase inhibitors. Bioorg. Med. Chem. Lett. 2001;11:1647–1650. doi: 10.1016/s0960-894x(01)00263-3. [DOI] [PubMed] [Google Scholar]
- 55.Rice CM. Virology. 1996. pp. 931–960. [Google Scholar]
- 56.Gallinari P, et al. Multiple enzymatic activities associated with recombinant NS3 Protein of hepatitis C virus. J. Virol. 1998;72:6758–6769. doi: 10.1128/jvi.72.8.6758-6769.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taremi SS, et al. Construction, expression, and characterization of a novel fully activated recombinant single-chain hepatitis C virus protease. Protein Sci. 1998;7:2143–2149. doi: 10.1002/pro.5560071011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hong Z, et al. Enzymatic characterization of hepatitis C virus NS3/4A complexes expressed in mammalian cells by using the herpes simplex virus amplicon system. J. Virol. 1996;70:4261–4268. doi: 10.1128/jvi.70.7.4261-4268.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tackett AJ, Chen Y, Cameron CE, Raney KD. Multiple full-length NS3 molecules are required for optimal unwinding of oligonucleotide DNA in vitro. J. Biol. Chem. 2005;280:10797–10806. doi: 10.1074/jbc.M407971200. [DOI] [PubMed] [Google Scholar]
- 60.Frick DN, Rypma RS, Lam AM, Gu B. The nonstructural protein 3 protease/helicase requires an intact protease domain to unwind duplex RNA efficiently. J. Biol. Chem. 2004;279:1269–1280. doi: 10.1074/jbc.M310630200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuang WF, et al. Hepatitis C virus NS3 RNA helicase activity is modulated by the two domains of NS3 and NS4A. Biochem. Biophys. Res. Commun. 2004;317:211–217. doi: 10.1016/j.bbrc.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 62.Morgenstern KA, et al. Polynucleotide modulation of the protease, nucleoside triphosphatase, and helicase activities of a hepatitis C virus NS3–NS4A complex isolated from transfected COS cells. J. Virol. 1997;71:3767–3775. doi: 10.1128/jvi.71.5.3767-3775.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gallinari P, et al. Modulation of hepatitis C virus NS3 protease and helicase activities through the interaction with NS4A. Biochemistry. 1999;38:5620–5632. doi: 10.1021/bi982892+. [DOI] [PubMed] [Google Scholar]
- 64.Porter DJ. A kinetic analysis of the oligonucleotide-modulated ATPase activity of the helicase domain of the NS3 protein from hepatitis C virus. The first cycle of interaction of ATP with the enzyme is unique. J. Biol. Chem. 1998;273:14247–14253. doi: 10.1074/jbc.273.23.14247. [DOI] [PubMed] [Google Scholar]
- 65.Porter DJ. Inhibition of the hepatitis C virus helicase-associated ATPase activity by the combination of ADP, NaF, MgCl2, and poly(rU). Two ADP binding sites on the enzyme-nucleic acid complex. J. Biol. Chem. 1998;273:7390–7396. doi: 10.1074/jbc.273.13.7390. [DOI] [PubMed] [Google Scholar]
- 66.Porter DJ, et al. Product release is the major contributor to kcat for the hepatitis C virus helicase-catalyzed strand separation of short duplex DNA. J. Biol. Chem. 1998;273:18906–18914. doi: 10.1074/jbc.273.30.18906. [DOI] [PubMed] [Google Scholar]
- 67.Levin MK, Gurjar MM, Patel SS. ATP binding modulates the nucleic acid affinity of hepatitis C virus helicase. J. Biol. Chem. 2003;278:23311–23316. doi: 10.1074/jbc.M301283200. [DOI] [PubMed] [Google Scholar]
- 68.Serebrov V, Pyle AM. Periodic cycles of RNA unwinding and pausing by hepatitis C virus NS3 helicase. Nature. 2004;430:476–480. doi: 10.1038/nature02704. [DOI] [PubMed] [Google Scholar]
- 69.Borowski P, et al. ATP-binding domain of NTPase/helicase as a target for hepatitis C antiviral therapy. Acta Biochim. Pol. 2000;47:173–180. [PubMed] [Google Scholar]
- 70.Borowski P, Schalinski S, Schmitz H. Nucleotide triphosphatase/helicase of hepatitis C virus as a target for antiviral therapy. Antiviral Res. 2002;55:397–412. doi: 10.1016/s0166-3542(02)00096-7. [DOI] [PubMed] [Google Scholar]
- 71.Yao N, Weber PC. Helicase, a target for novel inhibitors of hepatitis C virus. Antiviral Ther. 1998;3:93–97. [PubMed] [Google Scholar]
- 72.Lin C, Kim JL. Structure-based mutagenesis study of hepatitis C virus NS3 helicase. J. Virol. 1999;73:8798–8807. doi: 10.1128/jvi.73.10.8798-8807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frick DN, Rypma RS, Lam AM, Frenz CM. Electrostatic analysis of the hepatitis C virus NS3 helicase reveals both active and allosteric site locations. Nucleic Acids Res. 2004;32:5519–5528. doi: 10.1093/nar/gkh891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lam AM, Keeney D, Frick DN. Two novel conserved motifs in the hepatitis C virus NS3 protein critical for helicase action. J. Biol. Chem. 2003;278:44514–44524. doi: 10.1074/jbc.M306444200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prabhu R, et al. Inhibition of hepatitis C virus nonstructural protein, helicase activity, and viral replication by a recombinant human antibody clone. Am. J. Pathol. 2004;165:1163–1173. doi: 10.1016/S0002-9440(10)63377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Artsaenko O, Tessmann K, Sack M, Haussinger D, Heintges T. Abrogation of hepatitis C virus NS3 helicase enzymatic activity by recombinant human antibodies. J. Gen. Virol. 2003;84:2323–2332. doi: 10.1099/vir.0.19299-0. [DOI] [PubMed] [Google Scholar]
- 77.Hwang B, et al. Isolation of specific and high-affinity RNA aptamers against NS3 helicase domain of hepatitis C virus. RNA. 2004;10:1277–1290. doi: 10.1261/rna.7100904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nishikawa F, Funaji K, Fukuda K, Nishikawa S. In vitro selection of RNA aptamers against the HCV NS3 helicase domain. Oligonucleotides. 2004;14:114–129. doi: 10.1089/1545457041526335. [DOI] [PubMed] [Google Scholar]
- 79.Kwong AD, Risano C. Methods in Molecular Medicine. 1998. pp. 97–116. [DOI] [PubMed] [Google Scholar]
- 80.Kyono K, Miyashiro M, Taguchi I. Expression and purification of a hepatitis C virus NS3/4A complex, and characterization of its helicase activity with the scintillation proximity assay system. J Biochem (Tokyo) 2004;135:245–252. doi: 10.1093/jb/mvh029. [DOI] [PubMed] [Google Scholar]
- 81.Boguszewska-Chachulska AM, et al. Direct fluorometric measurement of hepatitis C virus helicase activity. FEBS Lett. 2004;567:253–258. doi: 10.1016/j.febslet.2004.04.072. [DOI] [PubMed] [Google Scholar]
- 82.Lamarre D, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- 83.Hinrichsen H, et al. Short-term antiviral efficacy of BILN 2061, a hepatitis C virus serine protease inhibitor, in hepatitis C genotype 1 patients. Gastroenterology. 2004;127:1347–1355. doi: 10.1053/j.gastro.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 84.Reesink H, et al. Digestive Disease Week. 2005. Initial results of a Phase 1b multiple dose study of VX-950, a hepatitis C virus protease inhibitor. [Google Scholar]
- 85.Godofsky E, et al. Digestive Disease Week. 2004. Phase I/II dose escalation trial assessing tolerance, pharmacokinetics, and antiviral activity of NM283, a novel antiviral treatment for hepatitis C. [Google Scholar]
- 86.Bretner M, et al. Synthesis and evaluation of ATP-binding site directed potential inhibitors of nucleoside triphosphatases/helicases and polymerases of hepatitis C and other selected Flaviviridae viruses. Antivir. Chem. Chemother. 2004;15:35–42. doi: 10.1177/095632020401500104. [DOI] [PubMed] [Google Scholar]
- 87.Locatelli GA, Spadari S, Maga G. Hepatitis C virus NS3 ATPase/helicase: an ATP switch regulates the cooperativity among the different substrate binding sites. Biochemistry. 2002;41:10332–10342. doi: 10.1021/bi026082g. [DOI] [PubMed] [Google Scholar]
- 88.Liu D, Windsor WT, Wyss DF. Double-stranded DNA-induced localized unfolding of HCV NS3 helicase subdomain 2. Protein Sci. 2003;12:2757–2767. doi: 10.1110/ps.03280803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Menard J, Patchett AA. Angiotensin-converting enzyme inhibitors. Adv. Protein Chem. 2001;56:13–75. doi: 10.1016/s0065-3233(01)56002-7. [DOI] [PubMed] [Google Scholar]
- 90.Zhang N, et al. Potent inhibition of NTPase/helicase of the West Nile Virus by ring-expanded (“fat”) nucleoside analogues. J. Med. Chem. 2003;46:4776–4789. doi: 10.1021/jm030277k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.