

Key Points
Our body contains many different protease and proteolytic systems that are involved in the recycling of proteins into amino acids, and also in a multitude of regulatory events inside and outside cells. Proteases are prominent drug targets because of their well-defined chemistry and their implication in a large number of diseases, such as cancer, neurodegeneration, arteriosclerosis, inflammation and infection.
Fluorescent reporter substrates can be used to directly probe the activities of proteases in their natural environment — that is, in cells and organisms. Conceptually different strategies have been used for this purpose depending on the location and the nature of the protease of interest.
Fluorescent reporters for the ubiquitin–proteasome system have been generated by linking constitutively active degradation signals to green fluorescent protein (GFP). These GFP-based substrates can be used for functional analysis of the ubiquitin–proteasome system in cells and transgenic animals.
A collection of different proteases is involved in degradation of small peptide fragments. This process can be followed in real time in living cells by confocal laser scanning microscopy after microinjection of internally quenched peptide substrates.
Extracellular and lysosomal proteases have the advantage that they are accessible for membrane-impermeable reporter substrates. Near-infrared fluorescence (NIRF) substrates are quenched fluorescent peptides that, because of their near-infrared excitation, can be readily detected in living animals and used for in vivo monitoring of, for example, lysosomal cathepsins or surface matrix metalloproteinases.
By combining specific pairs of fluorescent proteins (GFP and its variants with shifted excitation and emission spectra) in fusion proteins, fluorescence energy transfer (FRET) reporter substrates have been generated for initiator and effector caspases.
A fluorescent intracellular reporter for human immunodeficiency virus (HIV-1) protease activity was constructed by fusing a protease precursor protein composed of HIV-1 protease and GFP. Cells will only survive and emit fluorescence when the toxic protease activity is sufficiently blocked by drugs.
The diffusion rate of the endoplasmic reticulum-resident peptide transporter complex TAP correlates with activity and thus cytosolic peptide levels. By measuring the diffusion of TAP–GFP fusions with fluorescence recovery after photobleaching (FRAP), the kinetics of peptide generation can be followed in living cells.
Abstract
Cells contain numerous proteases, which are found at many different locations. These proteases recognize an even larger number of different substrates and are involved in almost every process in the cell. Aberrations in proteolysis are linked to a plethora of diseases, such as cancer, inflammation, arteriosclerosis, neurodegeneration and infection. Because of their well-defined chemistry and key role in pathologies, proteases have been important targets for drug development. Recent progress in the development of fluorescent probes has opened up the possibility of visualizing protease activities in the natural environment of the cell. We will describe various strategies to follow protease activities in cells and organisms.
Main
Proteolysis is a process crucial to the normal turnover of proteins in the cell. Proteases are not only involved in protein destruction, but also in activation, growth, cell division, differentiation, migration, signalling and other cellular processes. In addition, many viruses use cellular or viral proteases for various purposes, such as activation of their surface proteins or membrane fusion. Consequently, specific protease inhibitors are attractive drug candidates (Table 1). Although the different specificities of proteases have been used during recent years to assess their activities in vitro, their actions have been difficult to determine under more-or-less physiological conditions.
Table 1.
A few inhibitors and (potential) applications

Proteases can be divided into a number of classes: the serine, aspartic, cysteine, threonine and metalloproteases. This classification scheme basically refers to the amino acid or metal that catalyses the nucleophilic attack on the substrate's peptide bonds. Enzymes with proteolytic activities have frequently evolved to carry out important tasks, which has resulted in large and divergent families of proteases. These often unrelated enzymes differ in many respects, but one characteristic unites them: they hydrolyse peptide bonds and thereby cut proteins into small pieces or a few distinct fragments. It is precisely this chemical reaction that allows the design of probes to selectively monitor protease activities.
One generally recognized function of proteases is the recycling of proteins into amino acids. Proteins can be singled out for a number of reasons, including signs of malfunctioning or misfolding1. Although the vast majority of proteins face destruction when old and expired, a substantial fraction of newly synthesized proteins undergoes a similar fate2,3, possibly because they fail to adopt a proper conformation4. Because misfolded proteins tend to aggregate, the rapid recognition and degradation of such aberrant proteins is crucial for the survival of the cell5,6. Moreover, in many cases the destruction of proteins is crucial for the correct control of cellular regulatory switches. In this case, their degradation is not a result of malfunctioning, and is in fact determined by specific spatially and temporally controlled signalling events7.
The route that leads from a full-length intracellular protein to its constituent amino acids is a multistep process (Fig. 1). First, the processive degradation of proteins by the PROTEASOME generates small peptide fragments8. This initial cleavage stage is generally sufficient to abolish protein function and activity. Second, a collection of proteases — among which are tripeptidyl peptidase-II (TPP-II)9, leucine aminopeptidase (LAP)10 and thimet oligopeptidase (TOP)11 — trim these small peptide fragments step-wise into single amino acids that can be reused in the synthesis of new proteins. Endocytosed proteins do not usually enter the cytoplasm but are instead directed to lysosomal compartments (Fig. 1). In the lysosomes, another group of proteases, the cathepsins11, degrades the internalized protein into pieces. These fragments are subsequently reduced to single amino acids by lysosomal aminopeptidases like TPP-I and dipeptidyl peptidase-II (DPP-II)9. Given the broad substrate specificity of the proteasome and cathepsins, these proteases have to be compartmentalized to prevent the random degradation of 'innocent bystanders'. This is achieved by placing the active sites of the proteases within large multisubunit complexes, in the case of the proteasome, or within lysosomes, in the case of cathepsins. In addition to these broadly acting proteases, there are other proteases involved in many more specific processes, such as blood clotting12, apoptosis13, extracellular matrix degradation14, cleavage of transmembrane proteins15 and precursor processing in the secretory pathway16.
Figure 1. Recycling of intracellular and internalized proteins.

Proteins present in the cytosol and nucleus are degraded by the proteasome into peptide fragments that are further degraded by TPP-II, TOP, LAP and other peptidases into single amino acids. Internalized proteins are transported to the lysosomes, where cathepsins, TPP-I, DPP-II and other proteases digest the proteins to single amino acids. Not shown in this scheme are the more dedicated proteases, as discussed in the text. DPP, dipeptidyl peptidase; LAP, leucine aminopeptidase; TOP, thimet oligopeptidase; TPP, tripeptidyl peptidase.
Clearly, proteases not only destroy proteins but also convert proteins into active moieties. In addition, proteolytic cascades regulate important cellular switches. It is therefore not surprising that proteases get a lot of scientific attention. A recent literature search revealed that roughly 240,000 papers were published about proteases during the past 20 years. And this is yet another one, but one describing a timely issue: how can protease activity be monitored in vivo? Given the chemistry of proteases, various fluorescent substrates have been designed during the past few years to test their activities as well as the effects of drug-mediated inhibition of these systems in cells or animals. Understanding the physiology of proteolytic processes inside the living organism is of crucial importance in assessing the roles of proteases in diseases and for developing drugs that target these systems.
Fluorescent protein substrates
The ubiquitin–proteasome system is important in regulating the cell cycle and the induction of apoptosis by the degradation of key regulatory molecules7. In addition, this proteolytic system is responsible for keeping the cell free of misfolded proteins17. These important functions explain why the ubiquitin–proteasome system is implicated in a large number of diverse pathologies5,17,18. For example, aberrant proteasomal degradation contributes to malignant transformation19,20,21. The immune system relies on the ubiquitin–proteasome system for the generation of antigenic peptides that are presented by MAJOR HISTOCOMPATIBILITY COMPLEX (MHC) CLASS I molecules22,23. The proteasome also has a crucial role in the inflammatory response, as it degrades inhibitors of nuclear factor-κB24. Given these functions, it is not surprising that proteasome inhibitors have anticancer25, anti-inflammatory26, angiostatic27 and immuno-modulating activity28. Recently, the first proteasome inhibitor, bortezomib (Velcade; Millennium) — a boronic acid inhibitor (Table 1 and Fig. 2) — was approved29 after successful clinical trials in patients suffering from multiple myeloma29,30.
Figure 2. Inhibitors of the proteasome.

Structure of proteasome inhibitors. a | Peptide aldehyde. b | Peptide boronic acid. c | Peptide vinyl sulphone. d | Epoxomicin. e | β-lactone. Note that a–c are carboxy-modified peptides.
Notably, two drugs that affect proteasomal degradation have been used in patients for some time without being recognized as such. These drugs are the human immunodeficiency virus (HIV)-1 protease inhibitors ritonavir (Norvir; Abbot Laboratories) and saquinavir (Fortovase; Roche), and have recently been shown to affect proteasome activity as well31. It has been suggested that inhibition of the proteasome by these compounds could account for increased hyperlipidaemia32 and the reduced incidence of Kaposi's sarcoma33 in patients treated with these drugs. Moreover, HIV-1 inhibitors can radiosensitize tumour cells for ionizing radiation by blocking the proteasome34. Specific proteasome inhibitors, such as peptide aldehydes1, boronic acids27, vinyl sulfones35, epoxyketones26 or β-lactones36, have been used for many years in the laboratory (Fig. 2). So whereas drugs that target the proteasome are relatively new and unexplored, proteasome inhibitors and modulators hold great promise as potential drugs for multiple disorders37.
During the past few years several groups have developed fluorescent probes for the ubiquitin–proteasome system that behave as authentic protein substrates (Fig. 3a). Only proteins, which are specifically recruited to the proteasome, and subsequently unfolded and threaded into the core, can be degraded by this complex. The proteasome generally recognizes substrates by the presence of a poly-ubiquitin chain conjugated to an internal lysine residue of the substrate38 or, less commonly, conjugated to the free amino terminus of the substrate39,40. Proteins are modified by the addition of these poly-ubiquitin chains by specific ubiquitylation enzymes41 that physically interact with degradation signals (such as misfolded domains, small motifs or post-translational modifications) that are detected in the substrate42. A crucial observation regarding protein processing was that the degradation signals that target proteins for ubiquitylation and proteasomal degradation behave as MODULAR DOMAINS42. These domains can often be functionally transferred to other proteins. With the identification of fluorescent proteins in jellyfish43 and soft corals128, the possibility emerged of using fluorescent protein substrates resembling authentic protein substrates of the proteasome instead of the small fluorescent peptides commonly used in in vitro assays.
Figure 3. Fluorescent probes for proteolysis.

a | Destruction. Coupling of a degradation signal (DEG) to GFP will result in targeting of the GFP for ubiquitylation followed by proteasomal degradation. Degradation of the GFP substrate results in loss of fluorescence. b | Fluorescence resonance energy transfer (FRET) fluorophore peptide substrate. A FRET peptide substrate contains a donor and acceptor fluorophore with spectral overlap. When the FRET probe is intact and excited with the optimal wavelength for the donor, acceptor fluorescence will be emitted at the expense of donor fluorescence. Cleavage of the FRET probe results in separation of donor and acceptor fluorophores and increased emission from the donor with a concomitant loss of acceptor fluorescence. c | Quenching with fluorophore/quencher pair in peptide substrate. Small peptide fragments containing a fluorophore and a corresponding quencher will be essentially non-fluorescent. After cleavage, when the quencher is spatially separated from the fluorophore, the fluorophore will emit fluorescence. d | Quenching with identical fluorophores in peptide substrate. Two identical fluorophores will quench each other. Principle as in b. e | FRET fluorescent proteins. With CFP and YFP as donor and acceptor fluorophore, respectively. Principle as in b. CFP, cyan fluorescent protein; GFP, green fluorescent protein; YFP, yellow fluorescent protein.
In the first set of fluorescent reporters generated for the ubiquitin–proteasome system, the N-end rule and ubiquitin fusion degradation (UFD) signals were fused to the green fluorescent protein (GFP)44. These degradation signals were originally identified by Varshavsky and co-workers45,46. According to the N-end rule, the identity of the N-terminal amino-acid residue can be used to time the degradation of proteins by ubiquitin–proteasome-dependent proteolysis47 (Fig. 4a). In the case of the UFD substrate, an N-terminal ubiquitin moiety fused to the GFP open reading frame functions as the acceptor for poly-ubiquitin chains (Fig. 4b). Stack and co-workers showed that insertion of multiple UFD signals could further accelerate protein turnover and facilitate the degradation of alternative reporters, such as β-lactamase or luciferase, which remained stable on insertion of a single UFD signal48.
Figure 4. GFP-based proteasome substrates.
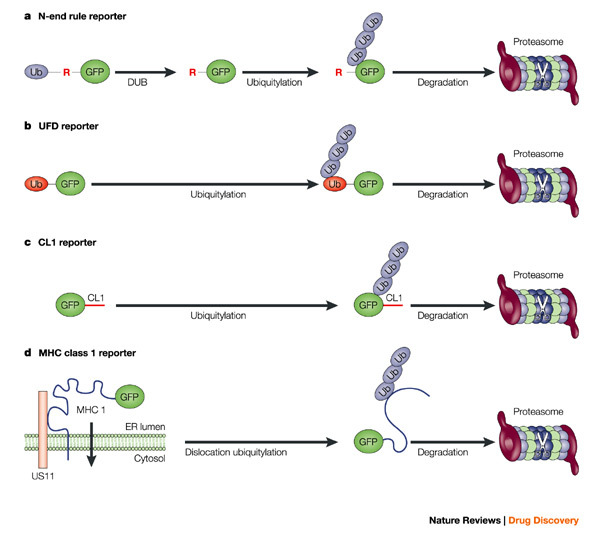
a | N-end rule GFP. A fusion of ubiquitin and GFP is expressed. The endogenous de-ubiquitylation enzymes (DUB) rapidly remove the N-terminal ubiquitin, which generates a GFP with an N-terminal arginine residue. This N-terminal Arg residue is recognized by ubiquitylation enzymes resulting in ubiquitylation close to the N terminus followed by proteasomal degradation. b | If the last amino acid of the ubiquitin moiety in the ubiquitin–GFP fusion is changed, the DUBs fail to remove the N-terminal ubiquitin. This results in ubiquitylation of the ubiquitin moiety and proteasomal degradation. c | Insertion of the small amino acid stretch CL1 directly targets GFP for ubiquitylation and proteasomal degradation. The degradation signals in a–c are marked in red. d | Co-expression of the viral US11 and GFP–MHC class I fusion results in rapid dislocation of GFP–MHC class I from the endoplasmic reticulum (ER) to the cytosol followed by proteasomal degradation. GFP, green fluorescent protein; MHC, major histocompatibility complex.
Another reporter substrate was generated with the help of the CL1 degradation signal49. The introduction of this small 16-amino-acid motif converted GFP into a short-lived proteasome substrate50 (Fig. 4c). It is noteworthy that the CL1 motif was originally identified in a random yeast screen for proteins that are targets for the ubiquitylation enzymes engaged in endoplasmic reticulum (ER)-ASSOCIATED DEGRADATION (ERAD)49,51. This ERAD pathway is used by aberrant ER proteins for release from the ER to access the cytosol proteasome for degradation52.
MHC class I molecules have been converted into a genuine ER-derived substrate of the ubiquitin–proteasome system. Several viruses frustrate antigen presentation by targeting MHC class I molecules53. For example, the human cytomegalovirus (HCMV) protein US11 binds MHC class I molecules in the ER and dislocates them to the cytosol where they are rapidly degraded by the ubiquitin–proteasome system54,55. Co-expressing the HCMV protein US11 and N-terminally GFP-tagged MHC class I in human cells resulted in a functional reporter system for proteasomal degradation of an ER protein56 (Fig. 4d).
The administration of proteasome inhibitors to reporter-expressing cells stabilized the GFP reporters and resulted in a 100–1,000-fold increase in fluorescent intensity44,50,56. The fact that these fluorescent proteins follow the same pathway as endogenous substrates allows functional analysis of the ubiquitin–proteasome system in cells. Indeed, accumulation of the UFD substrate in response to administration of proteasome inhibitors was shown to correlate with cell-cycle arrest and apoptosis44,57, which is a consequence of general blockage of proteasomal degradation58. Parallel evaluation of the individual proteolytic sites of the proteasome (with fluorigenic peptide substrates) and the functional status of the ubiquitin–proteasome system (with the GFP reporters) revealed that cells contain a large excess of proteasomal activity44,50, which is probably required to instantly clear misfolded proteins under stress conditions59. The UFD–GFP reporter was also used to study the individual contributions of the proteolytic active sites of the proteasome to protein degradation57.
The UFD and N-end rule GFP substrates can be used to analyse the effect of protein domains on proteasomal turnover after simple insertion of the domain in the reporter substrate. This approach has been used for detailed studies on a viral repetitive sequence60 and the expanded polyglutamine repeats61 that are the causative factor in a number of neurodegenerative disorders62. These two repetitive sequences had previously been shown to affect proteasomal degradation63,64,65. At present, these reporter substrates are further exploited for the identification of so-called stabilization signals66; that is, signals that enable proteins to resist proteasomal degradation (S. Heessen, M. G. Masucci and N. P. D., unpublished results).
Another convenient aspect of using protein substrate reporters, as opposed to peptide substrates, is that they can be readily applied to the generation of transgenic animals. It is notable that the expression of GFP is well tolerated in mice67,68, although some side effects have been reported in specific models69. Recently, the first transgenic mouse model of the ubiquitin–proteasome system was generated through ubiquitous expression of the UFD–GFP reporter70. As anticipated, the tissues of these mice did not emit GFP fluorescence despite the presence of reporter-encoding transcripts in all tissues analysed. The injection of proteasome inhibitors into these mice resulted in a dramatic increase in fluorescence intensities of affected tissues. In another animal model, a UFD bioluminescence reporter-expressing human cell line was implanted in nude mice. The human cells responded with a dose-dependent increase in bioluminescence after administration of the proteasome inhibitor bortezomib to the mice71.
In particular the transgenic mouse model opens a new dimension for the study of the ubiquitin–proteasome system in pathologies because of its ubiquitous expression72. It has long been suspected that impairment of the ubiquitin–proteasome system contributes to many neurodegenerative diseases and other CONFORMATIONAL DISORDERS5,17. Although it has been confirmed in cellular systems that some disease-associated proteins can indeed affect the ubiquitin–proteasome system50,73,74,75, these experimental approaches by no means reflect the complex pathology of such neurodegenerative diseases. This reporter mouse model can now be used, in conjunction with the excellent mouse models available for the study of these disorders76,77, to assess the functionality of the ubiquitin–proteasome system during disease progression. It should be emphasized, however, that impairment of the ubiquitin–proteasome system is just one of the many models put forward as an explanation for the pathology observed in conformational disorders78. Yet, if this model holds true, crosses between the GFP-reporter mice and disease model mice can also be used to evaluate the effect of drugs that counteract the effects of the impaired ubiquitin–proteasome system in these diseases.
Internally quenched substrates
Peptide degradation can be monitored using chemical FLUOROPHORES. A strategy for detecting peptidase activity in living cells uses peptides containing a fluorescence resonance energy transfer (FRET) fluorophore pair (Fig. 3b). FRET is only operational with 10–100 Å and not detectable when fluorophores are more than 100 Å apart. Therefore, spatial separation of two fluorophores as the result of protease activity can be easily assayed because this will result in increased emission from the donor fluorophore at the expense of emission from the acceptor fluorophore79 (Box 1). This strategy has been used with peptides containing a fluorescein (Fl) group and a tetramethylrhodamine (TMR) group separated by six amino acids. Energy transfer from Fl to TMR occurred with intact peptides but was lost when the peptides were separated as the result of degradation80. More dramatic differences were observed when instead of TMR, a QUENCHER molecule was used80. The quencher in these peptides very efficiently absorbs the emitted light from the Fl group, unless the two groups are separated (Fig. 3c).
Using more-or-less standard peptide chemistry, substrates can be synthesized for defined questions relating to peptide degradation in living cells. These peptides do not diffuse across membranes and therefore have to be introduced into living cells by microinjections. Detailed analysis of real-time degradation of such peptides in living cells revealed that peptides have a very short half-life of several seconds, solely as a result of aminopeptidase activity80. Some of these peptidases are crucial for trimming peptides to the correct size for MHC class I antigen presentation22. However, the individual contribution of peptidases to the total intracellular peptidase activities in a cell is unclear. Studies with specific inhibitors showed that the proteasome does not contribute to the digestion of small peptides in living cells even though in vitro peptide degradation can be readily measured and has been used as a standard method for quantifying proteasome activity80. A crucial role in peptide degradation was found for TPP-II. Although most aminopeptidases remove N-terminal sequences (of usually one to three amino acids) from their peptide substrate, TPP-II is able to remove longer fragments, including fragments for MHC class I (E. Reits and J. N., unpublished results). Along similar lines, RNA interference (RNAi) of defined peptidases might reveal the contribution of a specific peptidase to the total cellular peptidase activity.
Near-infrared fluorescence (NIRF) probes
Although small quenched peptide substrates have proven to be valuable tools for studying proteases in purified samples in vitro or in cell lines after microinjection, the fact that they cannot cross membranes limits their applications for monitoring intracellular proteolysis in vivo. However, for those proteases active at the cell surface or in the endosomal/lysosomal compartment, this is not a major limitation.
The cysteine, serine and aspartic proteases present in lysosomes have been implicated in many cellular processes. The lysosomal cathepsins are essential to the turnover of proteins that reach the lysosomal compartment from the extracellular space (by endocytosis), the secretory pathway (by sorting) or the cytosol (by autophagy) and have more subtle regulatory functions in MHC class II antigen presentation81. Lysosomal proteases are believed to be involved in diseases such as cancer, inflammation, autoimmune disorders, atherosclerosis and several genetic disorders82. It has been shown that these proteases can participate in the destruction of the extracellular matrix surrounding tumours, which is an important prerequisite for tumour invasion, metastasis and angiogenesis83. However, because most cathepsins have important roles in lysosomal protein turnover, caution should be taken in inhibiting these enzymes, as impairment of cathepsins can result in lysosomal storage diseases. More specialized lysosomal proteases, such as cathepsin S, which is expressed in the immune system and assists in MHC class II antigen presentation, are also important drug targets. In fact, selective inhibitors of cathepsin S have been developed84,85, and have been shown to prevent autoimmune responses in a mouse model of Sjögren syndrome85 (Table 1).
Another important class of drug targets that mediate the breakdown of the extracellular matrix is the extracellular matrix metalloproteases (MMPs)14,83, a large family of metalloproteases present at the cell surface and in the extracellular space. MMPs have been recognized as drug targets for cancer treatment because metastatic cells have to degrade the extracellular matrix for angiogenesis and invasion of tissue86. Several MMP inhibitors, such as BB-2516 (Marimastat; British Biotech) and BAY12-g566 (Tanomastat; Bayer) (Table 1), have been developed and tested in clinical trials. Although the clinical trials have not been without success, they did not live up to the high expectations for these compounds87. Still, there is ample reason to believe that more specific drugs or early interference in tumour progression might improve their efficacy87.
Weissleder and co-workers developed a near-infrared fluorescent (NIRF) peptide substrate that can be used for the in vivo monitoring of cathepsin activity88. They used a synthetic non-toxic and non-immunogenic co-polymer that had previously been tested in clinical trials89 and that was composed of a poly-lysine backbone, which was stabilized through the conjugation of multiple polyethylene glycol side chains. Fluorophores were coupled at a high density to these backbones80. Identical fluorophores will interfere with each other and increase their relaxation times when excited, similar to normal FRET pairs (Fig. 3d). Although the mechanism is not fully understood, this results in quenching unless the fluorophores are diluted by degradation. Indeed it was found that the emission signal was efficiently quenched in the intact co-polymer, rendering it almost non-fluorescent, whereas in vitro cleavage of the probe considerably increased emitted light88. To measure the fluorescence in living animals a dye was used that is excited with near-infrared light, which because of its long wavelength will hardly be scattered or absorbed by the tissue. This NIRF probe was expected to function as a general molecular probe for the lysosomal proteases that are able to cleave the poly-lysine backbone. Activation of the probe could be followed in cells, and inhibitor experiments confirmed that lysosomal cysteine and serine proteases were primarily responsible for activation of the NIRF reporter in cell lines88. Using xenotransplantation of human tumours in nude mice, it was shown that the NIRF probe was predominantly taken up and activated in the tumour tissues, possibly as a consequence of their relatively permeable neovasculature90,91. With the non-invasive near-infrared reflecting imaging technique, small tumours containing cleaved NIRF probes could be identified at a maximal tissue depth of 1 cm90.
The same NIRF reporter was also used to study the role of cathepsin B in atherosclerosis when administrated intravenously92. Cathepsin B activity was found to be upregulated in the lesions in a mouse model for atherosclerosis. NIRF tomographic imaging allowed three-dimensional imaging of the probes in deeper tissue and could localize lesions in the aortic arch in mice solely by the presence of the activated NIRF reporter.
Because the lysine composition of the co-polymer is important for the in vivo stability of this probe, the specificity cannot be changed by modifying the amino-acid sequence of the co-polymer itself. Instead, a strategy was used in which the fluorophores were connected to the copolymer backbone on peptide stalks conjugated by an isopeptide linkage to the lysine backbone. By changing the amino-acid sequence of the peptide stalk into protease cleavage sites, the probe could be converted into a substrate for specific proteases without losing in vivo stability. With this approach, specific NIRF reporters were generated for cathepsin D93 and MMP-2 (Refs 94,95). In vivo imaging of tumour-bearing mice after probe administration could be used to discriminate between tumours expressing low or high levels of the selected proteases90,93,94. More recently, similar probes were used for imaging lymph nodes in living mice96,97. Not only can these reporters be helpful for the detailed evaluation of the effect of potential drugs on lysosomal and extracellular proteases in vivo, they might also aid early diagnosis of tumours or atherosclerotic lesions, at least when imaging techniques will allow visualization of these probes deep in tissue.
In reality, the substrates of cathepsins or MMPs are not peptides but full-length proteins. It has been shown that protein substrates labelled with high-density fluorophores can also be used to study the activity of lysosomal proteases in cell lines98. Although it seems unlikely that this type of protein can be used in animals, important information about the combined action of tumour cells towards extracellular proteins can be deduced in cell culture systems.
FRET protein reporters
Since the introduction of recombinant DNA technology, many reporter substrates for proteases have been generated by the introduction of protease cleavage sites in proteins with easily detectable enzyme activities99,100. Proteolytic cleavage of such designed substrates inactivates their enzymatic activity, which forms a reliable and quantitative readout for protease activity. It seems tempting to apply the same approach to GFP or other fluorescent proteins, but unfortunately the presently available fluorescent proteins are not very well suited for this purpose. Although the presently available fluorescent proteins allow the generation of all kinds of N- and C-terminal fusion proteins, their tightly interwoven structure does not allow major modifications within the GFP open reading frame, as this generally results in a complete loss of fluorescence101.
Instead, researchers turned towards the FRET technique described above, which only became possible with the development of spectral GFP variants (Fig. 3e). By positioning a protease cleavage site between two fluorescent protein variants with overlapping emission (from the donor) and excitation spectra (from the acceptor), a FRET-based probe for proteases can be generated. The use of GFP variants instead of dyes enabled the construction of protease substrates that do not require any in vitro modification and which can therefore be directly expressed in target cells or organisms. Several FRET-based probes have been developed for caspases, a family of related cysteine proteases that are involved in the execution of apoptosis13. These enzymes are activated in a proteolytic cascade reaction that is tightly controlled. Caspases can be divided into two groups: initiator caspases, which set off the cascade by activating other caspases; and effector caspases, which cleave multiple cellular substrates and destroy the cellular protein content. Apoptosis is a key factor in many disease processes, including neuronal loss by excessive apoptosis in neurodegenerative disorders102 and escape from apoptosis in cancer103. Because caspases are the inducers as well as the executors of cell death, their potential as drug targets is evident104 (Table 1).
Xu and co-workers generated the first functional caspase FRET probe on the basis of fluorescent proteins by inserting a small peptide target sequence for the effector caspase-3 between GFP and the blue fluorescent protein (BFP)105 (Fig. 5a). Induction of apoptosis in cells expressing this fusion protein resulted in a loss of the FRET signal between the BFP donor and GFP acceptor as a consequence of caspase-3 activation. A major drawback of the BFP–GFP FRET pair is that the BFP donor is excited by ultraviolet light, which causes considerable cellular damage and is therefore far from optimal for studies with living cells. Several new spectral variants, based on modified GFP or fluorescent proteins from other sources, have been generated during the past few years106. Ultraviolet excitation is avoided in the more recent caspase-3 FRET probe, in which the cyan fluorescent protein (CFP) and the yellow fluorescent protein (YFP) are combined107 (Fig. 5b). By changing the connecting peptide sequence between the CFP–YFP combination, the FRET probe was converted to a probe specific for the initiator caspase-8 (Ref. 108). In this study, another advantage of the modular composition of the FRET probes was exploited. Instead of introducing a small cleavage site, larger protein fragments can be inserted because the probe is basically composed of three independent modular domains. In this reporter, the caspase-8 target, BH3-interacting domain death agonist (BID), was used to generate a functional YFP–BID–CFP reporter (Fig. 5c)108.
Figure 5. FRET/FLIM reporters for caspases.
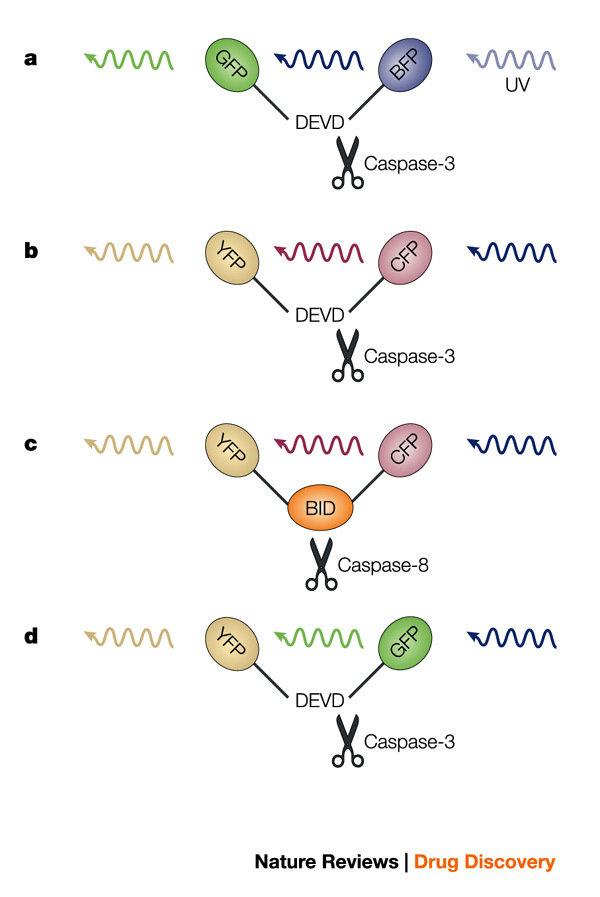
a | Caspase-3 fluorescence resonance energy transfer (FRET) probe. The DEVD caspase cleavage site is positioned between GFP and BFP. b | Caspase-3 FRET probe. The DEVD caspase cleavage site is positioned between YFP and CFP. c | Caspase-8 FRET probe. The caspase-8 target BID is positioned between YFP and CFP. d | Caspase-3 fluorescence lifetime imaging microscopy (FLIM) probe. The DEVD caspase cleavage site is positioned between the spectrally similar YFP and GFP. BFP, blue fluorescent protein; BID, BH3-interacting domain death agonist; GFP, green fluorescent protein; YFP, yellow fluorescent protein.
Unfortunately, all FRET probes containing the CFP or BFP variants have limited sensitivities due to the relatively poor QUANTUM YIELD of these variants. Recently, reporters for caspase-3 have been constructed by combining GFP and YFP, which do not suffer from these limitations109 (Fig. 5d). Optical filters cannot separate the largely overlapping excitation and emission spectra of these proteins and therefore energy transfer is difficult to quantify by sensitized emission FRET. An alternative quantitative method is fluorescence lifetime imaging microscopy (FLIM), which can measure energy transfer between probes with overlapping or even identical spectra109. FLIM measures the effect of two closely located fluorophores on the relaxation time of the donor and acceptor, which is a consequence of FRET. Like FRET, FLIM decays rapidly with distance and is detectable when the fluorophores are within 10–100 Å of one another. Using pulsed light sources, changes in the relaxation time can be determined. Although energy transfer might be more reliably determined by FLIM, the spatiotemporal resolution is considerably less than achieved with FRET due to longer sampling times.
Applications of fluorescent proteins in FRET reporters are not limited to caspases. Although the caspase studies provide a proof of principle, similar probes are feasible for virtually every protease for which a specific target sequence can be positioned between two fluorescent protein variants.
Other assays
The fact that the core of the fluorescent protein cannot generally be modified into probes for proteases limits the design of reporters to modular constructs in which the fluorescent protein is present as a functional domain. Besides the reporters for the ubiquitin–proteasome system, in which the fluorescent protein is provided with a degradation signal leading to processive degradation75, and the FRET106 and FLIM-based109 caspase reporters, a few other assays have been developed that use different concepts.
Lee and co-workers adapted the N-end rule GFP reporter for the ubiquitin–proteasome system44 into a probe for caspase activity110. In these reporters, a general caspase cleavage site was positioned directly after the variable N-terminus of a GFP fusion. Because caspases will cleave off the original N-terminus of this reporter and generate a new one, the fate of the GFP protein can be switched from efficient N-end rule proteasome substrate to an inherent stable GFP protein or vice versa, depending on the amino acids flanking the cleavage site. The stabilization or destabilization of the GFP reporter as a consequence of caspase activity could be quantified in living cells by flow cytometric or fluorimetric analyses110. This reporter requires only a single fluorescent protein and gives as a readout a simple decrease or increase in GFP fluorescence, two characteristics that make it favourable for use in high-throughput drug screens. However, one inherent problem is that compounds that interfere with the ubiquitin–proteasome system will affect the stability of these reporters as well.
Many infectious pathogens are dependent on proteases for their survival. Viruses often require virally encoded proteases for the processing of precursor proteins, which is important at various steps in the viral life cycle, and so viral proteases have been important targets for the development of antiviral drugs111. The HIV-1 genome encodes an aspartic protease that is essential for the generation of infectious virus particles112. The development of drugs that target this protease has been a successful endeavour, resulting in drugs such as ritonavir, saquinavir, indinavir (Crixivan; Merck), nelfinavir (Viracept; Agouron/Pfizer) and lopinavir (Kaletra; Abbot Laboratories) (Table 1). HIV-1 protease inhibitors are now routinely used as part of highly active antiretroviral therapy (HAART), a regime that can efficiently control HIV replication in patients113. Because HAART can successfully suppress but not eradicate the virus in patients, ongoing viral replication often results in the emergence of inhibitor-resistant viral strains114. There is therefore a need for novel HIV-1 protease inhibitors, as well as diagnostic tests to identify and anticipate inhibitor-resistant strains.
HIV-1 protease is generated by an autocatalytic event from a large precursor protein. The presence of HIV-1 protease is toxic for cells, which is not surprising considering that this protease has broad specificity and cleaves many cellular substrates115,116. Two considerations were taken into account when designing a GFP-based reporter for HIV-1 protease. First, the protease must be released from its precursor to become fully active; and second, the presence of fully active protease is toxic for cells. By generating an artificial precursor composed of GFP and the HIV-1 protease, a simple but robust reporter was made based on these two characteristics117. Transient transfection of cells with a plasmid encoding this non-toxic GFP–HIV-1 protease construct typically results in very low GFP levels, as cells that express this precursor will die because of the presence of the processed and fully active protease. The administration of different inhibitors to cells transfected with these reporters resulted in a dose-dependent increase in GFP fluorescence, due to accumulation of the unprocessed, non-toxic GFP–HIV-1 protease precursor. This non-infectious model is now being developed as a diagnostic assay for the analysis of inhibitor resistance of virus strains. Indeed, experiments with characterized mutant HIV-1 proteases from patient material showed that this probe can identify inhibitor-resistant HIV-1 proteases (T. Uhlikova, N. P. D., K. Lindsten, M. G. Masucci and J. Konvalinka, unpublished results). The HIV-1 protease inhibitors exemplify the successful approach of targeting viral proteases, and presently much effort is put into the development of similar compounds inhibiting proteases of other life-threatening viruses, such as the proteases of hepatitis C virus118 and the human corona virus that causes severe acute respiratory syndrome119 (Table 1).
Reits and co-workers used an indirect approach to monitor intracellular protein degradation in living cells. They followed the activity of a peptide transporter that is involved in antigen presentation by MHC class I molecules120. They showed that a GFP-tagged TRANSPORTER ASSOCIATED WITH ANTIGEN PROCESSING (TAP) could be used to indirectly monitor the generation of small peptide fragments by intracellular proteases2. Using fluorescence recovery after photobleaching (FRAP) techniques121 to visualize TAP diffusion in the ER membrane, it was shown that TAP diffuses rapidly when inactive, and slowly when in the process of pumping peptides. These differences in mobility probably reflect differences in shape or radius, as predicted by modifications of the Einstein–Stocks formula, in which diffusion is correlated with ln(1/r), where r is the radius of the protein in the membrane121. Recording of the mobility of TAP in living cells has been used to show that a large portion of peptides are generated from proteins that are immediately degraded after translation, to visualize the contribution of influenza infection to the intracellular peptide pool2 and to visualize the effect of γ-irradiation on intracellular peptide degradation (E. Reits and J. N., unpublished results). The disadvantage of this technique is that it is rather complicated; however, unlike the other reporters, this method visualizes the pool of intracellular peptides rather than the activity of selective proteases.
Conclusions
For understanding the mechanism of proteolysis and the effects of inhibitors in vivo, a variety of fluorescence-based assays have been developed. These assays allow the detection of protease activities in living cells and will help us to elucidate the individual roles of proteases in complex proteolytic systems, such as the living cell. Such assays can be used to monitor the effect of chemical inhibitors of protease activity37,58,87 or selective downregulation of proteases by RNAi122.
An interesting difference between following in vivo degradation of designed substrates, as compared with in vitro assays with purified components, is that the designed substrates, which are analogous to genuine substrates, can be subject to multiple cleavage events. For example, it has been shown that degradation of the GFP-based substrates for the ubiquitin–proteasome system can also be influenced by inhibitors of unrelated serine proteases that are apparently directly or indirectly involved in this proteolytic system123. The FRET caspase reporters and NIRF cathepsin reporters are also subject to cleavage by several caspases88,107. Although this might complicate the interpretation of the cleavage events, it reflects the in vivo situation of substrates and might aid the identification of novel proteases and unanticipated connections between proteolytic systems.
The complexity of proteolysis and the contribution of individual proteases to the total protease activity can only be fully appreciated in living cells or, even better, in vivo. The recently generated GFP-reporter mice allow monitoring of proteasome activities and the effect of proteasome inhibitors in vivo70. It is to be expected that other transgenic mice expressing fluorescent protein-based reporter systems will follow and aid the development of compounds targeting proteases.
Box 1 | Fluorescence resonance energy transfer (FRET).
FRET is the physical phenomenon of energy transfer between a donor and an acceptor fluorophore79. This will be possible only when the donor/acceptor pair are separated by a very short distance (<100 Å). This is usually one-quarter or less of the light wavelength used to excite the donor fluorophore. Because it is acting at such a short distance, the principle of energy transfer is not by emission of a photon that is captured by the acceptor fluorophore, but merely electromagnetic energy transfer as a result of dipole moments. FRET efficiency is highly dependent on distance and decays with distance, as formulated in Forster's law124. This makes FRET highly suitable for detecting conformational changes in a protein (when the distance between donor and acceptor changes) or degradation (when the donor and acceptor pair are separated after the cleavage of substrate).
In addition, the relative orientation of the fluorophores determines FRET. This so-called orientation factor is usually unknown and prevents the determination of real distances on the basis of FRET data.
Acknowledgements
We thank S. Heessen and E. Reits for helpful suggestions. N.P.D. is supported by a fellowship from the Swedish Research Council (VR). N.P.D. acknowledges the Netherlands Organization for Scientific Research (NWO) for a visitor grant.
Glossary
- PROTEASOME
A large barrel-shaped multi-subunit complex, which harbours several protease activities. Substrates are threaded into the barrel, where they are degraded by the proteases.
- MAJOR HISTOCOMPATIBILITY COMPLEX CLASS I
(MHC). A ubiquitously expressed cell-surface glycoprotein that presents a fragment of a protein to the immune system. This fragment, which usually comprises nine amino acids, is generally derived from intracellular proteins and MHC class I molecules, thereby allowing the immune system to assay the intracellular protein content, including pathogens.
- MODULAR DOMAINS
Domains that fold and function as independent elements. The insertion of modular domains in other proteins changes the behaviour of the protein accordingly.
- ER-ASSOCIATED DEGRADATION
(ERAD). Misfolded ER-resident proteins are translocated to the cytosol where they are degraded by the ubiquitin–proteasome system.
- CONFORMATIONAL DISORDERS
Diseases that are characterized by the accumulation of a conformationally abnormal protein that aggregates and forms insoluble protein deposits. Such disorders include Alzheimer's, Parkinson's and Huntington's diseases.
- FLUOROPHORES
Chemical structures that absorb light and emit light at a longer wavelength. A number of proteins, such as GFP, make an unnatural peptide bond, thereby creating a structure that acts as a fluorophore.
- QUENCHER
Chemical structures optimized for absorbing light without emitting detectable light.
- QUANTUM YIELD
The efficiency of fluorescence — basically, how much energy is emitted from the energy absorbed by the fluorophore.
- TRANSPORTER ASSOCIATED WITH ANTIGEN PROCESSING
(TAP). A specialized heterodimeric ATP-binding cassette transporter in the ER for peptide transfer into the ER lumen where peptides can bind to MHC class I molecules. TAP also associates with a dedicated chaperone called tapasin that recruits MHC class I molecules for peptide loading. This complex is called the MHC class I loading complex.
Biographies
Jacques Neefjes is head of the division of Tumor Biology at the Netherlands Cancer Institute in Amsterdam, The Netherlands. He is studying the cell biology of antigen presentation by major histocompatibility (MHC) class I and MHC class II molecules. Neefjes has designed methods to detect proteolysis in living cells using fluorescence loss in photobleaching (FLIP), fluorescence recovery after photobleaching (FRAP), fluorescence energy transfer (FRET) and other techniques.
Nico P. Dantuma received his Ph.D. from the Utrecht University in Utrecht, the Netherlands. He is presently at the Microbiology and Tumor Biology Center of the Karolinska Institute in Stockholm, Sweden. His research focuses on the role of the ubiquitin–proteasome system in diseases and regulation of proteasomal degradation. He has developed GFP-based reporters to study this system in cellular and animal models.
Related links
DATABASES
LocusLink
FURTHER INFORMATION
Encyclopedia of Life Sciences
Competing interests
The authors declare no competing financial interests.
References
- 1.Jensen TJ, et al. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 2.Reits EA, Vos JC, Gromme M, Neefjes J. The major substrates for TAP in vivo are derived from newly synthesized proteins. Nature. 2000;404:774–778. doi: 10.1038/35008103. [DOI] [PubMed] [Google Scholar]
- 3.Schubert U, et al. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- 4.Yewdell JW, Anton LC, Bennink JR. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J. Immunol. 1996;157:1823–1826. [PubMed] [Google Scholar]
- 5.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/S0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 6.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nature Rev. Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 7.Hershko A, Ciechanover A. The ubiquitin system. Annu. Rev. Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 8.Baumeister W, Walz J, Zuhl F, Seemuller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–380. doi: 10.1016/S0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 9.Tomkinson B. Tripeptidyl peptidases: enzymes that count. Trends Biochem. Sci. 1999;24:355–359. doi: 10.1016/S0968-0004(99)01435-8. [DOI] [PubMed] [Google Scholar]
- 10.Beninga J, Rock KL, Goldberg AL. Interferon-γ can stimulate post-proteasomal trimming of the N terminus of an antigenic peptide by inducing leucine aminopeptidase. J. Biol. Chem. 1998;273:18734–18742. doi: 10.1074/jbc.273.30.18734. [DOI] [PubMed] [Google Scholar]
- 11.York IA, et al. The cytosolic endopeptidase, thimet oligopeptidase, destroys antigenic peptides and limits the extent of MHC class I antigen presentation. Immunity. 2003;18:429–440. doi: 10.1016/S1074-7613(03)00058-X. [DOI] [PubMed] [Google Scholar]
- 12.Walsh PN, Ahmad SS. Proteases in blood clotting. Essays Biochem. 2002;38:95–111. doi: 10.1042/bse0380095. [DOI] [PubMed] [Google Scholar]
- 13.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 14.Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: a tail of a frog that became a prince. Nature Rev. Mol. Cell Biol. 2002;3:207–214. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- 15.Xia W, Wolfe MS. Intramembrane proteolysis by presenilin and presenilin-like proteases. J. Cell Sci. 2003;116:2839–2844. doi: 10.1242/jcs.00651. [DOI] [PubMed] [Google Scholar]
- 16.Taylor NA, Van De Ven WJ, Creemers JW. Curbing activation: proprotein convertases in homeostasis and pathology. FASEB J. 2003;17:1215–1227. doi: 10.1096/fj.02-0831rev. [DOI] [PubMed] [Google Scholar]
- 17.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/S0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz AL, Ciechanover A. The ubiquitin–proteasome pathway and pathogenesis of human diseases. Annu. Rev. Med. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- 19.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 20.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 21.Pagano M, Benmaamar R. When protein destruction runs amok, malignancy is on the loose. Cancer Cell. 2003;4:251–256. doi: 10.1016/S1535-6108(03)00243-5. [DOI] [PubMed] [Google Scholar]
- 22.Rock KL, Goldberg AL. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 1999;17:739–779. doi: 10.1146/annurev.immunol.17.1.739. [DOI] [PubMed] [Google Scholar]
- 23.Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nature Rev. Immunol. 2003;3:952–961. doi: 10.1038/nri1250. [DOI] [PubMed] [Google Scholar]
- 24.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 25.LeBlanc R, et al. Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002;62:4996–5000. [PubMed] [Google Scholar]
- 26.Meng L, et al. Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl Acad. Sci. USA. 1999;96:10403–10408. doi: 10.1073/pnas.96.18.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sunwoo JB, et al. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-κB, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 2001;7:1419–1428. [PubMed] [Google Scholar]
- 28.Schwarz K, et al. The selective proteasome inhibitors lactacystin and epoxomicin can be used to either up- or down-regulate antigen presentation at nontoxic doses. J. Immunol. 2000;164:6147–6157. doi: 10.4049/jimmunol.164.12.6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paramore A, Frantz S. Bortezomib. Nature Rev. Drug Discov. 2003;2:611–612. doi: 10.1038/nrd1159. [DOI] [PubMed] [Google Scholar]
- 30.Aghajanian C, et al. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin. Cancer Res. 2002;8:2505–2511. [PubMed] [Google Scholar]
- 31.Andre P, et al. An inhibitor of HIV-1 protease modulates proteasome activity, antigen presentation, and T cell responses. Proc. Natl Acad. Sci. USA. 1998;95:13120–13124. doi: 10.1073/pnas.95.22.13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang JS, et al. HIV protease inhibitors protect apolipoprotein B from degradation by the proteasome: a potential mechanism for protease inhibitor-induced hyperlipidemia. Nature Med. 2001;7:1327–1331. doi: 10.1038/nm1201-1327. [DOI] [PubMed] [Google Scholar]
- 33.Pati S, et al. Antitumorigenic effects of HIV protease inhibitor ritonavir: inhibition of Kaposi sarcoma. Blood. 2002;99:3771–3779. doi: 10.1182/blood.V99.10.3771. [DOI] [PubMed] [Google Scholar]
- 34.Pajonk F, Himmelsbach J, Riess K, Sommer A, McBride WH. The human immunodeficiency virus (HIV)-1 protease inhibitor saquinavir inhibits proteasome function and causes apoptosis and radiosensitization in non-HIV-associated human cancer cells. Cancer Res. 2002;62:5230–5235. [PubMed] [Google Scholar]
- 35.Bogyo M, et al. Covalent modification of the active site threonine of proteasomal β subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc. Natl Acad. Sci. USA. 1997;94:6629–6634. doi: 10.1073/pnas.94.13.6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fenteany G, et al. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 37.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem. Biol. 2001;8:739–758. doi: 10.1016/S1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 38.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breitschopf K, Bengal E, Ziv T, Admon A, Ciechanover A. A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998;17:5964–5973. doi: 10.1093/emboj/17.20.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003;115:71–82. doi: 10.1016/S0092-8674(03)00755-4. [DOI] [PubMed] [Google Scholar]
- 41.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev. Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 42.Laney J, Hochstrasser M. Substrate targeting in the ubiquitin system. Cell. 1999;97:427–430. doi: 10.1016/S0092-8674(00)80752-7. [DOI] [PubMed] [Google Scholar]
- 43.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 44.Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG. Short-lived green fluorescent proteins for quantification of ubiquitin/proteasome-dependent proteolysis in living cells. Nature Biotechnol. 2000;18:538–543. doi: 10.1038/75406. [DOI] [PubMed] [Google Scholar]
- 45.Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 46.Johnson ES, Ma PC, Ota IM, Varshavsky A. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J. Biol. Chem. 1995;270:17442–17456. doi: 10.1074/jbc.270.29.17442. [DOI] [PubMed] [Google Scholar]
- 47.Varshavsky A. The N-end rule: functions, mysteries, uses. Proc. Natl Acad. Sci. USA. 1996;93:12142–12149. doi: 10.1073/pnas.93.22.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stack JH, Whitney M, Rodems SM, Pollok BA. A ubiquitin-based tagging system for controlled modulation of protein stability. Nature Biotechnol. 2000;18:1298–1302. doi: 10.1038/82422. [DOI] [PubMed] [Google Scholar]
- 49.Gilon T, Chomsky O, Kulka RG. Degradation signals for ubiquitin system proteolysis in Saccharomyces cerevisiae. EMBO J. 1998;17:2759–2766. doi: 10.1093/emboj/17.10.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin–proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 51.Gilon T, Chomsky O, Kulka RG. Degradation signals recognized by the Ubc6p–Ubc7p ubiquitin-conjugating enzyme pair. Mol. Cell. Biol. 2000;20:7214–7219. doi: 10.1128/MCB.20.19.7214-7219.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plemper RK, Wolf DH. Retrograde protein translocation: ERADication of secretory proteins in health and disease. Trends Biochem. Sci. 1999;24:266–270. doi: 10.1016/S0968-0004(99)01420-6. [DOI] [PubMed] [Google Scholar]
- 53.Ploegh HL. Viral strategies of immune evasion. Science. 1998;280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- 54.Wiertz EJ, et al. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 55.Wiertz EJ, et al. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell. 1996;84:769–779. doi: 10.1016/S0092-8674(00)81054-5. [DOI] [PubMed] [Google Scholar]
- 56.Kessler BM, et al. Extended peptide-based inhibitors efficiently target the proteasome and reveal overlapping specificities of the catalytic β-subunits. Chem. Biol. 2001;8:913–929. doi: 10.1016/S1074-5521(01)00069-2. [DOI] [PubMed] [Google Scholar]
- 57.Myung J, Kim KB, Lindsten K, Dantuma NP, Crews CM. Lack of proteasome active site allostery as revealed by subunit-specific inhibitors. Mol. Cell. 2001;7:411–420. doi: 10.1016/S1097-2765(01)00188-5. [DOI] [PubMed] [Google Scholar]
- 58.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/S0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 59.Friant S, Meier KD, Riezman H. Increased ubiquitin-dependent degradation can replace the essential requirement for heat shock protein induction. EMBO J. 2003;22:3783–3791. doi: 10.1093/emboj/cdg375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dantuma N, Heessen S, Lindsten K, Jellne M, Masucci MG. Inhibition of proteasomal degradation by the Gly–Ala repeat of Epstein–Barr virus is influenced by the length of the repeat and the strength of the degradation signal. Proc. Natl Acad. Sci. USA. 2000;97:8381–8385. doi: 10.1073/pnas.140217397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verhoef LG, Lindsten K, Masucci MG, Dantuma NP. Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 2002;11:2689–2700. doi: 10.1093/hmg/11.22.2689. [DOI] [PubMed] [Google Scholar]
- 62.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 63.Levitskaya J, Sharipo A, Leonchiks A, Ciechanover A, Masucci MG. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly–Ala repeat domain of the Epstein–Barr virus nuclear antigen 1. Proc. Natl Acad. Sci. USA. 1997;94:12616–12621. doi: 10.1073/pnas.94.23.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharipo A, Imreh M, Leonchiks A, Imreh S, Masucci MG. A minimal glycine–alanine repeat prevents the interaction of ubiquitinated IκBα with the proteasome: a new mechanism for selective inhibition of proteolysis. Nature Med. 1998;4:939–944. doi: 10.1038/nm0898-939. [DOI] [PubMed] [Google Scholar]
- 65.Cummings CJ, et al. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron. 1999;24:879–892. doi: 10.1016/S0896-6273(00)81035-1. [DOI] [PubMed] [Google Scholar]
- 66.Dantuma NP, Masucci MG. Stabilization signals: a novel regulatory mechanism in the ubiquitin/proteasome system. FEBS Lett. 2002;529:22–26. doi: 10.1016/S0014-5793(02)03252-0. [DOI] [PubMed] [Google Scholar]
- 67.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. 'Green mice' as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/S0014-5793(97)00313-X. [DOI] [PubMed] [Google Scholar]
- 68.Hadjantonakis AK, Nagy A. The color of mice: in the light of GFP-variant reporters. Histochem. Cell Biol. 2001;115:49–58. doi: 10.1007/s004180000233. [DOI] [PubMed] [Google Scholar]
- 69.Huang WY, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nature Med. 2000;6:482–483. doi: 10.1038/74914. [DOI] [PubMed] [Google Scholar]
- 70.Lindsten K, Menendez-Benito V, Masucci MG, Dantuma NP. A transgenic mouse model of the ubiquitin/proteasome system. Nature Biotechnol. 2003;21:897–902. doi: 10.1038/nbt851. [DOI] [PubMed] [Google Scholar]
- 71.Luker GD, Pica CM, Song J, Luker KE, Piwnica-Worms D. Imaging 26S proteasome activity and inhibition in living mice. Nature Med. 2003;9:969–973. doi: 10.1038/nm894. [DOI] [PubMed] [Google Scholar]
- 72.Hernández, F., Díaz-Hernández, M., Avila, J. & Lucas, J. J. Testing the ubiquitin–proteasome hypothesis of neurodegeneration in vivo. Trends Neurosci. (in the press). [DOI] [PubMed]
- 73.Petrucelli L, et al. Parkin protects against the toxicity associated with mutant α-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/S0896-6273(02)01125-X. [DOI] [PubMed] [Google Scholar]
- 74.Lindsten K, et al. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J. Cell Biol. 2002;157:417–427. doi: 10.1083/jcb.200111034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lindsten K, Dantuma NP. Monitoring the ubiquitin/proteasome system in conformational diseases. Ageing Res. Rev. 2003;2:433–449. doi: 10.1016/S1568-1637(03)00031-X. [DOI] [PubMed] [Google Scholar]
- 76.Rubinsztein DC. Lessons from animal models of Huntington's disease. Trends Genet. 2002;18:202–209. doi: 10.1016/S0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- 77.Wong PC, Cai H, Borchelt DR, Price DL. Genetically engineered mouse models of neurodegenerative diseases. Nature Neurosci. 2002;5:633–639. doi: 10.1038/nn0702-633. [DOI] [PubMed] [Google Scholar]
- 78.Michalik A, Van Broeckhoven C. Pathogenesis of polyglutamine disorders: aggregation revisited. Hum. Mol. Genet. 2003;12:R173–186. doi: 10.1093/hmg/ddg295. [DOI] [PubMed] [Google Scholar]
- 79.Jares-Erijman EA, Jovin TM. FRET imaging. Nature Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 80.Reits E, et al. Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity. 2003;18:97–108. doi: 10.1016/S1074-7613(02)00511-3. [DOI] [PubMed] [Google Scholar]
- 81.Honey K, Rudensky AY. Lysosomal cysteine proteases regulate antigen presentation. Nature Rev. Immunol. 2003;3:472–482. doi: 10.1038/nri1110. [DOI] [PubMed] [Google Scholar]
- 82.Desnick RJ, Schuchman EH. Enzyme replacement and enhancement therapies: lessons from lysosomal disorders. Nature Rev. Genet. 2002;3:954–966. doi: 10.1038/nrg963. [DOI] [PubMed] [Google Scholar]
- 83.Edwards DR, Murphy G. Cancer. Proteases — invasion and more. Nature. 1998;394:527–528. doi: 10.1038/28961. [DOI] [PubMed] [Google Scholar]
- 84.Thurmond, R. L. et al. Identification of a potent and selective noncovalent cathepsin S inhibitor. J. Pharmacol. Exp. Ther. (2003) [epub ahead of print]. [DOI] [PubMed]
- 85.Saegusa K, et al. Cathepsin S inhibitor prevents autoantigen presentation and autoimmunity. J. Clin. Invest. 2002;110:361–369. doi: 10.1172/JCI0214682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 87.Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nature Rev. Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 88.Weissleder R, Tung CH, Mahmood U, Bogdanov A., Jr. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nature Biotechnol. 1999;17:375–378. doi: 10.1038/7933. [DOI] [PubMed] [Google Scholar]
- 89.Callahan RJ, Bogdanov A, Jr, Fischman AJ, Brady TJ, Weissleder R. Preclinical evaluation and phase I clinical trial of a 99mTc-labeled synthetic polymer used in blood pool imaging. Am. J. Roentgenol. 1998;171:137–143. doi: 10.2214/ajr.171.1.9648777. [DOI] [PubMed] [Google Scholar]
- 90.Mahmood U, Tung CH, Bogdanov A, Jr, Weissleder R. Near-infrared optical imaging of protease activity for tumor detection. Radiology. 1999;213:866–870. doi: 10.1148/radiology.213.3.r99dc14866. [DOI] [PubMed] [Google Scholar]
- 91.Bremer C, Tung CH, Bogdanov A, Jr, Weissleder R. Imaging of differential protease expression in breast cancers for detection of aggressive tumor phenotypes. Radiology. 2002;222:814–818. doi: 10.1148/radiol.2223010812. [DOI] [PubMed] [Google Scholar]
- 92.Chen J, et al. In vivo imaging of proteolytic activity in atherosclerosis. Circulation. 2002;105:2766–2771. doi: 10.1161/01.CIR.0000017860.20619.23. [DOI] [PubMed] [Google Scholar]
- 93.Tung CH, Mahmood U, Bredow S, Weissleder R. In vivo imaging of proteolytic enzyme activity using a novel molecular reporter. Cancer Res. 2000;60:4953–4958. [PubMed] [Google Scholar]
- 94.Bremer C, Tung CH, Weissleder R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nature Med. 2001;7:743–748. doi: 10.1038/89126. [DOI] [PubMed] [Google Scholar]
- 95.Zucker S, Cao J. Imaging metalloproteinase activity in vivo. Nature Med. 2001;7:655–656. doi: 10.1038/89016. [DOI] [PubMed] [Google Scholar]
- 96.Josephson L, Mahmood U, Wunderbaldinger P, Tang Y, Weissleder R. Pan and sentinel lymph node visualization using a near-infrared fluorescent probe. Mol. Imaging. 2003;2:18–23. doi: 10.1162/153535003765276255. [DOI] [PubMed] [Google Scholar]
- 97.Wunderbaldinger P, Turetschek K, Bremer C. Near-infrared fluorescence imaging of lymph nodes using a new enzyme sensing activatable macromolecular optical probe. Eur. Radiol. 2003;13:2206–2211. doi: 10.1007/s00330-003-1932-6. [DOI] [PubMed] [Google Scholar]
- 98.Sameni M, Moin K, Sloane BF. Imaging proteolysis by living human breast cancer cells. Neoplasia. 2000;2:496–504. doi: 10.1038/sj.neo.7900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baum EZ, Bebernitz GA, Gluzman Y. β-Galactosidase containing a human immunodeficiency virus protease cleavage site is cleaved and inactivated by human immunodeficiency virus protease. Proc. Natl Acad. Sci. USA. 1990;87:10023–10027. doi: 10.1073/pnas.87.24.10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Block TM, Grafstrom RH. Novel bacteriological assay for detection of potential antiviral agents. Antimicrob. Agents Chemother. 1990;34:2337–2341. doi: 10.1128/AAC.34.12.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsien RY. The green fluorescent protein. Annu Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 102.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 103.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/S0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 104.Reed JC. Apoptosis-regulating proteins as targets for drug discovery. Trends Mol. Med. 2001;7:314–319. doi: 10.1016/S1471-4914(01)02026-3. [DOI] [PubMed] [Google Scholar]
- 105.Xu X, et al. Detection of programmed cell death using fluorescence energy transfer. Nucleic Acids Res. 1998;26:2034–2035. doi: 10.1093/nar/26.8.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van Roessel P, Brand AH. Imaging into the future: visualizing gene expression and protein interactions with fluorescent proteins. Nature Cell Biol. 2002;4:E15–20. doi: 10.1038/ncb0102-e15. [DOI] [PubMed] [Google Scholar]
- 107.Rehm M, et al. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process. Role of caspase-3. J. Biol. Chem. 2002;277:24506–24514. doi: 10.1074/jbc.M110789200. [DOI] [PubMed] [Google Scholar]
- 108.Onuki R, et al. Confirmation by FRET in individual living cells of the absence of significant amyloid β-mediated caspase 8 activation. Proc. Natl Acad. Sci. USA. 2002;99:14716–14721. doi: 10.1073/pnas.232177599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Harpur AG, Wouters FS, Bastiaens PI. Imaging FRET between spectrally similar GFP molecules in single cells. Nature Biotechnol. 2001;19:167–169. doi: 10.1038/84443. [DOI] [PubMed] [Google Scholar]
- 110.Lee P, Beem E, Segal MS. Marker for real-time analysis of caspase activity in intact cells. Biotechniques. 2002;33:1284–1287. doi: 10.2144/02336rr02. [DOI] [PubMed] [Google Scholar]
- 111.Hellen CU. Assay methods for retroviral proteases. Methods Enzymol. 1994;241:46–58. doi: 10.1016/0076-6879(94)41058-5. [DOI] [PubMed] [Google Scholar]
- 112.Vogt VM. Proteolytic processing and particle maturation. Curr. Top. Microbiol. Immunol. 1996;214:95–131. doi: 10.1007/978-3-642-80145-7_4. [DOI] [PubMed] [Google Scholar]
- 113.Carr A. Toxicity of antiretroviral therapy and implications for drug development. Nature Rev. Drug Discov. 2003;2:624–634. doi: 10.1038/nrd1151. [DOI] [PubMed] [Google Scholar]
- 114.Molla A, et al. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nature Med. 1996;2:760–766. doi: 10.1038/nm0796-760. [DOI] [PubMed] [Google Scholar]
- 115.Strack PR, et al. Apoptosis mediated by HIV protease is preceded by cleavage of Bcl-2. Proc. Natl Acad. Sci. USA. 1996;93:9571–9576. doi: 10.1073/pnas.93.18.9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shoeman RL, et al. Human immunodeficiency virus type 1 protease cleaves the intermediate filament proteins vimentin, desmin, and glial fibrillary acidic protein. Proc. Natl Acad. Sci. USA. 1990;87:6336–6340. doi: 10.1073/pnas.87.16.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lindsten K, Uhlikova T, Konvalinka J, Masucci MG, Dantuma NP. Cell-based fluorescence assay for human immunodeficiency virus type 1 protease activity. Antimicrob. Agents Chemother. 2001;45:2616–2622. doi: 10.1128/AAC.45.9.2616-2622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lamarre D, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;26:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- 119.Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 120.Reits EA, Griekspoor AC, Neefjes J. How does TAP pump peptides? Insights from DNA repair and traffic ATPases. Immunol. Today. 2000;21:598–600. doi: 10.1016/S0167-5699(00)01720-5. [DOI] [PubMed] [Google Scholar]
- 121.Reits EA, Neefjes JJ. From fixed to FRAP: measuring protein mobility and activity in living cells. Nature Cell Biol. 2001;3:E145–147. doi: 10.1038/35078615. [DOI] [PubMed] [Google Scholar]
- 122.Dykxhoorn DM, Novina CD, Sharp PA. Killing the messenger: short RNAs that silence gene expression. Nature Rev. Mol. Cell Biol. 2003;4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 123.Kessler B, et al. Pathways accessory to proteasomal proteolysis are less efficient in major histocompatibility complex class I antigen production. J. Biol. Chem. 2003;278:10013–10021. doi: 10.1074/jbc.M211221200. [DOI] [PubMed] [Google Scholar]
- 124.Förster T. Zwischenmolekulare energiewandering und fluoreszenz. Annalen Physik. 1948;6:55–75. doi: 10.1002/andp.19484370105. [DOI] [Google Scholar]
- 125.Rudolphi K, Gerwin N, Verzijl N, van der Kraan P, van den Berg W. Pralnacasan, an inhibitor of interleukin-1β converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis Cartilage. 2003;11:738–746. doi: 10.1016/S1063-4584(03)00153-5. [DOI] [PubMed] [Google Scholar]
- 126.Whelan J. Caspase inhibitors for liver disease. Drug Discov. Today. 2002;7:444–445. doi: 10.1016/S1359-6446(02)02256-0. [DOI] [PubMed] [Google Scholar]
- 127.Menendez–Arias L. Targeting HIV: antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002;23:381–388. doi: 10.1016/S0165-6147(02)02054-0. [DOI] [PubMed] [Google Scholar]
- 128.Matz MV, et al. Fluorescent proteins from nonbioluminescent Anthozoa species. Nature Biotechnol. 1999;17:969–973. doi: 10.1038/13657. [DOI] [PubMed] [Google Scholar]