

Abstract
Ebola virus is a highly virulent pathogen capable of inducing a frequently lethal hemorrhagic fever syndrome. Accumulating evidence indicates that the virus actively subverts both innate and adaptive immune responses and triggers harmful inflammatory responses as it inflicts direct tissue damage. The host immune system is ultimately overwhelmed by a combination of inflammatory factors and virus-induced cell damage, particularly in the liver and vasculature, often leading to death from septic shock. We summarize the mechanisms of immune dysregulation and virus-mediated cell damage in Ebola virus–infected patients. Future approaches to prevention and treatment of infection will be guided by answers to unresolved questions about interspecies transmission, molecular mechanisms of pathogenesis, and protective adaptive and innate immune responses to Ebola virus.
Main
Ebola virus is an enveloped negative-strand RNA virus of the Filoviridae family, a group of viruses capable of inducing a severe hemorrhagic fever syndrome in humans and nonhuman primates. The virus was first recognized in 1976 during an outbreak in the Ebola River valley in Zaire (now the Democratic Republic of the Congo), Africa. A second outbreak caused by a distinct but related virus occurred in Sudan later the same year1,2. Since its discovery in central Africa, several outbreaks have recurred over the last 30 years, including a current confirmed outbreak (11 September 2007) in the Democratic Republic of the Congo (http://www.who.int/csr/don/2007_09_11/en/index.html). Although the reservoir of virus in nature and the range of intermediate hosts is not fully understood, recent studies have found that fruit bats may support replication of Ebola virus, indicating that these animals may be involved in the life cycle of the virus3. However, the natural host of Ebola virus in the absence of active outbreaks, together with the important question of how it is transmitted among various species, represents a continuing subject of investigation.
Human infections usually occur after direct contact with virus in dead or infected people or wildlife, with subsequent person-to-person transmission. Filoviruses enter the body through mucosal surfaces or skin abrasions or through the use of contaminated needles4 (Fig. 1a). The onset of Ebola virus–induced disease is sudden, with a 4 to 10 day incubation period. Patients initially show nonspecific flu-like symptoms such as fever, chills, malaise, muscle aches and headache. Abdominal pain, nausea and vomiting may follow, and a cough, sore throat or diarrhea may also be present. A rash often appears around day five and is a characteristic feature of filovirus infection. Systemic, gastrointestinal, respiratory, vascular and neurologic manifestations result from extensive viral replication, and necrosis is seen in many organs, including the liver, spleen, kidneys and gonads5. The terminal stage of the disease is characterized by coagulation disorders such as disseminated intravascular coagulation, fluid distribution problems, hypotension and hemorrhage due to liver inflammation and compromise, tissue disruption and a breakdown in endothelial barrier function that leads to increased vascular permeability. In fatal cases, death occurs typically between 7 and 16 days after infection, the result of multiple organ failure and the onset of a syndrome that resembles severe septic shock6. There are currently no antiviral drugs to treat infection and the mortality rates for the more virulent Zaire and Sudan species of the virus range from 40–90%7.
Figure 1. Infection, spread and target cell destruction by Ebola virus.
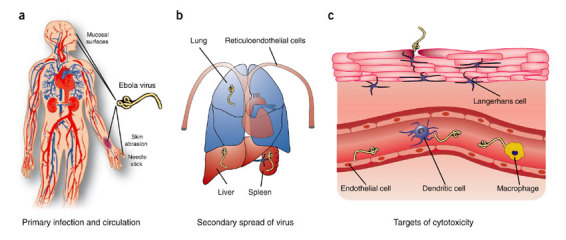
(a) Ebola virus (yellow) infects subjects through contact with body fluid or secretions from an infected patient and is distributed through the circulation. Entry can occur through abrasions in the skin during patient care, burial rituals and possibly contact with infected bushmeat, or across mucosal surfaces. Accidental needle stick is the primary route of occupational exposure. (b) Early targets of replication are reticuloendothelial cells, with high replication in several cell types within the lungs, liver and spleen. (c) Dendritic cells, macrophages and endothelium appear to be susceptible to cytopathic effects of Ebola virus gene products in vitro and possibly in vivo through disruption of cellular signaling pathways affected by virus binding, phagocytic uptake or both. Indirect damage may also be inflicted by circulating factors such as tumor necrosis factor and nitric oxide.
Host immune response to fatal Ebola infection
The uncontrolled viral replication of Ebola virus is central to its pathogenesis, both because of its cytopathic effects and because it induces prominent dysregulation of the host immune response. Virally induced immune system impairment occurs through a variety of mechanisms. Studies in nonhuman primates as well as guinea pigs raise the possibility that monocytes, macrophages and dendritic cells are early and preferred sites of viral replication8,9, though it remains possible that virus is present on these cells through binding to lectin receptors rather than active replication in vivo. It has been suggested that these cells act as vehicles for the transport of virus through the lymphatics10. Further viral replication ensues, followed by systemic spread to other organs and tissues (Fig. 1b). Although virus is observed in the reticuloendothelial system, little inflammation is seen within the lymphatics or in infected tissues during the course of the infection.
Infection of monocytes and macrophages leads to the release of proinflammatory cytokines and chemokines, including tumor necrosis factor, interleukin-1β, macrophage inflammatory protein-1α and reactive oxygen and nitrogen species11,12. The expression of these mediators is likely to attract more monocytes and macrophages to the sites of infection and may also attract neutrophils. Although recent data suggests that they are not productively infected, human neutrophils treated with filovirus in vitro show rapid activation of triggering receptor expressed on myeloid cells-1 (TREM-1)13; this results in the release of further inflammatory cytokines and chemokines that contribute to vasodilation and increased vascular permeability. In addition, infected monocytes and macrophages express cell surface tissue factor, which may be involved in the development of coagulopathies14. After productive infection, macrophages undergo cell lysis and apoptosis in large numbers15; thus, activated monocytes and macrophages do not seem to deter viral spread. Rather, they may contribute to dissemination by supporting viral replication or by transporting virus bound to cell surface lectin binding proteins within the lymphatic system. And like neutrophils, monocytes and macrophages may also secrete soluble factors that exacerbate pathogenic manifestations of the disease13.
Like monocytes and macrophages, immature dendritic cells (DCs) are 'targets' of Ebola virus, either by means of attachment of viral particles through interactions with DC-expressed C-type lectin DC-SIGN or by means of infection through interaction with other DC-expressed cell-surface receptors (Fig. 1c). Dendritic cells are among the most effective antigen-presenting cells of the immune system, and they secrete critical interleukins and cytokines that provide a critical link between innate and adaptive immune responses to many pathogens; DCs infected with Ebola virus are severely compromised in these critical functions. Human myeloid DCs infected with live virus in vitro, for example, fail to secrete the normal profile of proinflammatory cytokines and costimulatory molecules. These cells do not become mature or activated and are unable to upregulate major histocompatibility complex (MHC) molecules and thus to stimulate T cells16,17. By contrast, treatment with noninfectious Ebola virus–like particles (VLPs) activates DCs and stimulates a robust inflammatory response18, an effect dependent on the mucin-like domain of the envelope glycoprotein19. Inhibition of DC function with live or inactivated virus, but not with VLPs, indicates that the suppression of DC function and maturation is likely to be due to the presence of viral proteins or genomic material not present in the VLPs. Further studies are needed to clarify the detrimental effects of Ebola virus infection on other subpopulations of DCs, in particular on plasmacytoid DCs, which are important in antiviral interferon responses. The consequences of nonfunctional DCs include a diminished ability to stimulate humoral or cell-mediated immune responses, which may contribute to the lack of control of viral replication.
A principal determinant of the inhibitory effect on innate immune function is the resistance of Ebola virus to the antiviral effects of interferon, which is likely to be due to interruption of critical interferon response pathways by the virus itself20,21,22; interferon production is blocked in macrophages, peripheral blood mononuclear cells and DCs by Ebola virus infection in vitro and in vivo16,23. In addition, the expression of interferon-stimulated genes important in the type-I interferon response is decreased in Ebola virus–infected cells20,22,24. The interferon response has also been shown to be very important for disease outcome in mice. Immunocompetent mice are resistant to Ebola virus infection, but mice that lack the interferon-α/β receptor or the protein signal transducer and activator-1 (STAT1) or that are treated with antibody to interferon become susceptible to disease25, highlighting the critical role of interferon in protecting uninfected cells. Several mechanisms of Ebola virus–mediated resistance to the interferon response have been identified. Like some other viruses, Ebola encodes specific viral proteins that antagonize the interferon response. Two virally encoded proteins, VP24 and VP35, have been shown to interfere with the induction of the interferon response26,27,28. Nuclear accumulation of STAT1 is interrupted by VP24, which leads to a block in type-I interferon signaling and renders infected cells insensitive to this antiviral response27. Ebola VP35 blocks the activity of the interferon regulatory factor 3 (IRF-3), thus decreasing interferon responses26,28. More recently, VP35 was shown to counteract the activity of the double-stranded RNA–dependent protein kinase (PKR)29. In combination, these studies suggest that virus-induced inhibition of the interferon pathway not only decreases interferon-stimulated gene transcription to prevent an antiviral response state, but also contributes to lower numbers of mature and activated myeloid DCs, which in turn hinders the activation of the adaptive immune response.
Surprisingly, patients who succumb to Ebola virus infection show little evidence of an activated adaptive immune response. Adaptive immunity is severely compromised not only because of a lack of functional DCs and other important antigen-presenting cells, but also because lymphocytes undergo massive apoptosis in infected humans and nonhuman primates15,30,31. Although lymphocytes are not targets of the virus, substantial numbers—with the exception of B cells—undergo apoptosis during the illness32; as a result, numbers of CD4+ and CD8+ T cells are substantially reduced in fatal human and nonhuman primate infections before death30,31,33. Lymphocyte apoptosis is also a common manifestation of other viral hemorrhagic fevers and is frequently observed during septic shock34.
Studies with lymphocytes in vitro indicate that several molecules involved in triggering apoptosis are present on these cell populations, including TRAIL and Fas-FasL15. However, the mechanisms responsible for this 'bystander' apoptosis are still under investigation. It may be that inflammatory mediators and other factors, such as the proapoptotic soluble factor nitric oxide (NO) secreted by infected macrophages, are capable of inducing the observed lymphocyte apoptosis. Alternatively, impaired DC function and an overall immunosuppressive state may contribute to the phenomenon31. Yet another possibility is that cell death is actively triggered by direct interactions between lymphocytes and Ebola virus or soluble gene products. The importance of early responses involving cells of the innate immune system and/or a rapid adaptive antibody response is highlighted by a recent study showing protection of nonhuman primates with postexposure vaccine administration35.
Although filoviruses are among the most virulent and fatal pathogens known, some patients infected with Ebola virus recover from the infection. Identifying the differences in the immune response between fatal and nonfatal cases is important for future development of effective therapies and vaccines. Specific differences in clinical presentation and immune responses have been noted in those who succumb contrasted with those who recover from Ebola virus infection (Table 1). This comparison makes it clear that the development of an antigen-specific cell-mediated immune response correlates with clearance of the virus. Studies demonstrating antigen-specific cellular immune responses in vaccinated nonhuman primates that survived infectious Ebola virus challenge36,37,38 support this finding. Additionally, induction of a humoral and CD8+ T cell response was found to be required for protection in mice challenged with lethal Ebola virus infection39. However, the protective role of immunoglobulins remains uncertain, as a recent report indicates that the passive transfer of the neutralizing human monoclonal antibody KZ52 is unable to control infection in a macaque model40. Based on these considerations, it is becoming increasingly evident that an early and robust, but transient, innate immune response and the subsequent activation of adaptive immune response are necessary to protect against fatal infection. If such a host immune response is not generated, the virus evades immune control, and the infection progresses to end-stage disease.
Table 1.
Correlative differences in patients who survive versus patients who succumb to Ebola virus infection
| Nonlethal infection | Lethal infection | Refs. |
|---|---|---|
| Prominent CD8+ T cell activation | No CD8+ T cell activation | 30,33 |
| Above-normal numbers of T cells | Below-normal numbers of T cells | 33 |
| 107 viral genome copies/ml serum | 1010 viral genome copies/ml serum | 33,47 |
| Detectable antibodies in blood at onset of symptoms | No detectable antibodies in blood at onset of symptoms | 33,65 |
| Low NO | High NO | 33,65 |
Pathogenesis of infection
The pathological changes seen in patients dying from Ebola virus infection include coagulation abnormalities, vascular permeability, hemorrhage and organ necrosis and failure. The current hypothesis is that the fundamental mechanism of Ebola virus pathogenesis is vascular injury and damage secondary to coagulation abnormalities and increased vascular permeability, due to the release of inflammatory cytokines and chemokines by infected and activated monocytes and macrophages, and to direct endothelial cell damage from viral replication late in infection41,42. It is evident that in addition to the 'cytokine storm', the virus itself can also induce immunosuppression and damage host cells directly4,43,44. Thus, the deleterious manifestations of the infection stem in part from the factors secreted from dysfunctional immune cells and in part from virally induced damage of host tissues and organs.
Ebola virus shows tropism in vitro for cells of the innate immune system as well as endothelial cells, dendritic cells and several types of epithelial cells. Replication occurs at an unusually high rate in infected cells. The ability of virus to replicate in different cell types is less well characterized in vivo. In addition, viremia in infected patients is generally difficult to quantify6; however, a viral load exceeding 106 plaque-forming units per milliliter of serum (PFU/ml) was noted in at least one outbreak of the Zaire species of Ebola45. Viremia in infected nonhuman primates can reach up to 107 PFU/ml46. Humans with fatal infections have up to 1010 copies of viral RNA per milliliter, whereas much fewer (107 copies/ml) are found in the serum of those who survive Ebola virus infection (see Table 1)47. High rates of viral replication lead to lysis and necrosis in cells of many organs, including the liver, spleen, kidneys and gonads. Much of the observed necrosis is virally induced, as there is little infiltration within infected tissues, and extraordinary numbers of viral particles are present in the necrotic debris. In addition, microscopic examination of infected human tissues shows a correlation between tissue damage and the presence of viral antigens, nucleic acid and sites of viral replication4,43,44. This observation indicates that direct viral damage of tissues and organs may lead to organ failure and shock.
The infection of certain cell types plays an important role in Ebola virus pathogenesis. Infection of innate immune cells is thought to be pivotal to the systemic dissemination of the virus during human infection8,10. Infected monocytes and macrophages travel from the site of infection to lymph nodes, where more monocytes and macrophages are recruited and then become targets of infection. Infection of these cells leads to further amplification and dissemination of the virus through the lymphatic system12. In addition, infection and necrosis of hepatocytes causes impairment of liver function. Liver enzymes are elevated in most filovirus infections48,49,50, and decreased liver function could account for the decreased synthesis of coagulation factors and development of coagulation disorders prominent during fatal infection. Finally, the development of shock at later stages of the disease is multifactorial and, along with hemorrhage, may be due in part to the infection and resulting necrosis of cells of the adrenal cortex50, as these cells are important in the regulation of blood pressure.
Vascular impairment and Ebola glycoproteins
Endothelial impairment is a prominent feature of Ebola hemorrhagic fever. A loss of vascular integrity is often observed in humans and nonhuman primates during late stages of the disease and is associated with bleeding and the imbalance of fluid between tissue spaces. The complete mechanisms leading to endothelium permeability have not been elucidated. Several studies have shown that the virally induced release of inflammatory mediators increases vascular permeability in vitro11,51. However, endothelial cells are targets of infection during later stages of the disease and direct virus-induced cytotoxicity of endothelial cells cannot be ruled out as a contributory mechanism for increased hemorrhagic manifestations. Indeed, the viral envelope glycoprotein GP has been implicated as one of the major determinants of vascular cell injury.
GP is one of the most studied Ebola virus proteins because of its importance in viral entry and potential as a target for vaccine development. As mentioned, it is also under intense research because of its possible role in pathogenesis. The glycoprotein is responsible for targeting the virus to cells that are relevant to pathogenesis. GP is likely to have a role in immune suppression through its effects on downregulation of cell surface proteins essential for lymphocyte adhesion and antigen presentation52,53. Although some have suggested that soluble GP may compete for neutralizing antibodies that might otherwise target viruses or infected cells54,55, a protective role for such antibodies has not been demonstrated, and the biochemistry and antibody reactivity of soluble GP differ from those of the membrane-bound trimer spike56,57. Soluble GP does inhibit neutrophil activation57, providing a further mechanism by which viral immunity may affect the innate inflammatory response. Ebola virus entry depends also on endosomal cathepsins, enzymes critical for antigen presentation58,59, and release of cathepsins may contribute to virus-induced cell damage60.
Several groups have shown that GP has a direct cytotoxic effect. Yang and colleagues found that of the seven viral gene products, GP was responsible for cell rounding and detachment in endothelial cells both in vitro and ex vivo and that this can lead to a substantial increase in vascular permeability61. Expression of GP from all four species of Ebola virus induces variable degrees of cytotoxicity in cell lines and primary cells in vitro that are characterized by cell rounding and detachment, followed by cell death61. These effects are mediated by a heavily glycosylated mucin-like domain of the glycoprotein. Although there is some debate over the role of GP cytotoxicity during live viral infection62, differences in GP-induced cytotoxicity are correlated with the mortality rates of the different viral species52,61, suggesting that this gene product is important in the pathogenesis of the disease. Expression of membrane-bound GP seems to be precisely controlled during viral replication by a mechanism involving transcriptional editing by the viral polymerase63. This indicates that the glycoprotein may be a key viral determinant of pathogenicity during infection.
Thus, virus-induced and host factors combine to result in a destructive pathway in which a fatal response to Ebola virus infection invariably correlates with suppression of both B and T cell–mediated immunity. Patients who fail to recover have virtually no viral antigen–specific antibodies. Low amounts of specific immunoglobulin Ms are present in only 30% of fatally infected patients, and no specific immunoglobulin Gs are detected30,64,65. There seems to be limited initiation of cytotoxic T cell or CD4+ helper T cell responses, most likely owing to their depletion in fatal cases. Lymphocyte depletion is likely to exacerbate the uncontrolled viral replication within macrophages and other inflammatory cells. Therefore, fatal Ebola virus infection is characterized by broad immunosuppression typified by the development of an aberrant nonspecific and deleterious innate immune response and little or no stimulation of an antigen-specific adaptive response. This lack of response leads to an overwhelming viral burden and resultant immune- and virus-mediated pathology.
Relevance to other highly lethal pathogens and future research
Valuable insights into critical features of the host immune system can be gained from an examination of the immune response to highly virulent pathogens such as Ebola virus. One trend that seems to be emerging is that lethal, acute pathogens tend to kill rapidly, before the development of an adaptive immune response, whereas chronic pathogens can survive and replicate despite an adaptive immune response. In this regard, there are interesting parallels between Ebola virus infection and the highly pathogenic 1918 influenza virus (see the accompanying review by Ahmed and colleagues66). For example, Kobasa and co-workers found that a reconstituted 1918 strain of influenza shows high levels of viral replication, which correlates with macroscopic lesions in lung tissue of infected cynomolgus macaques67. Infection in this animal model culminated in acute respiratory distress and an overwhelmingly fatal outcome. Interestingly, infected animals were able to mount an immune response that was in many ways similar to the responses observed during Ebola infection in nonhuman primates. The immune response to 1918 influenza was characterized by an aberrant interferon response and the expression of abnormally high levels of cytokines and chemokines. The authors concluded that the high lethality of the 1918 influenza strain can be attributed in part to the generation of an atypical and harmful innate immune response that is insufficient for protection.
A comparison of the immune responses to Ebola and 1918 influenza viruses indicates that the high lethality of these viruses may stem from a combination of the deleterious effects of high viral titers and direct viral damage and a nonspecific and abnormally sustained innate immune response. A similar picture of overwhelming viremia, lack of control by the innate immune response and failure to develop adaptive immunity has been observed with other highly lethal viruses as well, including the severe acute respiratory syndrome (SARS) coronavirus, Marburg virus, Lassa fever virus and others. In each case, it would seem that the virus causes lethal infection through its overwhelming replication, though the specific receptors, cell and organ tropisms, mechanisms of evading inflammation and immunity and natural reservoir may differ.
Many questions remain unresolved regarding the mechanisms and full extent of virus-induced immune dysregulation. For example, the mechanism of lymphocyte apoptosis is unknown. Ebola virus does not target these cells directly, yet their numbers are rapidly exhausted once viral titers are measurable in the host. Do these cells enter an anergic terminal differentiation due to local cytokine imbalances, or is there aberrant target destruction by other immune cells? It is also unknown whether Ebola virus shows tropism for a particular DC that may enhance evasion of the antiviral response. The mechanism by which DC antigen presentation is impaired is unknown. The role of cathepsin in immunopathogenesis is also not completely understood; because cathepsins also contribute to antigen processing, it is possible they affect the adaptive immune response as well. Similar questions remain regarding the details of viral replication in vivo. Although Ebola virus can be found by immunostaining of a variety of cell types, including macrophages, DCs and endothelial cells, the virus binds to ubiquitous lectin receptors on many of these cells; thus, it is unclear whether the presence of the virus in a given cell represents active replication or merely binding to the cell surface. Finally, the role of cytokine storm versus direct viral cytotoxicity to endothelial cells remains the subject of much speculation but, unfortunately, very little data.
Ultimately, many of those questions, including the roles of specific parts of the immune system in protection, may be resolved with studies that use antibody depletion in vivo against cytokines, cytokine receptors and lymphocyte subsets in nonhuman primate models of infection. Until these important issues are addressed, the current hypotheses explain Ebola virus–induced pathophysiology in broad terms: a combination of factors, including uncontrolled and nonspecific inflammatory responses, virally induced immunosuppression and direct viral destruction of several cell types, collectively contribute to the collapse of the vascular system, multiorgan failure and shock-like syndrome of lethal Ebola virus infection.
Acknowledgements
We thank M. Cichanowski, A. Tislerics and T. Suhana for help with manuscript preparation and submission.
References
- 1.Bowen ET, et al. Viral haemorrhagic fever in southern Sudan and northern Zaire. Preliminary studies on the aetiological agent. Lancet. 1977;1:571–573. doi: 10.1016/S0140-6736(77)92001-3. [DOI] [PubMed] [Google Scholar]
- 2.Johnson KM, Lange JV, Webb PA, Murphy FA. Isolation and partial characterisation of a new virus causing acute haemorrhagic fever in Zaire. Lancet. 1977;1:569–571. doi: 10.1016/S0140-6736(77)92000-1. [DOI] [PubMed] [Google Scholar]
- 3.Leroy EM, et al. Fruit bats as reservoirs of Ebola virus. Nature. 2005;438:575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- 4.Zaki SR, Goldsmith CS. Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 1999;235:97–116. doi: 10.1007/978-3-642-59949-1_7. [DOI] [PubMed] [Google Scholar]
- 5.Bwaka MA, et al. Ebola hemorrhagic fever in Kikwit, Democratic Republic of the Congo: clinical observations in 103 patients. J. Infect. Dis. 1999;179(suppl. 1):S1–S7. doi: 10.1086/514308. [DOI] [PubMed] [Google Scholar]
- 6.Ksiazek TG, et al. Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J. Infect. Dis. 1999;179(suppl. 1):S177–S187. doi: 10.1086/514321. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez A, et al. Fields Virology. 2001. Filoviridae: Marburg and Ebola Viruses; pp. 1279–1304. [Google Scholar]
- 8.Geisbert TW, et al. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 2003;163:2347–2370. doi: 10.1016/S0002-9440(10)63591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connolly BM, et al. Pathogenesis of experimental Ebola virus infection in guinea pigs. J. Infect. Dis. 1999;179:S203–S217. doi: 10.1086/514305. [DOI] [PubMed] [Google Scholar]
- 10.Hensley LE, Young HA, Jahrling PB, Geisbert TW. Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily. Immunol. Lett. 2002;80:169–179. doi: 10.1016/S0165-2478(01)00327-3. [DOI] [PubMed] [Google Scholar]
- 11.Feldmann H, et al. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol. 1996;70:2208–2214. doi: 10.1128/jvi.70.4.2208-2214.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bray M, Geisbert TW. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int. J. Biochem. Cell Biol. 2005;37:1560–1566. doi: 10.1016/j.biocel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 13.Mohamadzadeh M, et al. Activation of triggering receptor expressed on myeloid cells-1 on human neutrophils by Marburg and Ebola viruses. J. Virol. 2006;80:7235–7244. doi: 10.1128/JVI.00543-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geisbert TW, et al. Mechanisms underlying coagulation abnormalities in Ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 2003;188:1618–1629. doi: 10.1086/379724. [DOI] [PubMed] [Google Scholar]
- 15.Gupta M, Spiropoulou C, Rollin PE. Ebola virus infection of human PBMCs causes massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Virology. 2007;364:45–54. doi: 10.1016/j.virol.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Bosio CM, et al. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J. Infect. Dis. 2003;188:1630–1638. doi: 10.1086/379199. [DOI] [PubMed] [Google Scholar]
- 17.Mahanty S, et al. Cutting edge: impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 2003;170:2797–2801. doi: 10.4049/jimmunol.170.6.2797. [DOI] [PubMed] [Google Scholar]
- 18.Bosio CM, et al. Ebola and Marburg virus-like particles activate human myeloid dendritic cells. Virology. 2004;326:280–287. doi: 10.1016/j.virol.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 19.Martinez O, Valmas C, Basler CF. Ebola virus-like particle-induced activation of NF-κB and Erk signaling in human dendritic cells requires the glycoprotein mucin domain. Virology. 2007;364:342–354. doi: 10.1016/j.virol.2007.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harcourt BH, Sanchez A, Offermann MK. Ebola virus inhibits induction of genes by double-stranded RNA in endothelial cells. Virology. 1998;252:179–188. doi: 10.1006/viro.1998.9446. [DOI] [PubMed] [Google Scholar]
- 21.Jahrling PB, et al. Evaluation of immune globulin and recombinant interferon-α2b for treatment of experimental Ebola virus infections. J. Infect. Dis. 1999;179(suppl. 1):S224–S234. doi: 10.1086/514310. [DOI] [PubMed] [Google Scholar]
- 22.Kash JC, et al. Global suppression of the host antiviral response by Ebola- and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006;80:3009–3020. doi: 10.1128/JVI.80.6.3009-3020.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahanty S, et al. Protection from lethal infection is determined by innate immune responses in a mouse model of Ebola virus infection. Virology. 2003;312:415–424. doi: 10.1016/S0042-6822(03)00233-2. [DOI] [PubMed] [Google Scholar]
- 24.Harcourt BH, Sanchez A, Offermann MK. Ebola virus selectively inhibits responses to interferons, but not to interleukin-1β, in endothelial cells. J. Virol. 1999;73:3491–3496. doi: 10.1128/jvi.73.4.3491-3496.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bray M. The role of the type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001;82:1365–1373. doi: 10.1099/0022-1317-82-6-1365. [DOI] [PubMed] [Google Scholar]
- 26.Basler CF, et al. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reid SP, et al. Ebola virus VP24 binds karyopherin α1 and blocks STAT1 nuclear accumulation. J. Virol. 2006;80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardenas WB, et al. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol. 2006;80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng Z, Cerveny M, Yan Z, He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J. Virol. 2007;81:182–192. doi: 10.1128/JVI.01006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baize S, et al. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 1999;5:423–426. doi: 10.1038/7422. [DOI] [PubMed] [Google Scholar]
- 31.Reed DS, Hensley LE, Geisbert JB, Jahrling PB, Geisbert TW. Depletion of peripheral blood T lymphocytes and NK cells during the course of Ebola hemorrhagic fever in cynomolgus macaques. Viral Immunol. 2004;17:390–400. doi: 10.1089/vim.2004.17.390. [DOI] [PubMed] [Google Scholar]
- 32.Geisbert TW, et al. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 2000;80:171–186. doi: 10.1038/labinvest.3780021. [DOI] [PubMed] [Google Scholar]
- 33.Sanchez A, et al. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: cellular responses, virus load, and nitric oxide levels. J. Virol. 2004;78:10370–10377. doi: 10.1128/JVI.78.19.10370-10377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geisbert TW, Jahrling PB. Exotic emerging viral diseases: progress and challenges. Nat. Med. 2004;10:S110–S121. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- 35.Feldmann H, et al. Effective post-exposure treatment of Ebola infection. PLoS Pathog. 2007;3:e2. doi: 10.1371/journal.ppat.0030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sullivan NJ, Sanchez A, Rollin PE, Yang Z-Y, Nabel GJ. Development of a preventive vaccine for Ebola virus infection in primates. Nature. 2000;408:605–609. doi: 10.1038/35046108. [DOI] [PubMed] [Google Scholar]
- 37.Sullivan NJ, et al. Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature. 2003;424:681–684. doi: 10.1038/nature01876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bukreyev A, et al. Successful topical respiratory tract immunization of primates against Ebola virus. J. Virol. 2007;81:6379–6388. doi: 10.1128/JVI.00105-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warfield KL, et al. Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection. J. Immunol. 2005;175:1184–1191. doi: 10.4049/jimmunol.175.2.1184. [DOI] [PubMed] [Google Scholar]
- 40.Oswald WB, et al. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathog. [online] 2007;3:e9. doi: 10.1371/journal.ppat.0030009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahanty S, Bray M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect. Dis. 2004;4:487–498. doi: 10.1016/S1473-3099(04)01103-X. [DOI] [PubMed] [Google Scholar]
- 42.Mohamadzadeh M, Chen L, Schmaljohn AL. How Ebola and Marburg viruses battle the immune system. Nat. Rev. Immunol. 2007;7:556–567. doi: 10.1038/nri2098. [DOI] [PubMed] [Google Scholar]
- 43.Dietrich M, Schumacher HH, Peters D, Knobloch J. Ebola Virus Haemorrhagic Fever. 1978. Human pathology of Ebola (Maridi) virus infection in the Sudan; pp. 37–42. [Google Scholar]
- 44.Murphy F, et al. Viral Pathogenesis. 1997. An atlas of viral disease pathogenesis; pp. 433–463. [Google Scholar]
- 45.Anonymous. Ebola haemorrhagic fever in Zaire, 1976. Bull. World Health Organ.56, 271–293 (1978). [PMC free article] [PubMed]
- 46.Jahrling PB, et al. Passive immunization of Ebola virus-infected cynomolgus monkeys with immunoglobulin from hyperimmune horses. Arch. Virol. Suppl. 1996;11:135–140. doi: 10.1007/978-3-7091-7482-1_12. [DOI] [PubMed] [Google Scholar]
- 47.Towner JS, et al. Rapid diagnosis of Ebola hemorrhagic fever by reverse transcription-PCR in an outbreak setting and assessment of patient viral load as a predictor of outcome. J. Virol. 2004;78:4330–4341. doi: 10.1128/JVI.78.8.4330-4341.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fisher-Hoch SP, et al. Pathogenic potential of filoviruses: role of geographic origin of primate host and virus strain. J. Infect. Dis. 1992;166:753–763. doi: 10.1093/infdis/166.4.753. [DOI] [PubMed] [Google Scholar]
- 49.Johnson E, Jaax N, White J, Jahrling P. Lethal experimental infections of rhesus monkeys by aerosolized Ebola virus. Int. J. Exp. Pathol. 1995;76:227–236. [PMC free article] [PubMed] [Google Scholar]
- 50.Ryabchikova EI, Kolesnikova LV, Luchko SV. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 1999;179(suppl. 1):S199–S202. doi: 10.1086/514293. [DOI] [PubMed] [Google Scholar]
- 51.Stolpen AH, Guinan EC, Fiers W, Pober JS. Recombinant tumor necrosis factor and immune interferon act singly and in combination to reorganize human vascular endothelial cell monolayers. Am. J. Pathol. 1986;123:16–24. [PMC free article] [PubMed] [Google Scholar]
- 52.Simmons G, Wool-Lewis RJ, Baribaud F, Netter RC, Bates P. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J. Virol. 2002;76:2518–2528. doi: 10.1128/jvi.76.5.2518-2528.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sullivan NJ, et al. Ebola virus glycoprotein toxicity is mediated by a dynamin-dependent protein-trafficking pathway. J. Virol. 2005;79:547–553. doi: 10.1128/JVI.79.1.547-553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ito H, et al. Ebola virus glycoprotein: proteolytic processing, acylation, cell tropism, and detection of neutralizing antibodies. J. Virol. 2001;75:1576–1580. doi: 10.1128/JVI.75.3.1576-1580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dolnik O, et al. Ectodomain shedding of the glycoprotein GP of Ebola virus. EMBO J. 2004;23:2175–2184. doi: 10.1038/sj.emboj.7600219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanchez A, et al. Biochemical analysis of the secreted and virion glycoproteins of Ebola virus. J. Virol. 1998;72:6442–6447. doi: 10.1128/jvi.72.8.6442-6447.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Z, et al. Distinct cellular interactions of secreted and transmembrane Ebola virus glycoproteins. Science. 1998;279:1034–1037. doi: 10.1126/science.279.5353.1034. [DOI] [PubMed] [Google Scholar]
- 58.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schornberg K, et al. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006;80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barrientos LG, Rollin PE. Release of cellular proteases into the acidic extracellular milieu exacerbates Ebola virus-induced cell damage. Virology. 2007;358:1–9. doi: 10.1016/j.virol.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 61.Yang Z-Y, et al. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000;6:886–889. doi: 10.1038/78654. [DOI] [PubMed] [Google Scholar]
- 62.Alazard-Dany N, et al. Ebola virus glycoprotein GP is not cytotoxic when expressed constitutively at a moderate level. J. Gen. Virol. 2006;87:1247–1257. doi: 10.1099/vir.0.81361-0. [DOI] [PubMed] [Google Scholar]
- 63.Volchkov VE, et al. Recovery of infectious Ebola virus from complementary DNA: RNA editing of the GP gene and viral cytotoxicity. Science. 2001;291:1965–1969. doi: 10.1126/science.1057269. [DOI] [PubMed] [Google Scholar]
- 64.Ksiazek TG, West CP, Rollin PE, Jahrling PB, Peters CJ. ELISA for the detection of antibodies to Ebola viruses. J. Infect. Dis. 1999;179:S192–S198. doi: 10.1086/514313. [DOI] [PubMed] [Google Scholar]
- 65.Baize S, et al. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 2002;128:163–168. doi: 10.1046/j.1365-2249.2002.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahmed R, Oldstone MBA, Palese P. Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat. Immunol. 2007;8:1188–1193. doi: 10.1038/ni1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kobasa D, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]