

Key Points
The hygiene hypothesis states that repeated exposure to diverse infections in childhood protects against asthma and allergy later in life.
The risk of developing asthma may be inversely correlated with microbial diversity, as suggested by studies of populations in rural environments (which have a low risk of asthma) and populations living in cities or towns (which have a high risk of asthma).
The human lower airway has a distinct microbiota and is not sterile. Different families of bacteria may be associated with airway diseases such as asthma. Changes in the microbiome may be involved in asthma pathogenesis and may be affected by asthma severity and perhaps also by treatment for the disease.
The respiratory viruses rhinoviruses (RVs) and human respiratory syncytial virus (RSV) are associated with asthma onset in young children, but the mechanisms responsible for this association are largely unknown.
Viruses and bacteria, including atypical bacteria in particular, are strongly associated with asthma exacerbations. The use of anti-infective drugs or boosting naturally occurring immunity to these pathogens may therefore be useful in controlling the burden of disease caused by asthma exacerbations.
Subject terms: Infectious diseases, Clinical microbiology, Virology, Pathogens, Pathogenesis, Asthma
Asthma is a heterogeneous, complex disease, and its causes have environmental, immunological, genetic and microbial components. In this Review, Edwardset al. describe how microorganisms can influence the risk, severity and pathogenesis of asthma, and protection against the disease.
Abstract
Asthma remains an important human disease that is responsible for substantial worldwide morbidity and mortality. The causes of asthma are multifactorial and include a complex mix of environmental, immunological and host genetic factors. In addition, epidemiological studies show strong associations between asthma and infection with respiratory pathogens, including common respiratory viruses such as rhinoviruses, human respiratory syncytial virus, adenoviruses, coronaviruses and influenza viruses, as well as bacteria (including atypical bacteria) and fungi. In this Review, we describe the many roles of microorganisms in the risk of developing asthma and in the pathogenesis of and protection against the disease, and we discuss the mechanisms by which infections affect the severity and prevalence of asthma.
Main
Asthma is a chronic airway disease for which the symptoms are wheezing, breathlessness, chest tightness and coughing, with variable and often reversible airway obstruction accompanied by airway hyper-reactivity. These symptoms result from a range of processes, including contraction of airway smooth muscle (ASM), airway inflammation, airway infections, immunoglobulin E-mediated allergic responses, exposure to pollutants, exercise, stress and the stimulation of cholinergic and sensory nerves1. A key feature of asthma is decreased lung function, measured as a reduction in either the peak expiratory flow (PEF) or the forced expiratory volume in 1 second (FEV1). Some of the immediate symptoms of asthma are reversed by a short-acting inhaled β2 agonist, but longer-term control requires inhaled anti-inflammatory glucocorticoids.
Worldwide, asthma affects ∼300 million people, including both adults and children. It is associated with onset early in life and is increasing in incidence in the developed world. An interesting aspect of asthma is its heterogeneous, complex nature and the fact that it can present as a chronic, stable disease and as asthma exacerbations. Asthma can be severe or mild, and can vary spontaneously in activity and greatly in terms of the time of onset or the response to therapy. Indeed, whether asthma is one disease or a spectrum of related but subtly different conditions is often debated2, and asthma has recently been divided into separate phenotypes, which are in turn subdivided into several endotypes3 (Table 1). Asthma exacerbations are often triggered by multiple factors that may work in an additive or synergistic manner. This complex pathogenesis has made it challenging to understand the cellular mechanisms that are responsible for asthma, to decipher candidate genetic predispositions and to identify causative agents. The causes of asthma are multifactorial, and epidemiological studies spanning three decades across five continents have proved that there is a strong association between respiratory infections and the risk and pathogenesis of asthma.
Table 1.
Asthma phenotypes
| Asthma phenotype | Description |
|---|---|
| Atopic (allergic) asthma |
•Reactivity to one or more known allergens (determined by skin prick test) •Higher than normal IgE levels •Specific IgE to allergen (determined by radioallergosorbent assay*) •Eosinophils in the airways (common but not always) |
| Non-allergic asthma |
•No known allergy •No or low serum IgE •No reactivity to allergen (determined by skin prick test or radioallergosorbent assay*) |
| Mild asthma | •Occasional symptoms, but well controlled by therapy (low-dose glucocorticoids and/or short-acting β2 agonists) |
| Stable asthma |
•Well-controlled symptoms •No dysregulated lung function or asthma exacerbations |
| Severe asthma | •Poor control of symptoms despite using maximal therapy (high-dose glucocorticoids and/or β2 agonists, or other therapies) |
| Asthma exacerbations | •Mild or severe asthma with sudden symptom onset, often requiring medical consultation or hospitalization and possible oral glucocorticoid intervention |
| Neutrophilic asthma | •Increased number of neutrophils in bronchoalveolar lavage or sputum samples (can be allergic or non-allergic asthma) |
| Allergic bronchopulmonary aspergillosis | •Invasive colonization of the airway with Aspergillus spp., an opportunistic pathogen; Aspergillus growth promotes allergic sensitization to at least 12 known fungal allergens |
| Virus-induced wheeze or bronchiolitis |
•Common in young children (<2 years of age) •Asthma caused by a viral infection, mucus and inflammation in young children, giving a typical 'wheezy' episode •Mild, severe, acute onset or febrile |
| High TH2-associated atopic asthma |
•TH2 cell-associated gene expression is typical (for example, expresses IL13 and IL-13-inducible genes) •Responds well to glucocorticoids •Prone to asthma exacerbations135 |
| Low TH2-associated atopic asthma |
•A mixed phenotype of asthma •Less prominent TH2 type cytokine responses •More refractory to glucocorticoid treatment than patients with high TH2-associated atopic asthma135 |
| IgE, immunoglobulin E; TH2, T helper 2. | |
| *A radioallergosorbent assay is a blood test that is used to quantify allergen-specific IgE antibodies. | |
In this Review, we summarize the association of microorganisms with asthma, discuss the importance of such organisms for the disease and consider the idea that part of the cause and at least some of the pathogenesis of asthma — in particular, of asthma exacerbations — resultfrom microorganisms and their components. We also discuss the potential therapeutic value of microorganisms and their products for treating asthma and argue that future treatment and management of the disease must carefully consider the role of microorganisms.
Microorganisms and asthma onset
The hygiene hypothesis. The hygiene hypothesis posits that repeated exposure to diverse common infections (in particular, with bacteria, food-borne and orofaecal parasites4, and hookworms5) and exposure to environmental microbiota during childhood6,7 are strongly associated with a healthy maturation of the immune system and with protection from the development of asthma and allergies later in life8,9 . Our understanding of the importance of microorganisms in protection from allergies and asthma has been recently enhanced by a molecular analysis of the microbiome. Populations that live in environments with diverse microbiological flora are associated with a low incidence of asthma, whereas populations that live in environments with low microbiological diversity are associated with a higher incidence of asthma7. The incidence of bacteria and their components in different environments has been studied using culture-dependent and molecular methods10,11,12, and both qualitative and quantitative differences are found between countries with a high incidence of atopy and those with a low incidence of atopy10. Evidence for the hygiene hypothesis is also supported by experiments in mice, whereby intranasal exposure to Escherichia coli13 or Acinetobacter lwoffii str. F78 protects against allergic inflammation of the airways14. In addition, a link has been established between the fermentation of dietary fibre by the mouse intestinal microbiota and protection against inflammatory diseases, including asthma15. Although these studies provide experimental support for the hygiene hypothesis, our understanding of the mechanistic basis of this phenomenon is still in its infancy. These studies have also promoted the use of bacterial components such as Toll-like receptor (TLR) ligands and other pathogen-associated molecular patterns as potential therapies for asthma (Table 2).
Table 2.
Current and future treatments for asthma
| Treatment | Notes |
|---|---|
| Glucocorticoids | •Anti-inflammatory small molecules that bind the cytosolic glucocorticoid receptor and both suppress inflammation and induce anti-inflammatory pathways |
| β2 agonists |
•Widely used therapies that reduce contraction of airway smooth muscle and result in bronchodilation •Can be short or long acting |
| Leukotriene receptor antagonists |
•Prevent cysteinyl leukotrienes from binding the CysLT1 receptor and inducing airway smooth muscle contraction, mucus secretion and airway inflammation •For example, montelukast |
| Therapies against TH2 type cytokines |
•Monoclonal antibodies and soluble receptors against IL-4, IL-13 and IL-5 |
| IgE-specific antibodies |
•Monoclonal antibodies (for example, omalizumab) •Reduce levels of circulating and cell-bound IgE •A striking effect of this therapy on the incidence of asthma exacerbations has been recently reported139 |
| Therapies against TNF |
•Soluble TNF receptors or TNF-specific antibodies •Reduce levels of circulating TNF (a major pro-inflammatory cytokine implicated in asthma pathogenesis) •Have shown limited efficacy in asthma treatment, and there are some safety concerns140 |
| Vitamin D |
•A link between infection risk, asthma and vitamin D has received much attention recently141,142,143 •Data suggest an association between vitamin D and asthma risk, but a clear mechanism of how vitamin D prevents asthma is still being sought |
| Probiotics | •Probiotics with immunomodulatory potential have been applied to allergy and asthma144,145,146 as a result of the recent rise in the number of studies advocating a role for microorganisms in asthma and other inflammatory disorders |
| Pathogen-associated molecular pattern and TLR agonists | •Shown to have a good efficacy as a treatment in animal models of asthma147,148•Beginning to appear in clinical trials for allergic rhinitis but have yet to be applied to asthma in humans |
| IFNs |
•Human studies suggest that IFNα has a clinical benefit in stable asthma149 •IFNβ is currently in a clinical trial for asthma exacerbations •IFNλs reduce allergic airway inflammation in mice150 |
| IFN, interferon; IgE, immunoglobulin E; IL, interleukin; TLR, Toll-like receptor; TH2, T helper 2; TNF, tumour necrosis factor. | |
Respiratory syncytial virus and rhinovirus infection in infants and bronchiolitis. Much evidence points to a relationship between severe lower respiratory infections with human respiratory syncytial virus (human RSV; referred to hereafter as RSV) or human rhinoviruses (RVs) early in life and the development of recurrent wheeze followed by asthma diagnosis in later childhood. RSV is a negative-sense single-stranded RNA (ssRNA) virus of the Paramyxoviridae family. It is an important pathogen of young children (<2 years of age) and accounts for ∼70% of severe infantile viral bronchiolitis cases16. Serological monitoring shows that RSV infects nearly all children in their first 2 years of life; however, only ∼40% of children exhibit clinical signs of infection in the lower respiratory tract. For unknown reasons, some children with bronchiolitis develop recurrent respiratory symptoms. RVs are members of the family Picornaviridae, are composed of a positive-sense ssRNA genome of approximately 7.1–7.5 kb and can be grouped into the major and minor groups on the basis of their host receptors: major group viruses bind intercellular adhesion molecule 1 (ICAM1), whereas minor group viruses bind low-density-lipoprotein receptor (LDLR). RVs can also be classified into three groups on the basis of nucleotide sequence identity (RV-A, RV-B and RV-C). The recently proposed RV-C group viruses17,18 have unique sequences at the ICAM1- and LDLR-binding sites, suggesting that they use a different (but currently unknown) receptor19. It is estimated that there are more than 100 serotypes of RVs, meaning that multiple serotypes may infect an individual throughout the year20. Recent cohort studies suggest that lower respiratory infections by RVs are as important as infections by RSV in terms of the risk of later asthma development21,22,23.
Whether these viruses are true causative agents of asthma or (owing to shared risk factors) simply act as markers for an increased risk of asthma development, or even whether both possibilities are true, is unclear and much debated. RSV-mediated bronchiolitis is clearly associated with later asthma development24,25, perhaps owing to an inappropriate or overexuberant immune response to the infection26. Although associations between RSV, wheeze and asthma development are evident, birth cohort studies have shown that 90% of non-atopic wheezers lose this phenotype at school age and have normal lung function, indicating that asthma and virus-induced wheeze may be distinct diseases27. The association of RSV with asthma is most clearly demonstrated in a long-term (18 years) study of a cohort of Swedish children. The most recent update from this study28 reports that, compared with controls, children who have RSV-mediated bronchiolitis in infancy are more likely to be asthmatic at 18 years old (39% versus 9%), show increased sensitization to perennial allergens (41% versus 14%) and have striking persistent and recurrent wheeze (30% versus 1%). Children with a history of bronchiolitis have an enhanced interleukin-4 (IL-4) (that is, T helper 2 (TH2) type) response to RSV and cat allergens, whereas controls have an equally strong interferon-γ (IFNγ) (that is, TH1 type) response to RSV antigens29. However, it is not yet clear whether the inflammation caused by viruses causes long-term respiratory disease or is associated with wheeze and asthma diagnosis because of shared risk factors.
Animal studies have further contributed to our understanding of this phenomenon30, and although many clinical studies are compatible with a direct effect of RSV on the later development of recurrent childhood wheeze, it has been hard to prove this direct causal relationship. The strongest evidence that human RSV-mediated bronchiolitis has long-term effects comes from studies of palivizumab (Synagis; MedImmune), a monoclonal antibody that prevents infection by RSV in infancy. These studies suggest a causal link between infantile RSV-mediated disease and wheeze followed by asthma diagnosis in later childhood; in a study of 191 palivizumab-treated and 230 control (untreated) premature babies who were followed for 24 months, the rates of recurrent wheeze were about 50% lower in those who had prophylactic treatment with palivizumab31. More recently, palivizumab prophylaxis was shown to be associated with a similar substantial protection against recurrent wheeze in children aged 2–5 years32.
Much debate has focused on which of these viruses are more important to asthma development, RSV or the RVs. The answer is likely to be complicated and may involve host genetics and susceptibility factors as well as the nature of the viruses themselves. It is possible that RSV and RVs contribute to asthma development in different ways. A recent study has shown that allergic sensitization precedes or is a risk factor for symptomatic infection with RVs, and both sensitization and RV infection are strongly correlated with the occurrence of asthma at 6 years of age33. By contrast, the role of RSV infection in this study seemed to be different, as sensitization did not increase the risk of infection in this cohort.
In the absence of independently conducted, placebo-controlled prospective studies of specific prophylaxis measures, doubt will remain about the nature of the association between infections with these viruses in infancy and the later development of childhood asthma. However, the evidence obtained so far raises hope that preventing such infections will have long-term beneficial effects on asthma development.
Specific asthma phenotypes or endotypes
Stable asthma. Microorganisms are now also thought to be important in the development and pathogenesis of stable asthma. 16S ribosomal RNA gene sequencing was carried out on bronchial brushings or bronchoalveolar lavage (BAL) from patients with stable asthma, patients with chronic obstructive pulmonary disease (COPD) and healthy controls34, and revealed 5,054 bacterial sequences that were unique to the 43 subjects with asthma or COPD, with each bronchus containing a mean of 2,000 bacterial genomes per cm2 surface sampled. Pathogenic proteobacteria, particularly Haemophilus spp., were more frequent in the bronchi of adults with asthma or COPD than in the bronchi of controls. Similar increases in proteobacteria were found in children with asthma. In addition, there was a lack of members of the phylum Bacteroidetes in individuals with asthma when compared with controls, suggesting a potential role for commensals as well as pathogens in influencing asthma34. It is thought that such commensals may stimulate the immune system such that pathogenic species are eliminated if and when infection occurs (Fig. 1). Changes in the abundance and diversity of commensals can also affect subsequent infection by influenza viruses in mice35. The lower airway thus contains a characteristic microbiota that is quantitatively and qualitatively different in patients with stable asthma or COPD than in control subjects34. This study challenges the previously held belief that in healthy individuals the lower airway is a sterile environment that does not support a complex microbial ecology. The diversity of bacteria that has now been revealed suggests that clinically important microorganisms which are unidentifiable by culture have roles in airway diseases such as asthma.
Figure 1. The microbiome is influenced by and may determine factors affecting asthma.
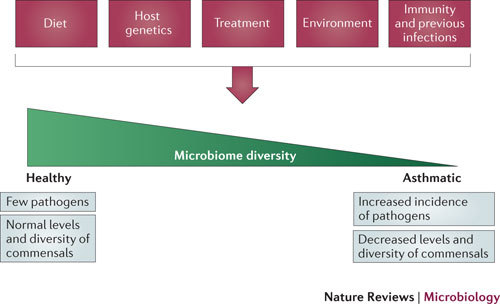
The microbiome is regulated by diet, environmental factors (such as smoking and pollution), treatment, host genetics, and previous infections and host immunity. The diversity of the microbiome may in turn affect asthma development, as suggested by the hygiene hypothesis, and may influence the pathogenic species that contribute to stable and severe asthma and asthma exacerbations.
Invasive pneumococcal infection (IPI), which is caused by the common pathogen Streptococcus pneumoniae, has also been linked to asthma. Adults with stable asthma have been shown to have a greater incidence of S. pneumoniae carriage36, and asthma has been shown to be a risk factor for IPI in both adults37,38 and children39. One study had a particularly important finding37: for low-risk asthma (as defined by the need for prescription medication for asthma) the odds ratio for IPI (versus no IPI) was 2.8-fold, whereas for high-risk asthma (as defined by hospitalization for asthma in the past 12 months) it was over 12-fold. Together, these studies show the importance of asthma in the risk of invasive S. pneumoniae infections, and combined with the other studies they strongly advocate the need for antipneumococcal vaccination for individuals with asthma.
Atopic (allergic) asthma. Atopic asthma is the most common form of asthma and is often characterized by eosinophils in BAL and sputum. Filamentous fungal species of the Aspergillus, Alternaria, Cladosporium, Penicillium and Didymella genera (in the phylum Ascomycota) produce spores that may act as allergens and initiate bronchial asthma in atopic individuals40. Comparative genomics studies have identified proteins that act as allergens in 22 fungal species41, many of which are ubiquitous in the environment and commonly found on grains and cereals. The release and abundance of fungal spores is affected by environmental factors, including season, humidity, rainfall and temperature42. Many fungal aeroallergens increase in abundance in the summer along with pollen levels and have been shown to be further increased by summer storms that include lightning strikes. Indeed, fungal aeroallergens have been associated with asthma epidemics that occur during summer lightning strikes43, possibly owing to spore aerosolization and fragmentation to produce finer particles that easily find their way to the lower respiratory tract.
Neutrophilic (non-eosinophilic) asthma. Neutrophilic asthma is a less common form of the disease that is characterized by neutrophils, with or without eosinophils, being the most abundant granulocyte in the airways of the patient44. Respiratory viral infections have been associated with airway neutrophilia: asthmatic adults experiencing asthma exacerbations show increased airway neutrophilia45, including neutrophil elastase in their sputum46. Respiratory viruses can induce neutrophil chemokine synthesis and release45,47, providing a mechanistic link between neutrophilic asthma and virus infections. Bacterial pathogens such as Haemophilus influenzae, Staphylococcus aureus, Moraxella catarrhalis and Pseudomonas aeruginosa have also been associated with neutrophilic asthma, with one study showing an association between airway neutrophils, increased bacterial load and the neutrophil chemokine IL-8 (also known as CXCL8)48.
Severe or poorly controlled asthma. Recently, a molecular approach was used to investigate the microbiota in poorly controlled asthma49, and the results suggest an association between the families Comamonadaceae, Sphingomonadaceae and Oxalobacteraceae and airway hyper-reactivity. Furthermore, this study shows that the bacteria which are found in the lung differ in health and disease and that the abundance of particular phylotypes, including members of the families Comamonadaceae, Sphingomonadaceae, Oxalobacteraceae and others, is highly correlated with the degree of bronchial hyper-responsiveness (disease severity).
Fungi have also been associated with severe asthma50,51 . In fact, severe asthma with fungal sensitization (SAFS) has been considered as an independent subgroup of severe asthma, and patients with SAFS may benefit from antifungal therapy. In a recent clinical trial of antifungal therapy in patients with SAFS, improvements were seen in antifungal-treated patients (versus placebo-treated patients) in terms of asthma-related quality of life, rhinitis score, lung function and total serum IgE52.
Allergic bronchopulmonary aspergillosis. Allergic bronchopulmonary aspergillosis (ABPA) is a unique form of asthma that is caused by colonization of the lower respiratory tract with Aspergillus spp. Chronic infection with Aspergillus fumigatus can lead to bronchiectasis53, a disease that can occur in parallel with asthma and is characterized by neutrophilic inflammation, epithelial destruction and degradation of the connective tissue matrix. In this disease, the fungus acts as both a source of allergen and a pathogen, and symptoms of ABPA can be attributed to both functions. Aspergillus spp. are widespread fungi and potent sources of allergens54, as shown by the reactivity of human IgE to at least 12 allergens derived from these species55. Recently, the genome of A. fumigatus was sequenced56, and nine new potential allergens were identified on the basis of sequence identity with other known allergens. The predicted and known allergens of A. fumigatus are diverse in nature and include secreted proteases, glucanases and cellulases. Recent work suggests that some A. fumigatus allergens are 'hidden' from the immune system; hydrophobin (RodA) is an immunologically inert surface protein on dormant conidia, and removal of RodA through chemical or genetic means produces conidia that are able to activate the immune system, potentially explaining why fungal species often do not evoke immune responses in healthy individuals57.
Colonization by A. fumigatus results in sensitization to fungal allergens and, subsequently, leads to allergic and/or asthma symptoms, eosinophilic and neutrophilic pneumonia53, and lung damage caused by the extracellular hyphae and lung inflammation. An infection model in mice using a bioluminescent A. fumigatus strain58 revealed that infection results in extensive haemorrhagic bronchopneumonia, characterized by inflammatory lesions on the bronchi and bronchioles, underscoring the potentially aggressive nature of this pathogen in affected individuals. Furthermore, A. fumigatus infection has been linked to IgE sensitization, airway neutrophilia and decreased lung function in ABPA, and antifungal therapy has also recently been shown to be effective in patients with ABPA59.
Microorganisms in asthma exacerbations. Respiratory viruses are the major viruses associated with asthma exacerbations, accounting for 50–60% of exacerbations in all age groups60,61 (Fig. 2). RV-C may represent a group that causes more severe exacerbations, although how this occurs is unknown18. Epidemiological studies have shown that, in the northern hemisphere, infections by RVs result in a marked increase in emergency room admissions owing to asthma exacerbations. In fact, this 'asthma epidemic' has been shown to coincide with Labour Day in Canada60, which is in the third week of September, ∼2 weeks after children return to school, highlighting the important role of school-age children as vectors of virus-induced asthma exacerbations. Mouse models of infection with major and minor group RVs62,63 and of RV-mediated exacerbations of airway allergen challenge62,64 have recently been developed and used to demonstrate the ability of RV infection to augment airway inflammation caused by allergen sensitization and challenge.
Figure 2. Viruses and bacteria associated with asthma exacerbations.
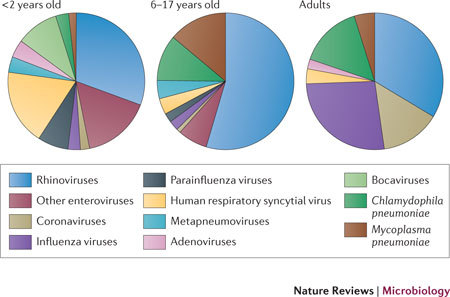
The prevalence of viruses and bacteria in young children (<2 years old), older children (6–17 years old) and adults, presented as median percentages from several studies (data were obtained from a recent review134). Enterovirus estimations in adults and bocavirus estimations in 6–17 year olds and in adults may be under-represented owing to that data not being available in published studies.
Despite common bacterial infections first being suggested to have a role in asthma exacerbations as early as the 1940s65, it was not thought that such infections were important in the pathogenesis of asthma exacerbations66 until recently. However, we now realize that patients with asthma have an increased susceptibility to respiratory bacterial infections37,38,39 and increased carriage of pathogenic respiratory bacteria (identified by both culture methods36 and molecular techniques34). Birth cohort studies have shown a link between early-life carriage of the respiratory bacteria S. pneumoniae, H. influenzae and M. catarrhalis and later development of recurrent wheeze in young children born to mothers with asthma67. A recent study of the same children showed that acute wheezing episodes during the first 3 years of life are equally associated with both bacterial and viral infections, showing that bacteria may contribute to asthma exacerbations independently of viruses68.
The atypical bacteria Mycoplasma pneumoniae and Chlamydophila pneumoniae are common respiratory pathogens and have also been associated with asthma, wheeze and asthma exacerbations in both adults and children69,70,71. Detection rates for these bacteria can be as high as 80% in patients with these conditions69, and serological positivity rates for these atypical bacteria are often 40–60%69,71, indicating that viral and atypical bacterial infections probably interact to increase the risk of asthma exacerbations. The macrolide antibiotic clarithromycin has previously shown efficacy in patients with stable asthma who are known to be carrying either M. pneumoniae or C. pneumoniae; in such patients, the drug improves lung function and reduces tumour necrosis factor (TNF) production72. More recently, the ketomacrolide telithromycin has shown efficacy in patients with asthma exacerbations, resulting in substantial reductions in the asthma symptom score and improvements in lung function71. However, the effects of telithromycin were found to be not necessarily related to C. pneumoniae or M. pneumoniae exposure, suggesting that telithromycin has additional protective properties that are independent of its antibacterial properties.
Microorganism-specific properties
The identification of key microorganisms that are linked with asthma development or asthma pathogenesis raises the important question of which properties of these microorganisms promote asthma. The microorganisms that commonly infect different areas of the lower airway are shown in Fig. 3. Some of these microorganisms may be found systemically in more severe disease, as has been shown for RVs73. A key feature of a microorganism in promoting asthma is the ability to induce lung inflammation, injury, or repair and remodelling. Many of the innate receptors that recognize respiratory fungi, viruses or bacteria, including TLRs, RIG-I-like helicases and NOD-like receptors, activate the nuclear factor-κB (NF-κB) family of transcription factors, which induce more than 100 pro-inflammatory and host response genes74. RSV and RVs induce a range of pro-inflammatory cytokines, chemokines, growth factors, adhesion molecules and mucins. Mucins cause mucous plugging of the airway and may provide a substrate for bacterial colonization, whereas cytokines facilitate cellular chemotaxis, and activation and proliferation of immune cells in the infected airway45,75 . Microorganisms may damage and compromise the integrity of the airway by infecting airway epithelial cells, causing cell death76 and shedding77. They can also affect epithelial permeability, leading to increased airway inflammation and creating opportunities for increased infection78, allergen uptake79 or exposure to environmental pollutants, all of which are important contributing factors in asthma (Fig. 4).
Figure 3. Microorganisms involved in asthma and their niches.
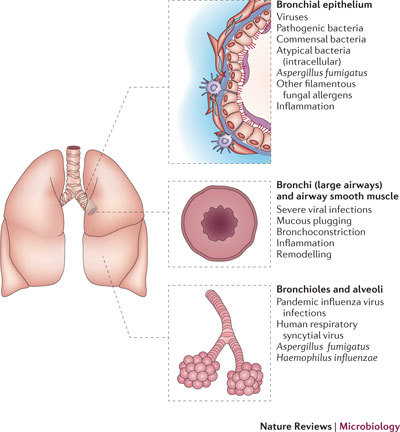
A cross-section of the human lower respiratory tract, showing sites of infection for different microorganisms and the effects that they have on airway function.
Figure 4. Viruses and bacteria induce airway inflammation.
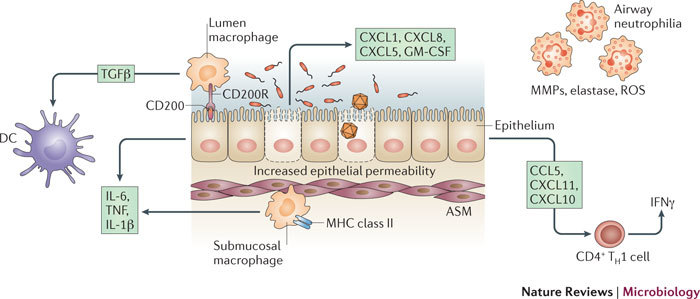
Bacterial and viral infections of the airways activate immune and structural cells, promoting inflammation and influencing responses to other pathogens, allergens and pollution. Infection of airway epithelium by both viruses and bacteria induces neutrophil chemokines (CXC-chemokine ligand 1 (CXCL1; also known as GROα), CXCL5 (also known as ENA78) and CXCL8 (also known as IL-8)), and T helper 1 (TH1) type chemokines (CC-chemokine ligand 5 (CCL5; also known as RANTES), CXCL10 (also known as IP10) and CXCL11 (also known as ITAC)), and increases epithelial permeability. Neutrophils secrete several mediators that can contribute to inflammation and the integrity of the airway, including matrix metalloproteinases (MMPs), elastase and reactive oxygen species (ROS). Epithelial cells normally express luminal CD200, which, when bound to its receptor, CD200R, on lumen macrophages, prevents the macrophage response to inflammatory stimuli. Lumen macrophages are normally inhibitory, as they produce transforming growth factor-β (TGFβ), which inhibits inflammatory airway dendritic cells (DCs). A higher epithelial permeability or epithelial damage as a result of infection allows the unrestrained submucosal macrophages access to the allergen and to other stimuli, and this can promote overzealous inflammation. Submucosal macrophages and epithelial cells are triggered by exposure to bacteria or viruses (and their products) to produce interleukin-1β (IL-1β), IL-6 and tumour necrosis factor (TNF), which promote inflammation. ASM, airway smooth muscle; GM-CSF, granulocyte–macrophage colony-stimulating factor; IFNγ, interferon-γ; MHC, major histocompatibility complex; TH1, T helper 1.
Respiratory pathogens can also induce mediators that are required for airway repair, resulting in airway scarring or remodelling. These processes include angiogenesis, increased ASM proliferation and hypertrophy, and thicker basement membranes owing to collagen and fibronectin deposition, as well as the generation of new lymphatic vessels that ultimately result in thicker airway walls and reduced lung function. Whether or not viral or bacterial infections directly initiate this process is unclear; however, what is clear is that respiratory infections, including by Mycoplasma pulmonis, RVs, RSV and A. fumigatus spores, can induce some of these pro-angiogenic mediators, including fibroblast growth factors and vascular endothelial growth factors80,81,82, and the structural proteins perlecan (also known as HSPG) and collagen83, the building blocks of airway remodelling.
Microorganisms and asthma treatment
Asthma treatment often requires daily treatment with a range of immunomodulatory or immunosuppressive agents, such as macrolide antibiotics or glucocorticoids, suggesting another way that microorganisms interact with asthma. Surprisingly, scant attention has been paid to the possible effects of such treatments on an individual's microbiota and their antimicrobial responses. However, some studies provide indirect evidence for an immunosuppressive effect. For example, increases in bacterial pneumonia have been noted in patients with COPD who are treated with glucocorticoid therapy84. Furthermore, an increased incidence of upper respiratory tract infections following treatment with the leukotriene receptor antagonist montelukast has been reported in young children with asthma85. It is also interesting to speculate whether the fact that asthma is a risk factor for IPI, as previously discussed, is directly attributable to asthma therapies such as glucocorticoids. Conversely, there is some experimental evidence suggesting that the action of glucocorticoids confers benefits to antimicrobial immunity. In vitro, glucocorticoids have been shown to induce a number of antimicrobial peptides, pentraxins, collectins and complement proteins86. Furthermore, clinical studies support the benefits of glucocorticoid use: in preschool children, the use of glucocorticoids for 2 years reduces asthma exacerbations (as measured by exacerbations that require systemic glucocorticoid administration and hospital admissions)87.
Although several studies support the use of macrolide antibiotics for the treatment of atypical pathogens in patients with asthma exacerbations88, it is important to consider how the microbiome is affected by macrolides or other antibiotics and how this might affect asthma and the host responses to infection. Treatment of mice with the antibiotics vancomycin, neomycin, metronidazole and ampicillin greatly changes their airway and gut microbiome and impairs their antiviral responses to influenza viruses35. In human studies, it is currently unclear whether the genera that seem to colonize the airways of patients with asthma or COPD and of individuals without asthma or COPD differ because of treatment-specific effects34. More research is needed to understand whether the microbiome of patients with asthma is actually a consequence of active therapy. Further large clinical trials with such treatments are required to understand the relationships between microorganisms and asthma treatments.
Host responses and asthma
Infections or environmental stresses may induce alterations in epithelial cells and underlying stromal cells, and these alterations may modify the response of these cells to further stimulation or may influence barrier function. For example, enhanced permeability caused by virus-induced cytopathology may lead to inhaled allergens, pollutants and other irritants having greater access to basal cells and the underlying airway tissue, which could be detrimental because submucosal antigen-presenting cells (APCs) are not restrained like their airway lumen counterparts. Epithelial cells orchestrate innate immune regulation in the airway lumen, preventing an inflammatory response to non-injurious stimuli by controlling TLR signalling and phagocytosis through the expression of surfactant proteins89. Epithelial cells also express mucin 1 (MUC1), which suppresses alveolar macrophage responses by binding to TLRs90.
IL-10 and transforming growth factor-β (TGFβ) from airway epithelial cells can influence the immune milieu by raising the threshold of activation of airway macrophages91 and driving the expression of the inhibitory receptor CD200R on alveolar macrophages. The ligand for CD200R, CD200 (also known as OX2), is expressed on the luminal aspect of the airway epithelium, and the interaction of these two proteins prevents the activation of alveolar macrophages in the presence or absence of inflammatory stimuli92. Thus, macrophages in the airspaces are less proficient at antigen presentation than submucosal macrophages, are poorly phagocytic and secrete prostaglandins and TGFβ to reduce T cell responses, leading to reversible T cell inactivation93. Alveolar macrophages also actively inhibit interdigitating dendritic cells (DCs) in the airways94, which in turn secrete IL-10. APCs in the submucosal tissues are less restrained by these factors simply because the cells are not exposed to these epithelium-derived innate suppressors. Should any of these pathways be altered through infection, the less restrained APCs may recognize innocuous antigens or allergens, ultimately contributing to inflammation and disease.
Recognition of respiratory microorganisms by pattern recognition receptors can inadvertently initiate or exacerbate an unrelated inflammatory condition. For instance, TLR agonists may act as microbial adjuvants that enhance the recognition of concurrent allergens95. The major cat allergen Fel d 1 is often contaminated with TLR ligands derived from microorganisms found in the feline oral cavity, and the house dust mite often contains the commensal fungus Aspergillus penicillioides, which activates TLR2 and TLR4. These allergen preparations can contain DNA from Gram-negative microorganisms96,97 . The relationship between TLRs and allergens has recently become more complex, with evidence showing that allergens themselves may be direct TLR agonists98,99 . Molecular mimicry between allergens and microbial components100 raises the idea that TLR-mediated recognition of allergens may be an important part of sensitization to these molecules.
Infection also causes the recruitment of APCs in high numbers and for long periods following pathogen clearance. For example, cells expressing CD11c (also known as integrin αX) display enhanced antigen presentation long after the resolution of an acute infection by influenza virus or RSV101,102 . Such cells are long lasting103, suggesting that an enhanced mature APC compartment assists immunological responses on subsequent exposure to environmental allergens. This would also be facilitated by an increase in tertiary lymphoid tissue in the lung parenchyma, which is a common feature following respiratory tract infection104, in human asthma105 and in experimental mouse models of asthma106.
Successive infections may also alter the lung environment and affect the delicate balance between host, pathogen and chronic airway disease. Bacterial infections are known to follow influenza virus infections; indeed, many of the individuals who died in the influenza pandemics exhibited coexisting bacterial pneumonia107. This has implications for asthma: what is assumed to be a virus-induced asthma exacerbation may in fact include a secondary bacterial infection. Data suggesting that this does occur have been reported for C. pneumoniae69, but further studies are required to determine whether secondary bacterial infections commonly follow viral infections in asthma. Thus, by causing barrier disruption, viruses may initiate a cascade that, as described above, allows environmental bacteria better access to the underlying tissue (Fig. 4).
Microorganism and host factor synergy
T helper 2 type immunity. TH2 cells are CD4+ T cells that produce a defined array of cytokines — classically, IL-4, IL-5, IL-9 and IL-13 — and home to sites of inflammation. TH2 cells are involved in defence against gastrointestinal nematodes and are also strongly implicated in the pathogenesis of asthma and asthma exacerbations. Case control studies show a clear link between respiratory virus infection together with allergen exposure in sensitized children108 and adults109 in increasing the risk of hospital admissions due to asthma exacerbations. Furthermore, experimental RV infection models have shown that patients with asthma have more severe lower respiratory tract symptoms and greater reductions in lung function than control patients, and have increases in bronchial hyper-reactivity75. Exacerbation severity is strongly correlated with both TH2 type cytokine production from BAL CD4+ T cells and viral load. RV infection in mouse asthma models supports these observations, as RVs enhance TH2 type cytokine production by sensitized mice that are simultaneously challenged with aeroallergen62. How viral infection can synergize with or act additively to TH2 type immunity is shown in Fig. 5.
Figure 5. Respiratory viruses interact with allergens in an additive or synergistic manner, promoting asthma.
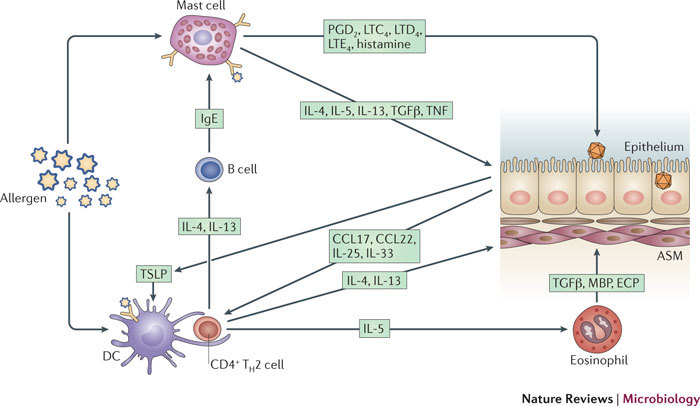
Following sensitization, allergen presentation by airway dendritic cells (DCs) facilitates the promotion of T helper 2 (TH2) cells. Viruses infect epithelial cells, stimulating the release of TH2 cell-promoting chemokines CC-chemokine ligand 17 (CCL17) and CCL22, and cytokines thymic stromal lymphopoietin (TSLP), interleukin-25 (IL-25) and IL-33. The TH2 type chemokines attract TH2 cells into the airway, and these in turn secrete IL-4, IL-5 and IL-13. IL-5 promotes eosinophilia, and the resultant eosinophils release the inflammatory mediators major basic protein (MBP), eosinophil cationic protein (ECP) and transforming growth factor-β (TGFβ), inducing inflammation in the airway smooth muscle (ASM). IL-4 and IL-13 cause antibody class switching to immunoglobulin E in B cells, so that B cells secrete allergen-specific IgE. This antibody then binds mast cells, and crosslinking of the allergen on mast cell-bound IgE causes mast cell degranulation and release of preformed mediators, including histamine, prostaglandin (PGD2) and leukotrienes (LTC4, LTD4 and LTE4). These mediators cause bronchoconstriction and further airway inflammation. Mast cells also produce the TH2 cytokines IL-4 and IL-13, as well as other cytokines, including TGFβ and tumour necrosis factor (TNF), promoting further TH2 type immune responses and inflammation.
Excessive TH2 type responses are implicated in the pathogenesis of RSV-mediated bronchiolitis110, and increased production of IL-5 by T cells at birth is associated with a greater risk of severe respiratory infection111. Genetic associations also link polymorphisms in the TH2 gene locus to severe RSV-mediated disease112,113 . Increased TH2 type responses are reported in peripheral blood mononuclear cells and sputum preceding acute asthma exacerbations in vivo and also in mixed TH1–TH2 cell populations during acute exacerbations in vivo114. TH2 type immunity encompasses structural cells in addition to TH2-polarized CD4+ T cells. Bronchial epithelial cells (BECs) express CC-chemokine ligand 17 (CCL17; also known as TARC), a chemokine that binds with high affinity to CC-chemokine receptor 4 (CCR4), which is abundantly expressed on TH2 cells. In mice, RSV was found to induce CCL17 production, and this finding was reinforced in a TH2-biasing model in which mice were first vaccinated with recombinant vaccinia virus expressing RSV major surface glycoprotein G and then infected with RSV115. In vitro, production of CCL17 by human BECs infected with human RSV requires the presence of TH2 type cytokines and is associated with activation of both NF-κB and STAT6 (signal transducer and activator of transcription 6)115. Recently, the signalling molecule endoplasmic reticulum IFN stimulator (ERIS; also known as TMEM173) has been shown to induce STAT6 activation in viral infections116, providing a potential mechanism for how viruses induce TH2 type cytokines or chemokines.
Thymic stromal lymphopoietin (TSLP) is a TH2 cell-promoting cytokine that is produced by BECs and can be induced by both bacterial (TLR2 and TLR9 activating) and viral (TLR3 and TLR8 activating) ligands117. This cytokine matures the DC network and, in humans, enhances the cytolytic activity of CD8+ T cells and the production of TH2 type cytokines, leading to enhanced airway hyper-reactivity. Furthermore, mast cells in the bronchial mucosa of patients with asthma express the TSLP receptor. Together with IL-1, supernatants from TLR2-activated airway epithelial cells induce mast cell production of IL-5 and IL-13, which have pivotal roles in the development and maintenance of asthma117. Both RV- and RSV-infected BECs produce TSLP. Human RSV-induced TSLP production stimulates DC expression of the TH2-associated molecules CCL17 and OX40 ligand (OX40L; also known as TNFSF4)118. RV-induced TSLP production is synergistically enhanced by IL-4 (Ref. 119). BECs from patients with asthma also produce higher levels of TSLP than BECs from healthy individuals when polyinosinic–polycytidylic acid stimulation is used as a surrogate for viral infection120. Thus, by producing TSLP in response to infection, the airway epithelium may orchestrate the pathophysiology of asthma.
IL-25 is structurally related to members of the IL-17 cytokine family but, like TSLP, is another TH2 cell-amplifying cytokine that is expressed by a number of different cells, including BECs. A recent study using a mouse RSV infection model demonstrated the importance of IL-25 in regulating airway disease following viral infection. Natural killer cell-depleted mice exhibit enhanced TH2 type airway inflammation following RSV infection. Using an IL-25-neutralizing antibody, this RSV-specific TH2 cell-skewed response was shown to be dependent on IL-25 production by airway epithelial cells121.
Allergen-specific IgE is a key mediator in allergic reactions and has a central role in the pathogenesis of asthma. IL-4 and IL-13 stimulate the production of IgE, which binds allergen, allowing crosslinking of IgE receptors on mast cells and basophils; this causes the release of mediators that have profound effects on airway inflammation and hyper-reactivity. Analysis of nasal secretions from RSV-infected infants revealed the presence of RSV-specific IgE, and IgE levels were found to positively correlate with wheeze and higher levels of histamine122, although these studies have been hard to replicate. In mice, primary RSV infection is usually dominated by TH1 type effects but may induce IL-4 and IL-13 expression in the lungs as well as the production of RSV-specific IgE in serum under some conditions. Transfer of RSV-specific IgE to naive mice increases airway hyper-reactivity following RSV infection, an effect that is dependent on expression of high-affinity IgE receptor123. RSV infection in neonatal mice also induces TH2 type cytokine-dependent RSV-specific IgE. Re-infection with RSV 5 weeks later induces a TH2 cell-skewed immune response that is reduced in magnitude by IgE-specific antibody therapy124. A mechanism has been proposed to explain how IgE against a related paramyxovirus, Sendai virus (SeV), enhances TH2 cell-associated airway disease in mice. This suggested mechanism involves infection-induced upregulation of high-affinity IgE receptor on conventional lung DCs. The authors speculate that the later appearance of SeV-specific IgE, after viral clearance, crosslinks IgE receptors (with viral antigen) on conventional DCs, stimulating the production of CCL28, which in turn recruits activated IL-13-producing TH2 cells to the lung125.
Although there is still much to learn, it has become clear that understanding how respiratory infections interact with TH2 type immunity, thereby increasing the severity of allergic airway inflammation, may provide opportunities for the development of novel strategies to treat asthma. Therapies targeting TH2 type immunity and IgE (Table 2) are already yielding encouraging results.
Increased susceptibility to viruses, bacteria or fungi in asthma. The idea that patients with asthma might be more susceptible to viral infections than healthy individuals first surfaced through the study of co-habiting spouse pairs with and without asthma126. Although individuals with and without asthma had the same incidence of RV infections, individuals with asthma had lower respiratory tract symptoms for longer than individuals without asthma, and had an increased severity of symptoms. Cultured primary BECs from adults with allergic asthma exhibit lower levels of RV-induced IFNβ (a type I IFN), lower levels of virus-induced apoptosis and higher RV loads than cells from control adults127. A deficiency in the antiviral type III IFNs, IFNλ1 (also known as IL-29), IFNλ2 (also known as IL-28A) and IFNλ3 (also known as IL-28B), has also been described128. IFNs may therefore be protective against RV-induced asthma exacerbations. However, two similar studies failed to see any difference between individuals with and without asthma regarding RV-induced IFN expression in similar model systems129,130, suggesting that impaired IFN expression is a feature of a subset of patients with asthma. These studies raise the possibility of using IFNβ and IFNλs as therapeutics against asthma exacerbations.
Although epidemiological data clearly show that asthma is a risk factor for IPI, there are few studies that have considered a mechanistic rationale for this. Genome-wide association studies in individuals with asthma or atopy do, however, highlight polymorphisms in innate immune genes such as those encoding TLRs and transcription factors, and such polymorphisms potentially explain the increased susceptibility of these individuals to viruses, bacteria and fungi131,132,133. Deficiencies in the TH1 type cytokines IFNγ and IL-12 in patients with asthma75 also suggest that impaired host defence responses to bacteria are probably implicated. An understanding of how these genetic and cytokine differences affect host susceptibility to infection may help in establishing new therapies for asthma.
Conclusion
Advances in molecular microbiology offer new avenues by which to discover the roles of microorganisms in asthma pathogenesis and exacerbation. There is now overwhelming evidence that asthma can be viewed as a chronic inflammatory condition which is initiated, triggered and maintained by the respiratory microbiota. Microorganisms have roles in the onset and development of asthma but can also be protective (the hygiene hypothesis). It is not yet clear whether some organisms (especially RSV) actually cause atopic disease or are instead associated with atopy because individuals who are prone to atopy suffer more from infantile bronchiolitis. Viruses undoubtedly cause most asthma exacerbations, but some non-viral pathogens may also play a part. Certain fungi and bacteria have diverse roles in driving the pathogenesis and increasing the severity of asthma, and various microorganisms play different parts in different asthma endophenotypes. More research is needed, but our ultimate goal is to identify microbial targets that are amenable to manipulation with vaccines or antimicrobial drugs and thus to reduce the burden of asthma on society.
Acknowledgements
The authors were supported by the Comprehensive Biomedical Research Centre (cBRC) of Imperial College London, UK. M.R.E. was supported by an Asthma UK Fellowship (RF-07_04), S.L.J. received UK Medical Research Council (MRC) project grant G0601236 and European Research Council FP7 Advanced grant 233015, and M.R.E. and S.L.J. were jointly supported by the Asthma UK Clinical Chair (CH1155) and MRC Centre grant G1000758. P.O. is supported by Wellcome Trust strategic and programme grants (090382/Z/09/Z and 083567/Z/07/Z) and the MRC.
Glossary
- Asthma exacerbations
Worsening or increased severity of asthma symptoms.
- Atopy
Reactivity to innocuous substances; caused by immunoglobulin E antibodies.
- Wheeze
Fluid or mucus in the airways, which gives rise to a typical 'wheezy' sound when breathing.
- Odds ratio
A measure of the ratio of the probability that an event will occur in one group versus another.
- Neutrophilia
An abnormal increase in the number of neutrophils in a body fluid such as bronchoalveolar lavage.
- Bronchiectasis
A chronic, irreversible inflammatory condition of the airways, involving tissue destruction and neutrophilic inflammation. Bronchiectasis has a wide range of causes and is associated with bacterial and fungal infections. It can occur in parallel with asthma.
- Cytopathology
Changes that occur to a tissue, at the single-cell level, during disease. Viral infections typically cause cytopathology in cultured cells, and this can be used as a diagnostic marker.
- Surfactant
A substance that interferes with the association of two liquids or a solid and a liquid. Detergents are surfactants that function by lowering the surface tension of liquids.
- Adjuvants
Substances that enhance adaptive immune responses (both B cell and T cell responses) to immunogens. Adjuvants are not recognized by T cells or B cells themselves but may function by activating specific cells or acting as a long-term depot for slow and effective immunogen release.
- Tertiary lymphoid tissue
Lymphoid tissue that develops only after stimulation with antigen, such as in the process of inflammation. The lymphocytes of tertiary lymphoid tissue are typically derived from primary or secondary lymphoid tissue.
- Parenchyma
The body or bulk of a tissue, such as the lungs. The lung parenchyma typically encompasses the alveoli and cells within.
Biographies
Michael R. Edwards is a lecturer in respiratory medicine and infections at Imperial College London, UK. He received his Ph.D. in immunology in 2001 from the University of New South Wales, Sydney, Australia, and then moved to Imperial College London. He was awarded an Asthma UK Fellowship in 2007 and is interested in host susceptibilities to rhinovirus infection in asthma exacerbations.
Nathan W. Bartlett is a lecturer in respiratory medicine. He initially worked in virology at the University of Oxford, UK, and then at Imperial College London, but then moved to the Department of Respiratory Medicine (Imperial), where he led the development of the first mouse models of rhinovirus infection and rhinovirus-induced asthma exacerbation. His current interests include mechanisms of virus–allergen interactions in asthma exacerbations.
Tracy Hussell is Professor of Inflammatory Disease at Imperial College London. She has recently been appointed as Director of the Manchester Collaborative Centre for Inflammation Research (MCCIR).Her work involves innate immunity in the lung in the absence of, presence of or following inflammation, focusing on bacterial susceptibility after acute and chronic inflammation and on therapeutic strategies to rectify prolonged alterations.
Peter Openshaw directs the Centre for Respiratory Infection at Imperial College London. He researches the regulation of immune responses in viral infection and leads a consortium to study influenza virus pathogenesis in hospitalized patients (MOSAIC). He is Vice-President for the European Scientific Working Group on Influenza (ESWI) and advises the UK government on influenza virus and human respiratory syncytial virus-mediated disease.
Sebastian L. Johnston is Professor of Respiratory Medicine & Allergy at Imperial College London and Director of the Medical Research Council (MRC) & Asthma UK Centre in Allergic Mechanisms of Asthma. Notable discoveries of his include establishing the viral aetiology of asthma and chronic obstructive pulmonary disease (COPD) exacerbations, and discovering novel mechanisms of susceptibility to and treatments for viral infection in patients with asthma and COPD.
Competing interests
The authors declare no competing financial interests.
References
- 1.Bateman ED, et al. Global strategy for asthma management and prevention: GINA executive summary. Eur. Respir. J. 2008;31:143–178. doi: 10.1183/09031936.00138707. [DOI] [PubMed] [Google Scholar]
- 2.Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;368:804–813. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- 3.Lotvall J, Svedmyr N. Salmeterol: an inhaled β2-agonist with prolonged duration of action. Lung. 1993;171:249–264. doi: 10.1007/BF03215869. [DOI] [PubMed] [Google Scholar]
- 4.Matricardi PM, et al. Exposure to foodborne and orofecal microbes versus airborne viruses in relation to atopy and allergic asthma: epidemiological study. BMJ. 2000;320:412–417. doi: 10.1136/bmj.320.7232.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leonardi-Bee J, Pritchard D, Britton J. Asthma and current intestinal parasite infection: systematic review and meta-analysis. Am. J. Respir. Crit. Care Med. 2006;174:514–523. doi: 10.1164/rccm.200603-331OC. [DOI] [PubMed] [Google Scholar]
- 6.Okada H, Kuhn C, Feillet H, Bach JF. The 'hygiene hypothesis' for autoimmune and allergic diseases: an update. Clin. Exp. Immunol. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ege MJ, et al. Exposure to environmental microorganisms and childhood asthma. N. Engl. J. Med. 2011;364:701–709. doi: 10.1056/NEJMoa1007302. [DOI] [PubMed] [Google Scholar]
- 8.Strachan DP. Family size, infection and atopy: the first decade of the “hygiene hypothesis”. Thorax. 2000;55(Suppl. 1):S2–S10. doi: 10.1136/thorax.55.suppl_1.S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez FD. The coming-of-age of the hygiene hypothesis. Respir. Res. 2001;2:129–132. doi: 10.1186/rr48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pakarinen J, et al. Predominance of Gram-positive bacteria in house dust in the low-allergy risk Russian Karelia. Environ. Microbiol. 2008;10:3317–3325. doi: 10.1111/j.1462-2920.2008.01723.x. [DOI] [PubMed] [Google Scholar]
- 11.Rintala H, Pitkaranta M, Toivola M, Paulin L, Nevalainen A. Diversity and seasonal dynamics of bacterial community in indoor environment. BMC Microbiol. 2008;8:56. doi: 10.1186/1471-2180-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee L, Tin S, Kelley ST. Culture-independent analysis of bacterial diversity in a child-care facility. BMC Microbiol. 2007;7:27. doi: 10.1186/1471-2180-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nembrini C, et al. Bacterial-induced protection against allergic inflammation through a multicomponent immunoregulatory mechanism. Thorax. 2011;66:755–763. doi: 10.1136/thx.2010.152512. [DOI] [PubMed] [Google Scholar]
- 14.Conrad ML, et al. Maternal TLR signaling is required for prenatal asthma protection by the nonpathogenic microbe Acinetobacter lwoffii F78. J. Exp. Med. 2009;206:2869–2877. doi: 10.1084/jem.20090845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maslowski KM, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall CB, et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009;360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmenberg AC, et al. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science. 2009;324:55–59. doi: 10.1126/science.1165557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lau SK, et al. Clinical features and complete genome characterization of a distinct human rhinovirus (HRV) genetic cluster, probably representing a previously undetected HRV species, HRV-C, associated with acute respiratory illness in children. J. Clin. Microbiol. 2007;45:3655–3664. doi: 10.1128/JCM.01254-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bochkov YA, et al. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nature Med. 2011;17:627–632. doi: 10.1038/nm.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makela MJ, et al. Viruses and bacteria in the etiology of the common cold. J. Clin. Microbiol. 1998;36:539–542. doi: 10.1128/jcm.36.2.539-542.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemanske RFJr, et al. Rhinovirus illnesses during infancy predict subsequent childhood wheezing. J. Allergy Clin. Immunol. 2005;116:571–577. doi: 10.1016/j.jaci.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 22.Guilbert TW, et al. Decreased lung function after preschool wheezing rhinovirus illnesses in children at risk to develop asthma. J. Allergy Clin. Immunol. 2011;128:532–538.e10. doi: 10.1016/j.jaci.2011.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson DJ, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008;178:667–672. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henderson J, et al. Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma, atopy and wheeze: a longitudinal birth cohort study. Pediatr. Allergy Immunol. 2005;16:386–392. doi: 10.1111/j.1399-3038.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 25.Carroll KN, et al. The severity-dependent relationship of infant bronchiolitis on the risk and morbidity of early childhood asthma. J. Allergy Clin. Immunol. 2009;123:1055–1061.e1. doi: 10.1016/j.jaci.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smyth RL, Openshaw PJ. Bronchiolitis. Lancet. 2006;368:312–322. doi: 10.1016/S0140-6736(06)69077-6. [DOI] [PubMed] [Google Scholar]
- 27.Illi S, et al. Perennial allergen sensitisation early in life and chronic asthma in children: a birth cohort study. Lancet. 2006;368:763–770. doi: 10.1016/S0140-6736(06)69286-6. [DOI] [PubMed] [Google Scholar]
- 28.Sigurs N, et al. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax. 2010;65:1045–1052. doi: 10.1136/thx.2009.121582. [DOI] [PubMed] [Google Scholar]
- 29.Pala P, et al. Enhanced IL-4 responses in children with a history of respiratory syncytial virus bronchiolitis in infancy. Eur. Respir. J. 2002;20:376–382. doi: 10.1183/09031936.02.00249902. [DOI] [PubMed] [Google Scholar]
- 30.Culley FJ, Pollott J, Openshaw PJ. Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J. Exp. Med. 2002;196:1381–1386. doi: 10.1084/jem.20020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simoes EA, et al. Palivizumab prophylaxis, respiratory syncytial virus, and subsequent recurrent wheezing. J. Pediatr. 2007;151:34–42. doi: 10.1016/j.jpeds.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 32.Simoes EA, et al. The effect of respiratory syncytial virus on subsequent recurrent wheezing in atopic and nonatopic children. J. Allergy Clin. Immunol. 2010;126:256–262. doi: 10.1016/j.jaci.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson DJ, et al. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am. J. Respir. Crit. Care Med. 2011;185:281–285. doi: 10.1164/rccm.201104-0660OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hilty M, et al. Disordered microbial communities in asthmatic airways. PLoS ONE. 2010;5:e8578. doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ichinohe T, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl Acad. Sci. USA. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jounio U, et al. Pneumococcal carriage is more common in asthmatic than in non-asthmatic young men. Clin. Respir. J. 2010;4:222–229. doi: 10.1111/j.1752-699X.2009.00179.x. [DOI] [PubMed] [Google Scholar]
- 37.Klemets P, et al. Risk of invasive pneumococcal infections among working age adults with asthma. Thorax. 2010;65:698–702. doi: 10.1136/thx.2009.132670. [DOI] [PubMed] [Google Scholar]
- 38.Talbot TR, et al. Asthma as a risk factor for invasive pneumococcal disease. N. Engl. J. Med. 2005;352:2082–2090. doi: 10.1056/NEJMoa044113. [DOI] [PubMed] [Google Scholar]
- 39.Pilishvili T, et al. Risk factors for invasive pneumococcal disease in children in the era of conjugate vaccine use. Pediatrics. 2010;126:e9–e17. doi: 10.1542/peds.2009-2150. [DOI] [PubMed] [Google Scholar]
- 40.O'Hollaren MT, et al. Exposure to an aeroallergen as a possible precipitating factor in respiratory arrest in young patients with asthma. N. Engl. J. Med. 1991;324:359–363. doi: 10.1056/NEJM199102073240602. [DOI] [PubMed] [Google Scholar]
- 41.Bowyer P, Fraczek M, Denning DW. Comparative genomics of fungal allergens and epitopes shows widespread distribution of closely related allergen and epitope orthologues. BMC Genomics. 2006;7:251. doi: 10.1186/1471-2164-7-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oliveira M, Ribeiro H, Delgado JL, Abreu I. The effects of meteorological factors on airborne fungal spore concentration in two areas differing in urbanisation level. Int. J. Biometeorol. 2009;53:61–73. doi: 10.1007/s00484-008-0191-2. [DOI] [PubMed] [Google Scholar]
- 43.Pulimood TB, Corden JM, Bryden C, Sharples L, Nasser SM. Epidemic asthma and the role of the fungal mold Alternaria alternata. J. Allergy Clin. Immunol. 2007;120:610–617. doi: 10.1016/j.jaci.2007.04.045. [DOI] [PubMed] [Google Scholar]
- 44.Macdowell AL, Peters SP. Neutrophils in asthma. Curr. Allergy Asthma Rep. 2007;7:464–468. doi: 10.1007/s11882-007-0071-6. [DOI] [PubMed] [Google Scholar]
- 45.Pizzichini MM, et al. Asthma and natural colds. Inflammatory indices in induced sputum: a feasibility study. Am. J. Respir. Crit. Care Med. 1998;158:1178–1184. doi: 10.1164/ajrccm.158.4.9712082. [DOI] [PubMed] [Google Scholar]
- 46.Wark PA, et al. Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur. Respir. J. 2002;19:68–75. doi: 10.1183/09031936.02.00226302. [DOI] [PubMed] [Google Scholar]
- 47.Johnston SL, et al. Low grade rhinovirus infection induces a prolonged release of IL-8 in pulmonary epithelium. J. Immunol. 1998;160:6172–6181. [PubMed] [Google Scholar]
- 48.Wood LG, Simpson JL, Hansbro PM, Gibson PG. Potentially pathogenic bacteria cultured from the sputum of stable asthmatics are associated with increased 8-isoprostane and airway neutrophilia. Free Rad. Res. 2009;44:146–154. doi: 10.3109/10715760903362576. [DOI] [PubMed] [Google Scholar]
- 49.Huang YJ, et al. Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J. Allergy Clin. Immunol. 2011;127:372–381.e3. doi: 10.1016/j.jaci.2010.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Driscoll BR, Hopkinson LC, Denning DW. Mold sensitization is common amongst patients with severe asthma requiring multiple hospital admissions. BMC Pulm. Med. 2005;5:4. doi: 10.1186/1471-2466-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Driscoll BR, et al. Comparison of skin prick tests with specific serum immunoglobulin E in the diagnosis of fungal sensitization in patients with severe asthma. Clin. Exp. Allergy. 2009;39:1677–1683. doi: 10.1111/j.1365-2222.2009.03339.x. [DOI] [PubMed] [Google Scholar]
- 52.Denning DW, et al. Randomized controlled trial of oral antifungal treatment for severe asthma with fungal sensitization: The Fungal Asthma Sensitization Trial (FAST) study. Am. J. Respir. Crit. Care Med. 2009;179:11–18. doi: 10.1164/rccm.200805-737OC. [DOI] [PubMed] [Google Scholar]
- 53.Wark PA, et al. Induced sputum eosinophils and neutrophils and bronchiectasis severity in allergic bronchopulmonary aspergillosis. Eur. Respir. J. 2000;16:1095–1101. doi: 10.1034/j.1399-3003.2000.16f13.x. [DOI] [PubMed] [Google Scholar]
- 54.Patterson R, Rosenberg M, Roberts M. Evidence that Aspergillus fumigatus growing in the airway of man can be a potent stimulus of specific and nonspecific IgE formation. Am. J. Med. 1977;63:257–262. doi: 10.1016/0002-9343(77)90240-6. [DOI] [PubMed] [Google Scholar]
- 55.Kodzius R, et al. Rapid identification of allergen-encoding cDNA clones by phage display and high-density arrays. Comb. Chem. High Throughput Screen. 2003;6:147–154. doi: 10.2174/1386207033329751. [DOI] [PubMed] [Google Scholar]
- 56.Nierman WC, et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature. 2005;438:1151–1156. doi: 10.1038/nature04332. [DOI] [PubMed] [Google Scholar]
- 57.Aimanianda V, et al. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature. 2009;460:1117–1121. doi: 10.1038/nature08264. [DOI] [PubMed] [Google Scholar]
- 58.Brock M, et al. Bioluminescent Aspergillus fumigatus, a new tool for drug efficiency testing and in vivo monitoring of invasive aspergillosis. Appl. Environ. Microbiol. 2008;74:7023–7035. doi: 10.1128/AEM.01288-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pasqualotto AC, Powell G, Niven R, Denning DW. The effects of antifungal therapy on severe asthma with fungal sensitization and allergic bronchopulmonary aspergillosis. Respirology. 2009;14:1121–1127. doi: 10.1111/j.1440-1843.2009.01640.x. [DOI] [PubMed] [Google Scholar]
- 60.Johnston NW, et al. The September epidemic of asthma exacerbations in children: a search for etiology. J. Allergy Clin. Immunol. 2005;115:132–138. doi: 10.1016/j.jaci.2004.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnston SL, et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bartlett NW, et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nature Med. 2008;14:199–204. doi: 10.1038/nm1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Newcomb DC, et al. Human rhinovirus 1B exposure induces phosphatidylinositol 3-kinase-dependent airway inflammation in mice. Am. J. Respir. Crit. Care Med. 2008;177:1111–1121. doi: 10.1164/rccm.200708-1243OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nagarkar DR, et al. Rhinovirus infection of allergen-sensitized and -challenged mice induces eotaxin release from functionally polarized macrophages. J. Immunol. 2010;185:2525–2535. doi: 10.4049/jimmunol.1000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heise HA. The role of streptococci in bronchial asthma; etiologic significance and treatment with Streptococcus filtrates. Ann. Allergy. 1949;7:240–249. [PubMed] [Google Scholar]
- 66.Pattemore PK, Johnston SL, Bardin PG. Viruses as precipitants of asthma symptoms. I. Epidemiology. Clin. Exp. Allergy. 1992;22:325–336. doi: 10.1111/j.1365-2222.1992.tb03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bisgaard H, et al. Childhood asthma after bacterial colonization of the airway in neonates. N. Engl. J. Med. 2007;357:1487–1495. doi: 10.1056/NEJMoa052632. [DOI] [PubMed] [Google Scholar]
- 68.Bisgaard H, et al. Association of bacteria and viruses with wheezy episodes in young children: prospective birth cohort study. BMJ. 2010;341:c4978. doi: 10.1136/bmj.c4978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wark PA, Johnston SL, Simpson JL, Hensley MJ, Gibson PG. Chlamydia pneumoniae immunoglobulin A reactivation and airway inflammation in acute asthma. Eur. Respir. J. 2002;20:834–840. doi: 10.1183/09031936.02.00192002. [DOI] [PubMed] [Google Scholar]
- 70.Cunningham AF, Johnston SL, Julious SA, Lampe FC, Ward ME. Chronic Chlamydia pneumoniae infection and asthma exacerbations in children. Eur. Respir. J. 1998;11:345–349. doi: 10.1183/09031936.98.11020345. [DOI] [PubMed] [Google Scholar]
- 71.Johnston SL, et al. The effect of telithromycin in acute exacerbations of asthma. N. Engl. J. Med. 2006;354:1589–1600. doi: 10.1056/NEJMoa044080. [DOI] [PubMed] [Google Scholar]
- 72.Kraft M, Cassell GH, Pak J, Martin RJ. Mycoplasma pneumoniae and Chlamydia pneumoniae in asthma: effect of clarithromycin. Chest. 2002;121:1782–1788. doi: 10.1378/chest.121.6.1782. [DOI] [PubMed] [Google Scholar]
- 73.Xatzipsalti M, et al. Rhinovirus viremia in children with respiratory infections. Am. J. Respir. Crit. Care Med. 2005;172:1037–1040. doi: 10.1164/rccm.200502-315OC. [DOI] [PubMed] [Google Scholar]
- 74.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Rev. Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 75.Message SD, et al. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc. Natl Acad. Sci. USA. 2008;105:13562–13567. doi: 10.1073/pnas.0804181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bossios A, et al. Rhinovirus infection induces cytotoxicity and delays wound healing in bronchial epithelial cells. Respir. Res. 2005;6:114. doi: 10.1186/1465-9921-6-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Turner RB, Hendley JO, Gwaltney JMJr. Shedding of infected ciliated epithelial cells in rhinovirus colds. J. Infect. Dis. 1982;145:849–853. doi: 10.1093/infdis/145.6.849. [DOI] [PubMed] [Google Scholar]
- 78.Bassetti S, et al. Dispersal of Staphylococcus aureus into the air associated with a rhinovirus infection. Infect. Control Hosp. Epidemiol. 2005;26:196–203. doi: 10.1086/502526. [DOI] [PubMed] [Google Scholar]
- 79.Marsland BJ, Scanga CB, Kopf M, Le Gros G. Allergic airway inflammation is exacerbated during acute influenza infection and correlates with increased allergen presentation and recruitment of allergen-specific T-helper type 2 cells. Clin. Exp. Allergy. 2004;34:1299–1306. doi: 10.1111/j.1365-2222.2004.02021.x. [DOI] [PubMed] [Google Scholar]
- 80.Volonaki E, et al. Synergistic effects of fluticasone propionate and salmeterol on inhibiting rhinovirus-induced epithelial production of remodelling-associated growth factors. Clin. Exp. Allergy. 2006;36:1268–1273. doi: 10.1111/j.1365-2222.2006.02566.x. [DOI] [PubMed] [Google Scholar]
- 81.Tourdot S, et al. Respiratory syncytial virus infection provokes airway remodelling in allergen-exposed mice in absence of prior allergen sensitization. Clin. Exp. Allergy. 2008;38:1016–1024. doi: 10.1111/j.1365-2222.2008.02974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Baluk P, et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Invest. 2005;115:247–257. doi: 10.1172/JCI200522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuo C, et al. Rhinovirus infection induces extracellular matrix protein deposition by primary bronchial epithelial cells and lung fibroblasts. Respirology. 2011;16:367–377. doi: 10.1111/j.1440-1843.2010.01918.x. [DOI] [PubMed] [Google Scholar]
- 84.Calverley PM, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N. Engl. J. Med. 2007;356:775–789. doi: 10.1056/NEJMoa063070. [DOI] [PubMed] [Google Scholar]
- 85.Bisgaard H, et al. Safety and tolerability of montelukast in placebo-controlled pediatric studies and their open-label extensions. Pediatr. Pulmonol. 2009;44:568–579. doi: 10.1002/ppul.21018. [DOI] [PubMed] [Google Scholar]
- 86.Zhang N, Truong-Tran QA, Tancowny B, Harris KE, Schleimer RP. Glucocorticoids enhance or spare innate immunity: effects in airway epithelium are mediated by CCAAT/enhancer binding proteins. J. Immunol. 2007;179:578–589. doi: 10.4049/jimmunol.179.1.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guilbert TW, et al. Long-term inhaled corticosteroids in preschool children at high risk for asthma. N. Engl. J. Med. 2006;354:1985–1997. doi: 10.1056/NEJMoa051378. [DOI] [PubMed] [Google Scholar]
- 88.Blasi F, Johnston SL. The role of antibiotics in asthma. Int. J. Antimicrob. Agents. 2007;29:485–493. doi: 10.1016/j.ijantimicag.2006.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Henning LN, et al. Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J. Immunol. 2008;180:7847–7858. doi: 10.4049/jimmunol.180.12.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ueno K, et al. MUC1 mucin is a negative regulator of Toll-like receptor signaling. Am. J. Respir. Cell. Mol. Biol. 2008;38:263–268. doi: 10.1165/rcmb.2007-0336RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fernandez S, Jose P, Avdiushko MG, Kaplan AM, Cohen DA. Inhibition of IL-10 receptor function in alveolar macrophages by Toll-like receptor agonists. J. Immunol. 2004;172:2613–2620. doi: 10.4049/jimmunol.172.4.2613. [DOI] [PubMed] [Google Scholar]
- 92.Snelgrove RJ, et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nature Immunol. 2008;9:1074–1083. doi: 10.1038/ni.1637. [DOI] [PubMed] [Google Scholar]
- 93.Balbo P, Silvestri M, Rossi GA, Crimi E, Burastero SE. Differential role of CD80 and CD86 on alveolar macrophages in the presentation of allergen to T lymphocytes in asthma. Clin. Exp. Allergy. 2001;31:625–636. doi: 10.1046/j.1365-2222.2001.01068.x. [DOI] [PubMed] [Google Scholar]
- 94.Holt PG, et al. Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J. Exp. Med. 1993;177:397–407. doi: 10.1084/jem.177.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Eisenbarth SC, et al. Lipopolysaccharide-enhanced, Toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valerio CR, Murray P, Arlian LG, Slater JE. Bacterial 16S ribosomal DNA in house dust mite cultures. J. Allergy Clin. Immunol. 2005;116:1296–1300. doi: 10.1016/j.jaci.2005.09.046. [DOI] [PubMed] [Google Scholar]
- 97.Phipps S, Lam CE, Foster PS, Matthaei KI. The contribution of Toll-like receptors to the pathogenesis of asthma. Immunol. Cell Biol. 2007;85:463–470. doi: 10.1038/sj.icb.7100104. [DOI] [PubMed] [Google Scholar]
- 98.Li DQ, et al. Short ragweed pollen triggers allergic inflammation through Toll-like receptor 4-dependent thymic stromal lymphopoietin/OX40 ligand/OX40 signaling pathways. J. Allergy Clin. Immunol. 2011;128:1318–1325. doi: 10.1016/j.jaci.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hammad H, et al. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nature Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trompette A, et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Beyer M, et al. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J. Allergy Clin. Immunol. 2004;113:127–133. doi: 10.1016/j.jaci.2003.10.057. [DOI] [PubMed] [Google Scholar]
- 102.Kirby AC, Newton DJ, Carding SR, Kaye PM. Pulmonary dendritic cells and alveolar macrophages are regulated by γδ T cells during the resolution of S. pneumoniae-induced inflammation. J. Pathol. 2007;212:29–37. doi: 10.1002/path.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Amano H, et al. Essential contribution of monocyte chemoattractant protein-1/C-C chemokine ligand-2 to resolution and repair processes in acute bacterial pneumonia. J. Immunol. 2004;172:398–409. doi: 10.4049/jimmunol.172.1.398. [DOI] [PubMed] [Google Scholar]
- 104.Moyron-Quiroz JE, et al. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nature Med. 2004;10:927–934. doi: 10.1038/nm1091. [DOI] [PubMed] [Google Scholar]
- 105.Elliot JG, et al. Aggregations of lymphoid cells in the airways of nonsmokers, smokers, and subjects with asthma. Am. J. Respir. Crit. Care Med. 2004;169:712–718. doi: 10.1164/rccm.200308-1167OC. [DOI] [PubMed] [Google Scholar]
- 106.Lee JJ, et al. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J. Exp. Med. 1997;185:2143–2156. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J. Infect. Dis. 2007;195:1018–1028. doi: 10.1086/511989. [DOI] [PubMed] [Google Scholar]
- 108.Murray CS, et al. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax. 2006;61:376–382. doi: 10.1136/thx.2005.042523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Green RM, et al. Synergism between allergens and viruses and risk of hospital admission with asthma: case-control study. BMJ. 2002;324:763. doi: 10.1136/bmj.324.7340.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Legg JP, Hussain IR, Warner JA, Johnston SL, Warner JO. Type 1 and type 2 cytokine imbalance in acute respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 2003;168:633–639. doi: 10.1164/rccm.200210-1148OC. [DOI] [PubMed] [Google Scholar]
- 111.Zhang G, et al. Interleukin-10/interleukin-5 responses at birth predict risk for respiratory infections in children with atopic family history. Am. J. Respir. Crit. Care Med. 2009;179:205–211. doi: 10.1164/rccm.200803-438OC. [DOI] [PubMed] [Google Scholar]
- 112.Bont L, et al. Peripheral blood cytokine responses and disease severity in respiratory syncytial virus bronchiolitis. Eur. Respir. J. 1999;14:144–149. doi: 10.1034/j.1399-3003.1999.14a24.x. [DOI] [PubMed] [Google Scholar]
- 113.Forton JT, et al. Genetic association study for RSV bronchiolitis in infancy at the 5q31 cytokine cluster. Thorax. 2009;64:345–352. doi: 10.1136/thx.2008.102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mamessier E, et al. T-cell activation during exacerbations: a longitudinal study in refractory asthma. Allergy. 2008;63:1202–1210. doi: 10.1111/j.1398-9995.2008.01687.x. [DOI] [PubMed] [Google Scholar]
- 115.Monick MM, et al. Respiratory syncytial virus synergizes with Th2 cytokines to induce optimal levels of TARC/CCL17. J. Immunol. 2007;179:1648–1658. doi: 10.4049/jimmunol.179.3.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen H, et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell. 2011;147:436–446. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 117.Allakhverdi Z, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 2007;204:253–258. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Qiao J, Li A, Jin X. TSLP from RSV-stimulated rat airway epithelial cells activates myeloid dendritic cells. Immunol. Cell Biol. 2010;89:231–238. doi: 10.1038/icb.2010.85. [DOI] [PubMed] [Google Scholar]
- 119.Kato A, Favoreto SJr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 2007;179:1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Uller L, et al. Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-λ in bronchial epithelial cells from donors with asthma. Thorax. 2010;65:626–632. doi: 10.1136/thx.2009.125930. [DOI] [PubMed] [Google Scholar]
- 121.Kaiko GE, Phipps S, Angkasekwinai P, Dong C, Foster PS. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J. Immunol. 2010;185:4681–4690. doi: 10.4049/jimmunol.1001758. [DOI] [PubMed] [Google Scholar]
- 122.Welliver RC, et al. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N. Engl. J. Med. 1981;305:841–846. doi: 10.1056/NEJM198110083051501. [DOI] [PubMed] [Google Scholar]
- 123.Dakhama A, et al. The role of virus-specific immunoglobulin E in airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2004;170:952–959. doi: 10.1164/rccm.200311-1610OC. [DOI] [PubMed] [Google Scholar]
- 124.Dakhama A, et al. Virus-specific IgE enhances airway responsiveness on reinfection with respiratory syncytial virus in newborn mice. J. Allergy Clin. Immunol. 2009;123:138–145. doi: 10.1016/j.jaci.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 125.Grayson MH, et al. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J. Exp. Med. 2007;204:2759–2769. doi: 10.1084/jem.20070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Corne JM, et al. Frequency, severity, and duration of rhinovirus infections in asthmatic and non-asthmatic individuals: a longitudinal cohort study. Lancet. 2002;359:831–834. doi: 10.1016/S0140-6736(02)07953-9. [DOI] [PubMed] [Google Scholar]
- 127.Wark PA, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Contoli M, et al. Role of deficient type III interferon-γ production in asthma exacerbations. Nature Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 129.Bochkov YA, et al. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010;3:69–80. doi: 10.1038/mi.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lopez-Souza N, et al. In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J. Allergy Clin. Immunol. 2009;123:1384–1390.e2. doi: 10.1016/j.jaci.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kormann MS, et al. Toll-like receptor heterodimer variants protect from childhood asthma. J. Allergy Clin. Immunol. 2008;122:86–92. doi: 10.1016/j.jaci.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 132.Schedel M, et al. IRF-1 gene variations influence IgE regulation and atopy. Am. J. Respir. Crit. Care Med. 2008;177:613–621. doi: 10.1164/rccm.200703-373OC. [DOI] [PubMed] [Google Scholar]
- 133.Carvalho A, et al. Polymorphisms in Toll-like receptor genes and susceptibility to pulmonary aspergillosis. J. Infect. Dis. 2008;197:618–621. doi: 10.1086/526500. [DOI] [PubMed] [Google Scholar]
- 134.Papadopoulos NG, et al. Viruses and bacteria in acute asthma exacerbations – a GA2LEN-DARE systematic review. Allergy. 2010;66:458–468. doi: 10.1111/j.1398-9995.2010.02505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Woodruff PG, et al. T helper type 2-driven inflammation defines major sub-phenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009;180:388–395. doi: 10.1164/rccm.200903-0392OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Corren J, et al. Lebrikizumab treatment in adults with asthma. N. Engl. J. Med. 2011;365:1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 137.Borish LC, et al. Interleukin-4 receptor in moderate atopic asthma. A phase I/II randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 1999;160:1816–1823. doi: 10.1164/ajrccm.160.6.9808146. [DOI] [PubMed] [Google Scholar]
- 138.Leckie MJ, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–2148. doi: 10.1016/S0140-6736(00)03496-6. [DOI] [PubMed] [Google Scholar]
- 139.Busse WW, et al. Randomized trial of omalizumab (anti-IgE) for asthma in inner-city children. N. Engl. J. Med. 2011;364:1005–1015. doi: 10.1056/NEJMoa1009705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Morjaria JB, et al. The role of a soluble TNFα receptor fusion protein (etanercept) in corticosteroid refractory asthma: a double blind, randomised, placebo controlled trial. Thorax. 2008;63:584–591. doi: 10.1136/thx.2007.086314. [DOI] [PubMed] [Google Scholar]
- 141.Brehm JM, et al. Serum vitamin D levels and severe asthma exacerbations in the Childhood Asthma Management Program study. J. Allergy Clin. Immunol. 2010;126:52–58.e5. doi: 10.1016/j.jaci.2010.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Urashima M, et al. Randomized trial of vitamin D supplementation to prevent seasonal influenza A in schoolchildren. Am. J. Clin. Nutr. 2009;91:1255–1260. doi: 10.3945/ajcn.2009.29094. [DOI] [PubMed] [Google Scholar]
- 143.Xystrakis E, et al. Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J. Clin. Invest. 2006;116:146–155. doi: 10.1172/JCI21759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kalliomaki M, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet. 2001;357:1076–1079. doi: 10.1016/S0140-6736(00)04259-8. [DOI] [PubMed] [Google Scholar]
- 145.Chen YS, Jan RL, Lin YL, Chen HH, Wang JY. Randomized placebo-controlled trial of lactobacillus on asthmatic children with allergic rhinitis. Pediatr. Pulmonol. 2010;45:1111–1120. doi: 10.1002/ppul.21296. [DOI] [PubMed] [Google Scholar]
- 146.Yoo J, Tcheurekdjian H, Lynch SV, Cabana M, Boushey HA. Microbial manipulation of immune function for asthma prevention: inferences from clinical trials. Proc. Am. Thorac. Soc. 2007;4:277–282. doi: 10.1513/pats.200702-033AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Van LP, et al. Treatment with the TLR7 agonist R848 induces regulatory T-cell-mediated suppression of established asthma symptoms. Eur. J. Immunol. 2011;41:1992–1999. doi: 10.1002/eji.201040914. [DOI] [PubMed] [Google Scholar]
- 148.Xirakia C, et al. Toll-like receptor 7-triggered immune response in the lung mediates acute and long-lasting suppression of experimental asthma. Am. J. Respir. Crit. Care Med. 2010;181:1207–1216. doi: 10.1164/rccm.200908-1255OC. [DOI] [PubMed] [Google Scholar]
- 149.Simon HU, Seelbach H, Ehmann R, Schmitz M. Clinical and immunological effects of low-dose IFN-α treatment in patients with corticosteroid-resistant asthma. Allergy. 2003;58:1250–1255. doi: 10.1046/j.1398-9995.2003.00424.x. [DOI] [PubMed] [Google Scholar]
- 150.Koltsida O, et al. IL-28A (IFN-λ2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol. Med. 2011;3:348–361. doi: 10.1002/emmm.201100142. [DOI] [PMC free article] [PubMed] [Google Scholar]