

Key Points
Adjuvant is a term used to describe a variety of components co-administered with a vaccine that enhance the immunogenicity of vaccine antigens. These components can be broadly divided into two subsets on the basis of their dominant mechanism of action: delivery systems and immune potentiators.
The cells, receptors and signalling pathways of the innate immune system are important targets for improved delivery systems and immune potentiators. The integrated model of host defence indicates that optimal generation of antigen-specific memory (the goal of vaccination) requires innate immune activation.
Recent advances in elucidating the mechanisms of innate immune activation and the links between innate and adaptive responses have shown that numerous natural and synthetic compounds possess specific immune-potentiating activities. These compounds have promise as next generation vaccine adjuvants.
The identification of the imidizaquinoline class of SMIPs (small-molecule immune potentiators) as Toll-like receptor-dependent indicate that the small-molecule platform could be exploited for immune potentiator design and discovery.
Vaccine delivery systems are used to ensure that the antigens and immune potentiators included in a vaccine are able to interact with the appropriate tissues or cells to promote the induction of a potent immune response. Biodegradable microparticles are proving to be attractive delivery systems for both vaccine antigens and adjuvants.
Mucosal vaccine delivery systems are specifically designed to allow vaccines to be successfully applied to mucosal surfaces, mostly involving oral or intranasal administration. Important elements of mucosal delivery systems include components designed to protect antigens from degradation and to promote their interaction with mucosal-associated lymphoid tissue.
Abstract
Adjuvant design has historically had a touch of alchemy at its heart due to its reliance on the complex biology of innate immune activation. However, a new mechanistic understanding of innate immunity, combined with new adjuvant and delivery platforms for exploiting this knowledge, has led to significant advances recently. Although many challenges remain, the field is moving rapidly and the proper tools and methodologies are in place for the use of traditional drug discovery engines in guiding the development of vaccine adjuvants. In this review, we outline the current trends in immune potentiator, delivery system and adjuvant design that will shape the vaccines of the future.
Main
During the past decade, a dramatic shift has occurred in immunological theory, which has found its most practical application in the discovery and design of novel vaccine ADJUVANTS. This change in thinking has been driven by an increased appreciation that activation of the INNATE IMMUNE SYSTEM initiates, amplifies and drives antigen-specific immune responses. Moreover, the identification of discrete cell types, specific receptors and the signalling pathways involved in activation of innate immunity has provided a multitude of new targets for exploitation by the development of novel adjuvants (IMMUNE POTENTIATORS and DELIVERY SYSTEMS).
Combined with these factors, there has been an increasing emphasis in the field of vaccines on the need for improved safety (for example, subunit vaccines), along with improved efficacy against particularly insidious pathogens (for example, human immunodeficiency virus (HIV) and hepatitis C virus (HCV)). More recently, the threat of a bio-terrorist attack has added an even greater sense of urgency to these efforts. As a result, there has been a dramatic increase in efforts to target and manipulate the innate immune response with improved vaccine adjuvants, which has coincided with a greater mechanistic understanding of how this might be optimally achieved.
Vaccines from practice to theory
Vaccination is considered by the World Health Organization to be the most cost-effective strategy for controlling infectious disease1. In terms of direct effects on public health, the development of the presently available vaccines is second only to the introduction of a clean water supply. Vaccination works by manipulating the body's immune system, preparing it for the rapid elimination of infectious agents and/or their toxic products. From a mechanistic perspective, vaccines select, activate and expand memory B and T cells of the immune system, which are then poised to respond rapidly and specifically to subsequent pathogen exposure.
Since Edward Jenner's first successful vaccination against smallpox, which can be considered the birth of immunology as a scientific discipline, vaccine design and discovery has been dominated by pragmatic considerations — one uses what works, regardless of understanding why it works. In recent years, however, basic immunological research has started to make a greater contribution to the vaccine development process. There are many factors driving this re-invigorated search for the theoretical and mechanistic underpinnings of vaccination, including the need for the development of safer, better defined and more effective vaccines against a number of important diseases for which vaccines do not presently exist (for example, HIV, HCV, Neisseria meningitides and severe acute respiratory syndrome (SARS)). Furthermore, recent breakthroughs in immunology have revealed numerous new targets and mechanisms of the innate arm of the immune system that can be manipulated by improved adjuvants to produce better vaccines.
The integrated model of host defence
Protective immunity against pathogen exposure is achieved by the integration of two distinct arms of the immune response, the innate and antigen-specific (also called adaptive) responses. The innate response acts early after infection (within minutes), detecting and responding to broad cues from invading pathogens. By contrast, the adaptive response takes time (days to weeks) to become effective, but provides the fine antigenic specificity required for complete elimination of the pathogen and the generation of IMMUNOLOGICAL MEMORY. Antigen-independent recognition of pathogens by the innate immune system leads to the immediate mobilization of immune effector and regulatory mechanisms that provide the host with three crucial survival advantages: rapid initiation of the immune responses (both innate and adaptive) and creation of the inflammatory and co-stimulatory context for antigen recognition; establishment of a first line of defence, which holds the pathogen in check during the maturation of the adaptive response; and steering of the adaptive immune response towards the cellular or humoural elements that are most appropriate for protection against the particular infectious agent. So, although the overall objective of vaccination is the activation of antigen-specific immunity, vaccines cannot optimally achieve this goal without effectively activating the pathogen-detection mechanisms of the innate immune response.
Vaccine adjuvants are broadly defined by their functional ability to enhance in vivo immunogenicity (that is, antigen-specific responses) of the antigens with which they are co-administered. As such, adjuvants represent important components of most of the successful vaccines, particularly those based on subunits of pathogens, including isolated fractions of the killed pathogens, or recombinant antigens. However, as a consequence of the growing appreciation of innate immune mechanisms, and the details of antigen processing and presentation, more sophisticated definitions of adjuvants are required. Therefore, we have begun to separate both traditional and novel adjuvants into two main categories (immune potentiators and delivery systems) on the basis of their dominant mechanism(s) of action2. Whereas delivery systems mainly function to localize vaccine components and to target vaccines to antigen-presenting cells (APCs), immune potentiators directly activate these cells through specific receptors (for example, TOLL-LIKE RECEPTORS (TLRs)). So, delivery systems are used to promote the interaction of both antigens and immune potentiators with the key cells of the innate immune system. Immune potentiators provide the inflammatory context necessary for optimal antigen-specific immune activation by activating APCs and amplifying the innate immune response (Fig. 1).
Figure 1. The three components of optimal subunit vaccines.

It is well known that subunit vaccines elicit more potent and durable antigen-specific immunity if combined with an adjuvant. The in vivo adjuvant effect can be divided into two principal components: delivery and immune potentiation. Delivery systems localize antigens and target them to the appropriate cell types of the innate immune system. Delivery can also be optimized for immune potentiator targeting. Immune potentiators directly activate innate immune cells providing the pro-inflammatory context for antigen recognition. Antigens provide the specific pathogen epitopes necessary to generate long-lived immunological memory. These three components are intrinsic to naturally occurring infections and whole-cell vaccines, whereas they must be combined in subunit vaccine formulations.
Innate defence strategies are designed to detect broad and conserved patterns which differ between pathogenic organisms and their multicellular hosts. This non-antigen-specific detection strategy is mediated by diverse and evolutionarily conserved families of pattern-recognition receptors (PRRs)3. An increasing number of PRRs are being identified, with the ten recently discovered members of the TLR family attracting the most interest at present. The pathogen-associated molecular patterns (PAMPs) which they recognize are structurally and chemically diverse compounds, but they share the common feature of being highly conserved in pathogens and absent from multicellular organisms4,5. This broad recognition strategy allows the innate immune system to respond rapidly to infection in advance of the more delayed activation of antigen-specific immunity. As such, the key advantage of an integrated system of pattern recognition and antigen-specific responses is that the immune system divides the responsibility of recognizing and eliminating infectious agents by fulfilling two mutually exclusive goals of the primary immune response: speed and specificity6. It is important to note that the optimal immune response required for long-term protective immunity — immunological memory — represents the merger of these two competing goals and that vaccination exploits this union with remarkable success (Fig. 2).
Figure 2. The integrated model of vaccine efficacy.

a | The division of labour between the innate and adaptive (antigen-specific) immune responses on first encounter with a vaccine or infectious agent maximizes the two mutually exclusive priorities of host defence (speed and specificity). The equal importance of these competing goals can be seen when they are merged in the remarkably efficient memory response. Generating rapid, specific and durable memory responses is the goal of vaccination. In general, immune potentiators and delivery systems target the innate arm, whereas vaccine antigens drive pathogen-specific responses by B and T cells. b | The arrows indicate cytokine networks and cross-talk between regulatory cells of both innate and antigen-specific immune systems, which are central to the initiation and amplification of specific T- and B-cell responses. Key cytokines that both amplify and steer effector cell generation include interleukin-2 (IL-2), IL-4, IL-12, type I and II interferons (IFN) and other pro-inflammatory mediators (for example, tumour-necrosis factor (TNF)). c | In the effector and memory stages, the elicited cell-mediated and humoral mechanisms potently and specifically target the pathogen and/or infected cells for destruction. Abs, antibodies; ADCC, antibody-dependent cell-mediated cytotoxicity; CTL, cytotoxic T lymphocyte; Imm-Pot, immune potentiator; mDC/pDC, myeloid/plasmacytoid dendritic cell; mono, monocyte; MΦ, macrophage; NC, natural cytotoxicity; NK, natural killer cell; NKT, natural killer T cell; Phag, phagocytosis; PMN, polymorphonuclear leukocyte (neutrophil); TH1, helper T cell 1 (cell-mediated immunity); TH2, helper T cell 2 (humoral immunity).
Targeting vaccines more effectively
Various cell types of the immune system are capable of presenting antigens to T cells, providing a crucial bridge between the innate and adaptive responses. Among these, dendritic cells (DCs) are considered 'professional APCs' because they are highly efficient and are specialized for this function7,8. DCs and other APCs constantly sample their environment through pinocytosis and/or phagocytosis, and use their PRRs to 'screen' for infectious agents. This constitutive process is thought to provide the mechanism by which the immune system not only mobilizes both innate and antigen-specific defences to infection or following vaccination, but also maintains tolerance (non-responsiveness) to autologous or benign agents9. The dual activation/tolerization function of DCs is mediated by their capacity to change the context of antigen presentation and to communicate to T cells the nature of the antigens they are presenting. They do this by responding to PAMPs or synthetic immune potentiators, and producing pro-inflammatory cytokines and expressing co-stimulatory molecules (for example, CD80/86) which induce full activation of antigen-specific T cells. In the absence of these pro-inflammatory cues, T-cell responses are shut down10. It is this better understanding of antigen presentation and APC biology that has provided opportunities for future improvements in vaccine development, as more selective and sophisticated immune potentiators and delivery systems are developed (Fig. 3).
Figure 3. Antigen-presenting cells serve as the bridge between innate and antigen-specific responses.
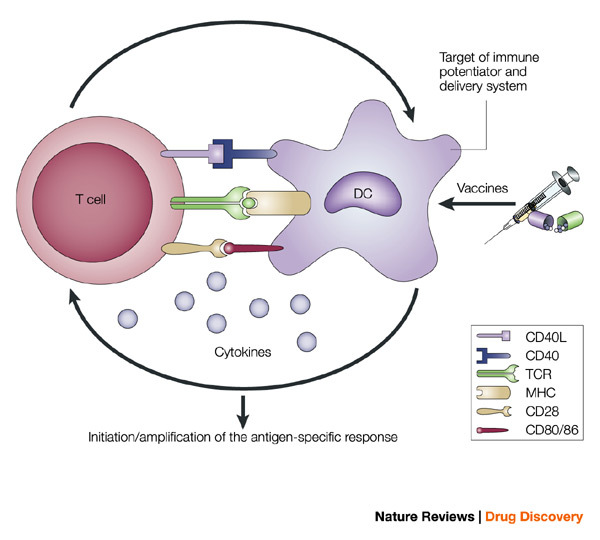
The intimate interactions (cognate recognition) between antigen-presenting cells (APCs) and antigen-specific T cells initiate and amplify pathogen-specific responses. A dendritic cell (DC) is depicted here. The key interaction is driven by the recognition of antigenic peptide–major histocompatibility complex (MHC) dimers by T cells bearing T-cell receptors (TCRs) with high affinity for the complex. However, this signal alone is not sufficient for initiation and amplification of specific T-cell responses. Co-stimulatory signals (that is, CD28 recognition of CD80/CD86) and the production of pro-inflammatory cytokines, provide the 'infectious context' by which the full activation of antigen-specific T cells is achieved. The expression of co-stimulatory molecules and cytokines by APCs is tightly regulated and induced only when the APC encounters antigens associated with pathogen-associated molecular patterns. From the perspective of adjuvant design, the APC is a high-priority target. Delivery systems increase antigen uptake and presentation, and can also target immune potentiators more efficiently to APCs. Immune potentiators induce co-stimulatory signals and cytokine production, and the antigens select the highly specific T cells leading to the initiation and amplification of antigen-specific immunity. CD40L, CD40 ligand.
Among other emerging targets of immune potentiators and delivery systems is the B cell. The direct adjuvant effects on B cells are particularly intriguing and seem to act at three levels. First, delivery systems, especially particulate ones, facilitate an ordered and repetitive array of B-cell epitopes, which results in more efficient B-cell activation through their antigen receptors11,12. Second, although B cells are considered part of the antigen-specific immune system, they also express PRRs (for example, TLRs), and, therefore, can be activated by PAMPs or immune potentiators in ways that modify both the quantity and quality of their antibody responses13,14. Last, B cells can also act as APCs, and B-cell activation by antigen and immune potentiators enhances their efficiency of antigen presentation to T cells, as well as inducing the production of cytokines. Given that antibody responses are traditionally considered the correlate of protection for most vaccines, further efforts to selectively target B cells with immune potentiators and delivery systems hold great promise.
It is important to keep in mind that infectious agents and most licensed vaccines, particularly live attenuated or killed whole-cell products, contain all of the components necessary for activating an integrated immune response. This is because the pathogens used in the vaccines possess all of the relevant antigens in a particulate form — a whole cell — which also contains many potent immune potentiators (that is, PAMPs). However, the trend in vaccine development is to move away from these whole-cell products towards safer and better-defined subunit vaccines, produced as highly purified recombinant proteins. Unfortunately, these recombinant antigens are often poorly immunogenic, because they lack intrinsic immune-potentiating activity. So, the challenge in subunit vaccine development is to reintroduce selective signals for activation of the innate immune response, which will be sufficient to mimic natural infection or traditional vaccination. The immune potentiators and delivery systems used to improve the potency of subunit vaccines will need to be low cost, but more effective and safer than whole-cell vaccines.
Innate immune-potentiating compounds
With the discovery of the TLR family of receptors, many pathogen products with known immune-potentiating activity (adjuvants) have now been shown to activate innate immunity via TLRs, or through an increasing number of non-TLR PRRs, including CD14, Dectin1, TREM1 and 2, RNA-dependent kinase (PKR) and CD91 (Refs 4,15–18). For example, bacterial lipopolysaccharide (LPS), the epitome of a pro-inflammatory agent, is now known to interact specifically with TLR4, which acts together with other receptors (for example, CD14) found on LPS-responsive cell types (for example, monocytes, macrophages, DCs and B cells). Other examples of pathogen products with corresponding TLR-dependence include tri-acyl lipopeptides (TLR1); lipidated peptides, proteins and carbohydrates (TLR2); double-stranded RNA (Poly I:C) (TLR3); flagellin (TLR5); di-acyl lipopeptides (TLR6); and CpG DNA (TLR9)4. Of the ten TLR family members, only TLR7, 8 and 10 have yet to have a natural pathogen-derived product identified. Cooperation between individual TLRs and other PRRs also seems to be a general rule for natural infections and traditional whole-cell vaccines19,20. Moreover, some TLRs seem to be highly promiscuous (for example, TLR2 and TLR4) and participate in responses to numerous and diverse pathogen products. Intriguingly, TLR2 and 4 have also been implicated in stress-induced responses to both pathogen- and host-derived heat-shock proteins4. These observations indicate that TLR2 and 4 play a more general role in initiation/amplification of the early immune response, whereas other TLR family members, and perhaps other PRRs, have evolved more specialized functions.
Given the plethora of natural products capable of activating innate immune mechanisms, research and development efforts to exploit these immune potentiators as adjuvants have been aggressive. Although in vivo proof of concept has been established for the use of many natural PAMPs as adjuvants, and a number of these have been advanced into clinical trials, the trend for the future indicates an increased reliance on synthetic analogues. This is due mainly to the lower manufacturing and regulatory hurdles associated with synthetic immune potentiators that are highly defined and standardized. In addition, a synthetic platform allows for a more rational approach to the optimization of next-generation compounds possessing greater potency and decreased toxicity. From this perspective, perhaps the most promising adjuvant/immune potentiator platform recently identified is based on a small-molecule approach. The identification of imidizaquinolines as TLR7- and 8-dependent small-molecule immune potentiators (SMIPs) indicates that more traditional pharmaceutical-based or drug-like molecules can be exploited as vaccine adjuvants21,22. Indeed, imidizaquinolines have been shown to enhance antigen-specific responses in mouse models and, therefore, have the potential to be developed as adjuvants for humans23.
An SMIP-based platform for adjuvant design and discovery holds significant potential for the future of vaccine development. There are numerous advantages that can be realized throughout the vaccine R&D pipeline if SMIPs rather than other natural or synthetic immune potentiators are chosen for optimization and development as adjuvants. For discovery efforts, the incredible diversity of scaffolds generated through combinatorial chemistry, the ability to target with exceptional selectivity innate immune mechanisms, and the tried and tested drug discovery engines of high-throughput screening and hit-to-lead optimization can now be applied to vaccine adjuvants. Later in development and manufacturing, an SMIP-based adjuvant provides a low cost, highly pure and standardized alternative to all other existing candidate immune potentiators. Given these advantages, and the likelihood that more and diverse families of SMIPs will be discovered, it seems that the imidizaquinoline TL7/8 agonists represent only the first in a long line of future small-molecule-based vaccine adjuvant candidates.
Development of vaccine delivery systems
Although the terms 'adjuvant' and 'delivery system' have often been used interchangeably in relation to vaccines, a clear distinction can often be made and the respective roles of each can be more clearly differentiated, particularly when a delivery system is used for a vaccine adjuvant. Included in lists of vaccine adjuvants are a number of particulate delivery systems (for example, emulsions, liposomes, iscoms, virus-like particles and MICROPARTICLES), whose principal mode of action is to promote the uptake of antigens into the key APC responsible for the induction of immune responses24. However, the potency of these delivery systems can be significantly improved by the addition of a vaccine adjuvant, or immune potentiator. Adjuvants are included in delivery systems to focus their effects onto the APCs, and to minimize effects on non-immune cells. Hence, delivery systems can improve the therapeutic ratio of adjuvants.
Delivery systems in vaccine design
If the geographical concept of immune reactivity, in which antigens that do not reach local lymph nodes do not induce responses25, is accepted, then the role and importance of delivery systems becomes clear. Delivery systems serve to maximize the concentration of antigen in local lymph nodes either by directly promoting antigen uptake into lymphatics, or by promoting antigen uptake into motile APCs that will then migrate to the lymph nodes. As such, the role of a vaccine delivery system is to enhance the amount of antigen reaching the cells or tissues responsible for immune response induction.
Alternative vaccine delivery systems
Particulate vaccine delivery systems (for example, emulsions, microparticles, iscoms, liposomes, virosomes and virus-like particles) have comparable dimensions to the pathogens that the immune system evolved to combat. Therefore, these particulates are normally taken up efficiently by APCs and function mainly to deliver associated antigen into these cells.
The main hurdle to the development of new and improved adjuvants has been safety, because vaccines that are to be used in healthy individuals will need to induce minimal adverse effects to prove acceptable for use. So, although many adjuvants have been extensively evaluated both preclinically and clinically, only aluminium salts (generically called 'alum') have been successfully licensed for use as vaccine adjuvants in North America26,27. However, an alternative particulate delivery system, the microemulsion MF59 (Ref. 28), was successfully introduced onto the market in Europe in 1997 to be used in conjunction with an influenza vaccine (Fluad; Chiron). MF59 seems to function as an adjuvant mainly through promoting the uptake of co-administered vaccine antigens into APCs29. Although MF59 has been shown to be safe and efficacious in a wide range of clinical trials30, its early clinical development yielded a number of important lessons. Originally, microemulsions such as MF59 were used as delivery systems for a potent adjuvant active molecule — a lipidated muramyl tripeptide (MTP-PE) — that was a novel synthetic derivative of mycobacterial cell wall31. However, the MF59/MTP-PE combination proved too reactogenic for routine clinical use, although MF59 alone was well-tolerated and had comparable immunogenicity to the MF59/MTP-PE combination31. Consequently, MF59 alone was used subsequently and proved sufficiently potent and safe to allow successful product development32. Our clinical experiences with microemulsions served to highlight the need for careful selection of immune potentiators to be included in vaccines. Overall, although MF59 is normally a more potent adjuvant than alum32, like alum, it cannot be expected to be appropriate for use with all vaccines. MF59 works well with a variety of antigens to enhance antibody and T-cell proliferative responses32. However, it is unable to induce potent T-cell responses of the TH1 type, defined by the production of the cytokine interferon-γ, which might be required to provide protective immunity against some intracellular pathogens. So, it is clear that additional adjuvant and delivery approaches will be needed for some vaccines.
In the recent past, we have focused on an alternative particulate delivery system for vaccines, comprising biodegradable microparticles prepared from the polymer POLY(LACTIDE CO-GLYCOLIDE) (PLG). Because PLG polymer has already been used for a variety of biomedical purposes, including the preparation of a controlled-release delivery system for the therapeutic protein human growth hormone33, it was an excellent choice as a vaccine delivery system. In addition, microparticles of the appropriate size (∼1 μm) have been shown to be taken up by APCs in vivo, which migrated to the T-cell area of local lymph nodes and differentiated into mature DCs34. Initial work focused on the use of microparticles for controlled release of trapped antigens, with the objective of making single-dose vaccines35. However, problems arose as a consequence of the degradation of antigens following microencapsulation and during release from PLG microparticles36. We therefore adopted the novel approach of adsorbing antigens onto the surface of microparticles to avoid exposing them to damaging conditions during encapsulation and release. Adsorption was enhanced by using charged surfactants for microparticle preparation, which promoted antigen interaction with the surface of the particles37. Consequently, the microparticles were designed to perform as a delivery system to promote the uptake of antigen into APCs, thereby resulting in the induction of potent antibody and T-cell responses in mice37 and non-human primates38. In addition, the adsorption of antigen on microparticles also serves to multimerize the antigens, and so facilitates direct recognition by B-cell antigen receptors. The approach of adsorbing antigens onto charged microparticles has proven sufficiently flexible to allow successful delivery of DNA vaccines, which are adsorbed onto the surface of cationic microparticles (Fig. 4). Using cationic microparticles as a delivery system for DNA vaccines resulted in significantly enhanced immune responses in comparison with immunization using naked DNA in mice39 and in non-human primates40. In mechanistic studies, we showed that the cationic PLG microparticles were able to deliver adsorbed DNA into DCs, whereas naked DNA was unable to transfect DCs41. Similar cationic microparticles can also be used as delivery systems for adsorbed adjuvants, including CpG DNA, whereas anionic microparticles have been used for adsorbed protein vaccines37,42. The potential of a broad range of particulate vaccine delivery systems to improve the potency of new generation vaccines has recently been reviewed in more detail2,43.
Figure 4. Representation of a cationic poly(lactide co-glycolide) (PLG) microparticle (1 μm mean size) with adsorbed DNA on the surface.
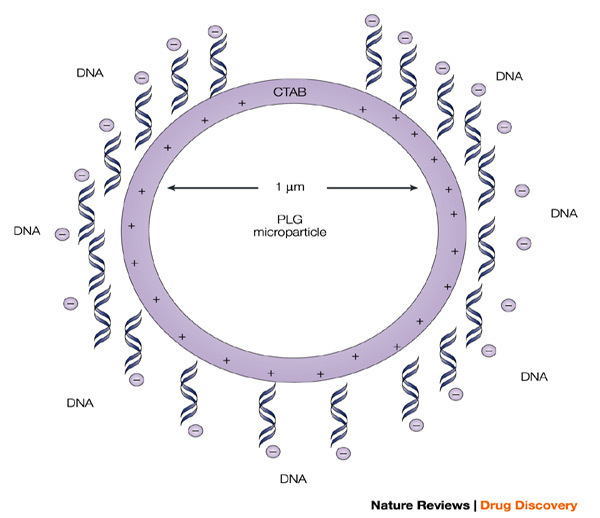
The microparticle is cationic because of the presence of the cationic detergent hexadecyltrimethyl ammonium bromide (CTAB), which binds to the surface during microparticle preparation, and allows the efficient adsorption of poly-anionic plasmids. This approach serves to significantly enhance the potency of the antigens encoded by the adsorbed DNA vaccines.
Delivery systems for mucosal immunization
Although the majority of vaccines have traditionally been administered by injection, mucosal administration of vaccines offers a number of important advantages, including easier administration, reduced adverse effects and the potential for frequent boosting. In addition, mucosal immunization induces local immunity at the sites where pathogens often establish infection. Oral immunization would be particularly advantageous in isolated communities, where access to health care professionals is problematic. Moreover, mucosal immunization would avoid the problem of infection due to the re-use of needles, which is common in the developing world. However, the difficulty of developing mucosal and particularly oral vaccines using non-living approaches should not be underestimated. Protein, peptide, polysaccharide and DNA immunogens are extremely labile and will be extensively degraded and damaged during passage through the gut if not adequately protected. Intranasal immunization is an attractive approach, owing to the absence of acidity and secreted enzymes in the nasal cavity, but also because of the ease of access to the nasal cavity offered by simple commercially available devices. Nevertheless, potent adjuvants and delivery systems will be required to facilitate the development of effective mucosal vaccines.
The most potent mucosal adjuvants available are the bacterial toxins secreted from Escherichia coli and Vibrio cholerae, called HEAT-LABILE ENTEROTOXIN (LT) (Fig. 5) and cholera toxin (CT), respectively. However, because these molecules are responsible for traveller's diarrhoea and cholera, respectively, the native molecules are clearly not appropriate for mucosal administration to humans. Therefore, these molecules have been manipulated to reduce toxicity, while retaining adjuvant activity44 (Fig. 6). One particular mutant of LT, LTK63, has recently been shown to be safe in a human clinical trial involving intranasal administration, following extensive preclinical toxicology testing30. In previous studies, we showed that the potency of LTK63 for intranasal administration could be enhanced by co-administration with bio-adhesive delivery systems in small45 and large animal models46. The bio-adhesive delivery systems were designed to retain the vaccine formulation in the nasal cavity for extended periods and to promote the interaction of the antigen and adjuvant with the mucosal epithelium. Recent data indicate that LTK63 might prove sufficiently potent to allow the mucosal administration of paediatric combination vaccines47.
Figure 5. A schematic representation of LT.
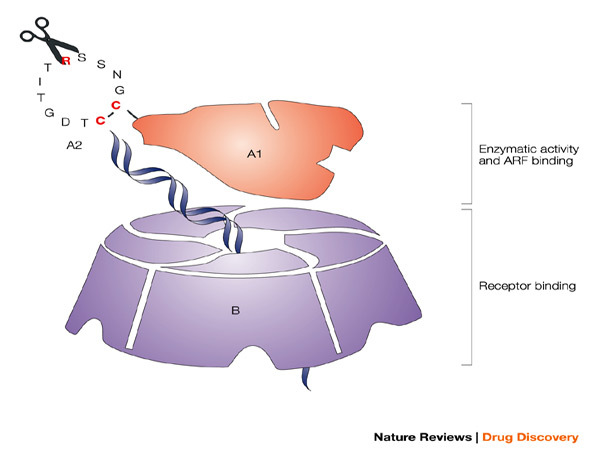
The potent mucosal immunogen and adjuvant heat-labile enterotoxin from Escherichia coli (LT), with the A and B subunits and their structural and functional activities, is shown. The B oligomer is a pentameric molecule of 55 kDa, which contains five identical polypeptide monomers and is responsible for binding to mucosal epithelial cells. The A1 subunit comprises a globular structure, which is enzymatically active and mediates ADP-ribosylation, resulting in permanent activation of adenylate cyclase, abnormal intracellular accumulation of cAMP and massive fluid loss, resulting in diarrhoea. The A1 subunit is linked to the B oligomer with the long helical A2 subunit, which must be proteolytically cleaved in addition to a disulphide being reduced to release and activate the A1 subunit. The ADP-ribosyltransferase activity of the A1 subunit is enhanced by interaction with intracellular ADP-ribosylation factors (ARF). Various groups have attempted to generate potent but safe mucosal adjuvants from LT, through a variety of genetic manipulations. Chiron scientists have focused on modifying or eliminating the enzymatic activity of LT (ADP-ribosylation) through single amino-acid substitutions in the active site of A1 (LTK63 and LTR72), while an alternative approach pursued by others was to modify the ability of the A1 subunit to be proteolytically cleaved from A2 by trypsin (LTR192G).
Figure 6. In vitro and in vivo toxicity of various mutants of heat-labile enterotoxin from Escherichia coli.

In vitro toxicity on Y1 adrenal cells is shown for heat-labile enterotoxin (LT) wild type (LTwt), for the mutants generated by Chiron, which have modifications in the enzymatic active site of the A1 subunit (LTK63 and LTR72), and for an alternative mutant, LTR192G, which is modified in its ability to be enzymatically cleaved and activated by trypsin. LTK63 has no enzyme activity in the A1 subunit and shows no toxicity on Y1 cells, whereas LTR72 has residual enzyme activity (about 0.6% of LTwt) and shows residual toxicity, but is 100,000-fold less toxic than LTwt. By contrast, LTR192G shows only a marginal reduction in toxicity. Similar results were obtained for in vivo toxicity in the standard rabbit ileal loop model, which shows significant fluid accumulation with LTwt. In contrast, LTK63 shows no fluid accumulation, even at the highest dose tested (1 mg), whereas LTR72 shows reduced fluid accumulation and the requirement for a higher dose to trigger this effect. LTR192G shows high levels of fluid accumulation, even at low doses, similar to LTwt, probably because there are alternative enzymes present that can cleave the molecule, in addition to trypsin. For further details on experimental methodology for these studies see Refs 65,66.
Although oral delivery of non-living vaccines remains extremely challenging, even with potent adjuvants, some success has been achieved in small animal models with non-toxic LT mutants48. Nevertheless, there is a requirement for the development of optimal delivery systems if successful oral vaccines are to be produced commercially. A variety of microparticle-based delivery systems can be designed to protect antigens against degradation in the gut, to promote interaction with the epithelium, or to be taken up by the mucosal-associated lymphoid tissues, and these offer significant promise as important components of oral vaccine delivery systems49. Recently, some success has been achieved in a human clinical trial with a vaccine trapped in PLG microparticles, but this approach is likely to require significant improvements to allow successful commercial development50.
Vaccine delivery devices
In its broadest sense, the concept of 'vaccine delivery systems' can be expanded to include a range of devices and physical delivery systems designed to allow immunization using novel non-invasive routes. Needle-free vaccination is attractive for a number of reasons. In the developed world, there are concerns about the number of vaccinations that are administered to infants and toddlers, and it would be preferable if these could be administered without the use of needles. In addition, 'needle phobia' is a significant problem that makes it difficult to convince adults to receive vaccinations. This problem might become particularly acute in the future if it proves necessary to immunize large numbers of individuals in response to a pandemic strain of influenza, or to the threat of a bio-terrorism attack. The problems related to vaccination with needles are even greater in the developing world, where the re-use of needles often results in the transmission of blood-borne pathogens, such as HIV and HCV. Although this problem has been partly remedied through the development of single-use 'auto-disposable' syringes, needle-based injections still represent an infectious threat which must be disposed of carefully.
One approach to obviate the problems associated with vaccines administered by needles involves a number of devices that are designed to deliver vaccines through or into the skin, but without the use of needles. Multiple-use jet injector devices that deliver liquid vaccines through the skin were widely used for mass immunization campaigns throughout the 1980s. However, these devices became discredited when it became apparent that they were capable of transmitting blood-borne pathogens such as hepatitis B51. More recently, single-dose jet injector devices have been developed, which are capable of delivering liquid52 or dry powder vaccines53 through the skin using high pressure. In addition, spring-powered liquid injection devices are available and have also been evaluated in the clinic54. However, none of these devices have yet attained broad acceptance in the medical community and their costs will make them difficult, if not impossible, to implement in the developing world. High-pressure-liquid, needle-free devices have also been used for the delivery of DNA vaccines in clinical trials55, as have new needle-free devices designed to deliver gold beads coated with DNA into the skin ('gene gun')56. An alternative needle-free approach to vaccine delivery, which could perhaps be described more accurately as 'needle-lite', involves the use of microprojection arrays designed to painlessly disrupt the outer layers of the skin to allow the vaccine access to the epidermis and associated Langerhans cells57,58.
Perhaps the most attractive needle-free approach to vaccine delivery being explored at present involves TRANSCUTANEOUS IMMUNIZATION through the topical application of vaccine patches59. This approach has recently provided promising observations in clinical trials60,61. In addition, the ability of a number of different adjuvants to function following topical application with vaccines is encouraging62, as is the ability of a topically applied 'immunostimulatory patch' to enhance the immunogenicity of a locally injected vaccine63.
In addition to the mucosal immunization approaches discussed above, which clearly fall into the needle-free category, some vaccines might also be administered mucosally via aerosols, including measles vaccine64. However, the available devices need to be made more robust and reliable to be broadly accepted for vaccine administration.
Conclusions
We are entering an exciting and dynamic time in vaccine research in which the principles governing the successful induction of potent and protective immune responses are becoming better understood. At the forefront of this work are discoveries relating to the presence of receptors on innate immune cells, which recognize the characteristic patterns and components present on pathogens. Recognition of the importance of activation of the innate immune system to the eventual induction of antigen-specific immunity has fired enthusiasm to identify pathogen-based ligands for these receptors as potential new-generation adjuvants to be used in combination with recombinant proteins. A significant amount of work is underway in this area and will likely lead to the development of whole new classes of vaccine adjuvants that are able to control and manipulate the immune response in a variety of ways. It is likely that these adjuvants could prove to be sufficiently potent to allow vaccines to be used to treat chronic infectious diseases, autoimmunity and cancer. However, the potency of these adjuvants needs to be carefully balanced with their potential to overactivate the immune system, with damaging consequences. In this regard, vaccine and adjuvant delivery systems might have a role to play in limiting the systemic distribution of adjuvants following administration. Moreover, the main role of vaccine delivery systems is to ensure that the antigens and adjuvants interact optimally with the appropriate immunocompetent cells to induce the desired responses. Nevertheless, for adjuvants capable of initiating potent inflammatory responses, limiting their ability to act on a broad range of cells could prove to be as important as focusing their effects on key immune cells. Overall, it is clear that pharmaceutical scientists have a unique and important role to play in the development of optimal vaccine and adjuvant delivery systems for systemic and mucosal administration.
Acknowledgements
We would like to acknowledge the contributions of our colleagues in Chiron for their research efforts and ideas. In particular, we thank M. Giulianni, M. Grazia Pizza, R. Rappuoli and G. Del Giudice for the toxicity data on the LT mutants. In addition, we acknowledge the contributions of our colleagues in the Vaccine Delivery Group at Chiron, Emeryville, USA. Finally, we thank N. Cronen for manuscript formatting.
Glossary
- ADJUVANT
The functional definition of diverse components co-administered with vaccine antigens that enhance antigen-specific immune responses in vivo.
- INNATE IMMUNITY
The system of rapid host defence that has evolved to respond to broad and highly conserved pathogen-associated molecular patterns (PAMPs).
- IMMUNE POTENTIATOR
A natural or synthetic compound which directly activates immune cells through specific receptors and/or pathways.
- DELIVERY SYSTEM
A vaccine formulation that localizes vaccine antigens and/or immune potentiators, and targets them to key immune cell types.
- IMMUNOLOGICAL MEMORY
The long-lived protective immunity elicited by vaccines and many naturally occurring infections, which is mediated by expanded populations of previously activated B and T cells selected for their expression of specific receptors for the antigens of a given pathogen.
- TOLL-LIKE RECEPTORS
(TLRs). An evolutionarily conserved family of pattern-recognition receptors that detect unique microbial products and allow rapid activation of innate immunity.
- MICROPARTICLES
Particulate carrier systems in the micron size range, normally prepared with synthetic polymers, which can be used as delivery systems for drugs or vaccines that are usually trapped within the particles.
- MF59
An oil-in-water microemulsion adjuvant which has been approved in Europe for use in combination with influenza vaccine in the elderly (Fluad).
- POLY(LACTIDE CO-GLYCOLIDE)
(PLG). A biodegradable and biocompatible synthetic polymer that has been used to prepare a number of successfully marketed drug delivery systems.
- HEAT-LABILE ENTEROTOXIN
(LT). A bacterial toxin secreted from Escherichia coli which is potently immuno-stimulatory when applied to mucosal surfaces and can also act as an adjuvant for co-administered antigens. However, the molecule is also the causative agent of traveller's diarrhoea.
- TRANSCUTANEOUS IMMUNIZATION
Involves the topical application of vaccines, usually in a specially designed patch, and mediates effective immunization without penetrating the protective layer of the skin with a device or needle.
Biographies
Derek O'Hagan has worked at Chiron Corporation in Emeryville, California, USA, since 1995 and is presently Director of Vaccine Adjuvants and Delivery Systems, a position he has held since 2000. He was originally trained as a formulation scientist at the Department of Pharmaceutical Sciences at the University of Nottingham, UK. He completed his thesis in 1987 on the use of 'Pharmaceutical formulations as immunological adjuvants' and has continued to work in this field ever since. He has made a number of contributions to the use of systemic and mucosal delivery systems for a wide range of vaccines, including proteins, peptides, DNA and protein polysaccharide conjugates. He was awarded the Conference Science medal of the Royal Pharmaceutical Society of Great Britain in 1997, and the Young Investigator Research Achievement Award of the Controlled Release Society in 1999. He is presently a member of the Board of Scientific Advisors of the Controlled Release Society.
Nicholas Valiante joined Chiron Corporation in Emeryville, California, USA, in 2002, and is presently Project Leader of the Adjuvant and Immune Potentiators programme. He obtained his PhD in immunology from the University of Pennsylvania in 1994. His worked focused on the mechanisms of natural killer (NK) cell activation and the biology of the then newly discovered cytokine, interleukin-12. After a postdoctoral fellowship at Stanford University where he continued to work on the mechanisms of innate immunity, Dr Valiante joined Chiron, Vaccines Research Institute in Siena, Italy in 1998. There he developed new systems for the study of innate immune lymphocytes in hepatitis C virus infections, and elucidated a cognate mechanism for the amplification and attenuation of innate immune responses via reciprocal regulation of NK cells and dendritic cells.
Related links
DATABASES
LocusLink
FURTHER INFORMATION
Encyclopedia of Life Sciences
References
- 1.WHO World Health Report (2002).
- 2.Singh M, O'Hagan DT. Recent advances in vaccine adjuvants. Pharm. Res. 2002;19:715–728. doi: 10.1023/A:1016104910582. [DOI] [PubMed] [Google Scholar]
- 3.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 4.Takeda K, Kaisho T, Akira S. Toll-like receptors. Ann. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 5.Medzhitov R, Janeway CA., Jr. An ancient system of host defense. Curr. Opin. Immunol. 1998;10:12–15. doi: 10.1016/S0952-7915(98)80024-1. [DOI] [PubMed] [Google Scholar]
- 6.O'Hagan, D. & Valiante, N. M. Recent advances in vaccine adjuvants and delivery systems. Pharmaceutical News (in the press).
- 7.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 8.Cella M, Sallusto F, Lanzavecchia A. Origin, maturation and antigen presenting function of dendritic cells. Curr. Opin. Immunol. 1997;9:10–16. doi: 10.1016/S0952-7915(97)80153-7. [DOI] [PubMed] [Google Scholar]
- 9.Banchereau J, et al. Immunobiology of dendritic cells. Ann. Rev. Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bachmann MF, et al. The influence of antigen organization on B cell responsiveness. Science. 1993;262:1448–1451. doi: 10.1126/science.8248784. [DOI] [PubMed] [Google Scholar]
- 12.Jegerlehner A, et al. A molecular assembly system that renders antigens of choice highly repetitive for induction of protective B cell responses. Vaccine. 2002;20:3104–3112. doi: 10.1016/S0264-410X(02)00266-9. [DOI] [PubMed] [Google Scholar]
- 13.Bernasconi NL, Traggiai E, Lanzavecchia A. Maintenance of serological memory by polyclonal activation of human memory B cells. Science. 2002;298:2199–2202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 14.Liu N, Ohnishi N, Ni L, Akira S, Bacon KB. CpG directly induces T-bet expression and inhibits IgG1 and IgE switching in B cells. Nature Immunol. 2003;4:687–693. doi: 10.1038/ni941. [DOI] [PubMed] [Google Scholar]
- 15.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by Dectin-1 and Toll-like receptor 2. J. Exp. Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouchon A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410:1103–1107. doi: 10.1038/35074114. [DOI] [PubMed] [Google Scholar]
- 17.Cella M, et al. Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J. Exp. Med. 1999;189:821–829. doi: 10.1084/jem.189.5.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/S1074-7613(01)00111-X. [DOI] [PubMed] [Google Scholar]
- 19.Royle MC, Totemeyer S, Alldridge LC, Maskell DJ, Bryant CE. Stimulation of Toll-Like receptor 4 by lipopolysaccharide during cellular invasion by live Salmonella typhimurium is a critical but not exclusive event leading to macrophage responses. J. Immunol. 2003;170:5445–5454. doi: 10.4049/jimmunol.170.11.5445. [DOI] [PubMed] [Google Scholar]
- 20.Nau GJ, Schlesinger A, Richmond JF, Young RA. Cumulative toll-like receptor activation in human macrophages treated with whole bacteria. J. Immunol. 2003;170:5203–5209. doi: 10.4049/jimmunol.170.10.5203. [DOI] [PubMed] [Google Scholar]
- 21.Hemmi H, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nature Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 22.Jurk M, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nature Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- 23.Vasilakos JP, et al. Adjuvant activities of immune response modifier R-848: comparison with CpG ODN. Cell. Immunol. 2000;204:64–74. doi: 10.1006/cimm.2000.1689. [DOI] [PubMed] [Google Scholar]
- 24.Vogel FR, Powell MF. Vaccine Design: The Subunit and Adjuvant Approach. 1995. pp. 141–228. [DOI] [PubMed] [Google Scholar]
- 25.Zinkernagel RM, et al. Antigen localisation regulates immune responses in a dose- and time-dependent fashion: a geographical view of immune reactivity. Immunol. Rev. 1997;156:199–209. doi: 10.1111/j.1600-065X.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]
- 26.Edelman R. New Generation Vaccines. 1997. pp. 173–192. [Google Scholar]
- 27.Kenney RT, Edelman R. Survey of human-use adjuvants. Expert Rev. Vaccines. 2003;2:165–188. doi: 10.1586/14760584.2.2.167. [DOI] [PubMed] [Google Scholar]
- 28.Ott G. Vaccine Adjuvants: Preparation Methods and Research Protocols. 2001. pp. 211–228. [Google Scholar]
- 29.Dupuis M, et al. Immunization with the adjuvant MF59 induces macrophage trafficking and apoptosis. Eur. J. Immunol. 2001;31:2910–2918. doi: 10.1002/1521-4141(2001010)31:10<2910::AID-IMMU2910>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Peppoloni S, et al. Mutants of the Escherichia coli heat-labile enterotoxin as safe and strong adjuvants for intranasal delivery of vaccines. Expert Rev. Vaccines. 2003;2:285–293. doi: 10.1586/14760584.2.2.285. [DOI] [PubMed] [Google Scholar]
- 31.Keefer MC, et al. Safety and immunogenicity of Env 2-3, a human immunodeficiency virus type 1 candidate vaccine, in combination with a novel adjuvant, MTP-PE/MF59. NIAID AIDS Vaccine Evaluation Group. AIDS Res. Hum. Retroviruses. 1996;12:683–693. doi: 10.1089/aid.1996.12.683. [DOI] [PubMed] [Google Scholar]
- 32.Ott G. Vaccine Design: The Subunit and Adjuvant Approach. 1995. pp. 229–248. [Google Scholar]
- 33.Johnson OL, et al. A month-long effect from a single injection of microencapsulated human growth hormone. Nature Med. 1996;2:795–799. doi: 10.1038/nm0796-795. [DOI] [PubMed] [Google Scholar]
- 34.Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/S1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 35.O'Hagan D, Singh M. Microparticles as vaccine adjuvants and delivery systems. Expert Rev. Vaccines. 2003;2:269–283. doi: 10.1586/14760584.2.2.269. [DOI] [PubMed] [Google Scholar]
- 36.Schwendeman SP. Recent advances in the stabilization of proteins encapsulated in injectable PLGA delivery systems. Crit. Rev. Ther. Drug Carrier Syst. 2002;19:73–98. doi: 10.1615/CritRevTherDrugCarrierSyst.v19.i1.20. [DOI] [PubMed] [Google Scholar]
- 37.Kazzaz J, Neidleman J, Singh M, Ott G, O'Hagan DT. Novel anionic microparticles are a potent adjuvant for the induction of cytotoxic T lymphocytes against recombinant p55 gag from HIV-1. J. Control Release. 2000;67:347–356. doi: 10.1016/S0168-3659(00)00226-1. [DOI] [PubMed] [Google Scholar]
- 38.Otten GR, et al. Induction of broad and potent anti-HIV immune responses in rhesus macaques by priming with a DNA vaccine and boosting with protein-adsorbed PLG microparticles. J. Virol. 2003;77:6087–6092. doi: 10.1128/JVI.77.10.6087-6092.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh M, Briones M, Ott G, O'Hagan D. Cationic microparticles: a potent delivery system for DNA vaccines. Proc. Natl Acad. Sci. USA. 2000;97:811–816. doi: 10.1073/pnas.97.2.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Hagan D, et al. Induction of potent immune responses by cationic microparticles with adsorbed HIV DNA vaccines. J. Virol. 2001;75:9037–9043. doi: 10.1128/JVI.75.19.9037-9043.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denis-Mize KS, et al. Plasmid DNA adsorbed onto cationic microparticles mediates target gene expression and antigen presentation by dendritic cells. Gene Ther. 2000;7:2105–2112. doi: 10.1038/sj.gt.3301347. [DOI] [PubMed] [Google Scholar]
- 42.Singh M, et al. Cationic microparticles are an effective delivery system for immune stimulatory CpG DNA. Pharm. Res. 2001;18:1476–1479. doi: 10.1023/A:1012269226066. [DOI] [PubMed] [Google Scholar]
- 43.Michalek SM, O'Hagan D, Gould-Fogerite S. Mucosal Immunology. 1999. pp. 759–778. [Google Scholar]
- 44.Rappuoli R, Pizza M, Douce G, Dougan G. Structure and mucosal adjuvanticity of cholera and Escherichia coli heat-labile enterotoxins. Immunol. Today. 1999;20:493–500. doi: 10.1016/S0167-5699(99)01523-6. [DOI] [PubMed] [Google Scholar]
- 45.Baudner BC, et al. Enhancement of protective efficacy following intranasal immunization with vaccine plus a nontoxic LTK63 mutant delivered with nanoparticles. Infect. Immun. 2002;70:4785–4790. doi: 10.1128/IAI.70.9.4785-4790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh M, Briones M, O'Hagan DT. A novel bioadhesive intranasal delivery system for inactivated influenza vaccines. J. Control Release. 2001;70:267–276. doi: 10.1016/S0168-3659(00)00330-8. [DOI] [PubMed] [Google Scholar]
- 47.Ugozzoli M, Mariani M, Del Giudice G, Soenawan E, O'Hagan DT. Combinations of protein polysaccharide conjugate vaccines for intranasal immunization. J. Infect. Dis. 2002;186:1358–1361. doi: 10.1086/344320. [DOI] [PubMed] [Google Scholar]
- 48.Barackman JD, Ott G, Pine S, O'Hagan DT. Oral administration of influenza vaccine in combination with the adjuvants LT-K63 and LT-R72 induces potent immune responses comparable to or stronger than traditional intramuscular immunization. Clin. Diagn. Lab. Immunol. 2001;8:652–657. doi: 10.1128/CDLI.8.3.652-657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O'Hagan D. Microparticles and polymers for the mucosal delivery of vaccines. Adv. Drug Deliv. Rev. 1998;34:305–320. doi: 10.1016/S0169-409X(98)00045-3. [DOI] [PubMed] [Google Scholar]
- 50.Katz DE, et al. Oral immunization of adult volunteers with microencapsulated enterotoxigenic Escherichia coli (ETEC) CS6 antigen. Vaccine. 2003;21:341–346. doi: 10.1016/S0264-410X(02)00613-8. [DOI] [PubMed] [Google Scholar]
- 51.Canter J, et al. An outbreak of hepatitis B associated with jet injections in a weight reduction clinic. Arch. Intern. Med. 1990;150:1923–1927. doi: 10.1001/archinte.1990.00390200105020. [DOI] [PubMed] [Google Scholar]
- 52.Jackson LA, et al. Safety and immunogenicity of varying dosages of trivalent inactivated influenza vaccine administered by needle-free jet injectors. Vaccine. 2001;19:4703–4709. doi: 10.1016/S0264-410X(01)00225-0. [DOI] [PubMed] [Google Scholar]
- 53.Chen D, et al. Epidermal immunization by a needle-free powder delivery technology: immunogenicity of influenza vaccine and protection in mice. Nature Med. 2000;6:1187–1190. doi: 10.1038/80538. [DOI] [PubMed] [Google Scholar]
- 54.Parent du Chatelet I, et al. Clinical immunogenicity and tolerance studies of liquid vaccines delivered by jet-injector and a new single-use cartridge (Imule): comparison with standard syringe injection. Imule Investigators Group. Vaccine. 1997;15:449–458. doi: 10.1016/S0264-410X(96)00173-9. [DOI] [PubMed] [Google Scholar]
- 55.Epstein JE, et al. Safety, tolerability, and lack of antibody responses after administration of a PfCSP DNA malaria vaccine via needle or needle-free jet injection, and comparison of intramuscular and combination intramuscular/intradermal routes. Hum. Gene Ther. 2002;13:1551–1560. doi: 10.1089/10430340260201644. [DOI] [PubMed] [Google Scholar]
- 56.Roy MJ, et al. Induction of antigen-specific CD8+ T cells, T helper cells, and protective levels of antibody in humans by particle-mediated administration of a hepatitis B virus DNA vaccine. Vaccine. 2000;19:764–778. doi: 10.1016/S0264-410X(00)00302-9. [DOI] [PubMed] [Google Scholar]
- 57.Mikszta JA, et al. Improved genetic immunization via micromechanical disruption of skin-barrier function and targeted epidermal delivery. Nature Med. 2002;8:415–419. doi: 10.1038/nm0402-415. [DOI] [PubMed] [Google Scholar]
- 58.Matriano JA, et al. Macroflux microprojection array patch technology: a new and efficient approach for intracutaneous immunization. Pharm. Res. 2002;19:63–70. doi: 10.1023/A:1013607400040. [DOI] [PubMed] [Google Scholar]
- 59.Glenn GM, Rao M, Matyas GR, Alving CR. Skin immunization made possible by cholera toxin. Nature. 1998;391:851. doi: 10.1038/36014. [DOI] [PubMed] [Google Scholar]
- 60.Glenn GM, et al. Transcutaneous immunization: a human vaccine delivery strategy using a patch. Nature Med. 2000;6:1403–1406. doi: 10.1038/82225. [DOI] [PubMed] [Google Scholar]
- 61.Guerena-Burgueno F, et al. Safety and immunogenicity of a prototype enterotoxigenic Escherichia coli vaccine administered transcutaneously. Infect. Immun. 2002;70:1874–1880. doi: 10.1128/IAI.70.4.1874-1880.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scharton-Kersten T, et al. Transcutaneous immunization with bacterial ADP-ribosylating exotoxins, subunits, and unrelated adjuvants. Infect. Immun. 2000;68:5306–5313. doi: 10.1128/IAI.68.9.5306-5313.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guebre-Xabier M, et al. Immunostimulant patch containing heat-labile enterotoxin from Escherichia coli enhances immune responses to injected influenza virus vaccine through activation of skin dendritic cells. J. Virol. 2003;77:5218–5225. doi: 10.1128/JVI.77.9.5218-5225.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dilraj A, et al. Response to different measles vaccine strains given by aerosol and subcutaneous routes to schoolchildren: a randomised trial. Lancet. 2000;355:798–803. doi: 10.1016/S0140-6736(99)95140-1. [DOI] [PubMed] [Google Scholar]
- 65.Giuliani MM, et al. Mucosal adjuvanticity and immunogenicity of LTR72, a novel mutant of Escherichia coli heat-labile enterotoxin with partial knockout of ADP-ribosyltransferase activity. J. Exp. Med. 1998;187:1123–1132. doi: 10.1084/jem.187.7.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giannelli V, et al. Protease susceptibility and toxicity of heat-labile enterotoxins with a mutation in the active site or in the protease-sensitive loop. Infect. Immun. 1997;65:331–334. doi: 10.1128/iai.65.1.331-334.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]