

Key Points
Immunomodulatory biologics are a class of biotechnology-derived therapeutic products that are designed to engage immune-relevant targets and are indicated in the treatment and management of a range of diseases, including immune-mediated inflammatory diseases and malignancies.
Despite their high specificity and therapeutic advantages, immmunomodulatory biologics have been associated with adverse reactions such as serious infections, malignancies and cytokine release syndrome, which arise owing to the on-target or exaggerated pharmacological effects of these drugs. Immunogenicity resulting in the generation of antidrug antibodies is another unwanted effect that leads to loss of efficacy and — rarely — hypersensitivity reactions.
For some adverse reactions, mitigating and preventive strategies are in place, such as stratifying patients on the basis of responsiveness to therapy and the risk of developing adverse reactions. These strategies depend on the availability of robust biomarkers for therapeutic efficacy and the risk of adverse reactions: for example, seropositivity for John Cunningham virus is a risk factor for progressive multifocal leukoencephalopathy. The development of effective biomarkers will greatly aid these strategies.
The development and design of safer immunomodulatory biologics is reliant on a detailed understanding of the nature of the disease, target biology, the interaction of the target with the immunomodulatory biologic and the inherent properties of the biologic that elicit unwanted effects.
The availability of in vitro and in vivo models that can be used to predict adverse reactions associated with immunomodulatory biologics is central to the development of safer immunomodulatory biologics. Some progress has been made in developing in vitro and in silico tests for predicting cytokine release syndrome and immunogenicity, but there is still a lack of models for effectively predicting infections and malignancies.
Two pathways can be followed in designing and developing safer immunomodulatory biologics. The first pathway involves generating a biologic that engages an alternative target or mechanism to produce the desired pharmacodynamic effect without the associated adverse reaction, and is followed when the adverse reaction cannot be dissociated from the target biology. The second pathway involves redesigning the biologic to 'engineer out' components within the biologic structure that trigger adverse effects or to alter the nature of the target–biologic interactions.
Supplementary information
The online version of this article (doi:10.1038/nrd3974) contains supplementary material, which is available to authorized users.
Subject terms: Biologics, Immunological disorders, Inflammation, Toxicology, Screening, Pharmaceutics, Drug safety, Drug discovery and development
Owing to their specificity, immunomodulatory biologics generally have better safety profiles than small-molecule drugs. However, adverse effects such as an increased risk of infections or cytokine release syndrome are of concern. Here, Park and colleagues discuss the current strategies used to predict and mitigate these adverse effects and consider how they can be used to inform the development of safer immunomodulatory biologics.
Supplementary information
The online version of this article (doi:10.1038/nrd3974) contains supplementary material, which is available to authorized users.
Abstract
Immunomodulatory biologics, which render their therapeutic effects by modulating or harnessing immune responses, have proven their therapeutic utility in several complex conditions including cancer and autoimmune diseases. However, unwanted adverse reactions — including serious infections, malignancy, cytokine release syndrome, anaphylaxis and hypersensitivity as well as immunogenicity — pose a challenge to the development of new (and safer) immunomodulatory biologics. In this article, we assess the safety issues associated with immunomodulatory biologics and discuss the current approaches for predicting and mitigating adverse reactions associated with their use. We also outline how these approaches can inform the development of safer immunomodulatory biologics.
Supplementary information
The online version of this article (doi:10.1038/nrd3974) contains supplementary material, which is available to authorized users.
Main
Biologics currently represent more than 30% of licensed pharmaceutical products and have expanded the therapeutic options available for a broad range of diseases, including immune-mediated inflammatory diseases and malignancies1,2. Between 1993 and 2011, 174 new biologics (that are not vaccines or blood products) were authorized by the European Medicines Agency (EMA) (see the European public assessment reports on the EMA website) and/or the US Food and Drug Administration (FDA) (see the Drugs@FDA page on the FDA website). A large proportion of these are immunomodulatory biologics, which can be grouped into three main categories according to their mode of action: cytokine immunomodulatory biologics that mimic, replace or augment endogenous cytokines (for example, granulocyte colony-stimulating factor (G-CSF) and interferon-β (IFNβ)) and are recombinant human proteins; antibody-based triggering immunomodulatory biologics that bind to relevant targets and trigger immune cell signalling pathways (for example, rituximab (Rituxan/Mabthera; Biogen Idec/Genentech/Roche) and muromonab-CD3); and blocking immunomodulatory biologics that bind to immune-relevant targets and block their function (for example, etanercept (Enbrel; Amgen/Pfizer) and omalizumab (Zolair; Genentech/Novartis)).
The high specificity of the interactions of immunomodulatory biologics with their relevant immune targets — the on-target effects — should theoretically abrogate off-target effects. Despite the generally superior safety profiles of immunomodulatory biologics compared with small-molecules, several clinical concerns have emerged relating to unwanted effects or adverse reactions associated with the use of immunomodulatory biologics (Tables 1,2,3). These adverse reactions can be broadly categorized into two groups. The first category includes adverse reactions that are due to on-target interactions (that is, exaggerated pharmacology). These include serious infections, malignancies, cytokine release syndrome (CRS), tumour lysis syndrome and autoimmunity. The second category includes adverse reactions that are due to the inherent property of the biologic, such as immunogenicity. Adverse reactions that are due to the inherent property of the biologic can result in the generation of neutralizing anti-drug antibodies (ADAs) and hypersensitivity reactions.
Table 1.
Clinically used recombinant cytokine immunomodulatory biologics
| Biologic | Type | Indications | Immune-related safety warnings |
|---|---|---|---|
| Aldesleukin | Recombinant human IL-2 | Metastatic renal cell carcinoma, metastatic melanoma |
• Boxed warnings for capillary leak syndrome and infection • Risk of hypersensitivity reported169 |
| Filgrastim | Recombinant methionyl human G-CSF | AML, bone marrow transplant in cancer, blood progenitor cell collection and therapy, chronic neutropaenia |
• No boxed or product label warnings issued • Risk of splenic rupture and ARDS indicated in product label • Additional monitoring required by the MHRA |
| Interferon alpha | Recombinant human IFNα | Chronic hepatitis C, hairy cell leukaemia, AIDS-related Kaposi's sarcoma, CML |
• Boxed warnings for autoimmune reactions and infections • Risk of cytopaenia indicated in product label • Risk of immunogenicity reported170 |
| Interferon beta-1a | Recombinant human IFNβ1a | Relapsing–remitting multiple sclerosis |
• No boxed or product label warnings issued • Anaphylaxis reported post-approval • Immunogenicity reported170 |
| Interferon beta-1b | Recombinant human IFNβ1b | Relapsing–remitting multiple sclerosis |
• No boxed warnings issued • Lymphopaenia risk indicated in product label • Risk of immunogenicity reported170 |
| Interferon gamma | Recombinant human IFNγ | Chronic granulomatous disease, malignant osteopetrosis | • No boxed or product label warnings issued |
| Peginterferon alfa-2a | Pegylated recombinant human IFNα2a | Chronic hepatitis C |
• Boxed warnings issued for autoimmune reactions and infections • Risk of cytopaenia indicated in product label |
| Peginterferon alfa-2b | Pegylated recombinant human IFNα2b | Chronic hepatitis C |
• Boxed warnings issued for autoimmune reactions and infections • Risk of cytopaenia indicated in product label |
| Sargramostim | Recombinant human GM-CSF | AML, non-Hodgkin's lymphoma, bone marrow transplant | • No boxed or product label warnings issued |
| AML, acute myeloid leukaemia; ARDS, acute respiratory distress syndrome; CML, chronic myeloid leukaemia; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte–macrophage CSF; IFNα, interferon-α; IL-2, interleukin-2; MHRA, Medicines and Healthcare products Regulatory Agency (UK). | |||
Table 2.
Clinically used antibody-based triggering immunomodulatory biologics
| Biologic | Type | Mechanism of action | Indications | Immune-related safety warnings |
|---|---|---|---|---|
| Alemtuzumab | Humanized mAb | Binds to CD52 on leukocytes and initiates antibody-dependent cell-mediated lysis | B cell chronic lymphocytic leukaemia |
• Boxed warnings for cytopaenias, infusion reactions and infections |
| Brentuximab vedotin | Chimeric mouse–human mAb | Antibody–drug conjugate (CD30-specific mAb conjugated to MMAE) that induces cell cycle arrest and apoptosis | Hodgkin's lymphoma, systemic anaplastic large cell lymphoma |
• Boxed warning for PML • Warning for neutropaenia and infection172 on product label |
| Catumaxomab | Rat–mouse hybrid mAb | Bispecific antibody; binds to CD3–EpCAM and forms a bridge between cancer cells and T cells | Cancer, malignant ascites |
• Boxed warnings for CRS and/or SIRS • Immunogenicity reported173 • Additional monitoring required by the MHRA |
| Denileukin diftitox | Fusion protein (IL-2 and Diphtheria toxin) | Binds to CD25 of IL-2R and triggers toxin-induced cell death | T cell lymphoma | • Boxed warnings for severe infusion reactions and capillary leak syndrome |
| Ibritumomab tiuxetan | Mouse mAb | Fab segment of the antibody targets CD20 on B cells, allowing covalently linked radioactive yttrium, which emits a β particle, to destroy the cell | Follicular non-Hodgkin's lymphoma |
• Boxed warnings for acute infusion reactions, severe cutaneous and mucocutaneous reactions, and prolonged and severe cytopaenia • Anaphylaxis and immunogenicity reported174 |
| Muromonab-CD3 | Mouse mAb | Kills CD3-positive cells by inducing antibody-dependent cell-mediated toxicity and complement-dependent cytotoxicity | Organ transplant rejection |
• Boxed warnings for CRS and anaphylactic reactions • Warnings for infections and malignancy on product label |
| Ofatumumab | Human mAb | Targets CD20 and facilitates cell lysis due to complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity of B cells | Chronic lymphocytic leukaemia |
• No boxed or product label warnings issued • Infections reported176 • Additional monitoring required by the MHRA |
| Rituximab | Chimeric mouse–human mAb | Binds to CD20 on B cells and triggers B cell lysis by complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity | Non-Hodgkin's lymphoma, chronic lymphocytic leukaemia, rheumatoid arthritis, Wegener's granulomatosis and microscopic polyangitis | • Boxed warnings for acute infusion reactions and/or CRS, tumour lysis syndrome, severe mucocutaneous reactions and PML |
| Tositumomab | Mouse mAb | Binds to CD20-positive cells and causes cell death through ionizing radiation when it is radiolabelled (131I); also induces complement-dependent and antibody-dependent cell-mediated cytotoxicity | Non-Hodgkin's lymphoma (CD20-positive, follicular) |
• Boxed warnings for severe allergic reactions and/or anaphylaxis, and prolonged and severe cytopaenia • Malignancy warning on product label • Immunogenicity reported177 |
| CRS, cytokine release syndrome; EpCAM, epithelial cell adhesion molecule; IL-2R, interleukin-2 receptor; mAb, monoclonal antibody; MHRA, Medicines and Healthcare products Regulatory Agency (UK); MMAE, monomethyl auristatin E; PML, progressive multifocal leukoencephalopathy; SIRS, systemic inflammatory response syndrome. | ||||
Table 3.
Clinically used blocking immunomodulatory biologics
| Biologic | Type | Mechanism of action | Indications | Immune-related safety warnings |
|---|---|---|---|---|
| Abatacept | Fusion protein (CTLA4–Fc–IgG1) | Binds to CD80 and/or CD86 to prevent interaction with CD28 and so inhibits selective T cell co-stimulation | Rheumatoid arthritis, JIA | • No boxed or product label warnings issued |
| Adalimumab | Human mAb | Binds to TNF and blocks its interaction with its receptors | Rheumatoid arthritis, JIA, psoriatic arthritis, ankylosing spondylitis, Crohn's disease |
• Boxed warnings for serious infections (tuberculosis and invasive fungal infections) and malignancies (lymphoma and leukaemia) • HSTCL reported in postmarketing studies • Immunogenicity reported178 |
| Alefacept | Fusion protein (soluble LFA3–Fc–IgG1) | Binds to CD2 and inhibits the interaction between CD2 and LFA3 during lymphocyte activation | Chronic plaque psoriasis |
• No boxed or product label warnings issued • Infection and malignancy reported in postmarketing studies |
| Anakinra | Recombinant human IL-1RA derived from Escherichia coli | Binds to IL-1 competitively and prevents the interaction between IL-1 and IL-1R1 | Rheumatoid arthritis |
• No boxed warnings issued • Warning for hypersensitivity to E. coli products on label |
| Basiliximab | Chimeric mouse–human mAb | Binds to IL-2Rα and prevents the interaction between IL-2 and IL-2R | Renal transplant rejection |
• Warnings for hypersensitivity and anaphylaxis20 on product label • Immunogenicity reported20 |
| Belatacept | Fusion protein (CTLA4–Fc–IgG1) | Binds to CD80 and/or CD86 to prevent the interaction with CD28 and so inhibits selective T cell co-stimulation | Renal transplant rejection |
• Boxed warnings for PTLD, malignancy and infection (EBV) • Additional monitoring required by the MHRA |
| Belimumab | Human mAb | BAFF-specific inhibitor | Systemic lupus erythematosus |
• No boxed or product label warnings issued • Hypersensitivity and anaphylaxis reported in postmarketing studies • Additional monitoring required by the MHRA |
| Canakinumab | Human mAb | Binds to IL-1β to prevent the interaction with IL-1R | Cryopyrin-associated periodic syndromes, FCAS, MWS |
• Warning for risk of infection on product label • Additional monitoring required by the MHRA |
| Certolizumab pegol | Pegylated Fab′ humanized mAb | Binds to TNF and blocks its interaction with TNFR | Crohn's disease, rheumatoid arthritis |
• Boxed warnings for serious infections (tuberculosis, invasive fungal infection, Legionella spp. and Listeria monocytogenes) and malignancy • Severe skin reactions reported in postmarketing studies • Additional monitoring required by the MHRA |
| Daclizumab (withdrawn from the market owing to non-safety reasons) | Humanized mAb | Binds to IL-2Rα and prevents the interaction between IL-2 and IL-2R | Renal transplant rejection | • No boxed or product label warnings issued |
| Denosumab | Human mAb | Prevents RANKL binding to receptors; inhibits osteoclast formation, function and survival | Osteoporosis |
• No boxed or product-label warnings issued • Hypersensitivity reported in postmarketing studies • Additional monitoring required by the MHRA |
| Eculizumab | Humanized mAb | Binds to complement protein C5 and inhibits its cleavage to C5a and C5b, and prevents formation of the terminal complement complex C5b–9 | Paroxysmal nocturnal haemoglobinuria, atypical haemolytic-uremic syndrome |
• Boxed warnings for serious infections (meningococcal) • Additional monitoring required by the MHRA |
| Efalizumab (withdrawn from the market owing to safety reasons) | Humanized mAb | Binds to CD11a: the alpha subunit of LFA1 | Psoriasis | • Boxed warnings for serious infections (PML) |
| Etanercept | Fusion protein (TNFR2–IgG1) | Binds to TNF and blocks its interaction with TNFR | Rheumatoid arthritis, JIA, ankylosing spondylitis, psoriasis | • Boxed warnings for serious infections (tuberculosis, invasive fungal infections, Legionella spp. and L. monocytogenes) and malignancy |
| Golimumab | Human mAb | Binds to TNF and blocks its interaction with TNFR | Rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis |
• Boxed warnings for serious infections and malignancy • Additional warnings for cytopaenia and hypersensitivity |
| Infliximab | Chimeric mouse–human mAb | Binds to TNF and blocks its interaction with TNFR | Crohn's disease, paediatric Crohn's disease, ulcerative colitis, paediatric ulcerative colitis, rheumatoid arthritis, ankylosing spondylitis |
• Boxed warnings for serious infections and malignancy • PML reported179 • HSTCL reported in postmarketing studies • Immunogenicity reported156 |
| Ipilimumab | Human mAb | Binds to CTLA4 to block the interaction of CTLA4 with its ligands (CD80 and CD86) | Melanoma (unresectable and/or metastatic) |
• Boxed warnings for immune-mediated adverse reactions • Additional monitoring required by the MHRA |
| Natalizumab | Humanized mAb | Binds to α4 subunit of α4β1 and α4β7 integrins, and inhibits α4 integrin-mediated adhesion of leukocytes (except in neutrophils) to their counter-receptors | Relapsing–remitting multiple sclerosis, Crohn's disease |
• Boxed warnings for serious infections (PML) • Immunogenicity reported180 • Additional monitoring required by the MHRA |
| Omalizumab | Humanized mAb IgG1κ | Binds to IgE and thus prevents IgE-mediated activation of mast cells and basophils | Asthma |
• Boxed warnings for anaphylaxis • Parasitic (helminth) infections reported181 • Additional monitoring required by the MHRA |
| Rilonacept | Fusion protein (IL-1R–Fc–IgG1) | Binds to IL-1β and neutralizes its activity by blocking its interaction with IL-1R | Cryopyrin-associated periodic syndromes | • No boxed or product label warnings issued |
| Tocilizumab | Humanized mAb | Binds to soluble and membrane-bound IL-6R and prevents IL-6 from binding to IL-6R | Rheumatoid arthritis, systemic JIA |
• Boxed warnings for serious infections (latent tuberculosis infection and opportunistic infections) • Risk of anaphylaxis reported in postmarketing studies • Immunogenicity reported182 • Additional monitoring required by the MHRA |
| Ustekinumab | Human mAb | Blocks the function of IL-12 and IL-23 by binding to the p40 subunit of these cytokines | Psoriasis |
• No boxed or product label warnings issued • Allergy and/or hypersensitivity reported in postmarketing studies |
| BAFF, B cell-activating factor; CTLA4, cytotoxic T lymphocyte antigen 4; EBV, Epstein–Barr virus; FCAS, familial cold autoinflammatory syndrome; HSTCL, hepatosplenic T cell lymphoma; IgG1, immunoglobulin G1; IL-1, interleukin-1; IL-1R1, IL-1 receptor 1; IL-1RA, IL-1R antagonist; JIA, juvenile idiopathic arthritis; LFA, lymphocyte function-associated antigen; mAb, monoclonal antibody; MHRA, Medicines and Healthcare products Regulatory Agency (UK); MWS, Muckle–Wells syndrome; PML, progressive multifocal leukoencephalopathy; PTLD, post-transplant lymphoproliferative disease; RANKL, receptor activator of NF-κB ligand; TNF, tumour necrosis factor; TNFR, TNF receptor. | ||||
Understanding the complex interactions among disease states, the host immune system, the target of immunomodulation and the immunomodulatory biologic is vital to predicting and mitigating the risk of unwanted effects (Fig. 1). Furthermore, such detailed understanding can contribute to the design and development of immunomodulatory biologics with a reduced risk of unwanted effects.
Figure 1. Complex interactions among the disease, the immune system and immunomodulatory biologics that influence safety and efficacy.
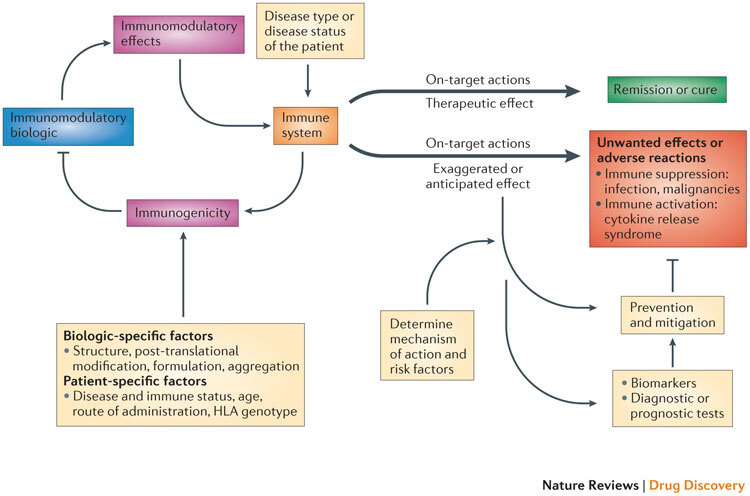
The interaction of the immunomodulatory biologic with the immune system and immune processes results in either the required (or intended) on-target therapeutic effect or unwanted reactions. Adverse reactions such as unwanted immunosuppression or immune activation are usually associated with the on-target exaggerated pharmacology of the biologic (for example, immunosuppression from a tumour necrosis factor (TNF)-specific therapy increases the risk of reactivation of tuberculosis or there is the risk of inducing cytokine release syndrome through the excessive activation of T cells with muromonab-CD3 therapy). The biologic also has the potential to induce a host immune response (termed immunogenicity), which results in the formation of drug-targeting antibodies that in turn can impede the therapeutic efficacy of the immunomodulatory biologic. The disease type and status of the patient can also influence the functional state of the immune system and thereby determine whether the interaction with the biologic leads to a therapeutic effect or unwanted adverse reactions (for example, chronic inflammation associated with diseases such as rheumatoid arthritis exposes the patient to an increased risk of malignancy). Other patient-specific factors such as human leukocyte antigen (HLA) type as well as the route and frequency of administration have a bearing on the propensity to develop immunogenicity to the biologic, as these factors contribute to antigen processing and presentation of immunogenic epitopes. Factors that are intrinsic to the property of the biologic (biologic-specific factors), such as the presence of immunogenic epitopes, glycosylation and aggregation, also affect the generation of an immunogenic response. The prevention and mitigation of these unwanted adverse reactions is predicated on a detailed knowledge and understanding of the mechanisms and risk factors that drive the adverse reactions and the use of effective biomarkers and diagnostic tests. For example, knowledge of the association of the John Cunningham virus (JCV) in the aetiopathology of progressive multifocal leukoencephalopathy (PML) observed in patients receiving natalizumab (Tysabri; Biogen Idec/Elan) led to the recognition of the presence of JCV as a risk factor for PML. Consequently, a diagnostic test for JCV seropositivity is now used to stratify patients before initiating natalizumab therapy.
In this Review, we first focus on the current understanding regarding immunomodulation leading to serious infections, malignancies and CRS, as well as the immunogenicity of immunomodulatory biologics. We also outline strategies that are being pursued to prevent, minimize or mitigate these unwanted effects. We discuss the current state of available tools to predict these adverse reactions. Finally, we present a scheme outlining how an increased understanding of target biology and the mechanisms that drive unwanted effects, along with predictive tools, can be used for developing immunomodulatory biologics that have improved safety profiles.
Current knowledge of adverse reactions
A comprehensive understanding of the nature and mechanisms of the adverse reactions associated with immunomodulatory biologics underpins the design and development of safer immunomodulatory biologics. Here, we focus on some of the current, frequently occurring adverse reactions such as serious infections, malignancies, CRS and immunogenicity. We have chosen to focus on these particular well-characterized adverse reactions to enable the emergence of meaningful ideas for informing the development of safer immunomodulatory biologics. Descriptions and discussions on other adverse reactions such as tumour lysis syndrome and autoimmunity have been comprehensively reviewed elsewhere3.
Immunomodulation leading to serious infections and malignancies. A major adverse reaction associated with immunomodulation is the occurrence of serious infections that result in mortality or in hospitalization, requiring management with intravenously administered antibiotics. Out of the 40 licensed immunomodulatory biologics, 18 have been associated with serious infections, including reactivation of bacterial infections (for example, mycobacterial, streptococcal, listerial or meningococcal infections), viral infections (for example, hepatitis B virus (HBV) or HCV), fungal infections (for example, histoplasmosis) or opportunistic infections (for example, John Cunningham virus (JCV) or Pseudomonas aeruginosa) (Tables 1,2,3).
The significance of these adverse reactions has been recognized by regulatory authorities (the FDA and the EMA) and is reflected in product labels that state this risk. For some of these biologics, there is a clear cause and effect, and with this knowledge (Box 1) it is possible to implement mitigating strategies. However, for other biologics the causes of the increased susceptibility to infections are less obvious and therefore further analyses are necessary before preventive strategies can be formulated (Box 1).
Natalizumab (Tysabri; Biogen Idec/Elan) is an example of an immunomodulatory biologic that has a clear cause and effect, and for which the implementation of a risk management strategy was possible. Natalizumab is indicated for the treatment of multiple sclerosis and Crohn's disease, and it exerts its therapeutic effects by binding to the α4 integrin subunit to interfere with T cell trafficking. However, it has been associated with progressive multifocal leukoencephalopathy (PML), a progressive sub-acute demyelinating neurological condition4 that is caused by the reactivation of latent JCV infection. The reduced T cell trafficking to the central nervous system (CNS) compromises the immune surveillance and control of JCV in susceptible patients, thus increasing the risk of developing PML. Indeed, up to 11 out of every 1,000 patients receiving natalizumab therapy may develop PML if they have all of the following risk factors: prior infection with JCV, presence of JCV antibodies and clinical factors such as increased duration of treatment and concomitant immunosuppressant therapy5. These risk factors have been formally taken into account by regulatory agencies and are recognized by boxed warnings. Moreover, the product label for natalizumab recommends testing patients for JCV-specific antibody status before and/or during treatment if the antibody status is unknown. Box 1 outlines other examples of cases that have clear causal relationships for an increased risk of infection, as well as those cases in which the causes are unclear.
Regarding the increased risk of malignancy, it is acknowledged that biologics with an immunosuppressive mechanism of action can indirectly contribute to an increased risk of malignancy owing to decreased immune competence and immune surveillance; however, this may be undetectable over the background incidence of malignancy induced by the disease states themselves (that is, through inflammation and autoimmunity).
For instance, malignancy has been reported as a serious adverse reaction associated with the long-term use of the tumour necrosis factor (TNF)-specific class of immunomodulatory biologics6,7; indeed, the FDA and EMA have stated this risk on boxed warnings (Table 3). This includes the observed incidence of hepatosplenic T cell lymphoma in young adults and children receiving anti-TNF treatment8. However, recent meta-analyses of data from randomized controlled clinical trials and safety registries have contested this previously held consensus9,10,11. The lack of compelling evidence for malignancies related to anti-TNF therapy suggests that the nature of the underlying disease and co-medications contribute to lowering immune surveillance mechanisms in some patients to a degree that causes malignancies to arise, irrespective of the immunomodulatory biologic intervention. This is particularly true of chronic inflammatory states that are themselves associated with the emergence of neoplasms12 (for example, lymphoma in autoimmune rheumatic diseases)13.
Nevertheless, some factors have been observed to clearly lead to an increased risk of malignancies, and therefore mitigation strategies can be implemented. For example, the functional suppression of T lymphocytes by belatacept (Nulojix; Bristol-Myers Squibb) is associated with an increased risk of post-transplant lymphoproliferative disease14. This risk was observed to be high in Epstein–Barr virus (EBV)-seronegative patients receiving belatacept15. Consequently, measurement of the EBV load and EBV-specific T lymphocytes are currently used for the risk management of patients who have undergone a transplant, and only those patients who are seropositive for EBV are selected for belatacept therapy.
In summary, regulatory agencies take a conservative approach regarding the labelling of immunomodulatory biologics, and in most cases the risk of serious infections and/or malignancies is stated in the product label or boxed warning. However, it is hoped that progress in the development of tools to better predict adverse consequences will lead to a refinement of labelling so that patients with a lack of suitable therapeutic options can be treated safely.
Exaggerated immune activation resulting in CRS. CRS is characterized by the uncontrolled release of pro-inflammatory cytokines — such as interleukin-6 (IL-6), TNF and IFNγ — induced by the immunomodulatory biologic16,17. The seriousness of CRS is exemplified by the Phase I trial of the CD28 super-agonist TGN1412 (an agonistic antibody that activates T cells through the co-stimulatory receptor CD28)18, in which six healthy volunteers became seriously ill within 90 minutes of receiving the antibody. The CRS in this instance was consistent with the biological and pharmacological activity of the immunomodulatory biologic: T cell activation (Box 2).
Several of the immunomodulatory biologics currently on the market have been reported to induce CRS in rare instances, including aldesleukin (Proleukin; Prometheus), basiliximab (Simulect; Novartis), alemtuzumab (Campath-1H; Genzyme), rituximab and muromonab-CD3 (Tables 1,2,3), and this has been recognized in the 'summary of product characteristics' documents and patient information leaflets of these biologics. Although the mode of action of the immunomodulatory biologic is a major cause of CRS induction, the contribution of patient-specific factors and disease states to the induction of CRS is not immediately apparent. Immunomodulatory biologics induce responses that are characterized by 'trigger pharmacology', or steep or bell-shaped dose–response curves. When these responses are combined with the varying immune activation thresholds of the patient, the therapeutic could either have a safe outcome or induce an exaggerated immune response: that is, CRS.
Other acute reactions that can be induced by immunomodulatory biologics include hypersensitivity, anaphylaxis and infusion-site reactions, which are difficult to predict from the pharmacology of the immunomodulatory biologic and may occasionally be fatal — for example, in the cases of tocilizumab (Actemra; Roche), omalizumab and belimumab (Benlysta; Human Genome Sciences/GlaxoSmithKline). In addition, preventive strategies for CRS rely on the development of predictive assays that are particularly challenging to design (Box 2). However, these challenges are currently being addressed, and the predictive value of recent in vitro assays (discussed below) has been encouraging.
Host immune responses leading to immunogenicity. Unwanted immunogenic responses have been reported with the majority of immunomodulatory biologics currently on the market, and these responses are characterized by ADA formation (Tables 1,2,3). Notably, the risk of immunogenicity is not just restricted to immunomodulatory biologics but is also applicable to other (non-immunomodulatory) biologics such as erythropoietin, thrombopoietin, abciximab and cetuximab (Erbitux; Bristol-Myers Squibb/Eli Lilly).
The development of ADAs against immunomodulatory biologics can result in loss of (or reduced) efficacy, altered pharmacokinetics, infusion reactions, crossreactivity to endogenous proteins and anaphylactic shock19,20. ADAs can be neutralizing (known as NAbs) or non-neutralizing antibodies (known as N-NAbs), depending on the sites on the immunomodulatory biologic to which they bind. N-NAbs bind to an immunomodulatory biologic without disrupting the interaction of the biologic with its intended target, whereas NAbs block the activity of the immunomodulatory biologic by binding to sites that are crucial for drug–target interactions. Both N-NAbs and NAbs can influence the pharmacokinetics of an immunomodulatory biologic either by enhancing its clearance or by prolonging its bioavailability21,22.
Various patient-specific factors also contribute towards an immunogenic response, including immunological state and disease type or severity. For example, disease activity has been positively correlated with increased ADA titres to infliximab (Remicade; Centocor Ortho Biotech) in rheumatoid arthritis23. It has been suggested that other patient-specific factors such as immunoglobulin G (IgG) allotype and IL10 gene polymorphisms also influence the predisposition for ADA generation24,25. In addition, the inherent properties of the biologic (biologic-specific factors), such as the degree of aggregation, glycosylation and oxidation, are involved in the induction of immunogenic responses (Box 3).
Regulatory authorities have recognized the potential impact that immunogenicity to biologics has on the prognosis and contribution to co-morbidity, and this has resulted in the publication of guidelines for immunogenicity testing26,27. Together with the identification of the patient-specific factors (for example, disease and immunological state) and biologic-specific factors (for example, degree of aggregation) that contribute to the risk of developing immunogenicity, the prediction and reduction of this adverse reaction can be minimized (Box 3).
Other factors to consider
There are various relevant factors to be considered in the area of immunomodulatory biologic development and use. These relate to the application of rational design strategies for immunomodulatory monoclonal antibodies (mAbs), biomarker development and patient stratification based on responsiveness and the known risk of adverse reactions (personalized medicine).
Modifications to Fc domains of immunomodulatory mAbs. Modifications to immunomodulatory mAbs — primarily by altering the Fc region of the antibody — are being pursued to favourably alter their pharmacokinetic/pharmacodynamic (PK/PD) profiles. However, such modifications can have potential consequences for the safety of immunomodulatory mAbs; for example, they can enhance the risk of developing immunogenicity. The Fc region mediates the effector function of therapeutic antibodies by eliciting antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) (for example, as observed with rituximab and alemtuzumab; see Table 2). The induction of CRS in some patients could arise from excessive triggering of these Fc-mediated effector functions, as seen with rituximab. In such cases, it is difficult to disconnect the potential for adverse and intended pharmacological reactions, because these are mediated by the same component of the immunomodulatory biologic (that is, the Fc domain).
Various approaches have been used for improving the half-life of mAbs, such as increasing their binding affinity to the neonatal Fc receptor (FcRn) through the introduction of mutations in specific amino acids within the Fc region28. Other approaches include enhancing the effector functions of mAbs (either ADCC or CDC) through afucosylation29,30,31, by adding N-acetylglucosamine32 and by altering the galactosylation status33,34.
Although such modifications can provide longer or tailored therapeutic effects for the same or reduced dose of a mAb, there are theoretical concerns regarding the impact of these modifications on safety profiles. These concerns include: immunogenicity, arising as a result of prolonged exposure to the mAb; immune perturbation through altering the breadth of the immune response and/or unfavourable drug disposition (including maternal–fetal transport via FcRn); and the increased risk of CRS.
Patient stratification and biomarkers. The response to an immunomodulatory biologic therapy, in terms of both efficacy and toxicity, differs among individuals. One approach to improve the benefit–risk profile and maximize the therapeutic benefit of immunomodulatory biologics is to use biomarkers to identify and stratify patients according to their responsiveness to the specific therapeutic (that is, whether they are responders, partial responders, low responders or non-responders) at an early stage of — or before — the treatment cycle.
This has been exemplified in patients with asthma receiving omalizumab (an IgE-specific antibody), in whom there is a strong correlation between clinical response and the suppression of serum IgE levels35. In this case, serum anti-IgE levels are a good biomarker for stratifying patients into responder and non-responder groups. Lebrikizumab (an IL-13-targeted therapy) is another example of a biologic that can be linked to a biomarker to identify which patients are most likely to respond to asthma treatment. In a clinical trial, improvement in lung function was higher in patients with higher serum levels of periostin36. The identification of potential non-responders to biologics based on biomarker status would therefore ensure that patients who have no prospect of receiving clinical benefit do not receive a potentially harmful drug.
In terms of avoiding adverse reactions with an immunomodulatory biologic therapy, there are currently few biomarkers available that can predict adverse reactions associated with an immunomodulatory biologic. These include JCV antibody status, EBV seropositivity and tuberculin tests for the risk of serious infections or general readouts of immune activation and suppression (for example, lymphocyte counts and activation markers).
Some human leukocyte antigen (HLA) association — albeit weak — has been identified for biologic-associated immunogenicity but this has limited value in patient stratification37. Biomarker development therefore represents an area of unmet need. Moreover, forward planning is needed to develop biomarkers and to facilitate stratified medicine (also known as personalized medicine); this will require the linking of detailed clinical data from patient studies to high-quality biobanks containing sera, plasma, DNA, RNA, urine, cells and diseased tissue samples to gain a more detailed understanding of the fundamental mechanisms by which immunomodulatory biologics interact with individual patients and elicit adverse reactions.
Current tools for predicting unwanted effects
Tests, tools and models for predicting unwanted effects are vital for developing safer immunomodulatory biologics. It must be recognized that although the current state of the art in this area is not very advanced, progress is being made in refining existing tools as well as in the development of new models to predict unwanted effects.
In general, the immune system of non-human primates (NHPs) is considered to be the most similar to the human immune system and is therefore thought of as the most appropriate model to evaluate immunomodulatory biologics for adverse reactions. However, marked differences exist between human and NHP immune processes, and therefore knowledge of inter-species similarities and differences in all aspects of the pharmacology and immunology of immunomodulatory biologics is crucial for the design of appropriate safety studies that can be effectively translated to humans (Box 4). A relevant model species should possess similar levels of tissue expression of the target, similar or identical target modulation, downstream signalling and effector function compared to humans, and it should also exhibit the intended functional potency of the biologic38,39.
Apart from NHPs, other models and test systems are being used to predict unwanted effects, and these are discussed briefly below.
Predicting serious infections and malignancies. Owing to the complexity of the interactions of pathogens and tumours with the host immune system, predictive tests for serious infections and malignancies require in vivo experimental systems.
General in vivo assessments of immunological status — such as total and differential leukocyte counts, globulin levels and the histopathology of lymphoid organs and tissues40,41 — can provide a broad indication of the nature of the immune perturbations that can be induced by an investigational immunomodulatory biologic. For a higher resolution on the nature of the biologic-induced immune effects, immunophenotyping of the affected tissues and of lymphocytes from blood and lymphoid organs (such as T cells, B cells and natural killer (NK) cells) can be performed using flow cytometry42. Although these assays help to provide detailed descriptions of the nature and extent of biologic-induced effects, they do not necessarily report on functional perturbations of the immune system.
The T cell-dependent antibody response (TDAR) assay is an immunological model that is used to assess the immunotoxicity of small-molecule compounds, but this assay is not as widely used with biologics. The TDAR assay provides an indication of the overall host response to a model antigen and could therefore be a useful tool in identifying biologic-induced immunosuppression. The currently used T cell-dependent antigen in a TDAR assay is keyhole limpet haemocyanin (KLH) in rodents, and KLH or tetanus toxoid in cynomolgus monkeys42,43. The TDAR assay evaluates both primary immunological responses (IgM and IgG) and secondary immunological responses (IgG) to an antigen. It provides an integrated readout of antigen processing and presentation, lymphocyte interaction and the antibody production capability of the host. This assay can therefore reveal biologic-induced effects on one or several aspects of the immune system and immune response.
Although preclinical TDARs may not be completely predictive with regard to human susceptibility to immunosuppresion-associated infection, they can be a useful tool for ranking biologics according to the degree of immunosuppression elicited and hence aid in candidate selection for drug development. The challenge now is to develop models that can report on immune parameters that are relevant to the specific biologic or to its mechanism of action; such parameters could replace or be used in conjunction with TDAR assays.
With regard to the selection of the most relevant in vivo models for evaluating the risk of developing serious infections upon receiving an immunomodulatory biologic, NHPs would be most appropriate. Opportunistic infections such as mycobacterial infections, malaria (Plasmodium spp.), gamma herpes virus, histoplasmosis, Salmonella spp. and Shigella spp. in NHPs are not unexpected following immunomodulation by some immunomodulatory biologics. As there is a very similar or comparable range of organisms associated with these kinds of infections in both NHPs and humans44, any knowledge on the incidence and type of such infections in NHPs following the administration of immunomodulatory biologics could be potentially translatable to humans44. However, because the incidence of serious infections is very low and studies in NHPs are not sufficiently statistically powered to detect this, the utility of NHPs in predicting these adverse reactions remains uncertain.
Models of host resistance to infection can also be utilized to assess the immunosuppressive effects of a biologic. These models can identify the effect of biologics on the host following challenge with several microorganisms, including bacteria, viruses or parasites. For example, the rodent influenza model can be used to assess the overall immune response as all the arms of the immune system are required for the clearance of the virus45. Further clarity in evaluating the immune status can be achieved using targeted models of host resistance, which can provide information on the specific dysregulation occurring in the immune system that is leading to the adverse reaction. For example, the murine model of latent cytomegalovirus infection can be used to assess the reactivation of latent viral infection caused by suppressed cellular or humoral immunity; models of Streptococcus pneumoniae or Listeria monocytogenes can be used to assess the innate immunity of phagocytes (macrophages or neutrophils); and models of S. pneumoniae or Haemophilus influenzae can be used to evaluate the status of marginal zone B cells45. The use of these models has been very limited so far, and more work is required to validate these models if they are to be considered as reliable tools for predicting serious infections.
Models that measure host resistance against tumours are used in routine immunotoxic assessments and can be adopted to predict the risk of malignancy arising from the use of immunomodulatory biologics. These models include syngeneicin vivo rodent tumour models (for example, EL4 lymphoma, B16F10 melanoma and PYB6 fibrosarcoma models) or the human peripheral blood lymphocyte severe combined immunodeficiency mouse model (for EBV-related lymphoproliferative disorders)46, which can be useful in determining host resistance against transplanted tumours45. Models that measure host resistance against tumours can be used on a case-by-case basis; if there is strong evidence that the biology of the target is associated with tumour resistance, then it may not be necessary to perform these assessments.
Lymphocryptovirus (LCV) in rhesus monkeys is similar to EBV in humans (in terms of cellular tropism, host immune response and lymphocyte stimulation potential); for this reason, it is being considered whether LCV load and/or LCV-specific T lymphocytes can be used as a surrogate marker for immunosuppression and/or virus-induced malignancy in NHPs47. However, the reliability and feasibility of LCV-associated surrogate biomarkers in NHPs needs to be established and standardized. The availability of such preclinical models will aid in the identification of immunomodulatory biologics that are associated with an increased risk of inducing EBV-related lymphomas.
Predicting CRS. In vitro toxicity testing with human blood cells is relevant to CRS and is generally considered to have good predictive value, particularly as inter-species differences severely limit the capacity of animal models to predict CRS (Box 2). Nevertheless, there are considerable ongoing efforts directed towards further development of human in vitro models for CRS as well as the optimization of the predictive value of these models16,48. For example, work is being carried out within the International Life Sciences Institute and the Health and Environmental Sciences Institute (ILSI/HESI) immunotoxicity group, and CRACK IT projects organized by the National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs) have aimed to refine in vitro cytokine release test models to improve their predictive capacity49 (see the CRACK IT 2011 challenge 4 on the CRACK IT website). In addition, other models being developed to assess mAb-mediated CRS include the 'optimized biomimetic cell culture system', which comprises multiple cells that are relevant for CRS together with the use of platelet-poor plasma (blood plasma with low platelet counts) to better mimic the in vivo environment50.
Although no in vitro cytokine release assay can be used to precisely quantify the risk of CRS, currently available methods are valuable in hazard identification and as sources of additional information on the mechanisms of potential CRS induction51. Furthermore, the outputs from such in vitro models are essential for the design of first-in-human (FIH) studies of immunomodulatory biologics, particularly with respect to the immunological parameters that should be monitored.
The predictive strength of in vivo models for CRS has been poor (Box 2). In addition, there are instances in which limitations in the predictive capacity of in vivo models are clear: for example, when there is no crossreactive species or when there are known major differences in the immune system or novel modes of action. However, if a relevant preclinical test species is available, it would be useful to supplement these safety data with observations from ex vivo studies using human cells designed and based on the target biology or pharmacology. These data can be valuable in FIH trials by providing an indication of potentially safe dose windows and in estimating the minimal anticipated biological effect level (MABEL)52,53,54.
Predicting immunogenicity. Progress has been made in the development of various test systems (in silico, in vitro (ex vivo) and in vivo) that have all been designed to predict the immunogenicity of biologics.
In silico methods are primarily designed to identify potentially immunogenic epitopes (T cell and B cells) in biologics. In silico prediction strategies used for designing effective vaccines and determining T cell epitopes in autoimmunity, as well as sequence alignment with major histocompatibility complex (MHC) class II gene products, could be adopted to predict the immunogenicity of immunological biologics55,56,57. Computational tools can also be used to identify immunogenic T cell epitopes (epitope mapping) and to predict peptide–MHC interactions that contribute to immunogenicity58,59,60. In addition, B cell immunogenic epitopes can be incorporated into immunogenicity prediction models. B cell epitopes can be either linear or conformational and are hence more difficult to predict than T cell epitopes. Although some in silico tools for predicting B cell epitopes have been developed61,62,63, these are not yet being utilized systematically to predict immunogenicity.
The presence of aggregates in the formulation of biologics has been associated with immunogenicity (Box 3). Recent efforts are therefore being directed towards evaluating the propensity for aggregation in biologics by predicting aggregation-prone regions contained within the T and B cell immunogenic epitopes64,65. In addition to their utility in the development of new biologics, these tests could be valuable in assessing the immunogenic potential of the modifications that have been made to existing immunomodulatory biologics.
The role of professional antigen-presenting cells (APCs) in mediating immunogenic responses by presenting immunogenic peptides on MHC molecules to T cells is well documented37. Therefore, in vitro assays have been modelled to predict the immunogenic potential of biologics by investigating the various stages of this process. In vitro HLA binding assays that directly measure the affinity of a peptide for a defined MHC class II molecule may assist in predicting immunogenic epitopes66. An 'MHC-associated peptide proteomics' (MAPPs) approach, which involves direct sequence analysis of peptide epitopes isolated from APCs, has been used to predict immunogenicity67. Ex vivo assays using human T cells have also been reported to be relatively accurate at predicting immunogenicity to biologics in humans68,69,70. The success of these predictive assays depends on testing relevant peptides in many individuals to ensure that the cohort covers a broad spectrum of the MHC II allotypes that are present in the general population. A variant of the ex vivo T cell assay that is being developed involves co-culturing dendritic cells (which are professional APCs) along with T cells to mimic the interactions of APCs with T cells in the immunogenic response.
Although animal models are frequently used in the in vivo assessment of the toxicological profile of investigational therapeutics during development, caution should be exercised when drawing conclusions from these assessments regarding the risk of developing immunogenicity in humans. Transgenic animal models that express the different prevalent human HLA types71,72 would be appropriate for testing some aspects of the immunogenic potential of the biologic. A transgenic animal expressing the human protein of interest, in which B cell tolerance has been disrupted by administering the therapeutic counterpart of the protein under investigation, could also be useful for evaluating the immunogenic potential of a biologic as well as physicochemical factors that skew the therapeutic proteins towards unwanted immunogenicity73,74.
The immune-system-humanized mouse model or the human immune system xenograft mouse model are other in vivo strategies being explored to study immunogenic responses75,76,77. These models are created by engrafting human haematopoietic stem cells or peripheral blood mononuclear cells into immunodeficient mice (NOD-SCID-Il2rg-null mice). However, this approach does have limitations when high-throughput profiling is required, as only a single person's immune phenotype can be grafted at a time. Therefore, the development of a similar model with a capacity to represent the various HLA types would be beneficial. Nevertheless, it must be recognized that not all aspects of the complex human immune system can be embedded within these transgenic animal models.
NHPs are considered to be more similar to the human immune system than rodents but NHPs can still mount immunogenic responses to human proteins. On balance, the various in vitro and ex vivo assays that can be used to evaluate the potential immunogenic contribution of biologics and the relevant immune cells have had more predictive value than the animal models. Strategies to meet the challenges of predicting immunogenicity should involve complementary approaches in which in silico tools are integrated with in vitro or ex vivo assays that utilize human T cells and APCs to identify immunogenic peptide sequences that may provide a more sensitive basis for the assessment and prediction of immunogenicity.
Developing safer immunomodulatory biologics
The process used for the discovery of small-molecule drugs is not entirely applicable to the discovery and design of immunomodulatory biologics. Knowledge regarding the role of the target in disease, the biology of the target and the preferred mode of biologic–target interactions is pivotal to the design of biologics with therapeutic efficacy and minimal safety risks.
The cumulative experience derived from studying the adverse reactions associated with immunomodulatory biologics, in both humans as well as preclinical tests, has provided much-needed insights into the mechanisms that underlie such adverse reactions. Valuable gains can be made by exploiting this increased understanding and knowledge for the discovery and design of new biologics. Furthermore, if they are tailored to suit the nature and biology of the relevant target or target cell and the intended mechanism (or mechanisms) of action, preclinical models for CRS (whole-blood models16,51, peripheral blood mononuclear cell models51 and biomimetic cell models50) and immunogenicity (immunogenic epitope mapping67, ex vivo T cell assays68, MAPPs67 and humanized in vivo models78) can be utilized in the selection of lead candidates that can be advanced to development.
In our opinion, a greater understanding of — and the ability to predict — adverse reactions can lead to two main pathways that can be followed for the discovery and design of safer biologics: the selection of new targets or the redesign of the existing immunomodulatory biologic (Fig. 2).
Figure 2. Pathways for the development of safer immunomodulatory biologics.
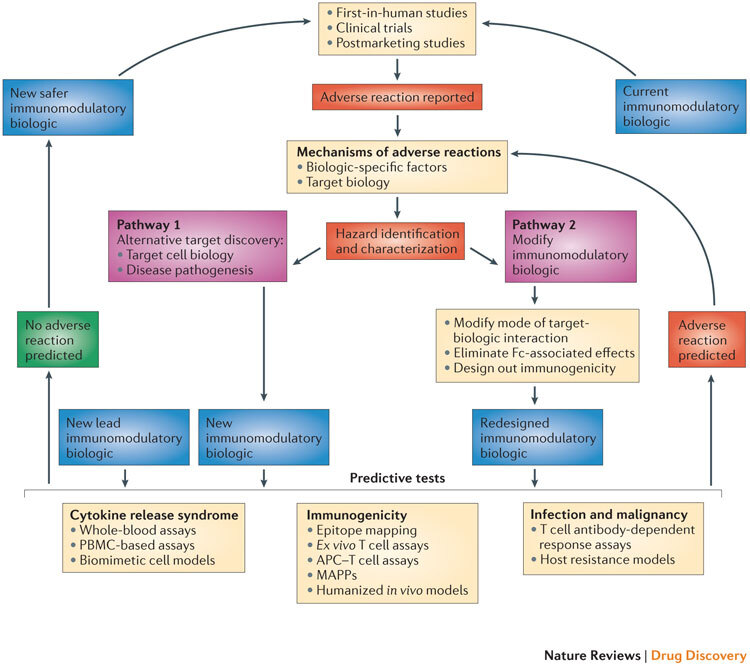
The iterative cycle for the development of safer immunomodulatory biologics incorporates two key pathways and depends on the collective knowledge obtained from adverse reactions observed in first-in-human studies, clinical trials and postmarketing pharmacovigilance analyses. These data provide the basis for understanding the frequency and nature of the adverse reactions associated with immunomodulatory biologic therapy, as well as the potential mechanisms by which these adverse reactions are induced. The next step in this process is the identification and characterization of hazards associated with the immunomodulatory biologic (for example, characterizing excessive Fc-mediated effector functions that result in cytokine release syndrome (CRS), overt or global immunosuppression leading to serious infections or the presence of immunogenic structures within the biologic that trigger immunogenicity). Pathways 1 and 2 are two different trajectories that utilize the understanding of the mechanism of adverse reactions to inform the design of safer and potentially more effective immunomodulatory biologics. Pathway 1 is followed when the adverse reaction cannot be dissociated from the target biology, and involves generating a biologic that engages an alternative target or mechanism to produce the desired pharmacodynamic effect without the associated adverse reaction. Pathway 2 involves redesigning the biologic to engineer out components within the biologic structure that trigger adverse effects, or to alter the nature of the target–biologic interactions. New immunomodulatory biologics from both of these pathways (and any other lead drug candidates) need to go through a panel of predictive tests until a safer biologic emerges. The selection of the predictive tests to be used is on a case-by-case basis, as it should take into account the nature of the target biology, the effector mechanisms that are engaged and any other factor or factors that influence the intended pharmacological effect and the risk of adverse reactions. Currently available tests that are suitable for determining the CRS-inducing risk of a new biologic with immunostimulatory properties could include whole-blood assays, peripheral blood mononuclear cell (PBMC)-based assays and biomimetic cell models. Predicting the risk of serious infections and malignancies for a new immunomodulatory biologic still remains a challenge, and limitations in T cell-dependent antibody response (TDAR) assays and host resistance models are due to species-specific variations both in target biology and in exposure to risk factors. Preclinical tools for predicting the risk of immunogenicity involve the use of in silico models (immunogenic epitope mapping), in vitro or ex vivo models (T cell assays, antigen-presenting cell (APC)–T cell assays and major histocompatibility complex (MHC)-associated peptide proteomics (MAPPs)) and in vivo models (humanized animals). The integration of in silico and in vitro or ex vivo assays increases the predictive value of these preclinical tools. A new or redesigned immunomodulatory biologic that is considered to be safe based on predictive preclinical assessments enters the cycle of testing in first-in-human studies and clinical trials, and then moves to the clinic with necessary safety precautions in place and with continuous monitoring for any potential adverse reaction. By contrast, any new immunomodulatory biologic that is flagged using predictive tests for its potential to cause adverse reactions (or observed to cause adverse reactions in clinical studies) will be passed through this iterative cycle again for the design and development of a safer immunomodulatory biologic.
Pathway 1: selection of new targets
The selection of new targets could be the preferred route for developing a new immunomodulatory biologic if the adverse reaction cannot be uncoupled from the intended pharmacological effect. This can be understood by examining the following four examples: ustekinumab (Stelara; Centocor Ortho Biotech), vedolizumab, epratuzumab and natalizumab.
Ustekinumab. One available option for treating psoriasis and inflammatory bowel disease (IBD) is the use of anti-TNF biologics79,80. However, the risks associated with long-term anti-TNF therapy (such as serious infections) cannot at present be overcome and they appear to be a class effect arising through interference with the functions of TNF. A functional IL-12–IL-23 pathway has been shown to be a key contributor in the pathogenesis of psoriasis and IBD81,82. This knowledge has been exploited in the design of a new biologic: ustekinumab is a mAb that targets the IL-12–IL-23 pathway with good therapeutic outcomes in psoriasis and IBD, and it has a favourable safety profile compared to anti-TNF biologics81,82.
Vedolizumab. Natalizumab has been used in the management of Crohn's disease but it carries the risk of inducing PML owing to reduced T cell immunosurveillance in treated patients. This appears to be due to the indiscriminate targeting of all α4 integrin-expressing T cells, including those in the CNS. In this case, designing an immunomodulatory biologic that could specifically suppress T cells that home in to the gut, without affecting those that are involved in CNS immune surveillance, is expected to be a safer approach. Indeed, a new biologic — vedolizumab — that preferentially targets gut-specific T cells is currently in clinical trials and proving to be efficacious in the treatment of IBD83. Vedolizumab selectively targets α4β7 integrin, which mediates the migration of T cells preferentially into gastrointestinal tissue without compromising immune surveillance in the CNS84, and this biologic has a potentially reduced risk of PML.
Epratuzumab. Rituximab, which is used for treating non-Hodgkin's lymphoma, is a CD20-targeting mAb that induces B cell-specific cytotoxicity through ADCC and CDC. Despite its therapeutic advantage, rituximab has been associated with serious infections and CRS. The serious infections are caused by the depletion of all B cell populations, including memory B cells. In this case, B cell depletion is the intended pharmacodynamic outcome, which could be potentially achieved by other means, with considerably lower safety risks, by targeting other molecules related to B cell survival. A potential alternative to CD20 is another B cell molecule, CD22, which can be targeted with the mAb epratuzumab for the treatment of non-Hodgkin's lymphoma85; clinical trials have indicated a good safety profile of epratuzumab in this setting.
Beyond natalizumab. A more detailed understanding of the cellular and molecular aetiopathology of disease processes can also open up pharmacological space for immunomodulatory biologics through the identification of new targets. Natalizumab, which blocks T cell entry into the CNS, is indicated for multiple sclerosis and is associated with the risk of PML. This adverse reaction is most likely to be related to the reduced T cell immunosurveillance in treated patients and cannot be dissociated from the pharmacodynamic action of natalizumab. A subset of T cells, T helper 17 (TH17) cells, has been implicated in the pathogenesis of multiple sclerosis86. However, the detailed mechanism and degree of contribution of TH17 cells to the disease is still unclear. If the weight of evidence is in favour of TH17 cells as key players in the pathology of multiple sclerosis, then targeting this particular subset of T cells could lead to therapeutic benefits without inducing the adverse reactions associated with the suppression of immune surveillance through the indiscriminate targeting of all α4 integrin-expressing T cells.
Pathway 2: redesign of the existing biologic
The second pathway that can be followed for the design of safer immunomodulatory biologics involves redesigning existing immunomodulatory biologics. This pathway can be followed when factors that are inherent to the biologic — rather than target-specific factors — are identified as the source of the adverse reactions, and this pathway is often the only way forward when alternative targets cannot be identified for the development of new biologics (Fig. 2).
There are three main strategies in this approach: changing the mode of the target–biologic interaction (for example, monovalent binding versus receptor clustering); avoiding Fc-mediated or other structure-dependent unwanted effects; and 'engineering out' immunogenic sites within the biologic structure (in instances where immunogenicity is the main unwanted effect).
Changing the mode of the target–biologic interaction. An example of this approach is the development of muromonab-CD3, which targets the CD3 receptor on T cells to induce cell death by ADCC and CDC, thereby controlling organ transplant rejection. However, as muromonab-CD3 works by clustering CD3 molecules on the cell surface, this often results in hyperimmune T cell activation. The interaction of a biologic with CD3 can be redesigned in such a way that this clustering does not occur, and small modular immune-pharmaceuticals (SMIPs) are being utilized to achieve this outcome87,88.
Avoiding Fc-mediated or other structure-dependent unwanted effects. The presence of an activating Fc domain in blocking immunomodulatory biologics is not necessary, and its presence can cause unfavourable responses by interacting with other immune cells expressing Fc receptors. Modifications to the Fc portion through mutations in specific amino acid residues could prevent the interaction of the Fc domain with other Fc receptors and complement components, and hence reduce these types of adverse reactions.
Complement activation that accompanies CD20 targeting by rituximab contributes to the triggering of CRS. This aspect of rituximab's mode of action has the potential to be designed out. Indeed, an SMIP targeting CD20 was reported to have attenuated complement activation compared to rituximab while providing a similar level of therapeutic efficacy89,90. Furthermore, it has been shown that the substitution of the amino acid residues Pro331 with Ser and Lys322 with Ala in the Fc region of the antibodies abrogated adverse reactions associated with complement activation in rats and monkeys91.
Restricting the target–biologic interaction to specific cell types and/or subsets can also reduce the risk of adverse reactions associated with the activation of accessory immune cells. Bispecific antibodies are designed to combine two domains: one that is specific for the target molecule and the other for a cell-specific molecule. This enables the homing of the biologic to the right target on the right cell and hence reduces nonspecific interactions of the biologic with other cells, which could possibly lead to increased safety outcomes. This can be exemplified by the bispecific T cell engager (BiTE) blinatumomab, which is a recombinant CD19- and CD3-specific mAb that combines the targeting of CD19 on B cells with direct activation of cytotoxic T cells (by engaging CD3 on the surface of T cells). Blinatumomab is currently being studied in the treatment of non-Hodgkin's lymphoma and chronic lymphocytic leukaemia (CLL)92.
CD20- and CD22-specific mAbs are another bispecific antibody format that have been tested preclinically. It has been reported that, in contrast to the therapeutic effect of rituximab, the antibody elicits its therapeutic effect via direct preferential and selective killing of tumour B cells without crosslinking and CDC93,94.
It is expected that these antibody formats would have favourable safety profiles but this will be confirmed after large-scale trial data become available.
Engineering out immunogenic sites within the biologic structure. Humanization of a biologic generally (but not always) reduces the risk of immunogenicity. For example, rituximab — a chimeric mAb (mouse and human) targeting CD20 — has reduced immunogenicity compared to tositumomab (Bexxar; GlaxoSmithKline), a mouse mAb targeting CD20. Immunogenicity arising from the presence of immunogenic epitopes and altered glycosylation can be rationally designed out (Box 3). Knowledge of pre-existing antibodies (in humans) to certain glycan structures will also enable drug developers to design a biologic without such unfavourable characteristics.
Irrespective of the pathway that is followed in developing new biologics, it is clear that the generation of safer biologics is reliant on greater knowledge and understanding of the disease, target biology and the availability of robust, reliable and reproducible predictive tests. New biologics that carry the risk of adverse reactions — as predicted through the utilization of these tests — will either undergo attrition or need to re-enter the iterative cycle described in Fig. 2 until a safer biologic is designed.
Conclusions and future directions
The remarkable success of immunomodulatory biologics in treating diseases that are either refractory to or not druggable by small-molecule drugs cannot be understated. Although concerns remain regarding the use of immunomodulatory biologics, it must be recognized that at present the clinical benefits, in most cases, outweigh the risk of developing adverse reactions.
Risk mitigation strategies along with robust pharmacovigilance frameworks (Supplementary information S1 (box)) can help in reducing and managing the impact of adverse reactions. However, continued progress in developing robust and reliable strategies in this area depends on overcoming several key challenges that are listed below:
Can the nature of the adverse reactions be predicted from a deeper understanding of the biology of the target and property of the product?
Would the ranking of immunomodulatory biologics based on their degree of hazard lead to a reasonable quantification of acceptable risk and align treatment regimens and pharmacovigilance strategies to anticipate a particular adverse reaction?
What exactly is the contribution of the patient and disease state to the induction or uncovering of a particular adverse reaction?
Can specific biomarkers be developed for predicting the risk of the induction of adverse reactions by immunomodulatory biologics?
Can patients be stratified based on efficacy as well as the degree of risk associated with developing an adverse reaction?
The identification of immunotoxic alerts based on the target molecule, target cell population (or subpopulation), mode of action (triggering, blocking or Fc-mediated activity) and structural information (glycosylation, Fc modifications, and so on) will be valuable in selecting lead candidates for development. However, this necessitates a detailed understanding of the immunology of the target, the disease and the interaction of the immunomodulatory biologic with these elements at the whole-organism, cellular and molecular levels. Considerable fundamental research efforts and investments are therefore a prerequisite to advance this cause.
Another major challenge that requires focused efforts is the ranking of immunomodulatory biologics within a particular class or among classes according to inherent hazard. Such a ranking of new immunomodulatory biologics — by degree of hazard — could be attempted from the cumulative experience of physicians, national registries and meta-analyses of clinical trials and case reports on current immunomodulatory biologics. This is clearly a retrospective approach that may have some predictive value but it requires the accumulation of many thousands of patient years of data before rankings can be assigned with any degree of confidence.
However, these efforts will contribute to a greater understanding of biologic-associated hazard that will inform the development of safer biologics. In the meantime, it is apparent that the decision to advance an immunomodulatory biologic therapy through to clinical evaluation must be based on an assessment of the balance between disease burden and the degree of predicted or estimated risk. The current gaps in our knowledge limit our ability to effectively predict adverse reactions to immunomodulatory biologics, so it is important that the risks are critically assessed and well explained, and that any decisions about therapy are both physician-led and patient-informed.
Box 1 | Estimating the risk of serious infection and malignancy.
The association of infections with tumour necrosis factor (TNF)-specific therapies is well documented95,96,97,98,99,100. Clinical trials are usually not sufficiently powered to capture a low incidence of serious adverse reactions and therefore meta-analyses performed on multiple studies are essential. Such meta-analysis of nine immunomodulatory biologics used for rheumatoid arthritis therapy indicated that abatacept (Orencia; Bristol-Myers Squibb) was the least likely to elicit serious infections101,102. Abatacept also reduced the incidence of side effects when it was used instead of a different TNF-specific biologic in the same patient103. For example, switching from adalimumab (Humira; Abbott) to abatacept eliminated adalimumab-associated autoimmune hepatitis104. However, there are case reports demonstrating the reactivation of infections such as hepatitis B virus in patients receiving abatacept105. Interestingly, increasing the on-target potency of abatacept through structural modifications (as in belatacept (Nulojix; Bristol-Myers Squibb))106 increased the risk of infections (Table 3). Certolizumab pegol (Cimzia; UCB) is clearly associated with an increased incidence of serious infections101 and there are emerging reports of Legionella spp. infections in patients receiving anti-TNF therapy107,108. In line with their immunosuppressive mode of action, all anti-TNF agents are contraindicated in patients with uncontrolled infections.
Although difficult, the identification of patient populations at risk of developing infections and malignancies is one approach for avoiding these adverse reactions — for example, by identifying patients carrying pathogens and applying vigilance and prophylactic antimicrobial strategies. Immunomodulatory biologics are usually given on top of the standard of care, which may involve other immunosuppressive or immunomodulatory drugs that can further increase the risk of serious infections. As yet, denosumab (Prolia/Xgeva; Amgen), an inhibitor of receptor activator of NF-κB ligand (RANKL; also known as TNFSF11) has not been associated with an increased incidence of serious infections109,110,111 but infective endocarditis and urinary tract infections can occur in rare instances112.
There are concerns about the risk of serious infections associated with other immunomodulatory biologics that do not target TNF, such as rituximab (Rituxan/Mabthera; Biogen Idec/Genentech/Roche), anakinra (Kineret; Amgen) and natalizumab (Tysabri; Biogen Idec/Elan). Rituximab (which targets the B cell molecule CD20) depletes CD20-positive B cells to reduce serum levels of immunoglobulin G (IgG) and B cell counts — effects that are consistent with the increased infection rates associated with this biologic113,114,115,116. Anakinra, an interleukin-1 receptor (IL-1R) antagonist, was associated with the incidence of serious infections in ∼2% of patients in a Phase III study117. However, this incidence may increase in real terms, as demonstrated by a 3-year extension study showing that the rate is 5.3 per 100 patient years118.
Efalizumab, which blocks integrins, is associated with the development of progressive progressive multifocal leukoencephalopathy (PML)119, an effect that is also associated with natalizumab (see main text). The high incidence of PML that occurred with efalizumab treatment led to its withdrawal from the market. This adverse reaction provides a potential insight into the importance of T cell trafficking in the control of John Cunningham virus (JCV)-induced PML. However, cases of PML associated with non-integrin-blocking immunomodulatory biologics such as rituximab (which targets CD20) and tocilizumab (which is an IL-6R antagonist) suggest that additional immune surveillance mechanisms are important in the control of JCV120,121.
The evidence for malignancy associated with the use of immunomodulatory biologics is reliant on meta-analyses, and is therefore not as strong as evidence obtained for serious infections6,7,9. Despite this, malignancy has been observed in clinical trials and in postmarketing surveillance, and boxed warnings have been given for anti-TNF agents (adalimumab, etanercept (Enbrel; Amgen/Pfizer), golimumab (Simponi; Centocor Ortho Biotech), certolizumab pegol and infliximab (Remicade; Centocor Ortho Biotech)) and belatacept (Table 3). The T cell-suppressive biologics belatacept14, muromonab-CD3 (Refs 122,123) and alefacept124 have been associated with malignancy.
Box 2 | Limitations of in vivo models for predicting cytokine release syndrome.
Despite the similarities between the immune systems of humans and non-human primates (NHPs), there are substantial functional inter-species differences in T cell activation, which have important consequences for safety assessments and the prediction of cytokine release syndrome (CRS)125. This became evident in early clinical trials of TGN1412 (Ref. 18).
Studies of TGN1412 in NHPs were unable to predict the potential risk of CRS, despite the fact that CD28 expression (the target of TGN1412) on T cell populations, the binding of TGN1412 to CD28 and receptor occupancy were identical in both NHPs and humans126,127. Despite these findings, before the trial was initiated no data had been made available to demonstrate that NHPs are a validated, pharmacologically relevant species for safety assessment (see the CIRCARE website for more information). In subsequent in vivo studies it was shown that the responding T cell population in humans was effector memory T cells, which are under-represented in human peripheral blood (in which the in vitro preclinical safety tests were performed). Furthermore, CD28 expression on cynomolgus memory T cells was very low or absent, and this contributed to the lack of immunostimulatory responses in NHPs128.
These fundamental differences were not taken into account in the experimental design of preclinical studies of TGN1412, primarily owing to limited knowledge of the biology of the target molecule and inter-species variability in immune cell subpopulations. But even without foreknowledge of these differences, CRS might have been anticipated on the basis of the known CD28 receptor biology, the intended pharmacological activity of TGN1412 and the overall knowledge of TGN1412 and surrogate molecules (including data in humanized mice)129,130,131,132,133.
Other immune-related variations that could cause discrepancies between preclinical studies in NHPs and clinical studies include species differences in complement pathway components134,135 and interactions between immunoglobulin G (IgG) and the Fcγ receptor136. For example, in cynomolgus monkeys there are differences in the expression of Fcγ receptor on granulocytes, allotypic variation of IgG4 Fab arm exchange and enhanced effector functions of IgG2 and IgG4 subclasses compared with humans136. Therefore, it is crucial to understand any limitations of the animal species selected for preclinical evaluation of novel therapeutics and to take these limitations into consideration in the overall safety assessment. Until reliable and accurate predictive tools become available, CRS-based immunotoxicity will continue to be treated with corticosteroids and cytokine-targeted drugs18,137.
Box 3 | Current approaches for reducing or mitigating the risk of immunogenicity.
Aggregates and immunogenicity
The formation of protein aggregates in immunomodulatory biologic formulations could trigger an unwanted immunogenic response138,139,140, and so preventing aggregate formation to reduce the incidence of immunogenic reactions to biologics is vital. The presence of multiple epitopes and/or the unique structural conformation of the aggregated molecule could initiate the immunogenic response138,139, as could oxidation of the biologic37,139. Aggregates could also induce immunogenicity by disrupting existing immune tolerance towards the monomeric version of the biologic141. The presence of antigenic epitopes or multimeric structures can directly stimulate B cells and/or increase the antigen uptake potential of antigen-presenting cells (APCs)37,74. Aggregation of biologics can trigger an innate and late-stage T cell response that is relevant to immunogenicity142. The qualitative and quantitative contribution of aggregates to immunogenicity is still unknown, so studies that are designed to define the size, type, amount and frequency of aggregates, and that are capable of inducing immunogenic responses, could help in reducing immunogenicity.
Protein sequence modifications
Modifications to the protein sequence of immunomodulatory biologics to remove non-human components (humanization) might reduce the immunogenicity that is associated with biologics of non-human origin (for example, murine or chimeric biologics). Removal of immunogenic T cell epitopes ('de-immunization') can also reduce immunogenicity143,144. Regulatory T cells (TReg cells) have the potential to suppress immunogenicity and so the inclusion of TReg epitopes ('Tregitopes') in the primary sequence of the immunomodulatory biologic could induce TReg cells and thereby suppress immunogenicity145. However, designing epitopes that are highly specific at expanding TReg cell populations without having any impact on other T cell populations remains a considerable challenge.
Structural modifications
Pegylation (covalent linkage of polyethylene glycol (PEG) to proteins) can reduce immunogenicity by interfering with antigen-processing mechanisms146. However, there is growing evidence that pegylation does not substantially decrease the immunogenic potential of biologics147. Although PEG is generally accepted to be non-immunogenic, there are reports of PEG-specific antibodies in patients treated with pegylated therapeutic enzymes148,149. The benefit ascribed to the pegylation of immunomodulatory biologics therefore requires re-evaluation.
Because biologics are produced in bacterial or mammalian cells, they can be glycosylated (as cells have several protein glycosylation systems). Glycosylated therapeutic monoclonal antibodies could react with pre-existing glycan-binding antibodies in humans. For example, the presence of N-glycolylneuraminic acid (Neu5Gc) modifications on cetuximab (Erbitux; Bristol-Myers Squibb/Eli Lilly) results in the formation of immune complexes between cetuximab and pre-existing Neu5Gc-specific antibodies in patient sera150. Engineering the addition of glycans to specific amino acids of the biologic leads to defective antigen processing151 and this can be one rational approach for reducing biologic-derived peptide presentation by APCs and subsequent immunogenic responses. A caveat to this strategy is that glycosylation may lead to the generation of neoantigenic peptides and can increase immunogenicity152, underlining the need for a better understanding of antigen processing and presentation of biologics.
Therapeutic approaches
The administration of immunomodulatory biologics at high concentrations or through mucosal surfaces can decrease immunogenicity through the induction of peripheral tolerance that is mediated by inducing tolerogenic dendritic cells and TReg cells153,154,155. Immune tolerance can be increased with the addition of methotrexate to the treatment regimen156 or by pretreatment with high doses of a non-antigen-binding variant of the therapeutic antibody, as demonstrated with alemtuzumab (Campath-1H; Genzyme)157. Moreover, inducing tolerance by increasing the number of TReg cells has shown potential in the treatment of psoriasis in a clinical trial of BT-061, a CD4-specific humanized monoclonal antibody158. Harnessing or boosting host tolerogenic mechanisms by expanding tolerogenic dendritic cells159 could have potential therapeutic applications in the management of T cell-mediated diseases in humans160.
Box 4 | Species selection for toxicology studies and models.
Recent advances in the whole-genome characterization of genetic variation within non-human primate (NHP) species161, combined with comparative immunoglobulin G (IgG)–Fcγ receptor (FcγR) structure–function relationships between humans and NHPs, have the potential to enhance preclinical safety assessments. In particular, captive cynomolgus monkeys that are used for the safety assessment of immunomodulatory biologics are often derived from distinct geographic origins162 and have diverse major histocompatibility complex (MHC) genetics163 and spontaneous pathologies164. Such intra-strain (that is, dependent on the geographic origin) genetic variation in cynomolgus monkeys could contribute to the differential phenotypic responses observed during in vivo and ex vivo safety assessment studies.
Therefore, the early assessment of intra-strain target genetic variation in cynomolgus monkeys should be considered during the development of immunomodulatory biologics. This will ensure the appropriate design of monoclonal antibody specificity, binding and potency assays based on the strain of cynomolgus monkey that will be used in toxicology studies to support the first dosing studies in humans. Systematic genotyping of NHPs is not yet standard practice but would facilitate both prospective and retrospective investigations of genotype–phenotype relationships.
Integration of known genetic variation of the human target into the selection of species for toxicology studies also needs to be considered. Identifying and characterizing functional differences in target and pathway biology — and understanding how these differences affect the safety assessment of immunomodulatory biologics — requires detailed and immediate attention on a case-by-case basis. One strategy that is currently followed when species-specific immunomodulatory biologics need to be assessed for safety is the use of surrogate or homologous molecules165,166. It is crucial that the surrogate molecule has a highly similar structure and pharmacological activity to the biologic being tested. Despite the sound rationale, surrogate molecules have either under-predicted, over-predicted or incorrectly predicted the effects seen in humans. The species-specific homologues that were used in the preclinical testing of infliximab (Remicade; Centocor Ortho Biotech) and efalizumab did not predict the adverse reactions (serious infections and malignancies) that subsequently arose in humans (efalizumab was withdrawn from the market because of this risk).
Disease models or transgenic animal models expressing human protein targets (for example, CD20 in models of diabetes or arthritis) that are used for evaluating the therapeutic efficacy of the immunomodulatory biologic therapy167,168 can also be used to assess the risk of adverse reactions. In the event of an adverse reaction, the same model could be exploited to identify the underlying mechanism (or mechanisms) for the observed adverse reaction. It is unlikely that any in vivo model can be developed that will recapitulate all of the effects of the biologic that are seen in large patient populations. Therefore, the research community needs to focus on developing novel alternative experimental models. The ultimate goal would be to design intelligent experimental systems based on human in vitro and ex vivo models that can recreate or uncover the potential for serious adverse reactions to an acceptable degree. In the meantime, preclinical data combined with detailed pharmacological investigations (including early-stage clinical data in a limited number of individuals) are most useful when they are used to understand the mechanisms that lead to adverse reactions (rather than as a tool to purely predict adverse reactions).
Supplementary information
Pharmacovigilance and the role of regulators (PDF 241 kb)
Acknowledgements
The authors thank F. Brennan for his helpful discussions.
Glossary
- Immunomodulatory biologics
Biotechnology-derived pharmaceutical agents (recombinant proteins and therapeutic monoclonal antibodies) that render their therapeutic effect primarily through modulating (augmenting or reducing) immune responses by targeting immune-relevant molecules.
- Adverse reactions
Any adverse events occurring in temporal association with a therapeutic for which causality (possible, probable or definite) has been determined.
- Boxed warnings
Warnings on a pharmaceutical product label indicating the risk of a serious or life-threatening adverse reaction (or reactions) associated with the use of the therapeutic agent.
- Post-transplant lymphoproliferative disease
A disorder characterized by the neoplastic proliferation of lymphocytes that usually occurs as a result of therapeutic immunosuppression induced to prevent transplant rejection.
- Fc region
A non-antigen-binding region of an antibody molecule that is involved in the binding of the antibody to Fc receptors and to the complement component C1q, which can lead to antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity.
- Antibody-dependent cellular cytotoxicity
(ADCC). An effector function mediated by the Fc region of an antibody that involves the death of a target cell owing to the interaction of the Fc region of the therapeutic antibody with activatory Fcγ receptors on phagocytic cells (such as neutrophils and macrophages) and natural killer cells. Target cell death is induced through either ingestion by phagocytic cells or via the secretion of cytotoxic molecules by natural killer cells.
- Complement-dependent cytotoxicity
(CDC). An effector function mediated by the Fc region of an antibody that involves complement activation induced by the engagement of C1q (a complement component) with the Fc region of the therapeutic antibody (which is bound to the target cell). This leads to the formation of a membrane attack complex, resulting in the destruction of the target cell membrane and cell death.
- Fc-mediated effector functions
A collective term referring to antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity; these processes result in target cell destruction but they may also result in adverse reactions such as cytokine release syndrome and tumour lysis syndrome.
- Neonatal Fc receptor
(FcRn). A type of Fc receptor that is structurally related to major histocompatibility complex (MHC) class 1 molecules and is involved in binding to and recycling circulating immunoglobulin G, thereby increasing the serum half-life of these immunoglobulins.
- Biomarkers
Readouts (usually biochemical in nature) obtained through measurements performed on humans or preclinical models that can report on and allow quantitative assessments to be made on one or a combination of the following: the disease state, immune status, pharmacological efficacy, pharmacological safety and the onset of adverse reactions.
- Human leukocyte antigen
(HLA). The human major histocompatibility complex (MHC) molecule involved in the presentation of antigenic peptides derived from intracellular (in the case of HLA class I) or extracellular (in the case of HLA class II) sources. The recognition of specific HLA–peptide complexes by antigen-specific T lymphocytes initiates the immune response.
- Humanized monoclonal antibody
An antibody that contains only the antigen-binding region derived from a non-human species (such as mice); the remaining regions of the antibody are of human origin.
- Primary immunological responses
The initial humoral immune responses that are generated following the exposure of naive B lymphocytes to antigens for the first time. This is a relatively weak response and it is predominantly associated with the production of the immunoglobulin M class of antibodies against the antigen.
- Secondary immunological responses
Rapid humoral immune responses that are triggered following the secondary or repeated exposure of antigen-experienced B lymphocytes to antigens. These responses are predominantly associated with the production of the immunoglobulin G class of antibodies with improved specificity and affinity for the antigens.
- Syngeneic
A term referring to animal models being genetically identical to the tumours or cells that they are to be transplanted with. This ensures that the immune system of the host animal does not react towards the transplanted cells.
- Minimal anticipated biological effect level
(MABEL). The anticipated dose level leading to a minimum biological effect level that can be ascribed to the pharmacological action of the immunomodulatory biologic. It is calculated using information obtained from all in vitro (using target cells from humans and relevant animal species) and in vivo (from relevant animal species) pharmacokinetic/pharmacodynamic (PK/PD) studies.
- B cell tolerance
Immunological processes (such as clonal deletion and anergy induction) that operate to prevent B cells from responding to self antigens.
- NOD-SCID-Il2rg-null mice
Non-obese diabetic (NOD) severe combined immunodeficient (SCID) mice lacking the gene encoding the γ-chain of the interleukin-2 receptor (Il2rg).
- Chimeric mAb
An antibody that contains regions derived from different species. For example, an antibody composed of an antigen-binding region of mouse origin and a constant region (Fc) of human origin.
Biographies
Jean G. Sathish is a lecturer in pharmacology at the University of Liverpool, UK, and works within the UK Medical Research Council (MRC) Centre for Drug Safety Science, where he leads the immune regulation group. He is trained in veterinary medicine and holds an M.Sc. in pharmacology and a Ph.D. in immunology. His research interests and expertise are in defining immune cell-regulatory mechanisms, immune cell signalling and immunotoxicology associated with biologics.
Swaminathan Sethu is currently a postdoctoral scientist at the MRC Centre for Drug Safety Science at the University of Liverpool, UK. He has a dentistry degree and a Ph.D. in immunology. His research interests are centred on immune cell and cytokine signalling, immunogenicity and adverse drug reactions to biologics.
Marie-Christine Bielsky studied medicine and after working in the pharmaceutical industry on the clinical development of a broad range of medicines she joined the Biologicals and Biotechnology Unit of the Licensing Division at the UK Medicines and Healthcare products Regulatory Agency (MHRA). As a medical assessor, she conducts the clinical evaluation of new biological medicines. She is also a member of the Biosimilar Medicinal Products Working Party at the European Medicines Agency (EMA).
Lolke de Haan is Director of Toxicology at MedImmune. After completing his Ph.D. in medical sciences and his postdoctoral training, he joined AstraZeneca as a discovery toxicologist. Following the acquisition of MedImmune by AstraZeneca, he became responsible for the toxicology team at MedImmune Cambridge, UK, which oversees the non-clinical development of a large portfolio of antibody- and non-antibody-based biologics in the following disease areas: respiratory disease, inflammation and autoimmunity, oncology, neuroscience and cardiometabolic disease.
Neil S. French is Senior Scientific Operations Manager at the MRC Centre for Drug Safety Science. With a background in molecular oncology and wound regeneration, he has experience of working in both the academic and biotechnology sectors. His current research interests include predictive models and biomarkers of drug-induced organ injury.
Karthik Govindappa is a Ph.D. student at the MRC Centre for Drug Safety Science, and is working on characterizing immunogenic responses to therapeutic interferon-β.
James Green is a Highly Distinguished Research Fellow at Boehringer Ingelheim, Sudbury, Massachusetts, USA. He is President of the Executive Board of the American Board of Toxicology (ABT) and Fellow of the Academy of Toxicological Sciences. He is currently vice-chairman of the Biotechnology Industry Association's non-clinical safety evaluation expert committee (BioSafe). He received his M.Sc. and Ph.D. in toxicology from New York University, USA. He has extensive publications in the area of safety evaluation and the development of biopharmaceutical products.
Christopher E. M. Griffiths is Foundation Professor of Dermatology at the University of Manchester, UK, and directs the Manchester Dermatopharmacology Unit. He runs a comprehensive research programme on psoriasis and has been involved in the development and clinical trials of many biologics and immunomodulatory therapies for this condition.
Stephen Holgate is an MRC Professor at the University of Southampton, UK. After studying medicine from the University of London, UK, he focused his research career on the underlying mechanisms of asthma and allied disorders using a range of approaches. He has participated in both the preclinical and clinical evaluation of many therapeutics in this disease space, including biologics targeting immunoglobulin E (IgE), interleukin-5 (IL-5), IL-13, IL-17 and tumour necrosis factor (TNF).
David Jones works as an expert pharmacotoxicologist within the licensing division of the MHRA. His current role involves assessing non-clinical data — both non-biological and biological — for clinical trial applications. He also offers regulatory advice to companies on behalf of the MHRA or the European Union (EU)'s Committee for Human Medicinal Products (CHMP), is the UK representative on the EU's Safety Working Party and has represented the EU in the International Conference on Harmonisation (ICH).
Ian Kimber is Professor of Toxicology at the University of Manchester. He has a Ph.D. in immunology and his research interests lie at the interface between toxicology and immunology, with particular emphasis on the initiation and regulation of immunogenic and allergic responses to chemicals, drugs and proteins. He also has interests in cutaneous immunology in health and disease, and in the activation and mobilization of dendritic cells.
Jonathan Moggs is Global Head of Molecular Toxicology at the Novartis Institutes for BioMedical Research, Basel, Switzerland, and has experience in preclinical safety assessment and drug development, with an emphasis on investigating mechanisms of toxicity at the molecular, biochemical and cellular levels. He is also Coordinator of the EU Innovative Medicines Initiative's MARCAR Consortium, which is focusing on mechanisms and biomarkers for non-genotoxic carcinogenesis.
Dean J. Naisbitt is a reader at the MRC Centre for Drug Safety Science. He completed his Ph.D. in chemistry with pharmacology, followed by postdoctoral training at the University of Liverpool and at the Institute of Allergy and Clinical Immunology, Bern, Switzerland. His laboratory focuses on the chemical and cellular basis of immunological drug reactions.
Munir Pirmohamed is Professor of Clinical Pharmacology and NHS (UK National Health Service) Chair of Pharmacogenetics. His main research interests are in drug safety, encompassing the epidemiology of adverse drug reactions, defining mechanisms of serious organ-based adverse reactions (including those due to biologics) and identifying genetic and non-genetic predictors of susceptibility to adverse drugs, with a view to develop biomarkers that can be translated into clinical practice.
Gabriele Reichmann obtained her Ph.D. in immunology and spent several years in academic research before joining the Paul-Ehrlich-Institute, where she is a non-clinical assessor with a special interest in the non-clinical development of monoclonal antibodies, immunomodulatory drugs and biosimilars.
Jennifer Sims is the President of the pharmaceutical consulting company Integrated Biologix, Basel, Switzerland, and before this she was Head of Translational Sciences and Safety for the Novartis Biologics Unit. She has broad experience in small-molecule and biological drug development and has also served as a senior preclinical assessor at the MHRA and as a UK delegate to the CHMP Safety Working Party.
Meena Subramanyam is the Vice President of Translational Medicine at Biogen Idec, Cambridge, Massachusetts, USA. Her scientific responsibilities involve the identification and development of bioanalytical methods using cutting-edge technologies to understand the in vivo pharmacology, mechanism of action and pharmacokinetics of therapeutics as well as the safety of biologics. Her research interests include understanding factors that contribute to the immunogenicity of biologics, as well as stratification markers of disease and therapeutic responses to advance personalized medicine efforts.
Marque D. Todd received her doctorate of veterinary medicine (D.V.M.) from Colorado State University, Fort Collins, Colorado, USA, and her M.Sc. in cancer biology from Harvard University, Cambridge, Massachusetts, USA. She works in Drug Safety Research and Development at Pfizer and is involved in non-clinical drug development and the regulatory strategy for late-stage small molecules and biologics. She is a Diplomate, on the American Board of Toxicology and is actively involved in several toxicology societies.
Jan Willem Van Der Laan is a senior pharmacology/toxicology assessor of the Medicines Evaluation Board in the Netherlands, and Chair of the Safety Working Party of the CHMP. As a member of the Expert Working Group on Carcinogenicity Testing, he has made significant contributions to the ICH on behalf of the EU. He was also the ICH Rapporteur for the 'Addendum for the Preclinical testing of Biotechnology-derived Proteins' (ICH S6(R1)).
Richard Weaver studied biochemistry and toxicology at the University of Aberdeen, Scotland, UK, and obtained an M.B.A. from Warwick Business School, Coventry, UK. After completing his postdoctoral research training, he joined the pharmaceutical industry where his experience spans drug discovery and development projects in several therapeutic research areas. He is currently the Scientific Director of Bioanalysis & New Technologies at the Drug Safety Assessment Centre, Biologie Servier, France, where he leads teams engaged in in vitro and in vivo research covering non-clinical pathology, genotoxicity, toxicogenomics, bioanalysis and toxicokinetics in support of both regulatory and non-regulatory studies.
B. Kevin Park is Professor of Pharmacology, Head of the Institute of Translational Medicine at the University of Liverpool, and Director of the MRC Centre for Drug Safety Science. He is a fellow of the Royal College of Physicians, a fellow of the Academy of Medical Sciences and a member of the Commission on Human Medicines (CHM). He studies the role of metabolism in human drug toxicity, with a particular interest in drug hypersensitivity and drug hepatotoxicity.
Related links
FURTHER INFORMATION
Citizens For Responsible Care and Research (CIRCARE) — Tegenero AG TGN1412 clinical trial
CRACK IT 2011 challenge 4 (“Improving the predictive capacity of in vitro cytokine release”)
Drugs@FDA — FDA-approved drug products
Competing interests
Lolke de Haan is an employee of Medimmune, Cambridge, UK.
James Green is an employee of Boehringer-Ingelheim, Sudbury, USA.
Jonathan Moggs is an employee of Novartis, Basel, Switzerland.
Jennifer Sims is an employee of Integrated Biologix, Basel, Switzerland.
Meena Subramanyam is an employee of Biogen Idec, Cambridge, Massachusetts, USA.
Marque Todd is an employee of Pfizer, California, USA.
Richard Weaver is an employee of Biologie Servier, Gidy, France.
All other authors declare no competing financial interests.
Footnotes
Jean G. Sathish and Swaminathan Sethu: These authors contributed equally to this work.
References
- 1.Swinney DC, Anthony J. How were new medicines discovered? Nature Rev. Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 2.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nature Rev. Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 3.Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nature Rev. Drug Discov. 2010;9:325–338. doi: 10.1038/nrd3003. [DOI] [PubMed] [Google Scholar]
- 4.Sorensen PS, et al. Risk stratification for progressive multifocal leukoencephalopathy in patients treated with natalizumab. Mult. Scler. 2012;18:143–152. doi: 10.1177/1352458511435105. [DOI] [PubMed] [Google Scholar]
- 5.Bloomgren G, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2012;366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 6.Bongartz T, et al. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295:2275–2285. doi: 10.1001/jama.295.19.2275. [DOI] [PubMed] [Google Scholar]
- 7.Bongartz T, et al. Etanercept therapy in rheumatoid arthritis and the risk of malignancies: a systematic review and individual patient data meta-analysis of randomised controlled trials. Ann. Rheum. Dis. 2009;68:1177–1183. doi: 10.1136/ard.2008.094904. [DOI] [PubMed] [Google Scholar]
- 8.US Food and Drug Administration. Tumor necrosis factor (TNF) blockers, azathioprine and/or mercaptopurine: update on reports of hepatosplenic T-cell lymphoma in adolescents and young adults. FDA website[online], (2011).
- 9.Dommasch ED, et al. The risk of infection and malignancy with tumor necrosis factor antagonists in adults with psoriatic disease: a systematic review and meta-analysis of randomized controlled trials. J. Am. Acad. Dermatol. 2011;64:1035–1050. doi: 10.1016/j.jaad.2010.09.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beukelman T, et al. Rates of malignancy associated with juvenile idiopathic arthritis and its treatment. Arthritis Rheum. 2012;64:1263–1271. doi: 10.1002/art.34348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Le Blay P, Mouterde G, Barnetche T, Morel J, Combe B. Short-term risk of total malignancy and nonmelanoma skin cancers with certolizumab and golimumab in patients with rheumatoid arthritis: metaanalysis of randomized controlled trials. J. Rheumatol. 2012;39:712–715. doi: 10.3899/jrheum.110982. [DOI] [PubMed] [Google Scholar]
- 12.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dias C, Isenberg DA. Susceptibility of patients with rheumatic diseases to B-cell non-Hodgkin lymphoma. Nature Rev. Rheumatol. 2011;7:360–368. doi: 10.1038/nrrheum.2011.62. [DOI] [PubMed] [Google Scholar]
- 14.Grinyo J, et al. An integrated safety profile analysis of belatacept in kidney transplant recipients. Transplantation. 2010;90:1521–1527. doi: 10.1097/TP.0b013e3182007b95. [DOI] [PubMed] [Google Scholar]
- 15.Arora S, Tangirala B, Osadchuk L, Sureshkumar KK. Belatacept: a new biological agent for maintenance immunosuppression in kidney transplantation. Expert Opin. Biol. Ther. 2012;12:965–979. doi: 10.1517/14712598.2012.683522. [DOI] [PubMed] [Google Scholar]
- 16.Walker MR, Makropoulos DA, Achuthanandam R, Van Arsdell S, Bugelski PJ. Development of a human whole blood assay for prediction of cytokine release similar to anti-CD28 superagonists using multiplex cytokine and hierarchical cluster analysis. Int. Immunopharmacol. 2011;11:1697–1705. doi: 10.1016/j.intimp.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Ponce R. Adverse consequences of immunostimulation. J. Immunotoxicol. 2008;5:33–41. doi: 10.1080/15476910801897920. [DOI] [PubMed] [Google Scholar]
- 18.Suntharalingam G, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 19.Abramowicz D, Crusiaux A, Goldman M. Anaphylactic shock after retreatment with OKT3 monoclonal antibody. N. Engl. J. Med. 1992;327:736. doi: 10.1056/NEJM199209033271018. [DOI] [PubMed] [Google Scholar]
- 20.Baudouin V, et al. Anaphylactic shock caused by immunoglobulin E sensitization after retreatment with the chimeric anti-interleukin-2 receptor monoclonal antibody basiliximab. Transplantation. 2003;76:459–463. doi: 10.1097/01.TP.0000073809.65502.8F. [DOI] [PubMed] [Google Scholar]
- 21.Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14:296–302. doi: 10.1208/s12248-012-9340-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ponce R, et al. Immunogenicity of biologically-derived therapeutics: assessment and interpretation of nonclinical safety studies. Regul. Toxicol. Pharmacol. 2009;54:164–182. doi: 10.1016/j.yrtph.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Bendtzen K, et al. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor α inhibitor infliximab. Arthritis Rheum. 2006;54:3782–3789. doi: 10.1002/art.22214. [DOI] [PubMed] [Google Scholar]
- 24.Bartelds GM, et al. Surprising negative association between IgG1 allotype disparity and anti-adalimumab formation: a cohort study. Arthritis Res. Ther. 2010;12:R221. doi: 10.1186/ar3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartelds GM, et al. Anti-adalimumab antibodies in rheumatoid arthritis patients are associated with interleukin-10 gene polymorphisms. Arthritis Rheum. 2009;60:2541–2542. doi: 10.1002/art.24709. [DOI] [PubMed] [Google Scholar]
- 26.European Medicines Agency. Guideline on immunogenicity assessment of biotechnology-derived therapeutic proteins. EMA website[online], (2008).
- 27.US Food and Drug Administration. Guidance for industry: assay development for immunogenicity testing of therapeutic proteins. FDA website[online], (2009).
- 28.Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn) J. Biol. Chem. 2006;281:23514–23524. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 29.Oganesyan V, Gao C, Shirinian L, Wu H, Dall'Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr. D Biol. Crystallogr. 2008;64:700–704. doi: 10.1107/S0907444908007877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mossner E, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115:4393–4402. doi: 10.1182/blood-2009-06-225979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salles G, et al. Phase 1 study results of the type II glycoengineered humanized anti-CD20 monoclonal antibody obinutuzumab (GA101) in B-cell lymphoma patients. Blood. 2012;119:5126–5132. doi: 10.1182/blood-2012-01-404368. [DOI] [PubMed] [Google Scholar]
- 32.Davies J, et al. Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FCγRIII. Biotechnol. Bioeng. 2001;74:288–294. doi: 10.1002/bit.1119. [DOI] [PubMed] [Google Scholar]
- 33.Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nature Rev. Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 34.Boyd PN, Lines AC, Patel AK. The effect of the removal of sialic acid, galactose and total carbohydrate on the functional activity of Campath-1H. Mol. Immunol. 1995;32:1311–1318. doi: 10.1016/0161-5890(95)00118-2. [DOI] [PubMed] [Google Scholar]
- 35.Slavin RG, et al. Asthma symptom re-emergence after omalizumab withdrawal correlates well with increasing IgE and decreasing pharmacokinetic concentrations. J. Allergy Clin. Immunol. 2009;123:107–113. doi: 10.1016/j.jaci.2008.09.050. [DOI] [PubMed] [Google Scholar]
- 36.Corren J, et al. Lebrikizumab treatment in adults with asthma. N. Engl. J. Med. 2011;365:1088–1098. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 37.Sethu S, et al. Immunogenicity to biologics: mechanisms, prediction and reduction. Arch. Immunol. Ther. Exp. 2012;60:331–344. doi: 10.1007/s00005-012-0189-7. [DOI] [PubMed] [Google Scholar]
- 38.Tabrizi MA, et al. Translational strategies for development of monoclonal antibodies from discovery to the clinic. Drug Discov. Today. 2009;14:298–305. doi: 10.1016/j.drudis.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Chapman K, et al. Preclinical development of monoclonal antibodies: considerations for the use of non-human primates. MAbs. 2009;1:505–516. doi: 10.4161/mabs.1.5.9676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haley P, et al. STP position paper: best practice guideline for the routine pathology evaluation of the immune system. Toxicol. Pathol. 2005;33:404–407. doi: 10.1080/01926230590934304. [DOI] [PubMed] [Google Scholar]
- 41.Kuper CF, Harleman JH, Richter-Reichelm HB, Vos JG. Histopathologic approaches to detect changes indicative of immunotoxicity. Toxicol. Pathol. 2000;28:454–466. doi: 10.1177/019262330002800317. [DOI] [PubMed] [Google Scholar]
- 42.Brennan FR, et al. Safety and immunotoxicity assessment of immunomodulatory monoclonal antibodies. MAbs. 2010;2:233–255. doi: 10.4161/mabs.2.3.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plitnick LM, Herzyk DJ. The T-dependent antibody response to keyhole limpet hemocyanin in rodents. Methods Mol. Biol. 2010;598:159–171. doi: 10.1007/978-1-60761-401-2_11. [DOI] [PubMed] [Google Scholar]
- 44.Hutto DL. Opportunistic infections in non-human primates exposed to immunomodulatory biotherapeutics: considerations and case examples. J. Immunotoxicol. 2010;7:120–127. doi: 10.3109/15476910903258252. [DOI] [PubMed] [Google Scholar]
- 45.Burleson FG, Burleson GR. Host resistance assays including bacterial challenge models. Methods Mol. Biol. 2010;598:97–108. doi: 10.1007/978-1-60761-401-2_7. [DOI] [PubMed] [Google Scholar]
- 46.Wagar EJ, et al. Regulation of human cell engraftment and development of EBV-related lymphoproliferative disorders in Hu-PBL-scid mice. J. Immunol. 2000;165:518–527. doi: 10.4049/jimmunol.165.1.518. [DOI] [PubMed] [Google Scholar]
- 47.Kawabata T, et al. Summary of roundtable discussion meeting: non-human primates to assess risk for EBV-related lymphomas in humans. J. Immunotoxicol. 2012;9:121–127. doi: 10.3109/1547691X.2011.635166. [DOI] [PubMed] [Google Scholar]
- 48.Kirton CM, Gliddon DR, Bannish G, Bembridge GP, Coney LA. In vitro cytokine release assays: reducing the risk of adverse events in man. Bioanalysis. 2011;3:2657–2663. doi: 10.4155/bio.11.272. [DOI] [PubMed] [Google Scholar]
- 49.The Health and Environmental Sciences Institute (HESI). HESI Technical Committee. Immunotoxicology: 2011–2012 Activities and Accomplishments. HESI Global[online], (2012).
- 50.Dhir V, et al. A predictive biomimetic model of cytokine release induced by TGN1412 and other therapeutic monoclonal antibodies. J. Immunotoxicol. 2012;9:34–42. doi: 10.3109/1547691X.2011.613419. [DOI] [PubMed] [Google Scholar]
- 51.Vidal JM, et al. In vitro cytokine release assays for predicting cytokine release syndrome: the current state-of-the-science. Report of a European Medicines Agency Workshop. Cytokine. 2010;51:213–215. doi: 10.1016/j.cyto.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 52.Muller PY, Milton M, Lloyd P, Sims J, Brennan FR. The minimum anticipated biological effect level (MABEL) for selection of first human dose in clinical trials with monoclonal antibodies. Curr. Opin. Biotechnol. 2009;20:722–729. doi: 10.1016/j.copbio.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 53.Muller PY, Brennan FR. Safety assessment and dose selection for first-in-human clinical trials with immunomodulatory monoclonal antibodies. Clin. Pharmacol. Ther. 2009;85:247–258. doi: 10.1038/clpt.2008.273. [DOI] [PubMed] [Google Scholar]
- 54.Milton MN, Horvath CJ. The EMEA guideline on first-in-human clinical trials and its impact on pharmaceutical development. Toxicol. Pathol. 2009;37:363–371. doi: 10.1177/0192623309332997. [DOI] [PubMed] [Google Scholar]
- 55.Inaba H, Martin W, De Groot AS, Qin S, De Groot LJ. Thyrotropin receptor epitopes and their relation to histocompatibility leukocyte antigen-DR molecules in Graves' disease. J. Clin. Endocrinol. Metab. 2006;91:2286–2294. doi: 10.1210/jc.2005-2537. [DOI] [PubMed] [Google Scholar]
- 56.Khan AM, et al. A systematic bioinformatics approach for selection of epitope-based vaccine targets. Cell. Immunol. 2006;244:141–147. doi: 10.1016/j.cellimm.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Groot AS, Berzofsky JA. From genome to vaccine — new immunoinformatics tools for vaccine design. Methods. 2004;34:425–428. doi: 10.1016/j.ymeth.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 58.De Groot AS, Moise L. Prediction of immunogenicity for therapeutic proteins: state of the art. Curr. Opin. Drug Discov. Devel. 2007;10:332–340. [PubMed] [Google Scholar]
- 59.Koren E, et al. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin. Immunol. 2007;124:26–32. doi: 10.1016/j.clim.2007.03.544. [DOI] [PubMed] [Google Scholar]
- 60.De Groot AS, Martin W. Reducing risk, improving outcomes: bioengineering less immunogenic protein therapeutics. Clin. Immunol. 2009;131:189–201. doi: 10.1016/j.clim.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 61.El-Manzalawy Y, Dobbs D, Honavar V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. 2008;21:243–255. doi: 10.1002/jmr.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sollner J, et al. Analysis and prediction of protective continuous B-cell epitopes on pathogen proteins. Immunome Res. 2008;4:1. doi: 10.1186/1745-7580-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kumar S, Mitchell MA, Rup B, Singh SK. Relationship between potential aggregation-prone regions and HLA-DR-binding T-cell immune epitopes: implications for rational design of novel and follow-on therapeutic antibodies. J. Pharm. Sci. 2012;101:2686–2701. doi: 10.1002/jps.23169. [DOI] [PubMed] [Google Scholar]
- 65.Agrawal NJ, et al. Aggregation in protein-based biotherapeutics: computational studies and tools to identify aggregation-prone regions. J. Pharm. Sci. 2011;100:5081–5095. doi: 10.1002/jps.22705. [DOI] [PubMed] [Google Scholar]
- 66.Barbosa MD, Vielmetter J, Chu S, Smith DD, Jacinto J. Clinical link between MHC class II haplotype and interferon-β (IFN-β) immunogenicity. Clin. Immunol. 2006;118:42–50. doi: 10.1016/j.clim.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 67.Kropshofer H, Singer T. Overview of cell-based tools for pre-clinical assessment of immunogenicity of biotherapeutics. J. Immunotoxicol. 2006;3:131–136. doi: 10.1080/15476910600845625. [DOI] [PubMed] [Google Scholar]
- 68.Jaber A, Baker M. Assessment of the immunogenicity of different interferon β-1a formulations using ex vivo T-cell assays. J. Pharm. Biomed. Anal. 2007;43:1256–1261. doi: 10.1016/j.jpba.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 69.Baker MP, Jones TD. Identification and removal of immunogenicity in therapeutic proteins. Curr. Opin. Drug Discov. Devel. 2007;10:219–227. [PubMed] [Google Scholar]
- 70.Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self Nonself. 2010;1:314–322. doi: 10.4161/self.1.4.13904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Depil S, et al. Peptide-binding assays and HLA II transgenic Aβ degrees mice are consistent and complementary tools for identifying HLA II-restricted peptides. Vaccine. 2006;24:2225–2229. doi: 10.1016/j.vaccine.2005.11.048. [DOI] [PubMed] [Google Scholar]
- 72.Pan S, Trejo T, Hansen J, Smart M, David CS. HLA-DR4 (DRB1*0401) transgenic mice expressing an altered CD4-binding site: specificity and magnitude of DR4-restricted T cell response. J. Immunol. 1998;161:2925–2929. [PubMed] [Google Scholar]
- 73.Palleroni AV, et al. Interferon immunogenicity: preclinical evaluation of interferon-α 2a. J. Interferon Cytokine Res. 1997;17:23–27. [PubMed] [Google Scholar]
- 74.Hermeling S, et al. Antibody response to aggregated human interferon α2b in wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J. Pharm. Sci. 2006;95:1084–1096. doi: 10.1002/jps.20599. [DOI] [PubMed] [Google Scholar]
- 75.Ishikawa F, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor γ chain(null) mice. Blood. 2005;106:1565–1573. doi: 10.1182/blood-2005-02-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pearson, T., Greiner, D. L. & Shultz, L. D. Creation of “humanized” mice to study human immunity. Curr. Protoc. Immunol. 1 May 2008 (doi:10.1002/0471142735.im1521s81). [DOI] [PMC free article] [PubMed]
- 77.King M, et al. A new Hu-PBL model for the study of human islet alloreactivity based on NOD-scid mice bearing a targeted mutation in the IL-2 receptor γ chain gene. Clin. Immunol. 2008;126:303–314. doi: 10.1016/j.clim.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 78.Brinks V, Jiskoot W, Schellekens H. Immunogenicity of therapeutic proteins: the use of animal models. Pharm. Res. 2011;28:2379–2385. doi: 10.1007/s11095-011-0523-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kircik LH, Del Rosso JQ. Anti-TNF agents for the treatment of psoriasis. J. Drugs Dermatol. 2009;8:546–559. [PubMed] [Google Scholar]
- 80.El Mourabet M, El-Hachem S, Harrison JR, Binion DG. Anti-TNF antibody therapy for inflammatory bowel disease during pregnancy: a clinical review. Curr. Drug Targets. 2010;11:234–241. doi: 10.2174/138945010790309885. [DOI] [PubMed] [Google Scholar]
- 81.Papp K.A., Griffiths C.E.M., Gordon K., Lebwohl M., Szapary P.O., Wasfi Y., Chan D., Hsu M.-C., Ho V., Ghislain P.D., Strober B., Reich K. Long-term safety of ustekinumab in patients with moderate-to-severe psoriasis: final results from 5 years of follow-up. British Journal of Dermatology. 2013;168(4):844–854. doi: 10.1111/bjd.12214. [DOI] [PubMed] [Google Scholar]
- 82.Ray K. IBD: ustekinumab shows promise in the treatment of refractory Crohn's disease. Nature Rev. Gastroenterol. Hepatol. 2012;9:690. doi: 10.1038/nrgastro.2012.213. [DOI] [PubMed] [Google Scholar]
- 83.Parikh A, et al. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflamm. Bowel Dis. 2012;18:1470–1479. doi: 10.1002/ibd.21896. [DOI] [PubMed] [Google Scholar]
- 84.Haanstra KG, et al. Antagonizing the α4β1 integrin, but not α4β7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J. Immunol. 2013;190:1961–1973. doi: 10.4049/jimmunol.1202490. [DOI] [PubMed] [Google Scholar]
- 85.Li S, et al. Pharmacokinetics and tolerability of human mouse chimeric anti-CD22 monoclonal antibody in Chinese patients with CD22-positive non-Hodgkin lymphoma. MAbs. 2012;4:256–266. doi: 10.4161/mabs.4.2.19136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang X, Markovic-Plese S. Interferon β inhibits the Th17 cell-mediated autoimmune response in patients with relapsing-remitting multiple sclerosis. Clin. Neurol. Neurosurg. 2010;112:641–645. doi: 10.1016/j.clineuro.2010.04.020. [DOI] [PubMed] [Google Scholar]
- 87.Wang C, et al. Small Modular ImmunoPharmaceutical (SMIPTM) molecules directed at the TCR complex (CD3) block T cell activation and cause minimal cytokine release in vitro. J. Immunol. 2010;184:96.25. [Google Scholar]
- 88.Beckett T, et al. Small Modular ImmunoPharmaceutical (SMIPTM) molecules directed at the TCR complex block acute graft versus host disease and cause minimal cytokine release in vivo. J. Immunol. 2010;184:145.30. [Google Scholar]
- 89.Burge DJ, et al. Pharmacokinetic and pharmacodynamic properties of TRU-015, a CD20-directed small modular immunopharmaceutical protein therapeutic, in patients with rheumatoid arthritis: a Phase I, open-label, dose-escalation clinical study. Clin. Ther. 2008;30:1806–1816. doi: 10.1016/j.clinthera.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 90.Lee S, Ballow M. Monoclonal antibodies and fusion proteins and their complications: targeting B cells in autoimmune diseases. J. Allergy Clin. Immunol. 2010;125:814–820. doi: 10.1016/j.jaci.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 91.Tawara T, et al. Complement activation plays a key role in antibody-induced infusion toxicity in monkeys and rats. J. Immunol. 2008;180:2294–2298. doi: 10.4049/jimmunol.180.4.2294. [DOI] [PubMed] [Google Scholar]
- 92.Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol. Ther. 2012;136:334–342. doi: 10.1016/j.pharmthera.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 93.Gupta P, Goldenberg DM, Rossi EA, Chang CH. Multiple signaling pathways induced by hexavalent, monospecific, anti-CD20 and hexavalent, bispecific, anti-CD20/CD22 humanized antibodies correlate with enhanced toxicity to B-cell lymphomas and leukemias. Blood. 2010;116:3258–3267. doi: 10.1182/blood-2010-03-276857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qu Z, et al. Bispecific anti-CD20/22 antibodies inhibit B-cell lymphoma proliferation by a unique mechanism of action. Blood. 2008;111:2211–2219. doi: 10.1182/blood-2007-08-110072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Furst DE. Development of TNF inhibitor therapies for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2010;28:S5–S12. [PubMed] [Google Scholar]
- 96.Greenberg JD, et al. Association of methotrexate and tumour necrosis factor antagonists with risk of infectious outcomes including opportunistic infections in the CORRONA registry. Ann. Rheum. Dis. 2010;69:380–386. doi: 10.1136/ard.2008.089276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Colombel JF, et al. The safety profile of infliximab in patients with Crohn's disease: the Mayo clinic experience in 500 patients. Gastroenterology. 2004;126:19–31. doi: 10.1053/j.gastro.2003.10.047. [DOI] [PubMed] [Google Scholar]
- 98.Zabana Y, et al. Infliximab safety profile and long-term applicability in inflammatory bowel disease: 9-year experience in clinical practice. Aliment. Pharmacol. Ther. 2010;31:553–560. doi: 10.1111/j.1365-2036.2009.04206.x. [DOI] [PubMed] [Google Scholar]
- 99.Lane MA, et al. TNF-α antagonist use and risk of hospitalization for infection in a national cohort of veterans with rheumatoid arthritis. Medicine (Baltimore) 2011;90:139–145. doi: 10.1097/MD.0b013e318211106a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Favalli EG, et al. Serious infections during anti-TNFα treatment in rheumatoid arthritis patients. Autoimmun. Rev. 2009;8:266–273. doi: 10.1016/j.autrev.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 101.Singh, J. A. et al. Adverse effects of biologics: a network meta-analysis and Cochrane overview. Cochrane Database Syst Rev 16 Feb 2011 (doi:10.1002/14651858.CD008794.pub2). This is a systematic review that enumerates the incidences of adverse reactions, particularly infections and malignancies, associated with immunomodulatory biologics. [DOI] [PMC free article] [PubMed]
- 102.Schoels Monika, Aletaha Daniel, Smolen Josef S, Wong John B. Comparative effectiveness and safety of biological treatment options after tumour necrosis factor α inhibitor failure in rheumatoid arthritis: systematic review and indirect pairwise meta-analysis. Annals of the Rheumatic Diseases. 2012;71(8):1303–1308. doi: 10.1136/annrheumdis-2011-200490. [DOI] [PubMed] [Google Scholar]
- 103.Curtis JR, Singh JA. Use of biologics in rheumatoid arthritis: current and emerging paradigms of care. Clin. Ther. 2011;33:679–707. doi: 10.1016/j.clinthera.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Grasland A, Sterpu R, Boussoukaya S, Mahe I. Autoimmune hepatitis induced by adalimumab with successful switch to abatacept. Eur. J. Clin. Pharmacol. 2012;68:895–898. doi: 10.1007/s00228-011-1191-4. [DOI] [PubMed] [Google Scholar]
- 105.Germanidis G, Hytiroglou P, Zakalka M, Settas L. Reactivation of occult hepatitis B virus infection, following treatment of refractory rheumatoid arthritis with abatacept. J. Hepatol. 2012;56:1420–1421. doi: 10.1016/j.jhep.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 106.Larsen CP, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am. J. Transplant. 2005;5:443–453. doi: 10.1111/j.1600-6143.2005.00749.x. [DOI] [PubMed] [Google Scholar]
- 107.Epping G, van der Valk PD, Hendrix R. Legionella pneumophila pneumonia in a pregnant woman treated with anti-TNF-α antibodies for Crohn's disease: a case report. J. Crohns Colitis. 2010;4:687–689. doi: 10.1016/j.crohns.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 108.US Food and Drug Administration. FDA drug safety communication: Drug labels for the tumor necrosis factor-α (TNFα) blockers now include warnings about infection with Legionella and Listeria bacteria. FDA website[online], (2011).
- 109.Kendler DL, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J. Bone Miner. Res. 2010;25:72–81. doi: 10.1359/jbmr.090716. [DOI] [PubMed] [Google Scholar]
- 110.Cohen SB, et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008;58:1299–1309. doi: 10.1002/art.23417. [DOI] [PubMed] [Google Scholar]
- 111.Askling J, et al. Time-dependent increase in risk of hospitalisation with infection among Swedish RA patients treated with TNF antagonists. Ann. Rheum. Dis. 2007;66:1339–1344. doi: 10.1136/ard.2006.062760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Watts NB, et al. Infections in postmenopausal women with osteoporosis treated with denosumab or placebo: coincidence or causal association? Osteoporos. Int. 2012;23:327–337. doi: 10.1007/s00198-011-1755-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Isvy A, et al. Safety of rituximab in rheumatoid arthritis: a long-term prospective single-center study of gammaglobulin concentrations and infections. Joint Bone Spine. 2012;79:365–369. doi: 10.1016/j.jbspin.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 114.Popa C, Leandro MJ, Cambridge G, Edwards JC. Repeated B lymphocyte depletion with rituximab in rheumatoid arthritis over 7 yrs. Rheumatology. 2007;46:626–630. doi: 10.1093/rheumatology/kel393. [DOI] [PubMed] [Google Scholar]
- 115.Emery P, et al. The efficacy and safety of rituximab in patients with active rheumatoid arthritis despite methotrexate treatment: results of a phase IIB randomized, double-blind, placebo-controlled, dose-ranging trial. Arthritis Rheum. 2006;54:1390–1400. doi: 10.1002/art.21778. [DOI] [PubMed] [Google Scholar]
- 116.Cohen SB, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793–2806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 117.Fleischmann RM, et al. Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: a large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 2003;48:927–934. doi: 10.1002/art.10870. [DOI] [PubMed] [Google Scholar]
- 118.Fleischmann RM, et al. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2006;65:1006–1012. doi: 10.1136/ard.2005.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Danilenko DM, Wang H. The yin and yang of immunomodulatory biologics: assessing the delicate balance between benefit and risk. Toxicol. Pathol. 2012;40:272–287. doi: 10.1177/0192623311430237. [DOI] [PubMed] [Google Scholar]
- 120.Boren EJ, Cheema GS, Naguwa SM, Ansari AA, Gershwin ME. The emergence of progressive multifocal leukoencephalopathy (PML) in rheumatic diseases. J. Autoimmun. 2008;30:90–98. doi: 10.1016/j.jaut.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 121.Carson KR, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 122.Barkholt L, Linde A, Falk KI. OKT3 and ganciclovir treatments are possibly related to the presence of Epstein-Barr virus in serum after liver transplantation. Transpl. Int. 2005;18:835–843. doi: 10.1111/j.1432-2277.2005.00145.x. [DOI] [PubMed] [Google Scholar]
- 123.Keay S, et al. Posttransplantation lymphoproliferative disorder associated with OKT3 and decreased antiviral prophylaxis in pancreas transplant recipients. Clin. Infect. Dis. 1998;26:596–600. doi: 10.1086/514579. [DOI] [PubMed] [Google Scholar]
- 124.Krueger GG, Gottlieb AB, Sterry W, Korman N, Van De Kerkhof P. A multicenter, open-label study of repeat courses of intramuscular alefacept in combination with other psoriasis therapies in patients with chronic plaque psoriasis. J. Dermatolog. Treat. 2008;19:146–155. doi: 10.1080/09546630701846103. [DOI] [PubMed] [Google Scholar]
- 125.Nguyen DH, Hurtado-Ziola N, Gagneux P, Varki A. Loss of Siglec expression on T lymphocytes during human evolution. Proc. Natl Acad. Sci. USA. 2006;103:7765–7770. doi: 10.1073/pnas.0510484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bugelski PJ, Martin PL. Concordance of preclinical and clinical pharmacology and toxicology of therapeutic monoclonal antibodies and fusion proteins: cell surface targets. Br. J. Pharmacol. 2012;166:823–846. doi: 10.1111/j.1476-5381.2011.01811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Martin PL, Bugelski PJ. Concordance of preclinical and clinical pharmacology and toxicology of monoclonal antibodies and fusion proteins: soluble targets. Br. J. Pharmacol. 2012;166:806–822. doi: 10.1111/j.1476-5381.2011.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Eastwood D, et al. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. Br. J. Pharmacol. 2010;161:512–526. doi: 10.1111/j.1476-5381.2010.00922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Legrand N, et al. Transient accumulation of human mature thymocytes and regulatory T cells with CD28 superagonist in “human immune system” Rag2−/−γc−/− mice. Blood. 2006;108:238–245. doi: 10.1182/blood-2006-01-0190. [DOI] [PubMed] [Google Scholar]
- 130.Luhder F, et al. Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. J. Exp. Med. 2003;197:955–966. doi: 10.1084/jem.20021024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tacke M, Hanke G, Hanke T, Hunig T. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: evidence for functionally distinct forms of CD28. Eur. J. Immunol. 1997;27:239–247. doi: 10.1002/eji.1830270136. [DOI] [PubMed] [Google Scholar]
- 132.Lin CH, Hunig T. Efficient expansion of regulatory T cells in vitro and in vivo with a CD28 superagonist. Eur. J. Immunol. 2003;33:626–638. doi: 10.1002/eji.200323570. [DOI] [PubMed] [Google Scholar]
- 133.Stebbings R, et al. “Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J. Immunol. 2007;179:3325–3331. doi: 10.4049/jimmunol.179.5.3325. [DOI] [PubMed] [Google Scholar]
- 134.Birmingham DJ, Hebert LA. CR1 and CR1-like: the primate immune adherence receptors. Immunol. Rev. 2001;180:100–111. doi: 10.1034/j.1600-065X.2001.1800109.x. [DOI] [PubMed] [Google Scholar]
- 135.Katschke KJ, Jr, et al. Structural and functional analysis of a C3b-specific antibody that selectively inhibits the alternative pathway of complement. J. Biol. Chem. 2009;284:10473–10479. doi: 10.1074/jbc.M809106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Warncke M, et al. Different adaptations of IgG effector function in human and nonhuman primates and implications for therapeutic antibody treatment. J. Immunol. 2012;188:4405–4411. doi: 10.4049/jimmunol.1200090. [DOI] [PubMed] [Google Scholar]
- 137.O'Day SJ, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann. Oncol. 2010;21:1712–1717. doi: 10.1093/annonc/mdq013. [DOI] [PubMed] [Google Scholar]
- 138.Kumar S, Singh SK, Wang X, Rup B, Gill D. Coupling of aggregation and immunogenicity in biotherapeutics: T- and B-cell immune epitopes may contain aggregation-prone regions. Pharm. Res. 2011;28:949–961. doi: 10.1007/s11095-011-0414-9. [DOI] [PubMed] [Google Scholar]
- 139.van Beers MM, Jiskoot W, Schellekens H. On the role of aggregates in the immunogenicity of recombinant human interferon β in patients with multiple sclerosis. J. Interferon Cytokine Res. 2010;30:767–775. doi: 10.1089/jir.2010.0086. [DOI] [PubMed] [Google Scholar]
- 140.van Beers MM, et al. Hybrid transgenic immune tolerant mouse model for assessing the breaking of B cell tolerance by human interferon β. J. Immunol. Methods. 2010;352:32–37. doi: 10.1016/j.jim.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 141.Braun A, Kwee L, Labow MA, Alsenz J. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon α (IFN-α) in normal and transgenic mice. Pharm. Res. 1997;14:1472–1478. doi: 10.1023/A:1012193326789. [DOI] [PubMed] [Google Scholar]
- 142.Joubert MK, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J. Biol. Chem. 2012;287:25266–25279. doi: 10.1074/jbc.M111.330902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.De Groot AS, Knopp PM, Martin W. De-immunization of therapeutic proteins by T-cell epitope modification. Dev. Biol. 2005;122:171–194. [PubMed] [Google Scholar]
- 144.Tangri S, et al. Rationally engineered therapeutic proteins with reduced immunogenicity. J. Immunol. 2005;174:3187–3196. doi: 10.4049/jimmunol.174.6.3187. [DOI] [PubMed] [Google Scholar]
- 145.De Groot AS, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood. 2008;112:3303–3311. doi: 10.1182/blood-2008-02-138073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Basu A, et al. Structure–function engineering of interferon-β-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug. Chem. 2006;17:618–630. doi: 10.1021/bc050322y. [DOI] [PubMed] [Google Scholar]
- 147.Jevsevar S, Kunstelj M, Porekar VG. PEGylation of therapeutic proteins. Biotechnol. J. 2010;5:113–128. doi: 10.1002/biot.200900218. [DOI] [PubMed] [Google Scholar]
- 148.Armstrong JK, et al. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer. 2007;110:103–111. doi: 10.1002/cncr.22739. [DOI] [PubMed] [Google Scholar]
- 149.Ganson NJ, Kelly SJ, Scarlett E, Sundy JS, Hershfield MS. Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res. Ther. 2006;8:R12. doi: 10.1186/ar1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ghaderi D, Taylor RE, Padler-Karavani V, Diaz S, Varki A. Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nature Biotech. 2010;28:863–867. doi: 10.1038/nbt.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.von Delwig A, et al. The impact of glycosylation on HLA-DR1-restricted T cell recognition of type II collagen in a mouse model. Arthritis Rheum. 2006;54:482–491. doi: 10.1002/art.21565. [DOI] [PubMed] [Google Scholar]
- 152.Singh SK. Impact of product-related factors on immunogenicity of biotherapeutics. J. Pharm. Sci. 2011;100:354–387. doi: 10.1002/jps.22276. [DOI] [PubMed] [Google Scholar]
- 153.Meritet JF, Maury C, Tovey MG. Induction of tolerance to recombinant therapeutic proteins. J. Interferon Cytokine Res. 2001;21:1031–1038. doi: 10.1089/107999001317205150. [DOI] [PubMed] [Google Scholar]
- 154.Meritet JF, Maury C, Tovey MG. Effect of oromucosal administration of IFN-α on allergic sensitization and the hypersensitive inflammatory response in animals sensitized to ragweed pollen. J. Interferon Cytokine Res. 2001;21:583–593. doi: 10.1089/10799900152547849. [DOI] [PubMed] [Google Scholar]
- 155.Nagler-Anderson C, Terhoust C, Bhan AK, Podolsky DK. Mucosal antigen presentation and the control of tolerance and immunity. Trends Immunol. 2001;22:120–122. doi: 10.1016/S1471-4906(00)01830-5. [DOI] [PubMed] [Google Scholar]
- 156.Baert F, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn's disease. N. Engl. J. Med. 2003;348:601–608. doi: 10.1056/NEJMoa020888. [DOI] [PubMed] [Google Scholar]
- 157.Somerfield J, et al. A novel strategy to reduce the immunogenicity of biological therapies. J. Immunol. 2010;185:763–768. doi: 10.4049/jimmunol.1000422. [DOI] [PubMed] [Google Scholar]
- 158.[No authors listed.] Deal watch: Boosting TRegs to target autoimmune disease. Nature Rev. Drug Discov.10, 566 (2011). [DOI] [PubMed]
- 159.Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv. Immunol. 2010;108:111–165. doi: 10.1016/B978-0-12-380995-7.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bluestone JA, Thomson AW, Shevach EM, Weiner HL. What does the future hold for cell-based tolerogenic therapy? Nature Rev. Immunol. 2007;7:650–654. doi: 10.1038/nri2137. [DOI] [PubMed] [Google Scholar]
- 161.Yan G, et al. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nature Biotech. 2011;29:1019–1023. doi: 10.1038/nbt.1992. [DOI] [PubMed] [Google Scholar]
- 162.Stevison LS, Kohn MH. Determining genetic background in captive stocks of cynomolgus macaques (Macaca fascicularis) J. Med. Primatol. 2008;37:311–317. doi: 10.1111/j.1600-0684.2008.00292.x. [DOI] [PubMed] [Google Scholar]
- 163.Saito Y, Naruse TK, Akari H, Matano T, Kimura A. Diversity of MHC class I haplotypes in cynomolgus macaques. Immunogenetics. 2012;64:131–141. doi: 10.1007/s00251-011-0568-y. [DOI] [PubMed] [Google Scholar]
- 164.Chamanza R, Marxfeld HA, Blanco AI, Naylor SW, Bradley AE. Incidences and range of spontaneous findings in control cynomolgus monkeys (Macaca fascicularis) used in toxicity studies. Toxicol. Pathol. 2010;38:642–657. doi: 10.1177/0192623310368981. [DOI] [PubMed] [Google Scholar]
- 165.Bussiere JL, et al. Alternative strategies for toxicity testing of species-specific biopharmaceuticals. Int. J. Toxicol. 2009;28:230–253. doi: 10.1177/1091581809337262. [DOI] [PubMed] [Google Scholar]
- 166.Clarke J, et al. Evaluation of a surrogate antibody for preclinical safety testing of an anti-CD11a monoclonal antibody. Regul. Toxicol. Pharmacol. 2004;40:219–226. doi: 10.1016/j.yrtph.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 167.Hu CY, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J. Clin. Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Huang H, Benoist C, Mathis D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc. Natl Acad. Sci. USA. 2010;107:4658–4663. doi: 10.1073/pnas.1001074107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Abraham D, McGrath KG. Hypersensitivity to aldesleukin (interleukin-2 and proleukin) presenting as facial angioedema and erythema. Allergy Asthma Proc. 2003;24:291–294. [PubMed] [Google Scholar]
- 170.Strayer DR, Carter WA. Recombinant and natural human interferons: analysis of the incidence and clinical impact of neutralizing antibodies. J. Interferon Cytokine Res. 2012;32:95–102. doi: 10.1089/jir.2011.0069. [DOI] [PubMed] [Google Scholar]
- 171.Investigators CT, et al. Alemtuzumab versus interferon β-1a in early multiple sclerosis. N. Engl. J. Med. 2008;359:1786–1801. doi: 10.1056/NEJMoa0802670. [DOI] [PubMed] [Google Scholar]
- 172.Gopal AK, et al. Safety and efficacy of brentuximab vedotin for Hodgkin lymphoma recurring after allogeneic stem cell transplant. Blood. 2012;120:560–568. doi: 10.1182/blood-2011-12-397893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ruf P, et al. Pharmacokinetics, immunogenicity and bioactivity of the therapeutic antibody catumaxomab intraperitoneally administered to cancer patients. Br. J. Clin. Pharmacol. 2010;69:617–625. doi: 10.1111/j.1365-2125.2010.03635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jankowitz R, Joyce J, Jacobs SA. Anaphylaxis after administration of ibritumomab tiuxetan for follicular non-hodgkin lymphoma. Clin. Nucl. Med. 2008;33:94–96. doi: 10.1097/RLU.0b013e31815ef825. [DOI] [PubMed] [Google Scholar]
- 175.Jaffers GJ, et al. Monoclonal antibody therapy. Anti-idiotypic and non-anti-idiotypic antibodies to OKT3 arising despite intense immunosuppression. Transplantation. 1986;41:572–578. doi: 10.1097/00007890-198605000-00004. [DOI] [PubMed] [Google Scholar]
- 176.Coiffier B, et al. Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: a phase 1–2 study. Blood. 2008;111:1094–1100. doi: 10.1182/blood-2007-09-111781. [DOI] [PubMed] [Google Scholar]
- 177.Buchegger F, et al. Long-term complete responses after 131I-tositumomab therapy for relapsed or refractory indolent non-Hodgkin's lymphoma. Br. J. Cancer. 2006;94:1770–1776. doi: 10.1038/sj.bjc.6603166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bartelds GM, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305:1460–1468. doi: 10.1001/jama.2011.406. [DOI] [PubMed] [Google Scholar]
- 179.Kumar D, Bouldin TW, Berger RG. A case of progressive multifocal leukoencephalopathy in a patient treated with infliximab. Arthritis Rheum. 2010;62:3191–3195. doi: 10.1002/art.27687. [DOI] [PubMed] [Google Scholar]
- 180.Sorensen PS, et al. Occurrence of antibodies against natalizumab in relapsing multiple sclerosis patients treated with natalizumab. Mult. Scler. 2011;17:1074–1078. doi: 10.1177/1352458511404271. [DOI] [PubMed] [Google Scholar]
- 181.Cruz AA, et al. Safety of anti-immunoglobulin E therapy with omalizumab in allergic patients at risk of geohelminth infection. Clin. Exp. Allergy. 2007;37:197–207. doi: 10.1111/j.1365-2222.2007.02650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stubenrauch K, et al. Subset analysis of patients experiencing clinical events of a potentially immunogenic nature in the pivotal clinical trials of tocilizumab for rheumatoid arthritis: evaluation of an antidrug antibody ELISA using clinical adverse event-driven immunogenicity testing. Clin. Ther. 2010;32:1597–1609. doi: 10.1016/j.clinthera.2010.07.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pharmacovigilance and the role of regulators (PDF 241 kb)