

Despite being protected by the blood-brain barrier, the CNS must constantly be monitored for insult or pathogen invasion. In this review, the authors illustrate the molecular and cellular players that preside over this surveillance of the brain and spinal cord.
Subject terms: Immunosuppression, Immunosurveillance, Central nervous system
Abstract
The CNS, which consists of the brain and spinal cord, is continuously monitored by resident microglia and blood-borne immune cells such as macrophages, dendritic cells and T cells to detect for damaging agents that would disrupt homeostasis and optimal functioning of these vital organs. Further, the CNS must balance between vigilantly detecting for potentially harmful factors and resolving any immunological responses that in themselves can create damage if left unabated. We discuss the physiological roles of the immune sentinels that patrol the CNS, the molecular markers that underlie their surveillance duties, and the consequences of interrupting their functions following injury and infection by viruses such as JC virus, human immunodeficiency virus, herpes simplex virus and West Nile virus.
Main
Although the CNS lacks lymphatics, displays low levels of major histocompatibility complex (MHC) molecules and is uniquely shielded from free diffusion of molecular and cellular components by the blood-brain barrier (BBB) and the blood–cerebrospinal fluid (CSF) barrier, the immune response in the brain can be impressive if not spectacular, as is the case in meningitis. By contrast, loss of immunity is often highlighted by cerebral infections. To protect this vital system, the healthy brain and spinal cord are under continual immune surveillance to detect and eliminate potential mediators of infection and damage. Both resident microglia and immune cells from the general circulation function as primary guardians of the CNS, and their sentinel duties contribute to the maintenance of homeostasis and thus optimal functioning of the brain and spinal cord.
Through their highly dynamic processes, nonactivated microglia relay environmental information to neurons and macroglia, which in turn advise microglial cells of their health status. In addition to microglia, peripheral immune cells can enter the uninflamed CNS through mechanisms similar to those in peripheral organs, albeit at a lower rate. For instance, the number of T cells that travel into the CNS is small, as these cells partake in intermittent, transient interactions with CNS endothelium through cell adhesion molecules that are themselves expressed at one-tenth of the level of that in other organs1. Basal interaction of leukocytes (rolling) in brain is also one-tenth of that seen in other organs1. Other immune cells of peripheral origin, such as perivascular macrophages and meningeal dendritic cells, are strategically located at the interface between the blood and brain, where they can sample the environment for foreign antigens and promote antigen presentation should a florid inflammatory response be necessary2. Here, we summarize knowledge of how microglia and peripheral immune cells participate in promoting physiological balance and protection of the normal CNS and how interruption of immune patrol of the brain and spinal cord can lead to and/or augment disease and injury.
Microglia mediate surveillance and homeostasis in the CNS
Even though they share surface markers with hematogenously derived macrophages3, it has recently been shown that one subset of microglia is separately derived from the yolk sac, with progenitors invading the CNS at approximately the tenth day of development in mice4,5,6. After birth, another subset of microglia appears to be bone marrow derived, with distinct function(s)7. A large body of work demonstrates that microglial cells are vital in maintaining homeostasis in the CNS. They are critical in synapse maintenance and turnover in the adult CNS8 and may provide trophic support to neurons and macroglia through their secretion of neurotrophins such as nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor, neurotrophin-3 (NT3) and glial cell line–derived neurotrophic factor (GDNF). These factors have been shown to protect and regulate the survival and regeneration of neuronal cells9. Microglia can take up neurotransmitters such as glutamate through cytokine-mediated mechnisms10 and are the primary cell type involved in phagocytosis and clearance of myelin inhibitors, debris and dead cells in the CNS. Even a function in the enhancement of remyelination, oligodendrogenesis and angiogenesis has been documented11,12. Together, these critical functions of microglia facilitate and promote equilibrium and optimal functioning of the CNS.
Another major role of microglia is to serve as first line defenders against infectious agents and injury-related products in the CNS parenchyma. These cells lie very close to one another, but every cell occupies its own niche, with minimal overlap of processes between neighboring cells13. The branches of resting microglia are rapidly and constantly moving, such that the entire extracellular space of the normal CNS is occupied and sampled by at least one microglial process over a few hours14. Nonactivated microglia, particularly in the white matter, constitutively express low levels of HLA-DR in the healthy human brain15 and MHC II in the rodent brain16, observations that imply a capability for antigen presentation and thus a function in immune surveillance of the brain. Indeed, the molecular markers of antigen presentation and activation, such as MHC II, CD80, CD86, CD40, CD11a, CD54 and CD58 (refs. 17,18), are rapidly augmented on microglia in response to even minor pathological changes in the CNS, and these cells are capable of presenting antigens to, and activating, T cells19. Specifically, in infectious models such as coronavirus-induced mouse hepatitis, upregulation of MHC I by microglia is postulated to reflect a vigilant state of these cells enabling them to present antigen to and engage CD8+ T cells early after infection20. Also, competent presentation of antigen to CD4+ T cells through MHC I and II on microglia21 during Theiler's murine encephalitis virus infection likely reflects an effort to clear the virus.
In addition to antigen presentation, microglia have all the machinery necessary to detect most microbes and noxious stimuli that access the CNS parenchyma and can rapidly mount a potent inflammatory response. Indeed, intraventricular administration of lipopolysaccharide rapidly induces a macrophage-like response with the release of cytokines (tumor necrosis factor (TNF), interleukin (IL)-1, interferons (IFNs) and others) and the production of many chemokines and chemoattractants that alert and recruit more immune cells to the brain. Bacterial meningitis, much like lipopolysaccharide administration, directly activates microglia, which release cytokines and chemokines that attract peripheral immune cells. The exuberant response that ensues likely contributes to the morbidity and mortality of this cerebral infection. However, some pathogens find refuge in the brain and do not appear to cause an immune response. As one example, Cryptococcus neoformans infiltrates the brains of HIV patients who have low T cell counts22. Although one could argue that this might be because the microglia are also immunocompromised, C. gattii, which has in the last 10 years emerged in North America, infiltrates the brains of immunocompetent humans with minimal signs of inflammation early in disease23.
Although a robust immune response can be mounted in the CNS, this vital organ is normally quiescent, with cellular components of the CNS mediating immunosuppression. For example, in the intact CNS, microglia are kept in a quiescent state by interactions between the neuronal CD200 receptor and the CD200 ligand on microglia. This was demonstrated directly in CD200-deficient mice, in which microglia spontaneously exhibit features of activation such as less ramified morphology and increase expression of CD11b and CD45 (ref. 24). Further, it has been reported that electrical activity of neurons may also suppress MHC expression in surrounding microglia and astrocytes, as paralysis of neuronal signaling by tetrodotoxin correlates with augmented interferon-γ-induced expression of MHC II on these glia25. In addition to neuronal activity, neuronally derived neurotrophins such as NGF, NT3 and BDNF have been demonstrated to dampen MHC I expression (critical for viral detection by CD8+ T cells) on microglia in brain slices or cultures26, and NGF inhibits expression of the costimulatory molecule B7-2 (CD86) and CD40 in cultured rat microglia27. Of note, resting microglia in old mice were recently found to possess less elaborate dendritic arbors concomitant with slower process movements and retarded migratory ability that may compromise their surveying ability and injury response28. This may explain the age-related increase in susceptibility to cerebral infection. In addition to keeping vigilance at low levels, suppressive mechanisms may also be ramped up to allow restitution of an immune response. However, the dominant mechanisms of restitution identified thus far involve molecules such as TGF-β secreted by astrocytes that downregulate the secretion of the proinflammatory molecules inducible nitric oxide synthase and IL-2 by activated microglia29,30. Increased production of microglial IL-10 also has been shown to dampen immune responses in the brain in an autoregulatory manner29.
In summary, microglia are vital in surveying the CNS environment for foreign and resident agents that can disrupt the homeostatic balance of the brain and spinal cord. They possess the molecular machinery to strongly activate the adaptive arm of the immune system if required. At the same time, these glia are kept in check by endogenous mechanisms, thus highlighting the tight control of immunity in the CNS.
Leukocytes patrol the healthy brain and spinal cord
Although present in small numbers relative to peripheral organs, peripherally derived T cells, macrophages and dendritic cells constitute another group of sentinels that inspect the healthy CNS for harmful agents. Whereas microglia are the primary watchmen in the parenchyma of the brain and spinal cord, peripheral immune cells patrol in specialized CNS compartments located outside the parenchyma31. Immune cells could gain access to the CNS via (1) the non-fenestrated vascularized stroma of the blood-CSF barrier that is surrounded by the choroid plexus epithelial cells, (2) the perivascular or Virchow-Robin space, where deep arteries are continuous with the subarachnoid space and (3) postcapillary venules that enter the parenchyma directly32 (Fig. 1). In all of these possible sites of extravasation, migration across the vascular wall and the glial limitans must occur for the cells to gain access to the parenchyma. The general conception is that surveying macrophages and dendritic cells are competently capable of presenting antigen to, and activating, patrolling T cells, and thus together these immune cells constitute a formidable line of defense against noxious agents in the CNS.
Figure 1. Possible sites of immune cell entry into the CNS.
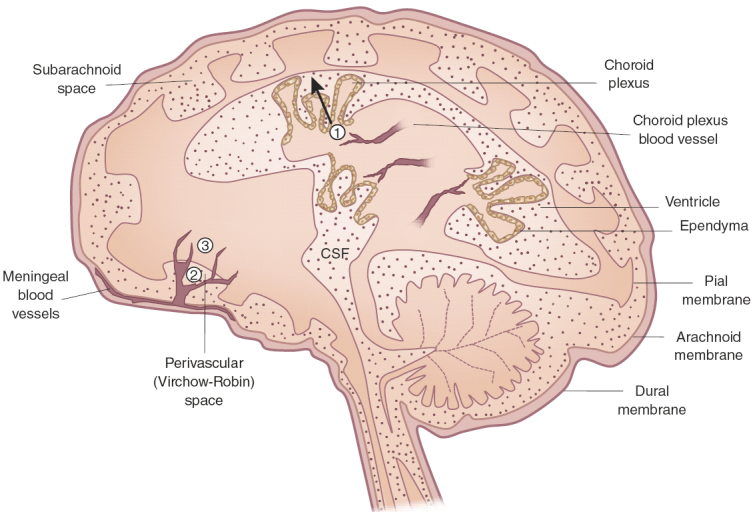
T cells and monocytes may gain access to the brain by crossing through the fenestrated blood vessels of the choroid plexus, across the ependymal layer and into the CSF (1); through the perivascular or Virchow-Robin space, where the meningeal blood vessels branch into the subarachnoid space (2); and directly into the CNS parenchyma through postcapillary venules (3).
Approximately 80% of immune cells found in the CSF of healthy individuals are T cells33 that have probably entered the CNS through the choroid plexus and meninges because in mice these areas are occupied by fluorescently labeled lymphocytes within 2 hours of intravenous injection of the labeled cells into healthy animals34. Kivisäkk and colleagues35,36 characterized the phenotype of CSF T cells in normal humans as predominantly CD4+CD45RA−CD27+CD69+ activated central memory T cells that expressed high levels of CCR7, CXCR3 and L-selectin. P-selectin has also been implicated in facilitating migration of T cells into the CSF of mice and healthy humans on the basis of the reduced CNS entry of leukocytes in P-selectin null mice34 and the high expression of P-selectin glycoprotein ligand 1 on CD4+ T cells in human CSF35,36. P-selectin, E-selectin and intercellular adhesion molecule 1 (ICAM-1) immunoreactivity has been detected in vessels of the choroid plexus and subarachnoid space in humans35, with other studies37 also implicating vascular cell adhesion molecule (VCAM)-1, mucosal addressin cell adhesion molecule (MadCAM)-1 and platelet endothelial cell adhesion molecule (PECAM)-1 in facilitating T cell entry into the healthy human brain. However, the latter observation is difficult to reconcile our understanding of MadCAM-1 as the intestinal homing receptor for T cells. Epithelial V-like antigen in human choroid plexus epithelial cells was recently implicated as a molecular target of a subset of CNS-surveying CD4+ T cells that are characterized by high secretion of IL-17 and IL-22 (ref. 38). Of interest, Loeffler and colleagues31 have showed that cellular immune surveillance in the healthy human brain differs among CNS regions. As expected, the highest numbers of immune cells are located in brain areas where the tight junction barrier of the BBB is reduced, such as the area postrema of the circumventricular organs and the ventro-rostral areas of the medulla oblongata. Not all studies agree with a population of CD4+ T cells in the healthy human brain. At least two separate groups have instead identified CD3+CD8+ granzymeB−perforin− lymphocytes and, to a limited extent, CD20+ B cells as the main cellular components39,40.
Peripherally activated T cells gain essential surface molecules necessary to traverse the BBB into the CNS parenchyma. Capture and adherence of activated TH1 T cells to CNS venules were demonstrated to occur via interactions between VLA-4 on TH1 cells and VCAM-1 on endothelial cells, whereas diapedesis of TH1 cells across the venule wall is facilitated by leukocyte function–associated antigen (LFA)-1 (ref. 41). Further, TH17 cell migration across the blood-CSF barrier in experimental allergic encephalomyelitis (EAE), a model of multiple sclerosis, has been shown to depend on the chemokine receptor CCR6 on TH17 T cells, which binds its ligand CCL20 that is constitutively produced by the choroid plexus epithelium42. However, other studies have shown enhanced EAE clinical symptoms in CCR6-null mice during the chronic disease phase that is associated with a lack of programmed cell death 1 ligand 1–expressing myeloid dendritic cells in spleen43 and reduced numbers of CD4+ T regulatory cells in the CNS44. It is likely that many chemokine receptors function in various immune cells in EAE and their action may vary depending on the stage of the disease. Very recently, a new pathway of T cell entry into the CNS of EAE mice was identified. Arima and colleagues45 have reported that the earliest CNS entry of CD4+ T cells in EAE occurs at a very specific site: the fifth lumbar level of the spinal cord. They observed high CCL20 chemokine expression in the dorsal root vessels. This was associated with sensory neurons that innervate the soleus and other leg muscles, and silencing of these neural pathways reduced chemokine expression and, ultimately, T cell recruitment into the spinal cord.
Under physiological conditions, the perivascular space that directly communicates with the CSF is inhabited by perivascular cells that are bone marrow derived. These cells are continuously replaced by monocytes, as demonstrated by bone marrow chimera studies in rodents46,47 and transplantation studies in humans48 that show a steady rate of perivascular cell turnover in the normal, uninflamed CNS. With their close association to the vessels in the CNS, perivascular cells have been speculated to continually scan and sample the CSF49,50 and may therefore be capable of detecting infectious agents early. They are capable of phagocytosing molecules from the CSF49, and in humans perivascular cells constitutively express MHC II, CD4, B7 and the chemokine receptors CCR3, CCR5 and CXCR4 (refs. 36,51), all of which likely contribute to the ability of these cells to potently and efficiently present antigen to T cells46. There is controversy as to whether perivascular cells re-enter the peripheral immune circuitry to instigate a peripheral immune reaction because direct implantation of antigens into the CNS has failed to elicit a peripheral immune response52. However, Cserr and colleagues53 demonstrated that CNS-derived antigens can drain to deep cervical lymph nodes that could potentially make CNS antigens available to the peripheral immune system. The recent finding that monocytes injected into the brain reach lymphatic organs by way of the cribroid plate54 lends credence to this idea. Perivascular cells may contribute to disease pathogenesis, such as that in multiple sclerosis and EAE, where they are implicated in reactivation of encephalitogenic T cells entering the CNS55. Indeed, specific elimination of monocytes markedly ameliorates clinical symptoms of EAE56.
Although dendritic cells have not been detected in the parenchyma of the normal CNS, these cells are abundant in the dura, arachnoid and pia mater and the choroid plexus of healthy rodents and humans57, and low numbers of myeloid (lin−CD11c+HLA-DRhiCD123lo) and plasmacytoid (lin−CD11c−HLA-DRmodCD123hi) dendritic cells58 that are CCR7+ (ref. 59) have been found in the CSF of healthy individuals58. Dendritic cells are potent antigen-presenting cells to, and activators of, CD4+ T cells55 and may therefore contribute to CNS immune surveillance. There is some evidence that dendritic cells can traffic to peripheral lymphoid organs and possibly present any antigens detected in the CNS to T cells in the periphery. Specifically, dendritic cells that were injected into CSF have been detected in B cell follicles of cervical lymph nodes60. The number of dendritic cells increases markedly in the perivascular space, as well as the parenchyma of the CNS in humans and animals, after CNS infection61; in diseases such as amyotrophic lateral sclerosis62, multiple sclerosis and EAE55; and after ischemic injury and kainic acid–induced excitotoxicity63. Although these cells help clear foreign antigen, the proinflammatory mediators secreted by dendritic cells, perivascular cells and T cells can contribute to disease progression or injury, and thus the function of these cells must remain under strict control. One mechanism that may contribute to suppressing immune reactions in the CNS is constitutive expression of Fas ligand on neurons that promote Fas-mediated death of CNS-infiltrating immune cells64.
Consequences of interrupting CNS immune surveillance
The importance of surveillance of the CNS is exemplified by the eruption of serious complications that arise when migration of immune cells to the brain and spinal cord is interrupted by endogenously produced or externally applied immunosuppressants. The prevailing view is that potentially pathogenic viruses such as JC polyoma virus (JCV) and herpes simplex virus (HSV) that are widespread in the human population, or acquired infections such as West Nile virus (WNV) and HIV, are held in check by constant immune pressure in the CNS. However, a disruption in the migration of immune cells to the brain and spinal cord allows unrestrained proliferation and dissemination of viral particles or deficient wound healing that can result in severe neuropathologies.
CNS infections.
The consequences of interrupting immune surveillance in the CNS have never been more evident than in the case of progressive multifocal leukoencephalopathy (PML), a usually fatal CNS infection caused by JCV. Approximately 80% of humans harbor JCV that resides latently in bone marrow and renal tubular epithelial cells and B cells. PML is often a consequence of therapies designed to prevent attacks by immune cells on the CNS. Upon suppression of CD4+ and CD8+ T cell mobilization, as occurs with HIV infection, chemotherapy or immunosuppressive therapy, the virus enters the brain, either within B cells or as cell-free virus, where it infects and kills oligodendrocytes, leading to demyelination65,66. There are no treatments for PML, and most patients either die or are left with severe disability. Armed with the knowledge that immune cells utilize cell adhesion molecules to infiltrate the CNS in inflammatory diseases, monoclonal antibodies against B cells (rituximab), VLA-4 (natalizumab) and LFA-1 (efalizumab) had been developed to treat multiple sclerosis, non-Hodgkin lymphoma, rheumatoid arthritis, Crohn's disease and systemic lupus erythematosus. These medications inhibit migration of B and T cells into the CNS67. As a result, the lack of surveillance normally imposed by the immune system allows unabated invasion of the pathogen into the brain.
Like JCV, HSV-1 is rampant in the human population but normally remains quiescent in neurons after initial infection of peripheral epithelial cells68. Various antiviral responses are mounted by the host's innate and adaptive immune systems to maintain the dormant state of the virus69. For example, recognition of HSV-1 pathogen-associated molecular patterns by toll-like receptor (TLR)-2 or TLR9 on microglial cells results in the secretion of type I IFNs, IL-15, TNF and the chemokine CCL2 (ref. 70), which recruits macrophages, and type I IFN production by neurons and microglia is further augmented in a TLR3-dependent mechanism. TLR3 expression by microglia and astrocytes may participate in controlling CNS invasion of HSV. Notably, children with a dominant negative TLR3 allele can develop HSV-1 encephalitis71. The IFNs contribute to an antiviral response by inducing RNase L and IFN-induced double-stranded RNA–activated protein kinase expression, which respectively result in degradation of mRNA and cessation of translation69. The adaptive immune system also helps control HSV-1 and maintain its latency. The production of TNF, nitric oxide and IFN-γ by CNS infiltrating macrophages and γδ T cells is associated with control of viral replication, and CD8+ T cells inhibit HSV-1 reactivation through MHC class I– and granzyme B–dependent mechanisms68,72. It is thus clear that the immune system is vital for suppressing reactivation of HSV in the CNS and that interruption of immune surveillance in the brain may lead to resurgence of the virus, as is often seen in immunosuppressed HIV-infected individuals73.
One complication of HIV infection is HIV-associated neurocognitive dysfunction, which comprises a range of neurological impairments that occur when the virus invades the CNS. Invading CD8+ cytotoxic T cells may eliminate virus-infected CNS cells and thus help control the virus74. Another negative relationship between lack of CNS immunity and disease is again highlighted in HIV infection, where survival from primary CNS lymphomas (PCNSL) in AIDS75 has been associated with infiltration of reactive T cells in the perivascular regions of the CNS76. PCNSL is diagnosed in 1.6–9.0% of patients with AIDS77,78, and susceptibility to PCNSL is inversely proportional to CD4+ T cell count79,80. Notably, some HIV-positive patients with PCNSL concomitantly develop Epstein-Barr virus (EBV) infection81, which is preceded by a loss of functional EBV-specific cytotoxic T cells, once again demonstrating the necessity of surveillance in the CNS as a vital immune response in preventing PCNSL development and EBV infection76,82.
In other acquired infections, such as WNV, innate and adaptive immunity is thought to limit dissemination of the virus within the brain. WNV is a mosquito-transmitted, single-stranded RNA virus that can infect and damage neurons, but most infections in humans are asymptomatic83,84, a phenomenon that is attributed to immunity in the brain that suppresses WNV infection. The current thinking is that, through TLR7 recognition of viral RNA, microglia initially sense that WNV has invaded the CNS. This results in secretion of IL-23, which promotes infiltration into the CNS of monocytes and macrophages, CD4+ T cells85,86, CD8+ T cells87, natural killer T cells, dendritic cells and γδ T cells88. The robust immune response participates in neutralizing the virus and limiting viral infectivity, likely in a CCR5-mediated mechanism, as CCR5-deficient mice are more susceptible than their wild-type counterparts to lethal WNV encephalitis, which correlates with increased brain viral burden and markedly reduced brain leukocyte traffic89. Other studies show that CXCL12-CXCR4 interactions appear to promote CNS migration of monocytes and CD8+ T cells, which is associated with neutralization of the virus and increased survival of the host90, and that CXCL10 secretion by infected cerebellar neurons recruits CXCR3-expressing CD8+ T cells that promote viral clearance91.
CNS trauma.
Infections are not the only potentially harmful situations against which the immune system needs to protect in the CNS. Trauma to, and diseases of, the brain and spinal cord can elicit or dampen immune responses that can determine whether the outcome is damaging or protective. For instance, the lack of long distance regeneration of injured axons after spinal cord injury has in part been attributed to an insubstantial immune reaction. Specifically, in comparison to the robust immune reaction following peripheral nerve injury, the response of innate immune cells such as microglia and monocytes in the damaged CNS is weak, and this likely contributes to the delay in phagocytosis of myelin debris and thus prolonged presence of myelin-associated inhibitors of neurite outgrowth. An augmentation of macrophage and microglial activation has been associated with enhanced regeneration of injured CNS axons that was attributed to increased phagocytosis of, and thus faster clearance, of myelin and its axon growth inhibitors, as well as the production by these innate cells of cytokines and growth factors that can support neuron survival, oligodendrogenesis, remyelination and angiogenesis11,12. In addition, enhancing CNS influx of T cells has been shown to promote CNS regeneration by stimulating microglial phagocytosis; buffering glutamate, thereby preventing excitoxicity92; and secreting neurotrophins to protect neurons from secondary degeneration93.
However, the early immune response to trauma and cell death likely involves neutrophils, microglia and macrophages. Endogenous intracellular molecules such as ATP, high mobility group protein B1 and DNA, when released into the extracellular milieu from dying or injured cells, function as danger-associated molecular patterns and as alarmins for the immune system. In addition, molecules modified by oxidation or proteolytic cleavage, such as collagen and hyaluronan, or precipitates such as uric acid crystals have been shown to activate the innate immune system. In the case of ATP, the activation occurs through the purinergic receptor P2X7, which activates NOD-like receptors, including NLRP-3, which activates caspase and IL-1 and induces inflammation. In addition to these molecules, mitochondrial peptides that have bacterial motifs (formylated peptides) activate the formylated peptide receptors found on neutrophils and other immune cells, inducing potent chemotaxis94. Although ATP has been shown to be detected by microglia when released from injured neurons, the role of most of the other aforementioned alarmins in immune response to cellular injury in the brain remains to be elucidated.
The resolution phase of cellular injury is also an area that remains poorly understood but also likely requires recruitment of phagocytic immune cells. Although controversial, a role for infiltrating monocytes in the clearance of β-amyloid in Alzheimer's disease has been suggested95, as amyloid burden is heavier when these cells are eliminated96 and when the immunosuppressive cytokine TGF-β is present at high levels in Alzheimer's disease–affected brains97, and a reduction in β-amyloid deposition is observed when monocyte invasion is stimulated98. More recently, Mildner and colleagues have shown that CCR2-positive perivascular myeloid cells, but not microglia or bone marrow–derived phagocytes, are associated with β-amyloid clearance99.
The type of immune response can, however, retard repair in the CNS, and thus homeostasis requires a constant balancing act. For example, in models of myocardial infarction, CD11b+Ly-6Chigh blood monocytes that possess enhanced phagocytic, proteolytic and inflammatory functions are recruited early to sites of pathology, and they can impair wound-healing, but at the same time they orchestrate the subsequent recruitment of CD11b+Ly-6Clow monocytes that promote tissue repair100.
Conclusion
Immunity is an active component of the CNS. Over the past decade, experiments in mice as well as naturally occurring experiments in humans have led us to conclude that surveillance is critical in the brain and that pathogens are likely breaching the barrier all the time and require immediate eradication. Immunosuppression, particularly with drugs that affect trafficking of immune cells, has highlighted the need for ongoing surveillance. Moreover, this immunological monitoring of the brain and spinal cord is dynamic, specific and tightly regulated.
Acknowledgements
This work was supported by the Canadian Institutes of Health Research, Alberta Innovates-Health Solutions, Canada Foundation for Innovation and the Multiple Sclerosis Society of Canada.
Competing interests
The authors declare no competing financial interests.
Contributor Information
Shalina S Ousman, Email: sousman@ucalgary.ca.
Paul Kubes, Email: pkubes@ucalgary.ca.
References
- 1.Carvalho-Tavares J, et al. A role for platelets and endothelial selectins in tumor necrosis factor-α–induced leukocyte recruitment in the brain microvasculature. Circ. Res. 2000;87:1141–1148. doi: 10.1161/01.RES.87.12.1141. [DOI] [PubMed] [Google Scholar]
- 2.Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia. 2001;36:118–124. doi: 10.1002/glia.1101. [DOI] [PubMed] [Google Scholar]
- 3.Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011;14:1227–1235. doi: 10.1038/nn.2923. [DOI] [PubMed] [Google Scholar]
- 4.Ginhoux F, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alliot F, Lecain E, Grima B, Pessac B. Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc. Natl. Acad. Sci. USA. 1991;88:1541–1545. doi: 10.1073/pnas.88.4.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulz C, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 7.Chen SK, et al. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. 2010;141:775–785. doi: 10.1016/j.cell.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tremblay MÈ, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010;8:e1000527. doi: 10.1371/journal.pbio.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmann T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007;81:1345–1351. doi: 10.1189/jlb.0207114. [DOI] [PubMed] [Google Scholar]
- 10.Shaked I, et al. Protective autoimmunity: interferon-γ enables microglia to remove glutamate without evoking inflammatory mediators. J. Neurochem. 2005;92:997–1009. doi: 10.1111/j.1471-4159.2004.02954.x. [DOI] [PubMed] [Google Scholar]
- 11.Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat. Rev. Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- 12.Li WW, Setzu A, Zhao C, Franklin RJ. Minocycline-mediated inhibition of microglia activation impairs oligodendrocyte progenitor cell responses and remyelination in a non-immune model of demyelination. J. Neuroimmunol. 2005;158:58–66. doi: 10.1016/j.jneuroim.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 13.Raivich G. Like cops on the beat: the active role of resting microglia. Trends Neurosci. 2005;28:571–573. doi: 10.1016/j.tins.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 15.Hayes GM, Woodroofe MN, Cuzner ML. Microglia are the major cell type expressing MHC class II in human white matter. J. Neurol. Sci. 1987;80:25–37. doi: 10.1016/0022-510X(87)90218-8. [DOI] [PubMed] [Google Scholar]
- 16.Akiyama H, Itagaki S, McGeer PL. Major histocompatibility complex antigen expression on rat microglia following epidural kainic acid lesions. J. Neurosci. Res. 1988;20:147–157. doi: 10.1002/jnr.490200202. [DOI] [PubMed] [Google Scholar]
- 17.Akiyama H, McGeer PL. Brain microglia constitutively express β-2 integrins. J. Neuroimmunol. 1990;30:81–93. doi: 10.1016/0165-5728(90)90055-R. [DOI] [PubMed] [Google Scholar]
- 18.Shrikant P, Benveniste EN. The central nervous system as an immunocompetent organ: role of glial cells in antigen presentation. J. Immunol. 1996;157:1819–1822. [PubMed] [Google Scholar]
- 19.Ford AL, Foulcher E, Lemckert FA, Sedgwick JD. Microglia induce CD4 T lymphocyte final effector function and death. J. Exp. Med. 1996;184:1737–1745. doi: 10.1084/jem.184.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malone KE, Stohlman SA, Ramakrishna C, Macklin W, Bergmann CC. Induction of class I antigen processing components in oligodendroglia and microglia during viral encephalomyelitis. Glia. 2008;56:426–435. doi: 10.1002/glia.20625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mack CL, Vanderlugt-Castaneda CL, Neville KL, Miller SD. Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler's virus model of multiple sclerosis. J. Neuroimmunol. 2003;144:68–79. doi: 10.1016/j.jneuroim.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 22.Kovacs JA, et al. Cryptococcosis in the acquired immunodeficiency syndrome. Ann. Intern. Med. 1985;103:533–538. doi: 10.7326/0003-4819-103-4-533. [DOI] [PubMed] [Google Scholar]
- 23.Cheng PY, Sham A, Kronstad JW. Cryptococcus gattii isolates from the British Columbia cryptococcosis outbreak induce less protective inflammation in a murine model of infection than Cryptococcus neoformans. Infect. Immun. 2009;77:4284–4294. doi: 10.1128/IAI.00628-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoek RM, et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- 25.Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- 26.Neumann H, Misgeld T, Matsumuro K, Wekerle H. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc. Natl. Acad. Sci. USA. 1998;95:5779–5784. doi: 10.1073/pnas.95.10.5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei R, Jonakait GM. Neurotrophins and the anti-inflammatory agents interleukin-4 (IL-4), IL-10, IL-11 and transforming growth factor-β1 (TGF-β1) down-regulate T cell costimulatory molecules B7 and CD40 on cultured rat microglia. J. Neuroimmunol. 1999;95:8–18. doi: 10.1016/S0165-5728(98)00248-3. [DOI] [PubMed] [Google Scholar]
- 28.Damani MR, et al. Age-related alterations in the dynamic behavior of microglia. Aging Cell. 2011;10:263–276. doi: 10.1111/j.1474-9726.2010.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aloisi F, Penna G, Cerase J, Menendez Iglesias B, Adorini L. IL-12 production by central nervous system microglia is inhibited by astrocytes. J. Immunol. 1997;159:1604–1612. [PubMed] [Google Scholar]
- 30.Vincent VA, Tilders FJ, Van Dam AM. Inhibition of endotoxin-induced nitric oxide synthase production in microglial cells by the presence of astroglial cells: a role for transforming growth factor β. Glia. 1997;19:190–198. doi: 10.1002/(SICI)1098-1136(199703)19:3<190::AID-GLIA2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 31.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 32.Ransohoff RM, Kivisäkk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 33.Svenningsson A, Andersen O, Edsbagge M, Stemme S. Lymphocyte phenotype and subset distribution in normal cerebrospinal fluid. J. Neuroimmunol. 1995;63:39–46. doi: 10.1016/0165-5728(95)00126-3. [DOI] [PubMed] [Google Scholar]
- 34.Carrithers MD, Visintin I, Viret C, Janeway CS., Jr. Role of genetic background in P selectin-dependent immune surveillance of the central nervous system. J. Neuroimmunol. 2002;129:51–57. doi: 10.1016/S0165-5728(02)00172-8. [DOI] [PubMed] [Google Scholar]
- 35.Kivisäkk P, et al. Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. USA. 2003;100:8389–8394. doi: 10.1073/pnas.1433000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kivisäkk P, et al. T-cells in the cerebrospinal fluid express a similar repertoire of inflammatory chemokine receptors in the absence or presence of CNS inflammation: implications for CNS trafficking. Clin. Exp. Immunol. 2002;129:510–518. doi: 10.1046/j.1365-2249.2002.01947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kleine TO, Benes L. Immune surveillance of the human central nervous system (CNS): different migration pathways of immune cells through the blood-brain barrier and blood-cerebrospinal fluid barrier in healthy persons. Cytometry A. 2006;69:147–151. doi: 10.1002/cyto.a.20225. [DOI] [PubMed] [Google Scholar]
- 38.Wojcik E, Carrithers LM, Carrithers MD. Characterization of epithelial V-like antigen in human choroid plexus epithelial cells: potential role in CNS immune surveillance. Neurosci. Lett. 2011;495:115–120. doi: 10.1016/j.neulet.2011.03.051. [DOI] [PubMed] [Google Scholar]
- 39.Loeffler C, et al. Immune surveillance of the normal human CNS takes place in dependence of the locoregional blood-brain barrier configuration and is mainly performed by CD3+/CD8+ lymphocytes. Neuropathology. 2011;31:230–238. doi: 10.1111/j.1440-1789.2010.01167.x. [DOI] [PubMed] [Google Scholar]
- 40.Schleich A, Stoll M, Bekure K, Meyermann R, Mittelbronn M. Lymphocyte distribution in normal and pathological human spinal cord. Acta Neuropathol. 2004;108:366. [Google Scholar]
- 41.Laschinger M, Vajkoczy P, Engelhardt B. Encephalitogenic T cells use LFA-1 for transendothelial migration but not during capture and initial adhesion strengthening in healthy spinal cord microvessels in vivo. Eur. J. Immunol. 2002;32:3598–3606. doi: 10.1002/1521-4141(200212)32:12<3598::AID-IMMU3598>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 42.Reboldi A, et al. C–C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- 43.Elhofy A, Depaolo RW, Lira SA, Lukacs NW, Karpus WJ. Mice deficient for CCR6 fail to control chronic experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2009;213:91–99. doi: 10.1016/j.jneuroim.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villares R, et al. CCR6 regulates EAE pathogenesis by controlling regulatory CD4+ T-cell recruitment to target tissues. Eur. J. Immunol. 2009;39:1671–1681. doi: 10.1002/eji.200839123. [DOI] [PubMed] [Google Scholar]
- 45.Arima Y, et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell. 2012;148:447–457. doi: 10.1016/j.cell.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 46.Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow–derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- 47.Bechmann I, et al. Turnover of rat brain perivascular cells. Exp. Neurol. 2001;168:242–249. doi: 10.1006/exnr.2000.7618. [DOI] [PubMed] [Google Scholar]
- 48.Unger ER, et al. Male donor-derived cells in the brains of female sex-mismatched bone marrow transplant recipients: a Y-chromosome specific in situ hybridization study. J. Neuropathol. Exp. Neurol. 1993;52:460–470. doi: 10.1097/00005072-199309000-00004. [DOI] [PubMed] [Google Scholar]
- 49.Kida S, Steart PV, Zhang ET, Weller RO. Perivascular cells act as scavengers in the cerebral perivascular spaces and remain distinct from pericytes, microglia and macrophages. Acta Neuropathol. 1993;85:646–652. doi: 10.1007/BF00334675. [DOI] [PubMed] [Google Scholar]
- 50.Hickey WF. Leukocyte traffic in the central nervous system: the participants and their roles. Semin. Immunol. 1999;11:125–137. doi: 10.1006/smim.1999.0168. [DOI] [PubMed] [Google Scholar]
- 51.Graeber MB, Streit WJ, Buringer D, Sparks DL, Kreutzberg GW. Ultrastructural location of major histocompatibility complex (MHC) class II positive perivascular cells in histologically normal human brain. J. Neuropathol. Exp. Neurol. 1992;51:303–311. doi: 10.1097/00005072-199205000-00009. [DOI] [PubMed] [Google Scholar]
- 52.Matyszak MK, Perry VH. Bacillus Calmette-Guerin sequestered in the brain parenchyma escapes immune recognition. J. Neuroimmunol. 1998;82:73–80. doi: 10.1016/S0165-5728(97)00190-2. [DOI] [PubMed] [Google Scholar]
- 53.Harling-Berg C, Knopf PM, Merriam J, Cserr HF. Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J. Neuroimmunol. 1989;25:185–193. doi: 10.1016/0165-5728(89)90136-7. [DOI] [PubMed] [Google Scholar]
- 54.Kaminski, M. et al. Migration of monocytes after intracerebral injection at entorhinal cortex lesion site. J. Leukoc. Biol. doi:10.1189/jlb.0511241 (30 January 2012). [DOI] [PubMed]
- 55.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 56.Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J. Immunol. 1998;161:3767–3775. [PubMed] [Google Scholar]
- 57.McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J. Comp. Neurol. 1999;405:553–562. doi: 10.1002/(SICI)1096-9861(19990322)405:4<553::AID-CNE8>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 58.Pashenkov M, et al. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001;124:480–492. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- 59.Kivisäkk P, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann. Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- 60.Hatterer E, et al. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood. 2006;107:806–812. doi: 10.1182/blood-2005-01-0154. [DOI] [PubMed] [Google Scholar]
- 61.Fischer HG, Bonifas U, Reichmann G. Phenotype and functions of brain dendritic cells emerging during chronic infection of mice with Toxoplasma gondii. J. Immunol. 2000;164:4826–4834. doi: 10.4049/jimmunol.164.9.4826. [DOI] [PubMed] [Google Scholar]
- 62.Henkel JS, et al. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- 63.Reichmann G, Schroeter M, Jander S, Fischer HG. Dendritic cells and dendritic-like microglia in focal cortical ischemia of the mouse brain. J. Neuroimmunol. 2002;129:125–132. doi: 10.1016/S0165-5728(02)00184-4. [DOI] [PubMed] [Google Scholar]
- 64.Flügel A, et al. Neuronal FasL induces cell death of encephalitogenic T lymphocytes. Brain Pathol. 2000;10:353–364. doi: 10.1111/j.1750-3639.2000.tb00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ransohoff RM. Natalizumab and PML. Nat. Neurosci. 2005;8:1275. doi: 10.1038/nn1005-1275. [DOI] [PubMed] [Google Scholar]
- 66.Houff SA, et al. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N. Engl. J. Med. 1988;318:301–305. doi: 10.1056/NEJM198802043180507. [DOI] [PubMed] [Google Scholar]
- 67.Carson KR, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) project. Lancet Oncol. 2009;10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 68.Khanna KM, Lepisto AJ, Decman V, Hendricks RL. Immune control of herpes simplex virus during latency. Curr. Opin. Immunol. 2004;16:463–469. doi: 10.1016/j.coi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 69.McGavern DB, Kang SS. Illuminating viral infections in the nervous system. Nat. Rev. Immunol. 2011;11:318–329. doi: 10.1038/nri2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conrady CD, Drevets DA, Carr DJ. Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J. Neuroimmunol. 2010;220:1–9. doi: 10.1016/j.jneuroim.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang SY, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–1527. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 72.Knickelbein JE, et al. Noncytotoxic lytic granule–mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008;322:268–271. doi: 10.1126/science.1164164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Celum CL. The interaction between herpes simplex virus and human immunodeficiency virus. Herpes. 2004;11(suppl. 1):36A–45A. [PubMed] [Google Scholar]
- 74.Poluektova LY, Munn DH, Persidsky Y, Gendelman HE. Generation of cytotoxic T cells against virus-infected human brain macrophages in a murine model of HIV-1 encephalitis. J. Immunol. 2002;168:3941–3949. doi: 10.4049/jimmunol.168.8.3941. [DOI] [PubMed] [Google Scholar]
- 75.Ponzoni M, et al. Reactive perivascular T-cell infiltrate predicts survival in primary central nervous system B-cell lymphomas. Br. J. Haematol. 2007;138:316–323. doi: 10.1111/j.1365-2141.2007.06661.x. [DOI] [PubMed] [Google Scholar]
- 76.Grovit-Ferbas K, Harris-White ME. Thinking about HIV: the intersection of virus, neuroinflammation and cognitive dysfunction. Immunol. Res. 2010;48:40–58. doi: 10.1007/s12026-010-8166-x. [DOI] [PubMed] [Google Scholar]
- 77.Rosenblum ML, et al. Primary central nervous system lymphomas in patients with AIDS. Ann. Neurol. 1988;23(suppl.):S13–S16. doi: 10.1002/ana.410230707. [DOI] [PubMed] [Google Scholar]
- 78.Welch K, et al. Autopsy findings in the acquired immune deficiency syndrome. J. Am. Med. Assoc. 1984;252:1152–1159. doi: 10.1001/jama.1984.03350090028018. [DOI] [PubMed] [Google Scholar]
- 79.Sparano JA, et al. Effect of highly active antiretroviral therapy on the incidence of HIV-associated malignancies at an urban medical center. J. Acquir. Immune Defic. Syndr. 1999;21(suppl. 1):S18–S22. [PubMed] [Google Scholar]
- 80.Inungu J, Melendez MF, Montgomery JP. AIDS-related primary brain lymphoma in Michigan, January 1990 to December 2000. AIDS Patient Care STDS. 2002;16:107–112. doi: 10.1089/108729102317330445. [DOI] [PubMed] [Google Scholar]
- 81.Tanner JE, Alfieri C. The Epstein-Barr virus and post-transplant lymphoproliferative disease: interplay of immunosuppression, EBV, and the immune system in disease pathogenesis. Transpl. Infect. Dis. 2001;3:60–69. doi: 10.1034/j.1399-3062.2001.003002060.x. [DOI] [PubMed] [Google Scholar]
- 82.Gasser O, et al. HIV patients developing primary CNS lymphoma lack EBV-specific CD4+ T cell function irrespective of absolute CD4+ T cell counts. PLoS Med. 2007;4:e96. doi: 10.1371/journal.pmed.0040096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Campbell GL, Marfin AA, Lanciotti RS, Gubler DJ. West Nile virus. Lancet Infect. Dis. 2002;2:519–529. doi: 10.1016/S1473-3099(02)00368-7. [DOI] [PubMed] [Google Scholar]
- 84.Davis LE, et al. West Nile virus neuroinvasive disease. Ann. Neurol. 2006;60:286–300. doi: 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- 85.Kulkarni AB, Müllbacher A, Blanden RV. Functional analysis of macrophages, B cells and splenic dendritic cells as antigen-presenting cells in West Nile virus-specific murine T lymphocyte proliferation. Immunol. Cell Biol. 1991;69:71–80. doi: 10.1038/icb.1991.12. [DOI] [PubMed] [Google Scholar]
- 86.Sitati EM, Diamond MS. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J. Virol. 2006;80:12060–12069. doi: 10.1128/JVI.01650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J. Virol. 2006;80:119–129. doi: 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang T, et al. IFN-γ-producing γδ T cells help control murine West Nile virus infection. J. Immunol. 2003;171:2524–2531. doi: 10.4049/jimmunol.171.5.2524. [DOI] [PubMed] [Google Scholar]
- 89.Glass WG, et al. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McCandless EE, Zhang B, Diamond MS, Klein RS. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc. Natl. Acad. Sci. USA. 2008;105:11270–11275. doi: 10.1073/pnas.0800898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang B, Chan YK, Lu B, Diamond MS, Klein RS. CXCR3 mediates region-specific antiviral T cell trafficking within the central nervous system during West Nile virus encephalitis. J. Immunol. 2008;180:2641–2649. doi: 10.4049/jimmunol.180.4.2641. [DOI] [PubMed] [Google Scholar]
- 92.Schwartz M. Sell Memorial Lecture. Helping the body to cure itself: immune modulation by therapeutic vaccination for spinal cord injury. J. Spinal Cord Med. 2003;26(suppl. 1):S6–S10. doi: 10.1080/10790268.2003.11753719. [DOI] [PubMed] [Google Scholar]
- 93.Kerschensteiner M, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J. Exp. Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McDonald B, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 95.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 96.Wyss-Coray T, et al. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 97.Town T, et al. Blocking TGF-β–Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of β-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA. 2009;106:1261–1266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mildner A, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer's disease. J. Neurosci. 2011;31:11159–11171. doi: 10.1523/JNEUROSCI.6209-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nahrendorf M, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]