

ABSTRACT
The proteasome is an essential regulator of protein homeostasis. In yeast and many mammalian cells, proteasomes strongly concentrate in the nucleus. Sts1 from the yeast Saccharomyces cerevisiae is an essential protein linked to proteasome nuclear localization. Here, we show that Sts1 contains a non-canonical bipartite nuclear localization signal (NLS) important for both nuclear localization of Sts1 itself and the proteasome. Sts1 binds the karyopherin-α import receptor (Srp1) stoichiometrically, and this requires the NLS. The NLS is essential for viability, and over-expressed Sts1 with an inactive NLS interferes with 26S proteasome import. The Sts1–Srp1 complex binds preferentially to fully assembled 26S proteasomes in vitro. Sts1 is itself a rapidly degraded 26S proteasome substrate; notably, this degradation is ubiquitin independent in cells and in vitro and is inhibited by Srp1 binding. Mutants of Sts1 are stabilized, suggesting that its degradation is tightly linked to its role in localizing proteasomes to the nucleus. We propose that Sts1 normally promotes nuclear import of fully assembled proteasomes and is directly degraded by proteasomes without prior ubiquitylation following karyopherin-α release in the nucleus.
KEY WORDS: Cut8, Karyopherin, NLS, Proteasome, Sts1, Yeast
Summary: Sts1 binds karyopherin α, and this complex associates preferentially with assembled 26S proteasomes, which degrade Sts1 without its prior ubiquitylation.
INTRODUCTION
Protein degradation is essential to cellular function, and most regulated protein degradation in eukaryotes is performed by the ubiquitin–proteasome system (Hochstrasser, 1996; Saeki and Tanaka, 2012; Wehmer and Sakata, 2016). Protein modification by ubiquitin and ubiquitin polymers is carried out by a series of enzymes in an ATP-dependent pathway (Lu et al., 2015; Saeki, 2017; Vittal et al., 2015). Polyubiquitin-modified proteins are frequently targeted to the proteasome for degradation. Proteasomes are found in all eukaryotes as well as archaea and some bacteria (Budenholzer et al., 2017). In eukaryotes, the primary form of the proteasome, called the 26S proteasome, consists of a cylindrical 20S core particle (CP) and a 19S regulatory particle (RP) bound to one or both ends of the CP (Tomko and Hochstrasser, 2013). The CP houses the proteasomal proteolytic sites within an internal chamber, while the RP is responsible for recognition of ubiquitylated substrates, their translocation into the CP, and removal of ubiquitin from substrates during translocation.
The RP is assembled from a pair of subcomplexes called the lid and base (Glickman et al., 1998; Tomko and Hochstrasser, 2013). Each subcomplex of the 26S proteasome – the lid, base and CP – can assemble independently in vivo. The lid assembles in a stepwise manner, first forming two smaller complexes, Module 1 (Rpn5,6,8,9,11) and LP3 (Rpn3,7, Sem1), which join to form the lid assembly intermediate LP2 (Tomko and Hochstrasser, 2011, 2014). Rpn12, the last lid subunit to be added, binds to LP2 and induces conformational changes that result in formation of the mature lid, which is competent to bind to the base (Dambacher et al., 2016; Tomko et al., 2015). Lid assembly does not require any dedicated assembly factors, in contrast to the RP base and CP, which both utilize multiple assembly chaperones (Funakoshi et al., 2009; Kaneko et al., 2009; Roelofs et al., 2009; Saeki et al., 2009).
Nuclear localization has been shown to be important in proteasome function, at least in yeast (Tsuchiya et al., 2013). Subunits of both the RP base and CP contain functional nuclear localization signals (NLSs) (Enenkel, 2014; Lehmann et al., 2002; Tanaka et al., 1990; Wendler et al., 2004); the lid apparently does not, yet it exhibits strong nuclear localization even in a mutant strain where it accumulates as free lid (Isono et al., 2007). The lid might normally be capable of piggybacking into the nucleus by binding to the RP base or it could bind to an adapter that contains an NLS, allowing direct engagement of the import pathway (Chen and Madura, 2014; Chen et al., 2011; Ha et al., 2014; Tabb et al., 2000). It is uncertain whether the RP and CP NLSs are accessible in the assembled 26S proteasome. The base can translocate into the nucleus in the absence of lid association, and the CP has been reported to be imported at assembly stages prior to full CP maturation (Wendler and Enenkel, 2019).
Nuclear protein import through the nuclear pore complex (NPC) utilizes import receptor proteins called karyopherins or importins (Kaffman and O'Shea, 1999; Lee et al., 2005). In the classical import pathway, a karyopherin α/β heterodimer mediates import (Bayliss et al., 2000; Enenkel et al., 1995; Suntharalingam and Wente, 2003). Karyopherin α (yeast Srp1/Kap60) binds directly to the NLS of cargo proteins; the classical NLS (cNLS) sequence is characterized by a short stretch of basic residues, typically near the N-terminus of the protein (Kalderon et al., 1984; Leung et al., 2003). Karyopherin β (yeast Kap95) binds to the karyopherin α-cargo complex and ferries the complex through the NPC.
Saccharomyces cerevisiae Sts1 (called Cut8 in Schizosaccharomyces pombe) has been linked to a variety of cellular processes (Amrani et al., 1996; Houman and Holm, 1994; Liang et al., 1993; Romero-Perez et al., 2007), including serving as a potential bridge between the karyopherin α-receptor and the proteasome, facilitating the latter's nuclear import (Chen and Madura, 2014; Chen et al., 2011; Ha et al., 2014). Sts1 can bind weakly to the individual lid subunit Rpn11 (Chen et al., 2011; Tabb et al., 2000) as well as the full 26S proteasome isolated from yeast (Ha et al., 2014). Subcellular localization of endogenous Sts1 was originally characterized as cytoplasmic (Amrani et al., 1996; Liang et al., 1993), but an over-expressed tagged form of Sts1 concentrated in the nucleus (Tabb et al., 2000), and Cut8, the S. pombe ortholog, is also nuclear (Takeda and Yanagida, 2005).
Sts1 is essential for viability (Liang et al., 1993). While no structural data exist for S. cerevisiae Sts1, a crystal structure for Cut8 (Takeda et al., 2011) allows provisional assignment of structural features (see Fig. 1A). The Cut8 structure displays an overall architecture that matches secondary structure predictions of Sts1; both proteins appear to contain only α-helices and unstructured regions. The unstructured N-terminal region of Cut8 contains an NLS, which is followed by a small helical domain that is probably responsible for dimerization, and a six-helix bundle of uncertain function (Tatebe and Yanagida, 2000). These and other data suggest that Sts1 may function as a homodimer (Amrani et al., 1996; Takeda et al., 2011) and that at least the first of the two basic patches towards the N-terminus may contribute to nuclear localization through binding to Srp1 (karyopherin α) (Chen et al., 2011; Tabb et al., 2000).
Fig. 1.

Sts1 contains an apparent bipartite NLS essential for cell viability. (A) Predicted domain architecture and selected functional elements of Sts1 based on sequence analysis and comparison with the crystal structure of the S. pombe homolog Cut8. The suggested NLS elements are indicated with the sequences shown below; the two mutations in the sts1-DD mutant are also shown. (B) Sts1 lacks a strong match to canonical NLS, but has substantial similarity to a confirmed bipartite NLS in the Ty1 integrase; in both cases the linker separating the two basic elements is unusually long. (C) Viability assay of Sts1 NLS mutants. The noted sts1 alleles, expressed under the endogenous STS1 promoter and terminator from pRS314-based plasmids, were transformed into MHY9580 yeast, in which the chromosomal sts1Δ allele is covered by pRS316-STS1. Transformed cells were struck on 5-FOA plates to evict the cover plasmid. EV, empty vector; WT, wild-type. (D) WT MHY500 yeast transformed with MET25 promoter-based plasmids expressing the indicated NLS sequences fused to 2GFP. Sts1 constructs expressed Sts1 residues 1–76 appended to the N-terminus of 2GFP. The NLS sequence from Ty1 integrase N-terminally tagged with 2GFP was used as a positive control for a bipartite NLS, and 2GFP without an NLS (‘No NLS’) was used as a negative control (both obtained from Anita Corbett). Transformants were grown to mid-log phase at 30°C prior to fluorescence imaging. Three biological replicates of at least 100 cells each were counted (right panel). A t-test was used to determine statistical significance of localization differences (****P<0.0001). Scale bar: 5 μm.
Here, we show that endogenous S. cerevisiae Sts1 is an extremely short-lived protein that is strongly stabilized by missense mutations, including those affecting its nuclear localization. Strikingly, Sts1 degradation in vivo occurs through the 26S proteasome but is largely if not entirely ubiquitin independent, and it can be directly degraded by purified 26S proteasomes when not bound by karyopherin α. We provide evidence that Sts1 has a non-canonical bipartite NLS that is essential both for cell viability and for proteasome and Sts1 nuclear localization. Sts1 binds preferentially to fully assembled 26S proteasomes in vitro, but in vivo data indicate that while it promotes nuclear import of 26S proteasomes, it can also stimulate transport of the lid subcomplex when the latter is not incorporated into full proteasomes. Sts1 provides a rare example of a regulatory protein that is degraded by the proteasome without ubiquitylation and is degraded in a way directly coupled to its function. Its preferential binding to fully assembled proteasomes and ability to promote nuclear import of 26S proteasomes suggests that it might serve as a checkpoint for accurate proteasome assembly by selectively transporting fully assembled proteasomes into the nucleus.
RESULTS
Evidence for a bipartite NLS in Sts1
Although Sts1 has been linked to proteasome nuclear localization, the precise mechanism by which it acts remains unclear. High-copy STS1 and RPN11 (an RP lid subunit) both suppress the srp1-49 allele of karyopherin α but not srp1-31, and srp1-49 cells are defective in proteasome nuclear localization (Tabb et al., 2000). Conversely, srp1-31 mutants have a general defect in nuclear protein localization, but nuclear concentration of proteasomes appears normal (Chen et al., 2011; Tabb et al., 2000). One hypothesis that could explain these differences is that Sts1 has an NLS distinct from the more thoroughly analysed cNLS and therefore interacts in a distinct fashion with karyopherin α. Sts1 has two N-terminal basic patches, called NLS1 and NLS2 (Tabb et al., 2000). A plasmid with STS1 lacking NLS2 complemented a sts1Δ strain, but if the 5-residue NLS1 sequence was deleted instead, viability was not restored (Tabb et al., 2000). The sts1ΔNLS1 protein also does not bind Srp1 in vitro (Chen et al., 2011; Tabb et al., 2000). No function has been ascribed to NLS2.
Most standard NLS prediction programs do not yield high-confidence NLS scores for Sts1 (Kosugi et al., 2009). We hypothesized that NLS1 and NLS2 comprise a bipartite NLS (Fig. 1A,B) in which the linker between the two basic regions is longer than the canonical 9- to 12-residue linker length used in most search algorithms. Lange et al. (2010) characterized a bipartite NLS in the yeast Ty1 integrase that has a 29-residue spacer separating a pair of KKR sequences. Sts1 has a 24-residue linker between a pair of KRR elements and also shares with the integrase a highly acidic segment just after the first basic element, which contributes to NLS function in the integrase (Fig. 1B). As a first test for a bipartite NLS in Sts1, we introduced single point mutations into each basic patch, R38D and R65D; a double mutant, sts1-R38D,R65D, was also constructed (hereafter called sts1-DD). An sts1Δ mutant carrying a plasmid expressing sts1-DD was inviable, but sts1Δ strains expressing either single mutant grew normally at 30°C (Fig. 1C). A strain expressing a Green Fluorescent Protein (GFP)-tagged sts1-R38D single mutant grew slightly slower at high temperature than either wild-type (WT) Sts1-GFP- or sts1-R65D-GFP-expressing cells (Fig. S1A). The former mutant also had a mild nuclear localization defect based on fluorescence microscopy (Fig. S1B). The first 116 residues of Sts1 bearing the NLS were sufficient for nuclear localization, although passive diffusion into the nucleus cannot be excluded for this fusion (Fig. S2).
To evaluate whether the apparent bipartite NLS was sufficient for nuclear localization, we fused the first 76 residues of Sts1, which are predicted to be largely disordered, to a duplicated GFP reporter (2GFP) to prevent passive diffusion into the nucleus (Lange et al., 2010). The WT Sts1 sequence caused strong nuclear localization of the reporter, comparable to the positive control bearing the Ty1 integrase NLS (Fig. 1D). The R38D and R65D single mutants both caused a partial reduction of relative reporter levels in the nucleus, whereas the double mutant no longer showed any detectable nuclear concentration of the reporter. We conclude that Sts1 contains a non-canonical bipartite NLS in its N-terminal domain.
We further characterized the localization of full-length sts1-DD and other sts1 mutant proteins fused to GFP. Yeast sts1Δ cells expressing the WT STS1-GFP fusion from either the MET25 or GPD promoter on low-copy plasmids were fully viable (not shown). The latter construct allows a mild over-expression of Sts1, which we found was necessary for reliable detection of the Sts1-GFP signal. We confirmed the nuclear concentration previously observed for Sts1 under strong over-expression conditions (Tabb et al., 2000). Sts1-GFP was enriched in the nucleus compared with the cytoplasm, and this was also true for the R38D and R65D single mutants expressed in WT cells (Fig. 2A). In contrast, sts1-DD-GFP localization was shifted to the cytoplasm relative to Sts1-GFP. While the reduction in nuclear localization was significant, the magnitude was smaller than expected as sts1-DD was predicted to have little or no NLS activity (Fig. 1). However, the localization experiments had to be performed in a strain expressing WT Sts1 because sts1-DD by itself was inviable (Fig. 1C). The predicted heterodimerization of the mutant GFP-tagged proteins with endogenous Sts1 could drive a fraction of the GFP fusions into the nucleus.
Fig. 2.

Nuclear localization is essential for Sts1 function. (A) Wild-type MHY500 yeast was transformed with p415GPD-based plasmids expressing the indicated sts1-GFP fusion alleles. ‘SV40’ denotes the cNLS from SV40T antigen fused to the N-terminus of the test protein. The transformants were grown at room temperature prior to imaging by fluorescence microscopy in mid-log phase. Three replicates of at least 100 cells were counted (right panel). A t-test was used to determine statistical significance of differences in localization (****P<0.0001). Scale bar: 5 μm. (B) Viability assay. MHY9580 cells (sts1Δ, pRS316-STS1) were transformed with the p415GPD-based alleles noted and grown at room temperature on 5-FOA to select against the original cover plasmid. (C) Sts1–Srp1 complex formation depends on the Sts1 bipartite NLS. The indicated recombinant proteins were co-expressed in E. coli, and binding was determined based on co-purification on a GST-binding glutathione resin or polyHis-binding TALON resin followed by SDS-PAGE. (D) The indicated recombinant proteins were co-expressed in E. coli, and binding was determined based on co-purification on either GSH beads or TALON beads. *Proteolytic fragments derived from Sts1–6His.
Importantly, both the reduction in nuclear localization and the loss of viability observed in sts1-DD cells were ameliorated by appending the well-studied cNLS from SV40 virus T-antigen to the N-terminus of sts1-DD (Fig. 2A,B). The SV40 NLS dramatically increased the nuclear localization of both Sts1 and sts1-DD (Fig. 2A), although the slower growth of SV40-sts1-DD compared with SV40-STS1 cells indicated that the SV40 NLS was an imperfect substitute for the natural NLS of Sts1 (Fig. 2B). This rescue analysis shows that an NLS is necessary for Sts1 function in vivo but that the natural NLS has features that are optimal for Sts1 activity.
Sts1 binds stoichiometrically to karyopherin α (Srp1)
As Sts1 has a functional NLS, it should interact with Srp1, and Sts1–Srp1 association has been reported (Tabb et al., 2000). To investigate the interaction between Sts1 and Srp1 in more detail, we co-expressed the two proteins in Escherichia coli as Sts1–6His and GST–Srp1 fusions. Binding of either protein on the appropriate affinity resin brought down the other protein in apparent stoichiometric amounts. For example, TALON resin binding of Sts1–6His also retrieved GST–Srp1 from bacterial extracts co-expressing the two proteins (Fig. 2C). Importantly, GST–Srp1 did not bind detectably to sts1-DD-6His, indicating the bipartite NLS in Sts1 is indeed important for interaction with the Srp1 karyopherin (Fig. 2C, right two lanes). Binding of sts1-DD (+GST–Srp1) to the glutathione (GSH) column was still observed, but the ratio of Sts1 protein to GST–Srp1 eluted from the column was strongly reduced (Fig. 2C, left two lanes); when Sts1 is not bound to Srp1 it is prone to aggregate (see below), which might account for this latter association. We also determined that Sts11-116-6His, which included the unstructured N-terminal domain with the NLS and the dimerization domain (Fig. 1A), bound to GST–Srp1, again in an apparently stoichiometric manner (Fig. 2D). Thus, a fragment of Sts1 lacking the six-helix bundle and C-terminal domain is sufficient for interaction with Srp1.
Sts1 is rapidly degraded but stabilized by mutations that impair its function
While examining the steady-state levels of the Sts1–GFP proteins expressed from various deletion constructs, we noticed that the levels varied considerably, with most of the sts1 mutant proteins being much more abundant than WT Sts1 (Fig. S2). Subsequent cycloheximide-chase experiments showed that NLS mutants of Sts1–GFP were longer lived than WT Sts1–GFP (Fig. 3A, and not shown), consistent with the much higher steady-state level of Sts1(Δ4-57)–GFP, which lacks the NLS1 element (Fig. S2B). Tagged Sts1 – as well as Cut8 – have previously been reported to be short-lived proteins (Chen et al., 2011; Tatebe and Yanagida, 2000).
Fig. 3.
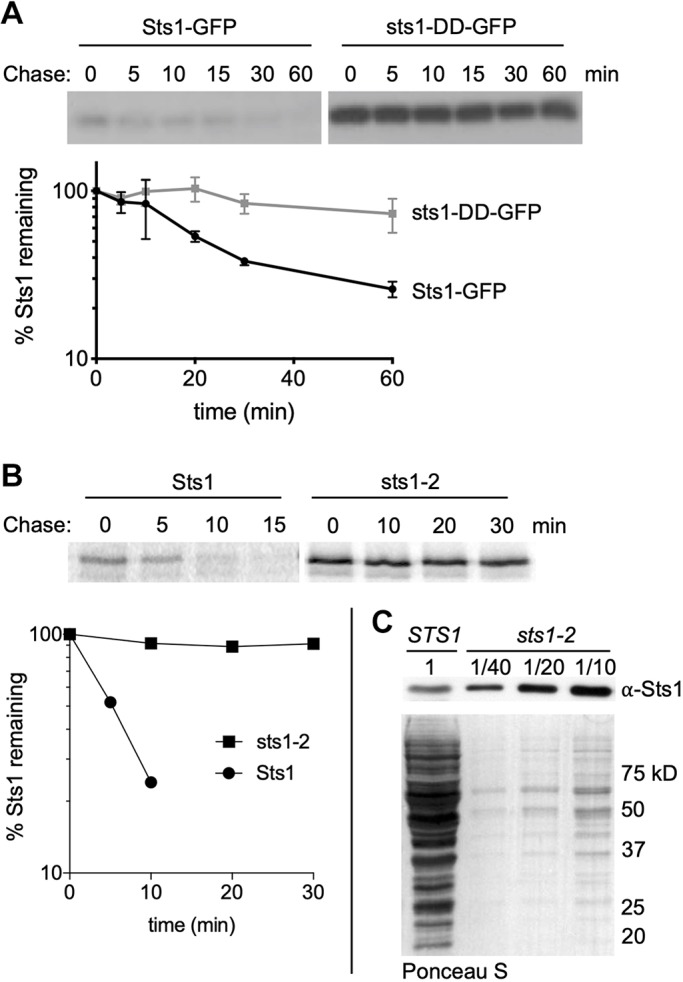
Degradation of wild-type and mutant forms of Sts1 in cells. (A) Cycloheximide-chase analysis was performed to determine the degradation rates of Sts1-GFP and sts1-DD-GFP using anti-GFP immunoblotting. MHY500 cells carried either p415MET25-STS1-GFP or p415MET25-sts1-DD-GFP and were grown at 30°C; cycloheximide was added at time 0 to block further protein synthesis. For quantitation, Sts1-GFP and sts1-DD-GFP levels were normalized to a PGK loading control. (B) Radioactive pulse-chase analysis of Sts1 degradation in MHY9692 (STS1) and MHY9693 (sts1-2) cells at 30°C. Proteins were immunoprecipitated using affinity-purified anti-Sts1 antibodies. Bottom panel shows the phosphorimager quantification of degradation rates. (C) Cell extracts from MHY9692 and MHY9693 were separated by SDS-PAGE and immunoblotted with anti-Sts1 antibodies. Extracts from MHY9693 were diluted to the indicated fraction of extract loaded for MHY9692. Ponceau S-stained membrane shows relative amounts of total protein extract.
These experiments were conducted using plasmid constructs expressing tagged versions of Sts1 under the control of a MET25 promoter (as expression from this promoter closely approximated the levels observed for the endogenous protein). We wanted to know whether untagged Sts1 expressed from its normal chromosomal context is also short-lived. To determine this, we generated an antibody to Sts1 and used it in radioactive pulse-chase experiments. Indeed, endogenous Sts1 was very rapidly degraded, with a half-life of ∼5 min (Fig. 3B).
We tested whether mutations in Sts1 besides those in the NLS region might alter protein half-life. The most utilized temperature-sensitive allele of STS1, called sts1-2 (Tabb et al., 2000), expresses the sts1-C194Y protein. Strikingly, sts1-C194Y was completely stable over a 30 min chase (Fig. 3B). This contrasts with an earlier report using Flag-tagged Sts1 and sts1-2 proteins, which had concluded that the mutant protein was even more short-lived than WT (Chen et al., 2011). Immunoblotting for steady-state levels of Sts1 in the WT and sts1-2 strains revealed that sts1-C194Y was present at more than 40 times the concentration of the WT protein, even at 30°C, a permissive temperature for growth of the sts1-2 strain (Fig. 3C). The function of the six-helix bundle where Cys194 resides remains to be fully defined, but appears to include proteasome binding. A fusion of sts1-C194Y to GFP concentrates in the nucleus at 30°C (Fig. S3A), a temperature at which proteasome binding in vitro is compromised (Fig. S3B). As Sts11-116 is sufficient for binding Srp1, it is unlikely that sts1-C194Y mutation affects Srp1 interaction. Thus, our data show that impairing two functionally distinct regions of Sts1 leads to stabilization of this normally short-lived protein.
Sts1 degradation depends on the 26S proteasome but not ubiquitin
The rapid in vivo degradation of Sts1 and its unexpected stabilization by loss-of-function mutations led us to examine the mechanism of its turnover. Surprisingly, when a temperature-sensitive mutant of the E1 ubiquitin-activating enzyme, uba1-204, was tested at non-permissive temperature, little or no reduction in endogenous Sts1 degradation rate was observed (Fig. 4A,B). Strong inhibition of cellular ubiquitylation in the uba1-204 mutant was reflected in the complete block to the degradation of a known ubiquitin-dependent substrate, Deg1-Flag-Ura3 (Zattas et al., 2013), in the same cells (Fig. 4B). Ubiquitin-independent degradation of specific proteins can depend on the full 26S proteasome (RP–CP) (Murakami et al., 1992) or the CP alone (Kumar Deshmukh et al., 2019). To distinguish between these mechanisms, we tested Sts1 degradation in an RP mutant, cim3-1 (Ghislain et al., 1993). Severe inhibition of Sts1 degradation was observed in the mutant at restrictive temperature, implying that the full 26S proteasome was required (Fig. 4C).
Fig. 4.
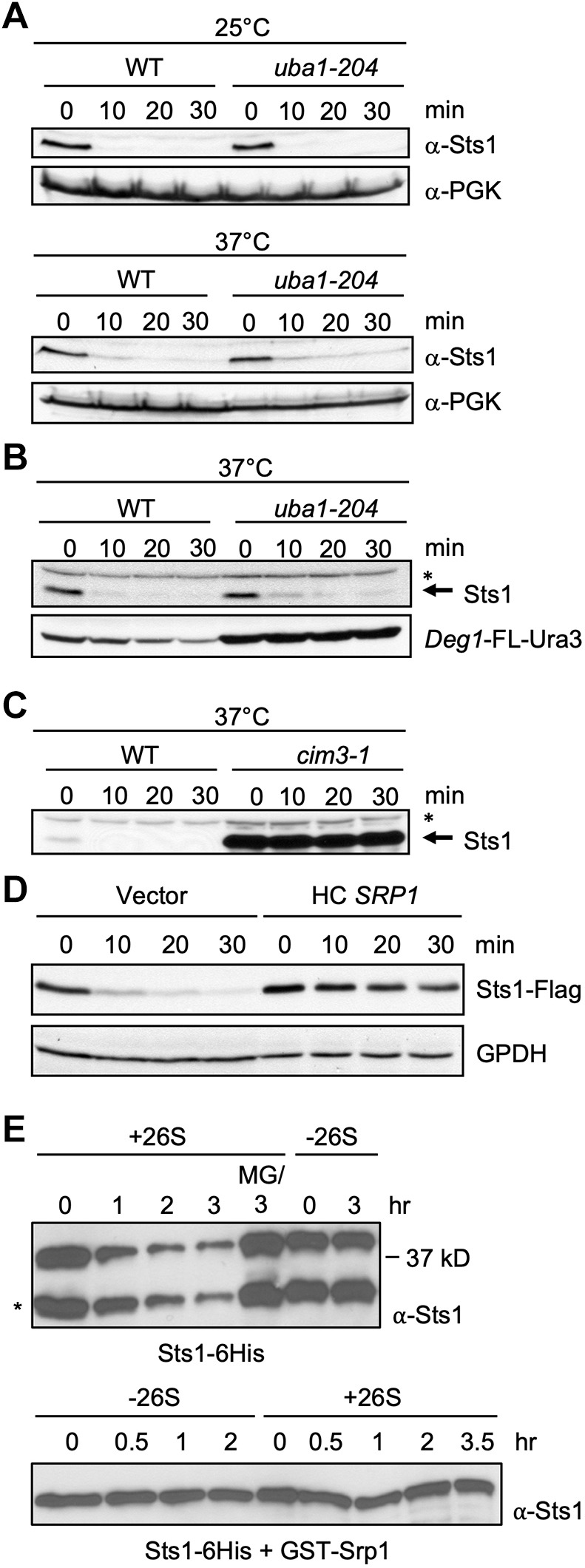
Sts1 is degraded by the proteasome in a ubiquitin-independent manner. (A) Cycloheximide-chase analysis to determine the degradation rates of endogenous Sts1 in the indicated strains at both the permissive (25°C) and restrictive (37°C) temperatures for the uba1-204 strain (RJD3269). Immunoblot for PGK serves as a loading control. (B) Cycloheximide-chase analysis of endogenous Sts1 and plasmid-expressed Deg1-FLAG-Ura3 in the indicated strains at the restrictive temperature (37°C) for the uba1-204 strain. A cross-reactive band in the immunoblot for Sts1, indicated by an asterisk, shows unchanging levels of a protein that is not degraded over the 30 min chase. (C) Cycloheximide-chase analysis of endogenous Sts1 in the indicated strains at the restrictive temperature (37°C) for the cim3-1 strain (MHY4464, which carries a mutation in the Rpt6 subunit of the proteasome). (D) Cycloheximide-chase analysis of Flag-tagged Sts1 in WT cells carrying either an empty high-copy (HC) vector or the same plasmid with SRP1. GPDH served as a loading control. Threefold less protein was loaded for the latter extracts to achieve roughly equal Sts1-Flag levels for the zero-minute samples. The endogenous STS1 locus was 3′-tagged with 6xGly-3xFLAG. (E) In vitro degradation of purified recombinant Sts1–6His by 26S proteasomes purified from yeast. For the ‘MG/3 hr’ sample, proteasomes were treated with 50 µM MG132 inhibitor for 10 min prior to addition of Sts1–6His. Degradation was measured at room temperature. *Sts1 fragment that is often generated during purification from E. coli when Sts1 is not co-expressed with Srp1 (see Fig. 2C,D) and is also a substrate for the proteasome in vitro. In the lower samples, recombinant Sts1–6His was co-purified from bacteria with GST-Srp1 and used as substrate.
We also asked whether increasing cellular Srp1/karyopherin α levels would change Sts1 degradation kinetics. A significant inhibition of degradation was observed when Srp1 was expressed from a high-copy plasmid (Fig. 4D). This would appear to contradict the finding that the sts1-DD protein, which is impaired for Srp1 binding, is much longer lived than WT Sts1 (Fig. 3). These findings could be reconciled if Sts1 were selectively degraded in vivo only following separation of Sts1 and Srp1, which is expected to occur in the nucleus following import of the transport receptor–substrate complex (see Discussion). We cannot, however, rule out indirect effects of Srp1 over-expression on the Sts1 proteolytic pathway.
If Sts1 degradation were truly ubiquitin independent but 26S proteasome dependent, it might be possible to reconstitute degradation in vitro with purified components. Full proteasomes were affinity-purified from yeast and recombinant Sts1–6His substrate was purified from bacteria. The substrate needed to be used immediately in these assays because it tends to aggregate with time. Strikingly, in the presence of ATP and 26S proteasomes but without ubiquitin, Sts1 was degraded (Fig. 4E, upper panel). This degradation required the proteasome and was blocked by pre-incubation with the MG132 proteasome inhibitor. When the same experiment was done with Srp1 bound to Sts1, degradation was severely inhibited (Fig. 4E, lower panel). This finding is consistent with the in vivo inhibition of Sts1 degradation by high levels of Srp1/karyopherin α (Fig. 4D).
Sts1 effects on WT and mutant proteasome localization
As Sts1 function is closely intertwined with that of the proteasome, particularly the RP lid, we examined its genetic interactions with mutations in different lid subunits. We used three lid mutants: sem1Δ, rpn11-1 and rpn12-234Δ. These were chosen because they are non-lethal mutants and accumulate distinct assembly intermediates. To follow proteasome localization, we used yeast strains expressing fluorescently tagged proteasome subunits from their endogenous loci. We used Rpn5-GFP (lid), Rpn2-GFP (base), Rpn2-mCherry (base) and Pre1-mCherry (CP) proteins, which are known to be fully functional or nearly so (Fukunaga et al., 2010).
By fluorescence microscopy, sem1Δ and especially rpn12-234Δ cells exhibited reduced nuclear localization of lid subcomplexes; concomitantly, slightly decreased nuclear concentration of base along with mildly increased nuclear localization of the CP was observed (Fig. 5). Proteasome nuclear localization in rpn11-1 was not reduced (Fig. 5A; Fig. S4). The LP2 lid precursor (containing all lid subunits except Rpn12) accumulates to high levels in rpn12-234Δ cells, and this intermediate fails to bind the RP base (Tomko et al., 2015); the data in Fig. 5A indicate that LP2 also fails to enter the nucleus efficiently. The Module 1 intermediate (Rpn5,6,8,9,11) that accumulates in sem1Δ (tracked by Rpn5–GFP) also appears to be retarded in its nuclear import but not to the same extent as LP2.
Fig. 5.

Select lid mutants alter proteasome subparticle localization. Lid mutants were grown in minimal medium to log phase and imaged by fluorescence microscopy; three replicates of at least 100 cells were counted. Two-way ANOVA was used to determine statistical significance of differences in localization (****P<0.0001; ns, not significant). Scale bar: 5 μm. Yeast strains used: MHY6956, MHY6954, MHY6377, MHY9583, MHY6938, MHY9581 and MHY9172. Pre1-mCherry (quantified here) and Rpn2-mCherry images are shown in Fig. S4.
Nuclear localization of WT proteasomes increases upon over-expression of Sts1 (Chen et al., 2011), but the impact of Sts1 on the nuclear localization of incomplete proteasome subcomplexes has not yet been explored. We tested whether over-expression of Sts1 altered localization of such subcomplexes, focusing primarily on lid intermediates (Fig. 6A). In the corresponding base and CP analyses, the cytoplasmic signal was often so low that reliable quantification of the nucleus-to-cytoplasmic (N/C) ratios was precluded (representative images in Fig. S5). In WT cells where most of the tagged subunits are in 26S proteasome complexes, Sts1 over-expression drives a higher percentage of proteasomes into the nucleus (Fig. 6A; only the quantitations are shown), as observed previously. In both rpn12-234Δ and rpn11-1, Sts1 over-expression also increased the nuclear localization of the Rpn5 lid subunit, suggesting that intermediates such as LP2 are probably more concentrated there as well. By contrast, there was no change in localization of Rpn5–GFP-containing species in sem1Δ. Interestingly, Sts1 over-expression had parallel effects on growth in these same mutants (Fig. 6B). Both rpn12-234Δ and rpn11-1 growth defects were partially suppressed by extra Sts1, whereas sem1Δ cells showed the opposite, with mutant cells becoming strongly inhibited in growth. These observations support the hypothesis that Sts1 stimulates nuclear import of 26S proteasomes as well as some, but not all, lid assembly intermediates.
Fig. 6.

High levels of Sts1 rescue certain lid mutant mislocalization and growth defects. (A) Yeast was grown in SD–Trp and imaged by fluorescence microscopy; three replicates of at least 100 cells were counted. Two-ANOVA was used to determine statistical significance of differences in localization (****P<0.0001; ns, not significant). Scale bar: 5 μm. The indicated mutant yeast expressed Rpn5-GFP and was transformed with either p414GPD (empty vector) or p414GPD-STS1. Strains used: MHY6964, MHY9583 and MHY9174. (B) Serial dilution growth assays. Matched wild-type (WT) and mutant transformants were grown as follows, from left to right: SD –Trp –Arg+1 µM canavanine, 34°C (5 days); SD–Trp, 30°C (5 days); and SD–Trp, 34°C (2 days). Strains used: MHY500, MHY5748, MHY9509 and MHY9134. (C) Recombinant GST-Srp1–Sts1-6His complex (1 μM) was immobilized on glutathione (GSH) resin and incubated with 1 μM purified CP, RP, 26S proteasome or 26S proteasomes reconstituted from RP and CP (‘RP+CP’) to detect interactions. All input complexes were isolated from yeast using anti-Flag affinity purifications. Proteins from the GST pulldowns (PD) were examined by anti-Flag immunoblotting.
Sts1–Srp1 binds preferentially to full 26S proteasomes
This led us to investigate to which proteasomal species Sts1 binds in vitro. It is known that a ternary complex of Srp1–Sts1–26S can form (Ha et al., 2014), but the relative binding of Sts1 to proteasome subcomplexes has not been examined. We chose to compare RP, CP and 26S proteasomes, all of which were purified from yeast. Additionally, we tested a reconstituted 26S proteasome species produced by incubating purified RP and CP in the presence of ATP. Recombinant Sts1–6His/GST–Srp1 bound to 26S (both directly purified and reconstituted) but not detectably to RP or CP alone under the same reaction conditions (Fig. 6C). The level of 26S proteasome binding was relatively modest, at best about 5% of input (Fig. 6C; Fig. S6). This could be due to only a small subset of proteasomes being in a conformation competent for binding, or the failure of in vitro conditions to mimic those in vivo. When assessing the interaction with GST–Srp1 alone, there appeared to be very weak binding to RP, CP and reconstituted RP–CP, but not to directly purified 26S; this could be due to exposure of the known cNLS sequences in these subparticles (Fig. S6B). Binding of free RP and CP was not seen for Sts1–6His/GST–Srp1.
Recombinant bacterially expressed Sts1 alone showed no binding to any of the proteasomal particles over background levels, but this is difficult to interpret because of the tendency of Sts1 to aggregate by itself (not shown). In an earlier study, proteasome binding by Sts1 alone was reported (Romero-Perez et al., 2007); however, Sts1 had been purified from yeast, and it is possible that Srp1 co-purified with it. Taken together, our results suggest that the Sts1–Srp1 complex exhibits a binding preference for full 26S proteasomes, and that Srp1 by itself has much lower binding to the proteasome.
Sts1-DD interferes with nuclear import of proteasomes
To address whether the Sts1 bipartite NLS was important for the concentration of proteasomes in the nucleus, we over-expressed the NLS mutant sts1-DD in yeast and tracked the localization of fluorescently tagged proteasome subunits. The strains had the intact endogenous STS1 locus to maintain viability. Over-expressed sts1-DD induced significant redistribution of proteasomes to the cytoplasm in WT cells (Fig. 7A,B). This is opposite to the effect of over-expressing WT Sts1, which enhanced proteasome nuclear localization (Fig. 6). In some cells, localization at the nuclear periphery was also observed. In these cells, the fluorescently tagged lid subunit showed the same localization pattern as the tagged subunits from the RP base or CP, suggesting that intact 26S holoenzymes were being visualized.
Fig. 7.

The sts1-DD mutant induces 26S proteasome mislocalization in vivo. Microscopy was performed as in Fig. 5A. Yeast was transformed with either empty vector (p414GPD) or p414GPD-sts1-DD. Lid and base were examined in panel A, while lid and CP were imaged in panel B. Two-ANOVA was used to determine statistical significance of differences (****P<0.0001; ns, not significant). Scale bars: 5 μm. Strains used were MHY6966, MHY6964, MHY6956 and MHY6954.
Remarkably, upon over-expression of sts1-DD in rpn12-234Δ cells, the base was no longer diverted to the cytoplasm and the CP showed a much more limited redistribution. A plausible interpretation of these data is that sts1-DD functions in a dominant-negative fashion, interfering with the ability of endogenous Sts1 to promote nuclear import of full 26S proteasomes; in rpn12-234Δ cells, which accumulate high levels of the LP2 lid precursor that binds poorly to the base (Tomko and Hochstrasser, 2011), the free RP base and CP can now enter the nucleus by mechanisms independent of Sts1. This interpretation implies that a high fraction of 26S proteasomes normally assemble in the cytoplasm prior to import, consistent with previous data (Pack et al., 2014).
DISCUSSION
The results presented here address several outstanding issues regarding nuclear localization of proteasomes and the function of Sts1 as an adapter protein that links the lid and 26S proteasome to the karyopherin α/β nuclear import pathway. We provide evidence for a bipartite NLS in the N-terminal domain of Sts1, demonstrating its importance for Sts1 and proteasome nuclear import and yeast growth. A mutant with point mutations in both basic elements, sts1-DD, is inviable. The NLS is required for Srp1/karyopherin α binding (Fig. 2C) and is probably sufficient for this (Fig. 2D).
Over-expressed sts1-DD protein fails to localize to the nucleus and acts as a dominant-negative inhibitor of nuclear import of the fully assembled 26S proteasome in otherwise WT cells. If association of the RP lid and base is strongly impaired, however, using the rpn12-234Δ mutation, sts1-DD now has far less impact on base and CP trafficking to the nucleus (Fig. 7). These data argue for the importance of Sts1 not only for import of the free lid, which may normally occur only to a limited degree in WT cells, but also for importing fully assembled 26S proteasomes, despite the presence of several intrinsic NLSs on their RP base and CP subcomplexes. The vast majority of proteasome subunits are in full 26S proteasomes in actively dividing yeast cells (Pack et al., 2014).
The physiological reasons for the striking concentration of proteasomes in the nuclei of yeast and many metazoan cells are not understood. It could reflect the high number of nuclear substrates that must be degraded by proteasomes or the documented binding of proteasomes to many chromatin sites (McCann and Tansey, 2014). Similarly, the mechanisms by which proteasomes translocate into the nucleus have been uncertain. A handful of proteasomal subunits have intrinsic NLSs, in particular, several α-subunits in the CP and the Rpt2 and Rpn2 subunits of the RP base (Lehmann et al., 2002; Wendler and Enenkel, 2019). The lid apparently has none. Not all of these NLSs appear to be necessary for nuclear import. For example, a large deletion encompassing the Rpn2 NLS impairs RP base import, while deletion of the Rpt2 NLS does not (Wendler et al., 2004). Moreover, it is not known whether these NLS elements are accessible in the intact 26S proteasome in vivo.
Sts1 and Cut8 orthologs are broadly conserved in diverse eukaryotes, indicating the presence of a related protein in the last eukaryotic common ancestor (LECA) (Takeda et al., 2011). Interestingly, more recent genome sequencing data have revealed an even broader distribution, including certain vertebrates (our observations). In particular, orthologues can be recognized in many bony fishes as well as lancelets. The gene appears to have been lost in the lineage leading to tetrapods. Whether a functional equivalent exists in species that lack Sts1 and Cut8 but still have high nuclear concentrations of proteasomes remains to be determined.
The full 26S proteasome is capable of being imported into the nucleus (Pack et al., 2014), but when mutations are used to force accumulation of separate lid or base RP subcomplexes, these subcomplexes still concentrate in the nucleus (Isono et al., 2007). Our data with Sts1 and sts1-DD over-expression, and the loss of Srp1 binding by sts1-DD, argue that the free lid/LP2 can be imported by a Sts1 and Srp1-dependent mechanism. We note that Isono et al. (2007) had found that the free lid still concentrated in the nucleus in a srp1-49 import mutant shifted to non-permissive temperature. However, a large Rpn2 base subunit deletion that strongly limited base assembly was used in their analysis, and the double mutant was severely growth impaired, potentially leading to altered nuclear permeability under high physiological stress.
Cells carrying the rpn12-234Δ mutation, which limits completion of lid assembly and lid–base joining (Tomko et al., 2015), accumulate the penultimate lid precursor, LP2, in the cytoplasm (Fig. 5A). Sts1 and Srp1 may bind LP2 more poorly than the full proteasome, but high levels of Sts1 can potentially overcome this and drive LP2 into the nucleus; Sts1 over-expression also partially suppresses the rpn12-234Δ growth defect (Fig. 6B). Our in vitro binding data for Sts1 with LP2, RP and full 26S proteasome are consistent with this idea (Fig. 6C; and not shown). Sts1 has been shown previously to interact with the Rpn11 lid subunit by yeast two-hybrid and pulldown assays, but this interaction appears to be very weak (Chen et al., 2011; Tabb et al., 2000). The preference of the Sts1 import factor to bind the fully assembled forms of the proteasome might limit nuclear import of defective or incomplete proteasomal subcomplexes.
Rapid degradation of native Sts1 has been shown here for the first time. Remarkably, many tested mutant Sts1 proteins, including sts1-2, are far more long lived in vivo (e.g. Fig. 3); this contrasts with most defective mutant proteins, which tend to be less metabolically stable than their WT counterparts due to misfolding. Using an epitope-tagged form of Sts1, others had previously found Sts1 to be short lived, but argued that the sts1-2 protein was even less stable and that this instability was responsible for its phenotypic defects (Chen et al., 2011). Our data indicate that the sts1-2 and sts1-DD proteins are in fact both strongly stabilized but apparently for different reasons. The sts1-DD protein cannot bind Srp1 (Fig. 2C), whereas sts1-2 protein binds the karyopherin as well as WT protein (not shown) and is imported into the nucleus but is impaired for proteasome binding (Fig. S3).
Another unexpected finding here is the independence of Sts1 degradation by the 26S proteasome from ubiquitin conjugation (Fig. 4). It is likely that Sts1 degradation is mediated by direct proteasome binding and exposure of its disordered N-terminal region to initiate degradation, as has been described previously for other proteins (Yu et al., 2016). Few examples exist of regulatory proteins being degraded directly by 26S. The most thoroughly documented is ornithine decarboxylase, the rate-limiting enzyme of polyamine biosynthesis, the degradation of which is stimulated by its binding to a protein called antizyme (Murakami et al., 1992). Degradation of the yeast Rpn4 transcription factor also has a ubiquitin-independent component (Ha et al., 2014). Rpn4 regulates the transcription of almost all proteasome genes. Ubiquitin-independent degradation of Rpn4 has been reported to occur on ribosomes by a mechanism that requires Srp1 binding to the nascent polypeptide and Sts1-mediated recruitment of the proteasome (Ha et al., 2014). It is thus possible that the regulation of Sts1 and Rpn4 degradation is coupled in some way to control proteasome levels and localization.
To explain these surprising results, we propose a model for Sts1 degradation in which Sts1 is specifically degraded following its entry into the nucleus (Fig. 8). As such, it would act as a single-turnover proteasome import factor. The Sts1-karypherin α/β complex should be disassociated by RanGTP binding to the karyopherin β-subunit; RanGTP is highly enriched in the nucleus (Goldfarb et al., 2004). In this model, Srp1-free Sts1 would remain bound to the 26S proteasome, causing it to become a direct substrate for the protease, potentially using the now unbound, disordered N-terminal domain as a degron to initiate proteolysis. If Sts1 cannot bind Srp1, it will be expected to remain in the cytoplasm and will probably not associate efficiently with the proteasome (Fig. 6C; and not shown).
Fig. 8.
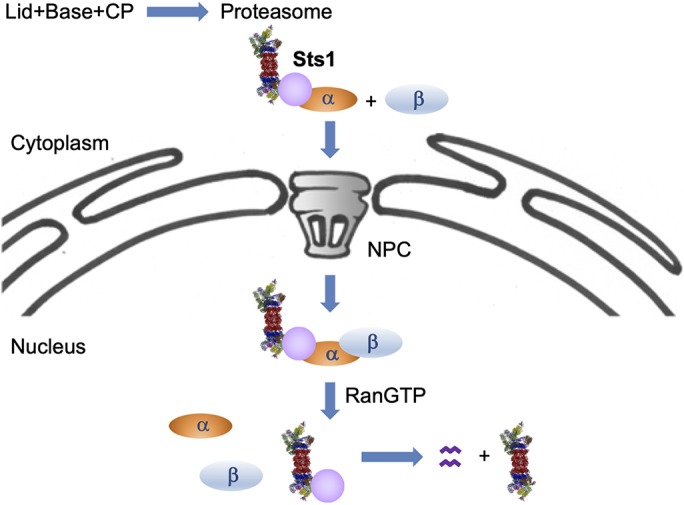
Model of Sts1 degradation and function in proteasome nuclear import. Cytoplasmic 26S proteasomes are transported into the nucleus via Sts1. The bipartite NLS in Sts1 binds Srp1 (karyopherin α), and the complex of proteasome–Sts1–Srp1 can then interact with karyopherin β (Kap95), which promotes import of the complex through the NPC. Sts1, in complex with Srp1, appears to bind more efficiently to fully assembled 26S proteasomes than to proteasomal subparticles, although Sts1 can also promote lid import when Sts1 is overproduced. In the nucleus, RanGTP binding breaks up the import complex. It is possible that this is insufficient for Sts1 release from Srp1, requiring the Cdc48Ubx4 segregase (Chien and Chen, 2013) or other factors to fully dissociate it. When Srp1 is removed from the disordered Sts1 N-terminal domain, Sts1 is degraded by the proteasome to which it is bound.
Sts1 mutants demonstrate a tight correlation between Sts1 function and its degradation, a highly unusual but not unprecedented circumstance. Many transcriptional activators, for example, are rapidly degraded, and their degradation appears to be intimately connected to their ability to promote transcription (Howard and Tansey, 2016). The model presented in Fig. 8 makes a number of predictions about Sts1 function, degradation and its means of promoting proteasomal import into the nucleus. We are actively testing several of these.
MATERIALS AND METHODS
Yeast strain construction and growth
Table S1 includes the complete list of the yeast strains used in this study. Yeast strains were created and maintained following standard techniques (Guthrie and Fink, 2002). All strains are based on the MHY500 WT background unless otherwise noted. Modified proteasome alleles (fusions with fluorescent proteins or mutations) were created at the normal chromosomal loci and were expressed from their endogenous promoters unless otherwise specified. As STS1 is essential, the endogenous gene was typically retained, and the various constructs for Sts1 mutant expression from plasmids were as described in the individual assays. Viability assays for sts1 mutants were done by plasmid shuffle in sts1Δ/pRS316-STS1 cells transformed with plasmids carrying the mutant alleles of interest.
To create a haploid strain lacking the chromosomal copy of STS1, the WT diploid strain MHY606 was transformed with an hphMX cassette amplified from pAG32, replacing one of the two STS1 loci with an sts1Δ::hphMX allele (Goldstein and McCusker, 1999). The heterozyote was transformed with URA3-marked plasmid pRS316-STS1 and then sporulated; tetrads were dissected, and a segregant from a full tetrad showing 2:2 segregation of hygromycin-B resistance was isolated. This strain fails to survive on 5-fluoroorotic acid (5-FOA) medium, which selects against the URA3 marker, demonstrating that STS1 is essential in vegetatively growing cells, as previously reported (Houman and Holm, 1994).
Yeast were grown on rich yeast–peptone–dextrose (YPD) or minimal (synthetic defined, SD) medium at room temperature or 30°C unless otherwise noted. Other than the plate-based growth and viability assays, all experiments were performed using exponentially growing yeast harvested at an optical density measured at 600 nm wavelength (OD600) between 0.8 and 1.2.
Plasmid constructions
A list of the plasmids used in this study is provided in Table S2. The WT STS1 open reading frame (ORF) was amplified from pGEX-2TK-STS1, a gift from Kiran Madura (Department of Pharmacology, Rutgers University, Piscataway, NJ), and cloned into various expression vectors. Other STS1-based plasmids were created by standard cloning methods from other plasmids in the Hochstrasser laboratory database, or by PCR amplification from yeast genomic DNA. Point mutants of STS1 were created using one or two iterations of site-directed mutagenesis (QuikChange; Thermo Fisher Scientific). The STS1–GFP plasmid expression construct was made by removing the Deg2 element from p415MET25-Deg2-GFP (Hickey et al., 2018) and subcloning in the STS1 ORF (creating plasmid 91-2-5). Other Sts1–GFP-expressing plasmids were created using site-directed mutagenesis and/or subcloning. STS1 and sts1-DD gene fusions with the SV40T antigen NLS were created by amplifying the desired STS1 allele with primers encoding an N-terminal SV40 sequence of MPKKKRKV. The sts1–2GFP expression constructs were made by PCR amplifying sequences encoding Sts1 N-terminal residues 1–76 from plasmids 91-2-6 (WT), 91-3-9 (R38D), 91-9-8 (R65D) or 91-8-5 (DD), and subcloning in-frame with the GFP–GFP reporter in pAC1056, a gift from Anita Corbett (Department of Biochemistry, Emory University, Atlanta, GA), using the XbaI and HindIII restriction sites.
Antibodies and immunoblotting
An antibody to Sts1 was generated in rabbits by contract with Cocalico Biologicals (Reamstown, PA). Antigen was derived from 6His-MBP-Sts1 protein expressed in E. coli Rosetta 2 cells. After purification of 6His-MBP-Sts1 using HisPur Cobalt resin, the 6His-MBP tag was removed by TEV protease cleavage (in phosphate-buffered saline, PBS) between the MBP and Sts1 moieties. Upon TEV addition, the Sts1 protein almost immediately dropped out of solution; the precipitate was collected by centrifugation at 21,000 g for 20 min at 4°C. The supernatant protein consisted almost entirely of 6His-MBP and TEV protease and was discarded. The pellet containing Sts1 was washed in PBS and re-centrifuged as above. The washed pellet was then resuspended in PBS plus 6 M urea and incubated with a small amount of HisPur cobalt resin to remove any residual 6His-MBP-Sts1. The unbound fraction from this incubation was used as immunogen at approximately 1 mg/ml. A fraction of the rabbit serum containing anti-Sts1 reactivity was used to affinity purify anti-Sts1 antibodies on a column generated by coupling GST-Sts1 to SulfoLink beads (Pierce). Matrix coupling and antibody purification were carried out according to the protocol provided with the SulfoLink beads.
Immunoblotting was performed using the following primary antibodies: anti-GFP (JL8 antibody catalogue no. 632380, Takara; 1:1000), anti-PGK (catalogue no. 459250, Invitrogen; 1:20,000), anti-FLAG (F3165, Sigma; 1:10,000) and anti-Sts1 (described above; 1: 1000). Either donkey anti-rabbit IgG linked to horseradish peroxidase or sheep anti-mouse IgG linked to horseradish peroxidase (catalogue nos NA934V and NXA931V, respectively, GE Healthcare; 1:5000) was used as the secondary antibody. Proteins were visualized on film (catalogue no. E3018, Thermo Fisher Scientific) or with a Syngene G-box (for quantification) using enhanced chemiluminescence (ECL).
Cycloheximide-chase immunoblot and radioactive pulse-chase analyses
Protein degradation rates were determined following previously described protocols but with slight modifications (Hickey and Hochstrasser, 2015). Cells were grown overnight at room temperature in 5 ml cultures in synthetic medium and diluted to 0.2 OD600 in 20 ml of fresh medium. Once cells reached mid-log phase (OD600=0.8–1.2), 2.5 OD600 equivalents per time point were collected by centrifugation and resuspended in 7.5 ml culture medium and allowed to recover for 5 min. Aliquots (1 ml) were harvested from each sample, followed immediately by addition of cycloheximide to 0.25 mg/ml. Subsequent 1 ml aliquots were harvested at various intervals. Each sample was added to 1 ml of ice-cold stop solution (30 mM sodium azide in water), followed by washing, cell lysis and protein extraction. Pulse-chases with [35S]Met were done exactly as described previously except using anti-Sts1 for immunoprecipitation (Hickey and Hochstrasser, 2015).
Degradation assays with purified proteins
Recombinant Sts1–6His or Sts1–6His/GST–Srp1 were incubated with or without purified 26S proteasomes [assay buffer conditions: 50 mM HEPES, pH 7.0, 150 mM NaCl, 10% glycerol, 6 mM MgCl2, 5 mM ATP, 0.1 mg/ml bovine serum albumin (BSA)]. Reactions were incubated at room temperature with 20 μl fractions removed at the indicated intervals. Fractions were spun down and supernatant was separated from any precipitated material; pellets were resuspended in 20 μl of assay buffer. Both pellet and supernatant fractions were placed on ice until the experiment was completed. A separate reaction of Sts1–6His was tested in the presence of 26S proteasomes that were pre-incubated for 10 min with 50 μM MG132, a proteasome inhibitor. Supernatant fractions were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted for Sts1.
Protein expression level analysis
Cells were grown overnight in 5 ml of the appropriate medium, diluted to 0.2 OD600 or below in 5 ml fresh medium, and grown to mid-log phase (OD600 between 0.8 and 1.2). Cells were lysed using the NaOH/SDS boiling method (Kushnirov, 2000). Cells equivalent to 2.0 OD600 units were harvested, pelleted and washed with sterile water. Yeast cells were resuspended in 0.4 ml of 0.1 M NaOH, incubated at room temperature for 5 min, pelleted and resuspended in 100 μl of 1X Laemmli loading buffer (5X stock is 10% SDS, 0.04% Bromophenol Blue, 600 mM dithiothreitol, 50% glycerol, 300 mM Tris HCl, pH 6.8). The samples were heated at 100°C for 5 min and resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) or frozen at −20°C until use. Yeast proteins from 0.1 OD600 units were resolved by 10% SDS-PAGE followed by immunoblotting with antibodies against Sts1, GFP and the loading control Pgk1 (phosphoglycerate kinase or PGK).
Yeast growth analysis with serial dilution assays
Cells were grown overnight in YPD, diluted to 0.1 OD600 equivalents per millilitre in sterile water, and serially diluted in 6-fold steps; diluted cells (4–5 μl/drop) were then spotted onto the appropriate plates. Growth was followed at various temperatures for up to 10 days, as noted in each figure legend. Visualization of certain defects and their suppression was enhanced by growth under proteasome stress conditions, including high temperature and addition of 1 µM canavanine, an arginine analog that causes protein misfolding, as noted in the figure legends.
Live cell microscopy and nuclear and cytoplasmic signal quantification
Cells were grown overnight in synthetic medium at either 30°C or room temperature, diluted to below 0.2 and grown to mid-log phase (OD600 between 0.8 and 1.2). Culture aliquots of 500 μl were centrifuged, and cells were resuspended in 50 μl of the appropriate synthetic medium. Slides were spotted with 8 μl of cell suspension and immediately imaged.
Imaging was performed on an Axioskop epifluorescence microscope (Carl Zeiss, Thornwood, NY) using a 100× objective lens (plan-Apochromat 100×/1.40 oil DIC) and an AxioCam MRm CCD camera (Carl Zeiss) with AxioVision software. All fluorescent images were captured using auto-exposure. After capture, the background was subtracted in ImageJ (Schneider et al., 2012), followed by quantification.
Quantification was performed using ImageJ (Schneider et al., 2012). The summed signal intensities in equal-sized regions in the nucleus (N) and cytoplasm (C) of the same yeast cell were measured and the N/C ratio was determined using Microsoft Excel. Only cells with an identifiable nucleus (excluding cells that were clearly sick or dying, or cells where the nucleus was not in the plane of focus) were counted. In all images where the vacuole was visualized, this region was avoided when taking measurements of cytoplasmic signal intensity.
Every experiment was repeated with three independent liquid growth cultures of each strain, or three independent plasmid transformants per strain. At least 100 yeast cells were quantified from each replicate. The difference in ratio of the nuclear to cytoplasmic signals between different strains or conditions was analysed for statistical significance in GraphPad Prism8 by two-way ANOVA, unless otherwise noted.
Protein purification
Recombinant glutathione-S-transferase (GST) and hexahistidine (6His) protein fusions, specifically Sts1–6His, sts1–DD-6His, sts11-116–6His, GST–Srp1 and Sts1–6His/GST–Srp1, were expressed and purified from Rosetta E. coli cells by their respective affinity tags by standard methods. In the case of the co-expressed Sts1–6His and GST–Srp1 polypeptides, complexes were purified using 6His-tag binding to a TALON resin (Takara). 26S proteasomes, CP and RP were purified from yeast, as described previously (Li et al., 2015).
Analytical binding assays
Recombinant GST or GST–Srp1/Sts1–6His species were immobilized on 20 μl (packed volume) of glutathione (GSH) beads (equilibrated in buffer SSB, 20 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 10% glycerol, 0.1 mg/ml BSA). Bait proteins and GSH beads were mixed and rotated for 1 h at 4°C to bind the bait species to the resin. Beads were spun down and the supernatant fractions discarded. Beads bound to bait protein were resuspended in 1 ml buffer SSB and centrifuged, and the supernatant was aspirated and discarded. Bait-bound beads were resuspended in 150 μl of buffer SSBA (buffer SSB supplemented with an additional 4 mM MgCl2 and 2 mM ATP), and 50 μl of 1 μM purified 26S proteasome or proteasomal subcomplex was added to the beads. Samples were rotated for 2 h at 4°C to bind the prey complexes to the immobilized bait. Samples were centrifuged, and supernatant fractions were removed. The beads were washed by four cycles of resuspending in 1 ml buffer SSBA, centrifuging and removing the supernatant fraction. Bound protein species were eluted from the GSH beads in 50 μl Laemmli sample buffer, heated to 100°C for 5 min, and resolved by 11% SDS-PAGE followed by immunoblotting for Rpn3 and Pre6.
Viability assay (plasmid shuffle) for sts1 mutants
Yeast strains MHY9579 or MHY9580 with chromosomal knockouts of STS1 and carrying the URA3-marked pRS316-STS1 plasmid to maintain viability were transformed with the desired sts1 plasmids, always including an empty vector control. After selection for the plasmid, transformants were streaked onto plates containing 5-FOA at 1 mg/ml. Inclusion of 5-FOA is incompatible with expression of the URA3 gene, allowing growth only of cells that had lost pRS316-STS1. Growth on 5-FOA plates indicates that the sts1 allele on the plasmid in question maintains the essential function of STS1. This plasmid shuffle technique was also used in generating certain proteasome mutant strains for our experiments (rpn11-1).
Supplementary Material
Acknowledgements
We would like to thank Jason Berk for helpful suggestions for experiments and figures. We also thank Robb Tomko and Judy Ronau for strains or plasmids made in M.H.’s laboratory, Kiran Madura for the pGEX-2TK-Sts1 plasmid and Anita Corbett for the 2GFP reporter plasmids.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: L.B., C.H., M.H.; Methodology: L.B., C.B., C.H.; Investigation: L.B., C.B., C.H.; Data curation: L.B.; Writing - original draft: L.B.; Writing - review & editing: L.B., C.B., C.H., M.H.; Supervision: M.H.; Project administration: M.H.; Funding acquisition: L.B., M.H.
Funding
This research was supported by National Institutes of Health (NIH) grants GM046904 and GM083050 (to M.H.). Support was also provided in part by a National Science Foundation predoctoral fellowship (to L.B.) and NIH predoctoral training program T32 GM007223 (to L.B. and C.B.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.236158.supplemental
References
- Amrani N., Dufour M. E., Bonneaud N. and Lacroute F. (1996). Mutations in STS1 suppress the defect in 3′ mRNA processing caused by the rna15-2 mutation in Saccharomyces cerevisiae. Mol. Gen. Genet. 252, 552-562. 10.1007/BF02172401 [DOI] [PubMed] [Google Scholar]
- Bayliss R., Littlewood T. and Stewart M. (2000). Structural basis for the interaction between FxFG nucleoporin repeats and importin-beta in nuclear trafficking. Cell 102, 99-108. 10.1016/S0092-8674(00)00014-3 [DOI] [PubMed] [Google Scholar]
- Budenholzer L., Cheng C. L., Li Y. and Hochstrasser M. (2017). Proteasome structure and assembly. J. Mol. Bio. 429, 3500-3524. 10.1016/j.jmb.2017.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. and Madura K. (2014). Degradation of specific nuclear proteins occurs in the cytoplasm in Saccharomyces cerevisiae. Genetics 197, 193-197. 10.1534/genetics.114.163824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Romero L., Chuang S.-M., Tournier V., Joshi K. K., Lee J. A., Kovvali G. and Madura K. (2011). Sts1 plays a key role in targeting proteasomes to the nucleus. J. Biol. Chem. 286, 3104-3118. 10.1074/jbc.M110.135863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien C.-Y. and Chen R.-H. (2013). Cdc48 chaperone and adaptor Ubx4 distribute the proteasome in the nucleus for anaphase proteolysis. J. Biol. Chem. 288, 37180-37191. 10.1074/jbc.M113.513598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambacher C. M., Worden E. J., Herzik M. A., Martin A. and Lander G. C. (2016). Atomic structure of the 26S proteasome lid reveals the mechanism of deubiquitinase inhibition. eLife Sciences 5, e13027 10.7554/eLife.13027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enenkel C. (2014). Nuclear transport of yeast proteasomes. Biomolecules 4, 940-955. 10.3390/biom4040940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enenkel C., Blobel G. and Rexach M. (1995). Identification of a yeast karyopherin heterodimer that targets import substrate to mammalian nuclear pore complexes. J. Biol. Chem. 270, 16499-16502. 10.1074/jbc.270.28.16499 [DOI] [PubMed] [Google Scholar]
- Fukunaga K., Kudo T., Toh-e A., Tanaka K. and Saeki Y. (2010). Dissection of the assembly pathway of the proteasome lid in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 396, 1048-1053. 10.1016/j.bbrc.2010.05.061 [DOI] [PubMed] [Google Scholar]
- Funakoshi M., Tomko R. J., Kobayashi H. and Hochstrasser M. (2009). Multiple assembly chaperones govern biogenesis of the proteasome regulatory particle base. Cell 137, 887-899. 10.1016/j.cell.2009.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghislain M., Udvardy A. and Mann C. (1993). S. cerevisiae 26S protease mutants arrest cell division in G2/metaphase. Nature 366, 358-362. 10.1038/366358a0 [DOI] [PubMed] [Google Scholar]
- Glickman M. H., Rubin D. M., Fried V. A. and Finley D. (1998). The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell. Biol. 18, 3149-3162. 10.1128/MCB.18.6.3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb D. S., Corbett A. H., Mason D. A., Harreman M. T. and Adam S. A. (2004). Importin alpha: a multipurpose nuclear-transport receptor. Trends Cell Biol. 14, 505-514. 10.1016/j.tcb.2004.07.016 [DOI] [PubMed] [Google Scholar]
- Goldstein A. L. and McCusker J. H. (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541-1553. 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K [DOI] [PubMed] [Google Scholar]
- Guthrie C. and Fink G. R. (eds) (2004). Guide to Yeast Genetics and Molecular and Cell Biology, pp. 1-933. Elsevier. [Google Scholar]
- Ha S.-W., Ju D. and Xie Y. (2014). Nuclear import factor Srp1 and its associated protein Sts1 couple ribosome-bound nascent polypeptides to proteasomes for cotranslational degradation. J. Biol. Chem. 289, 2701-2710. 10.1074/jbc.M113.524926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey C. M. and Hochstrasser M. (2015). STUbL-mediated degradation of the transcription factor MATα2 requires degradation elements that coincide with corepressor binding sites. Mol. Biol. Cell 26, 3401-3412. 10.1091/mbc.E15-06-0436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey C. M., Xie Y. and Hochstrasser M. (2018). DNA binding by the MATα2 transcription factor controls its access to alternative ubiquitin-modification pathways. Mol. Biol. Cell 29, 542-556. 10.1091/mbc.E17-10-0589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M. (1996). Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 30, 405-439. 10.1146/annurev.genet.30.1.405 [DOI] [PubMed] [Google Scholar]
- Houman F. and Holm C. (1994). DBF8, an essential gene required for efficient chromosome segregation in Saccharomyces cerevisiae. Mol. Cell. Biol. 14, 6350-6360. 10.1128/MCB.14.9.6350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard G. C. and Tansey W. P. (2016). Interaction of Gcn4 with target gene chromatin is modulated by proteasome function. Mol. Biol. Cell 27, 2735-2741. 10.1091/mbc.e16-03-0192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isono E., Nishihara K., Saeki Y., Yashiroda H., Kamata N., Ge L., Ueda T., Kikuchi Y., Tanaka K., Nakano A. et al. (2007). The assembly pathway of the 19S regulatory particle of the yeast 26S proteasome. Mol. Biol. Cell 18, 569-580. 10.1091/mbc.e06-07-0635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffman A. and O'Shea E. K. (1999). Regulation of nuclear localization: a key to a door. Annu. Rev. Cell Dev. Biol. 15, 291-339. 10.1146/annurev.cellbio.15.1.291 [DOI] [PubMed] [Google Scholar]
- Kalderon D., Roberts B. L., Richardson W. D. and Smith A. E. (1984). A short amino acid sequence able to specify nuclear location. Cell 39, 499-509. 10.1016/0092-8674(84)90457-4 [DOI] [PubMed] [Google Scholar]
- Kaneko T., Hamazaki J., Iemura S.-I., Sasaki K., Furuyama K., Natsume T., Tanaka K. and Murata S. (2009). Assembly pathway of the mammalian proteasome base subcomplex is mediated by multiple specific chaperones. Cell 137, 914-925. 10.1016/j.cell.2009.05.008 [DOI] [PubMed] [Google Scholar]
- Kosugi S., Hasebe M., Tomita M. and Yanagawa H. (2009). Systematic identification of cell cycle-dependent yeast nucleocytoplasmic shuttling proteins by prediction of composite motifs. Proc. Natl. Acad. Sci. USA 106, 10171-10176. 10.1073/pnas.0900604106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar Deshmukh F., Yaffe D., Olshina M. A., Ben-Nissan G. and Sharon M. (2019). The contribution of the 20S proteasome to proteostasis. Biomolecules 9, 190 10.3390/biom9050190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov V. V. (2000). Rapid and reliable protein extraction from yeast. Yeast 16, 857-860. 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- Lange A., McLane L. M., Mills R. E., Devine S. E. and Corbett A. H. (2010). Expanding the definition of the classical bipartite nuclear localization signal. Traffic 11, 311-323. 10.1111/j.1600-0854.2009.01028.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J., Matsuura Y., Liu S. M. and Stewart M. (2005). Structural basis for nuclear import complex dissociation by RanGTP. Nature 435, 693-696. 10.1038/nature03578 [DOI] [PubMed] [Google Scholar]
- Lehmann A., Janek K., Braun B., Kloetzel P.-M. and Enenkel C. (2002). 20 S proteasomes are imported as precursor complexes into the nucleus of yeast. J. Mol. Biol. 317, 401-413. 10.1006/jmbi.2002.5443 [DOI] [PubMed] [Google Scholar]
- Leung S. W., Harreman M. T., Hodel M. R., Hodel A. E. and Corbett A. H. (2003). Dissection of the karyopherin α nuclear localization signal (NLS)-binding groove: functional requirements for NLS binding. J. Biol. Chem. 278, 41947-41953. 10.1074/jbc.M307162200 [DOI] [PubMed] [Google Scholar]
- Li Y., Tomko R. J. and Hochstrasser M. (2015). Proteasomes: isolation and activity assays. Curr. Protoc. Cell Biol. 67, 3.43.1-3.43.20. 10.1002/0471143030.cb0343s67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S., Lacroute F. and Képès F. (1993). Multicopy STS1 restores both protein transport and ribosomal RNA stability in a new yeast sec23 mutant allele. Eur. J. Cell Biol. 62, 270-281. [PubMed] [Google Scholar]
- Lu Y., Lee B.-H., King R. W., Finley D. and Kirschner M. W. (2015). Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science 348, 1250834-1250834. 10.1126/science.1250834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann T. S. and Tansey W. P. (2014). Functions of the proteasome on chromatin. Biomolecules 4, 1026-1044. 10.3390/biom4041026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y., Matsufuji S., Kameji T., Hayashi S., Igarashi K., Tamura T., Tanaka K. and Ichihara A. (1992). Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 360, 597-599. 10.1038/360597a0 [DOI] [PubMed] [Google Scholar]
- Pack C.-G., Yukii H., Toh-e A., Kudo T., Tsuchiya H., Kaiho A., Sakata E., Murata S., Yokosawa H., Sako Y. et al. (2014). Quantitative live-cell imaging reveals spatio-temporal dynamics and cytoplasmic assembly of the 26S proteasome. Nat. Commun. 5, 3396 10.1038/ncomms4396 [DOI] [PubMed] [Google Scholar]
- Roelofs J., Park S., Haas W., Tian G., McAllister F. E., Huo Y., Lee B.-H., Zhang F., Shi Y., Gygi S. P. et al. (2009). Chaperone-mediated pathway of proteasome regulatory particle assembly. Nature 459, 861-865. 10.1038/nature08063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Perez L., Chen L., Lambertson D. and Madura K. (2007). Sts1 can overcome the loss of Rad23 and Rpn10 and represents a novel regulator of the ubiquitin/proteasome pathway. J. Biol. Chem. 282, 35574-35582. 10.1074/jbc.M704857200 [DOI] [PubMed] [Google Scholar]
- Saeki Y. (2017). Ubiquitin recognition by the proteasome. J. Biochem. 161, 113-124. 10.1093/jb/mvw091 [DOI] [PubMed] [Google Scholar]
- Saeki Y. and Tanaka K. (2012). Assembly and function of the proteasome. Methods Mol. Biol. 832, 315-337. 10.1007/978-1-61779-474-2_22 [DOI] [PubMed] [Google Scholar]
- Saeki Y., Toh-e A., Kudo T., Kawamura H. and Tanaka K. (2009). Multiple proteasome-interacting proteins assist the assembly of the yeast 19S regulatory particle. Cell 137, 900-913. 10.1016/j.cell.2009.05.005 [DOI] [PubMed] [Google Scholar]
- Schneider C. A., Rasband W. S. and Eliceiri K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671-675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suntharalingam M. and Wente S. R. (2003). Peering through the pore: nuclear pore complex structure, assembly, and function. Dev. Cell. 4, 775-789. 10.1016/S1534-5807(03)00162-X [DOI] [PubMed] [Google Scholar]
- Tabb M. M., Tongaonkar P., Vu L. and Nomura M. (2000). Evidence for separable functions of Srp1p, the yeast homolog of importin alpha (Karyopherin alpha): role for Srp1p and Sts1p in protein degradation. Mol. Cell. Biol. 20, 6062-6073. 10.1128/MCB.20.16.6062-6073.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K. and Yanagida M. (2005). Regulation of nuclear proteasome by Rhp6/Ubc2 through ubiquitination and destruction of the sensor and anchor Cut8. Cell 122, 393-405. 10.1016/j.cell.2005.05.023 [DOI] [PubMed] [Google Scholar]
- Takeda K., Tonthat N. K., Glover T., Xu W., Koonin E. V., Yanagida M. and Schumacher M. A. (2011). Implications for proteasome nuclear localization revealed by the structure of the nuclear proteasome tether protein Cut8. Proc. Natl. Acad. Sci. USA 108, 16950-16955. 10.1073/pnas.1103617108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K., Yoshimura T., Tamura T., Fujiwara T., Kumatori A. and Ichihara A. (1990). Possible mechanism of nuclear translocation of proteasomes. FEBS Lett. 271, 41-46. 10.1016/0014-5793(90)80367-R [DOI] [PubMed] [Google Scholar]
- Tatebe H. and Yanagida M. (2000). Cut8, essential for anaphase, controls localization of 26S proteasome, facilitating destruction of cyclin and Cut2. Curr. Biol. 10, 1329-1338. 10.1016/S0960-9822(00)00773-9 [DOI] [PubMed] [Google Scholar]
- Tomko R. J. and Hochstrasser M. (2011). Incorporation of the Rpn12 subunit couples completion of proteasome regulatory particle lid assembly to lid-base joining. Mol. Cell 44, 907-917. 10.1016/j.molcel.2011.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko R. J. and Hochstrasser M. (2014). The intrinsically disordered Sem1 protein functions as a molecular tether during proteasome lid biogenesis. Mol. Cell 53, 433-443. 10.1016/j.molcel.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko R. J. Jr and Hochstrasser M. (2013). Molecular architecture and assembly of the eukaryotic proteasome. Annu. Rev. Biochem. 82, 415-445. 10.1146/annurev-biochem-060410-150257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko R. J., Taylor D. W., Chen Z. A., Wang H.-W., Rappsilber J. and Hochstrasser M. (2015). A single α helix drives extensive remodeling of the proteasome lid and completion of regulatory particle assembly. Cell 163, 432-444. 10.1016/j.cell.2015.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya H., Arai N., Tanaka K. and Saeki Y. (2013). Cytoplasmic proteasomes are not indispensable for cell growth in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 436, 372-376. 10.1016/j.bbrc.2013.05.105 [DOI] [PubMed] [Google Scholar]
- Vittal V., Stewart M. D., Brzovic P. S. and Klevit R. E. (2015). Regulating the regulators: recent revelations in the control of E3 ubiquitin ligases. J. Biol. Chem. 290, 21244-21251. 10.1074/jbc.R115.675165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehmer M. and Sakata E. (2016). Recent advances in the structural biology of the 26S proteasome. Int. J. Biochem. Cell Biol. 79, 437-442. 10.1016/j.biocel.2016.08.008 [DOI] [PubMed] [Google Scholar]
- Wendler P. and Enenkel C. (2019). Nuclear transport of yeast proteasomes. Front. Mol. Biosci. 6, 34 10.3389/fmolb.2019.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendler P., Lehmann A., Janek K., Baumgart S. and Enenkel C. (2004). The bipartite nuclear localization sequence of Rpn2 is required for nuclear import of proteasomal base complexes via karyopherin alphabeta and proteasome functions. J. Biol. Chem. 279, 37751-37762. 10.1074/jbc.M403551200 [DOI] [PubMed] [Google Scholar]
- Yu H., Kago G., Yellman C. M. and Matouschek A. (2016). Ubiquitin-like domains can target to the proteasome but proteolysis requires a disordered region. EMBO J. 35, e201593147-e1536. 10.15252/embj.201593147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zattas D., Adle D. J., Rubenstein E. M. and Hochstrasser M. (2013). N-terminal acetylation of the yeast Derlin Der1 is essential for Hrd1 ubiquitin-ligase activity toward luminal ER substrates. Mol. Biol. Cell 24, 890-900. 10.1091/mbc.e12-11-0838 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.