

The spread of influenza virus strains resistant to the current generation of anti-viral drugs makes the identification of new druggable targets and lead compounds of prime importance. Kao et al. show that the influenza A nucleoprotein can be targeted by a small molecule that protects mice from lethal viral challenges.
Supplementary information
The online version of this article (doi:10.1038/nbt.1638) contains supplementary material, which is available to authorized users.
Subject terms: Target identification, Antiviral agents, Chemical genetics, Influenza virus
Abstract
Influenza A remains a significant public health challenge because of the emergence of antigenically shifted or highly virulent strains1,2,3,4,5. Antiviral resistance to available drugs such as adamantanes or neuraminidase inhibitors has appeared rapidly6,7,8,9, creating a need for new antiviral targets and new drugs for influenza virus infections. Using forward chemical genetics, we have identified influenza A nucleoprotein (NP) as a druggable target and found a small-molecule compound, nucleozin, that triggers the aggregation of NP and inhibits its nuclear accumulation. Nucleozin impeded influenza A virus replication in vitro with a nanomolar median effective concentration (EC50) and protected mice challenged with lethal doses of avian influenza A H5N1. Our results demonstrate that viral NP is a valid target for the development of small-molecule therapies.
Supplementary information
The online version of this article (doi:10.1038/nbt.1638) contains supplementary material, which is available to authorized users.
Main
The propensity of influenza virus to develop resistance to commonly used drugs requires continued development of new therapeutics. In the 2008–2009 flu season, almost 100% of the seasonal influenza H1N1 viruses circulating in the United States were resistant to the neuraminidase inhibitor oseltamivir (Tamiflu), and all isolates of the H3N2 viruses were resistant to adamantanes1,2,3,4,5,6. Over half of the individuals infected by the H5N1 subtype died irrespective of treatment with both classes of drug7,8,9. In our previous studies on SARS coronavirus, we demonstrated that a forward chemical genetics approach using a chemical library of 50,240 compounds with diverse structures could interrogate most known targets for viral infection10,11. Here we screened the same library using Madin-Darby canine kidney (MDCK) cell–based influenza A infection assays and identified 950 compounds that showed protective effects as primary hits.
We evaluated the selected compounds in a secondary screen using a cytopathic effect assay and selected 39 compounds for further studies based on their potency. To investigate the modes of action of these bioactive compounds, we focused on processes crucial for successful establishment of influenza infection.
Influenza NP is the most abundantly expressed protein during the course of infection with multiple functionalities12. NP accumulates in the nucleus in the early phases of infection and is exclusively distributed in cytoplasm later during viral assembly and maturation12,13,14,15. We examined the effects of the 39 compounds on NP nuclear trafficking by fluorescence microscopy and identified 5 compounds that blocked the nuclear accumulation of NP. Compound FA-1 showed the best potency with EC50 < 1 μM in a plaque reduction assay (PRA) on MDCK cells infected with influenza A/WSN/33 (H1N1) virus. The schematic representation of the procedures and results of the primary, secondary and subsequent fluorescence microscopy screens are summarized in Supplementary Figure 1.
Based on the structural information of compound FA-1, four structurally similar analogs (Fig. 1a) obtained from commercial sources were shown to have EC50 against influenza A/WSN/33 virus at sub-micromolar levels in PRA. We selected compound FA-4 (nucleozin) for further characterization based on its better solubility in aqueous solutions (unpublished observations) and potent antiviral activities.
Figure 1. Chemical structures and biological activities of nucleozin and related compounds.
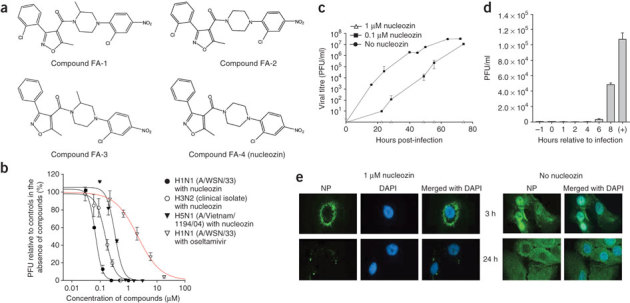
(a) Chemical structures of compound FA-1, FA-2, FA-3 and FA-4. (b) Nucleozin is effective against human H1N1, H3N2 and H5N1 influenza viruses. MDCK cells were infected with different strains of virus and antiviral activities determined by PRA. Oseltamivir (curve in red) was included for comparisons of in vitro efficacies. (c) Antiviral activity of nucleozin in multicycle growth assays. MDCK cells were infected with A/WSN/33 virus at 0.001 MOI in the presence or absence of nucleozin (0.1 or 1 μM). Zanamivir at 1 μM has stopped viral growth but data were omitted for clarity. Viral titers were determined by plaque assay at the time indicated. (d) Efficacies of nucleozin added at various time points. MDCK cells were infected at an MOI of 2 and nucleozin (1 μM) was added before infection (−1 h), at the time of infection (0 h) and at 1, 2, 4, 6 and 8 h after infection as indicated. +, controls with no nucleozin added. Viral titers were determined at 12 h after infection by plaque assay. The experiments were carried out in triplicate and repeated twice for confirmation. The mean value is shown with s.d. (e) Nucleozin blocked nuclear accumulation of influenza A NP in virus-infected A549 cells. Cells were infected with A/WSN/33 virus (10 MOI) in the presence or absence of 1 μM nucleozin. Influenza A NP accumulated in the nucleus at early-infection stage and was distributed exclusively in the cytoplasm at late-infection stage in the absence of nucleozin. At the indicated time point, cells were fixed and DAPI staining and mouse anti-influenza A NP antibodies were used to define the locations of the nucleus and viral NP, respectively. PFU, plaque forming unit.
Nucleozin inhibited infection of MDCK cells by the viruses influenza A/WSN/33, H3N2 (clinical isolate) and Vietnam/1194/04 (H5N1) with an EC50 of 0.069 ± 0.003 μM, 0.16 ± 0.01 μM and 0.33 ± 0.04 μM in PRA, respectively (Fig. 1b), severely suppressed viral growth at 0.1 μM and totally inhibited virus production at 1 μM in multicycle growth assays (Fig. 1c). The compound has a TC50 (50% toxic concentration) >250 μM as demonstrated by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay, suggesting a wide therapeutic window (Supplementary Fig. 2). Furthermore, nucleozin effectively inhibited viral growth even when added within 6 h after inoculation of the MDCK cells with the virus (Fig. 1d), indicating that the antiviral activities of nucleozin reside on post-entry and post-nuclear events, suggesting that multiple processes involving NP may be affected, although only the nuclear import process of NP can be readily observed.
Detailed fluorescence microscopy studies using human alveolar basal epithelial (A549) cells as the host for influenza A/WSN/33 virus infection showed that nucleozin is a potent antagonist of NP accumulation in the nucleus, leading to the formation of a halo of dense NP surrounding the perinuclear region in the cytoplasm at 3 h after infection (Fig. 1e). Because NP failed to enter the nucleus in the presence of nucleozin, NP trapped in the cytoplasm was seen scattered randomly in host cells at 24 h after infection (Fig. 1e). Nucleozin also inhibited the nuclear accumulation of NP in virus-infected MDCK cells (Supplementary Fig. 3), suggesting that the mechanism of action of nucleozin is not restricted to particular cell types.
Using the published crystal structure of influenza A/WSN/33 NP16, we carried out molecular docking studies trying to see if NP is a direct molecular target of nucleozin. For unbiased predictive virtual dockings, we set the docking potential gradient box to cover the entire NP monomer to probe all potential binding sites. We identified three potential binding sites of nucleozin (Fig. 2a): a previously unidentified small groove (binding site 1, Supplementary Fig. 4) in the back of the body of NP, the arginine-rich groove (binding site 2, Supplementary Fig. 5) proposed to be the RNA binding domain and the proposed tail loop groove (binding site 3, Supplementary Fig. 6).
Figure 2. Influenza A NP is the molecular target of nucleozin.
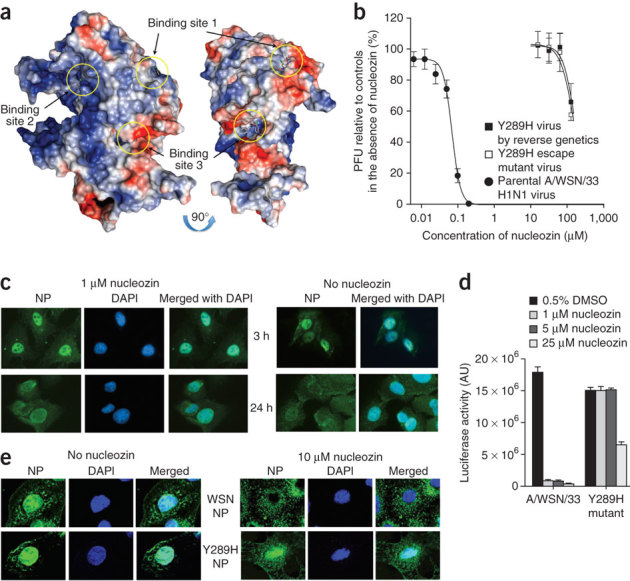
(a) Three potential binding sites of nucleozin on the influenza A NP crystal structure as predicted by molecular docking models. Electrostatic surface representation of influenza A NP is color-coded (red, negative; blue, positive; light gray, neutral). Potential binding sites of nucleozin are highlighted by yellow circles. (b) Escape mutant virus and recombinant virus carrying the Y289H substitution in influenza A NP confer resistance to high concentrations of nucleozin. MDCK cells were infected with A/WSN/33 virus, Y289H escape mutant virus or Y289H variant virus generated by reverse genetics. Antiviral activities determined by PRA. The highest concentration of nucleozin used was limited to 125 μM as fine precipitates appeared at higher concentration that interfered with the determination of plaques. All virus strains were tested in the same experiments for comparisons of in vitro resistance profiles. (c) MDCK cells were infected with the Y289H escape mutant virus (MOI = 5) in the presence or absence of 1 μM nucleozin. The addition of nucleozin did not block the nuclear accumulation of the viral NP. At the indicated time points, cells were fixed and DAPI staining and mouse anti-influenza A NP antibodies were used to define the locations of the nucleus and viral NP, respectively. (d) Nucleozin inhibits the parental virus NP activity but not theY289H variant virus NP in a luciferase reporter assay. 0, 1, 5 or 25 μM of nucleozin was added to 293T cells transfected with minigenomes containing A/WSN/33 virus NP or Y289H variant NP. Luciferase activities were measured 24 h post-transfection. The experiments were carried out in triplicate and repeated twice. The mean value is shown with s.d. (e) Nucleozin inhibits the nuclear import of exogenously added A/WSN/33 NP but not the Y289H variant NP. Purified recombinant NP or Y289H variant NP at 25 μM was added to digitonin-treated MDCK cells in the presence or absence of 10 μM nucleozin. Nuclear import of proteins was allowed for 30 min and followed by cell fixation and immunostaining for the presence of NP in the nucleus. DAPI was used to indicate the location of the nucleus. Images were visualized by confocal microscopy.
As the docking method treats the nucleoprotein as a rigid molecule and thus might not perfectly resemble the binding of nucleozin to nucleoprotein in vivo, the docking results should be considered as preliminary indications of such potential binding sites, to be subjected to further validations by biochemical and mutational studies. To localize the binding site of nucleozin to NP, escape virus mutants resistant to nucleozin were selected with increasing concentrations of nucleozin. We were not able to obtain escape mutants by using nucleozin as the selecting agent but an escape mutant showing cross-resistance to nucleozin was obtained after five passages of selection using compound FA-1. The resistant viral clone was plaque purified and selected for further studies. It is noteworthy that in the absence of nucleozin, the viral titer of the resistant mutant was only slightly lower than the parental virus under low (0.001) multiplicity of infection (MOI) conditions (Supplementary Fig. 7) but about an order of magnitude lower than the wild-type virus when a high MOI of 7 was used (unpublished observations). We speculate that the escape mutant virus may produce more defective interfering particles at high MOI but this observation may be applicable only to this Y289H mutant and not be generalized to other strains of influenza virus. Furthermore, although the mutant virus seemed to be less fit for infection in MDCK cells when compared with wild-type WSN virus, we caution that the fitness of a laboratory-induced mutant virus could only be meaningfully evaluated in the context of relevant animal models.
Sequencing of all eight gene segments of the escape virus revealed only a single T-to-C mutation at nucleotide position 865 of the NP gene in segment 5 of the influenza A genome; this mutation translates into a single amino acid substitution of tyrosine to histidine in residue 289 of NP. To further confirm that the Y289H substitution was indeed the only mutation contributing to the resistant phenotype of the selected mutant virus, we used reverse genetics to create an influenza A/WSN/33 recombinant virus with a single nucleotide substitution of T with C at position 865 of the NP gene. After co-transfection of eight pHW2000-based plasmids (replacing the pHW2000-NP with the mutant plasmid) encoding the gene segments of the virus into co-cultured 293T/MDCK cells17, we plaque purified and isolated the resulting recombinant virus that had a single Y289H substitution in NP and demonstrated that the recombinant virus was resistant to high concentrations of nucleozin and had a resistance profile indistinguishable from the originally isolated resistant escape viral clone (Fig. 2b).
Fluorescence microscopy studies showed that NP of the escape mutant accumulated in the nucleus despite the presence of nucleozin (Fig. 2c), indicating that the Y289H mutation overcame the observed antagonistic effects of nucleozin against the nuclear accumulation of NP and supporting our notion that nucleozin inhibits viral infection by halting the nuclear accumulation of viral NP. Results from the luciferase reporter–based mini-genome assay18 further showed that concentrations of nucleozin that effectively abolished the replication of the virus had little effect on the Y289H variant NP (Fig. 2d). The results from mini-genome assays also indicated that the replication activity of the Y289H was about 20% lower than the wild-type NP, which might explain the lower NP signal in Y289H immunofluorescence studies (Fig. 2c). When we used purified recombinant wild-type NP or Y289H NP in a nuclear import assay19, the nuclear import of the wild-type NP was abolished in the presence of nucleozin (Fig. 2e), indicating that nucleozin acts on NP in the absence of other viral components. The inability of nucleozin to inhibit Y289H variant NP in nuclear import showed again that Y289H is a crucial mutation for nucleozin resistance.
Influenza A NP has six tryptophan residues scattered throughout the protein. Binding of exogenous molecules is likely to induce a change in the microenvironments of tryptophan molecules leading to a change in intrinsic fluorescence. Fluorescence spectroscopy showed that incubation of nucleozin with purified recombinant NP elicited a clear dose-dependent fluorescence-quenching effect, indicating that nucleozin binds to influenza A NP and that this binding may induce conformational changes in the protein. Fluorescence titration using the Y289H variant NP resulted in a much lower fluorescence-quenching effect, suggesting that the binding of nucleozin to the Y289H variant NP is much weakened but not totally abolished (Fig. 3a). The docking model (Supplementary Fig. 4) suggests that residue N309 of NP forms a hydrogen bond with nucleozin, and residue Y289 forms hydrophobic interactions with nucleozin by aromatic ring stacking. Replacement of tyrosine by histidine at position 289 presumably disrupts the ring-stacking effect and destabilizes the binding of the nucleozin to NP. As the experimental results with the Y289H mutant virus agreed with the predicted binding of nucleozin to NP, the small groove behind the body (Fig. 2a and Supplementary Fig. 4) of NP is likely to be critical to the binding of nucleozin. Sequencing the NP gene of ten independent nucleozin-resistant clones from our mutant-raising experiment has shown that nine clones are Y289H mutants. One clone carried a N309K instead of the Y289H mutation. This second resistance mutation reconciles well with the original predictions of the docking model (Fig. 2a and Supplementary Fig. 4), suggesting that N309 may stabilize the binding of nucleozin through hydrogen bonds.
Figure 3. Nucleozin interacts with NP and causes NP aggregation.
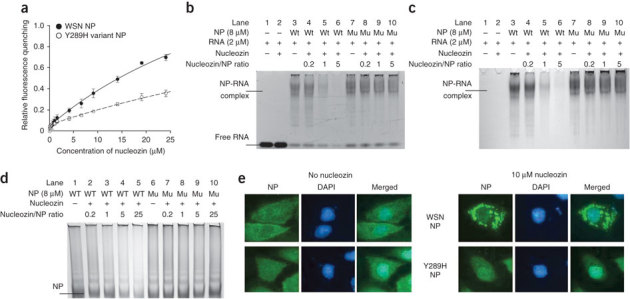
(a) Binding of nucleozin to NP induced fluorescence quenching. We used 4 μM wild-type WSN NP or Y289H variant NP and 0–25 μM of nucleozin for fluorescence titrations. Internal fluorescence-quenching effect was due to micro-conformational changes upon the binding of nucleozin to the NP. The experiments were carried out in duplicate and repeated three times for confirmation. (b–d) Nucleozin does not inhibit RNA binding but mediates formation of large NP-RNA complexes not resolvable by gradient gel electrophoresis. Free RNA oligomer and NP-RNA complex were visualized by ethidium bromide staining in b and NP was visualized by Coomassie brilliant blue G-250 staining in c. RNA incubated with 40 μM of nucleozin in the absence of NP (lane 2) was used as a control. (d) Visualization and comparison of the effects of nucleozin on recombinant wild-type and Y289H NP (in the absence of RNA); native gradient gel conditions, stained by Coomassie brilliant blue G-250. WT, purified recombinant A/WSN/33 NP; Mu, purified recombinant Y289H variant NP; +, in the presence; −, in the absence. Positions of free RNA, NP-RNA complex and NP are indicated. (e) Visualization of nucleozin-induced NP aggregates in cells. MDCK cells transfected with plasmids expressing wild-type NP or Y289H variant NP were treated with nucleozin for 4 h. DAPI staining and mouse anti-influenza A NP antibodies were used to define the locations of the nucleus and NP respectively.
Y289 is not in close proximity to the identified nuclear localization signal (residues 3–13 and 198–216) of NP making, it unlikely that direct interactions of nucleozin with elements related to the nuclear localization signal are responsible for the inhibitory effect. We speculate that the binding of nucleozin to influenza NP may induce conformational changes in the protein that render NP unsuitable for nuclear trafficking. Further evidence supporting our proposal that nucleozin alters the conformations of NP comes from the RNA binding experiment showing that nucleozin did not inhibit NP-RNA binding (Fig. 3b) but caused a dose-dependent reduction of NP-RNA complex that could be run into the 4–12% polyacrylamide gradient gel (Fig. 3b,c; lanes 3–6). The result suggested that nucleozin induced the formation of very large NP-RNA aggregates that were too big to get into the gradient gel during electrophoresis. The NP in the assay was not lost on account of degradation; this was apparent when we separated the reaction content by denaturing SDS-PAGE under reducing conditions and detected intact NP (Supplementary Fig. 8), indicating the presence of NP aggregates that could be dissolved in denaturing conditions. In the absence of RNA, the nucleozin treatment could also reduce the amount of NP running into the native gradient gel in a dose-dependent manner as judged by the intensities of Coomassie blue–stained NP in each lane (Fig. 3d, lanes 1–5), presumably due to formation of very large NP complexes. Separating the reaction content by denaturing SDS-PAGE demonstrated that the NP was present at comparable levels in treated and untreated samples and detectable under denaturing conditions (Supplementary Fig. 9). The data suggest that although nucleozin alone induced NP aggregate formation, the presence of RNA in the system greatly enhanced the formation of very large NP-RNA complexes nonresolvable by native electrophoresis. The Y289H variant NP, on the other hand, was inert to the addition of nucleozin, in the absence or presence of RNA (Fig. 3c, lanes 7–10; Fig. 3d, lanes 6–10).
To investigate if nucleozin also induces NP aggregate formation in cells, we expressed NP in MDCK cells until sufficient amounts of NP were present for immunofluorescence microscopy detection. We then treated the cells with cycloheximide to stop further protein translation, added nucleozin to the cells and monitored NP aggregate formation by immunofluorescence microscopy. The formation of nucleozin-induced NP aggregates could be detected 1 h after the addition of nucleozin (unpublished observation) and was readily observed 4 h after the addition of nucleozin (Fig. 3e). We speculate that the halo of dense NP surrounding the perinuclear region of infected cells were nucleozin-induced NP aggregates (Fig. 1e).
Based on the immunofluorescence and biochemical data, we propose that nucleozin inactivates NP by inducing the formation of very large NP complexes that aggregate with RNA and possibly other cellular components yet to be identified, leading to a complete halt in NP nuclear import. As NP is involved in different steps crucial for viral replication in many phases of the replication cycle20,21, we speculate that other processes involving NP will also be disrupted by the nucleozin-mediated formation of NP complexes.
With respect to in vivo antiviral efficacy, mice treated with nucleozin had a considerably higher survival rate after inoculation by influenza A virus H5N1 strain A/Vietnam/1194/04 than untreated controls. The protection against this highly pathogenic strain highlights the efficacy of this compound, as the H5N1 virus rapidly disseminates to multiple organs, killing the host soon after infection22. Without any treatment, all mice had died 7 d after inoculation. All mice survived when treated with the potent neuraminidase inhibitor zanamivir (Relenza). In the nucleozin-treated group, 50% of those receiving two doses of nucleozin (100 μl of 2.3 mg/ml nucleozin) per day for 7 d survived for more than 21 d (Fig. 4a). Three mice were euthanized from each treated and untreated group on the 6th day after infection and their lungs tested for the presence of live virus by plaque assay. There was about a tenfold reduction of viral load in the lungs of the nucleozin-treated mice when compared to the untreated control group (Fig. 4b). The animal study results show that nucleozin protected mice against hypervirulent influenza A H5N1 virus in vivo and thus has the potential to be developed into useful anti-influenza therapeutics.
Figure 4. Efficacies of nucleozin in a mice H5N1 virus infection model.
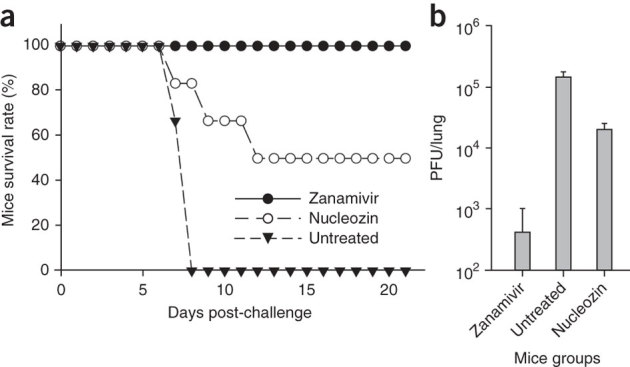
(a) Nucleozin protected the mice infected with highly pathogenic influenza H5N1 virus. Mice (nine per group) infected with 5 LD50 (median lethal dose) of A/Vietnam/1194/04 H5N1 virus received 100 μl of 20 mg/ml zanamivir, 2.3 mg/ml nucleozin or PBS twice daily intraperitoneally. Treatments stopped at day 7 after infection. Conditions of the mice were monitored for 21 d. (b) Zanamivir and nucleozin reduced the viral load in the lungs of infected mice when compared to the control (untreated mice). Three mice from each group were euthanized at day 6 and lungs removed for viral load determination by standard plaque assay. Shown are the mean values with s.d.
While this manuscript was in preparation, the novel swine-origin influenza A (S-OIV) H1N1 virus appeared. Analyzing all the publicly available NP sequence of human H1, H2, H3, H5, H7, H9 influenza A subtypes revealed that although Y289H is a rare polymorphism in influenza A viruses, the newly emerged S-OIV is a natural Y289H variant. Among 525 Y289H variants detected in H1N1 viruses, 512 belong to the S-OIV (Supplementary Tables 1,2,3). The S-OIV has high resistance to nucleozin (EC50 > 50 μM; data not shown) in MDCK cell infection models but a close analog, compound FA-10, of nucleozin has shown promising antiviral activity against the Y289H variant and the S-OIV, with EC50 = 11.3 ± 0.9 μM and 5.0 ± 0.5 μM, respectively. FA-10, however, has reduced activity toward the wild-type WSN virus, with EC50 = 15.3 ± 2 μM (Supplementary Fig. 10). Molecular modeling suggests that FA-10 may bind favorably into binding site 1 of nucleozin (Supplementary Fig. 11) but the hydrogen bond formation, with NP residue N309 observed in the original nucleozin-NP binding model (Supplementary Fig. 4), is abolished, presumably leading to the reduced activity of FA-10 toward wild-type WSN virus when compared with nucleozin. Further mutational studies of the binding site of nucleozin and derivatives in conjunction with structure-activity relationship are warranted to generate useful small-molecule compounds targeting the NP of a variety of viral strains.
In summary, using a forward chemical genetics approach23,24, we have identified the influenza A NP as a druggable target. We also describe a lead compound, nucleozin, with efficacy in vitro and in animal studies. Nucleozin induces the formation of NP aggregates and antagonizes its nuclear accumulation, leading to cessation of viral replication. Investigation of other nucleoproteins of other viruses as direct druggable targets for small-molecule therapeutics is warranted.
Methods
Virus and chemical reagents.
Influenza A/WSN/33, H3N2 and swine-origin influenza A (H1N1) virus S-OIV (A/HK/415742/09) were propagated in MDCK cells. After full cytopathic effects developed in cultures in infected MDCK cell cultures, the viral particles were harvested and stored in −70 °C freezers for further studies. The influenza A virus strain A/Vietnam/1194/04 was grown in embryonated eggs and the virus-containing allantoic fluid was harvested and stored in aliquots at −70 °C. A total of 50,240 structurally diverse small-molecule compounds (ChemBridge) was screened. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) was purchased from Sigma-Aldrich. RNA oligomer (5′-UUUGUUACACACACACACGCUGUG-3′) used for RNA binding assays was synthesized by IDT (Integrated DNA Technologies).
Cell-based high-throughput screening (HTS) in 384-well microtiter plates.
The primary HTS was carried out in a fully automated Beckman Coulter Core System (Fullerton) integrated with a Kendro robotics CO2 incubator (Thermo Fisher Scientific) at Chemical Genetics Unit, Department of Microbiology, Research Center of Infection and Immunology, LKS Faculty of Medicine, the University of Hong Kong. Compounds were arrayed in 384-well microtiter plates (Greiner Bio-One) in triplicate with a final concentration of 20 μg/ml and 5,000 MDCK cells per well in 50 μl complete Eagle's minimal essential medium (EMEM) supplemented with 1% heat-inactivated FBS. Cells were then infected with influenza virus (A/WSN/33) at an MOI of 0.01. After infection, plates were incubated at 37 °C with 5% CO2. At 3 d post-infection, 20 μl of 0.625 mg/ml of MTT was added into each well followed by an additional incubation time of 8 h at 37 °C with 5% CO2. At the end of the incubation, 30 μl SDS with 0.01 M HCl was added to solubilize the formazan, and after overnight incubation, MTT readings were recorded in a DTX 880 multimode detector (Beckman Coulter) at 570 nm with 640 nm as the reference wavelength.
HTS data analysis.
The HTS data was transferred to a Dell Precision 500 Workstation and normalized by custom designed data analysis software in two stages. For stage one normalization, each well was divided by the median of each plate and a normalized value was obtained for each well. As the HTS was carried out in triplicate, the second stage normalization took into account the variation between each well and the final reading was calculated by averaging the two closest readings. This two-stage normalization method minimized potential experimental errors, which are usually random and sporadic in nature.
Secondary screening.
Secondary screening was carried out in triplicate in 96-well tissue culture plate (TPP) at 10 μg/ml. Selected compounds were first dispensed in the wells, followed by the addition of 20,000 MDCK cells and 200 PFU of influenza A/WSN/33 (H1N1) virus into each well. The plates were incubated at 37 °C with 5% CO2 and monitored daily using a Leica DM inverted light microscope for virus-induced cytopathic effect. Compounds that gave full protection of MDCK cells (no cytopathic effect) were selected for further studies. The cytotoxicity of selected compounds was determined by MTT assay according to manufacturer's instructions.
Plaque reduction assay.
The PRA assay was performed in triplicate in 24-well tissue culture plates (TPP). The MDCK cells were seeded at 1 × 105 cells/well in EMEM (Invitrogen) with 10% FBS on the day before carrying out the assay. After 16–24 h, 40–50 PFU of influenza virus were added to the cell monolayer with or without the addition of compounds and the plates further incubated for 2 h at 37 °C with 5% CO2 before removal of unbound viral particles by aspiration. The cell monolayer was washed once with EMEM before being overlaid with 1% low melting agarose (Cambrex) in EMEM containing 1% FBS, 1 μg/ml TPCK trypsin (Invitrogen) and appropriate amounts of compounds. The plates were incubated at 37 °C with 5% CO2 for 72 h. At 72 h after infection, the wells were fixed with 10% formaldehyde (BDH). After removal of the agarose plugs, the monolayers were stained with 0.7% crystal violet (BDH) and the plaques counted. The percentage of plaque inhibition relative to the control (without the addition of compound) plates were determined for each compound concentration and the EC50 was calculated using Sigma plot (SPSS). The PRA were carried out in triplicate and repeated twice for confirmation. For multicycle growth experiments for the evaluation of antiviral activities of compounds, 0.001 MOI was used accordingly25 and viral yield determined by plaque assay.
Immunofluorescence microscopy.
A549 and MDCK cells were grown to 70–80% confluency on coverslips. Cells were infected for 2 h at MOI = 10 and MOI = 5 for A549 and MDCK cells, respectively in the presence or absence of 1 μM nucleozin and washed. Nucleozin was maintained in culture throughout the experiment. Infections were stopped at indicated time points by fixation in 4% paraformaldehyde (Electron Microscopy Sciences) for 15 min. Cells were permeabilized in 0.1% Triton-X100 for 5 min and then were incubated for 1 h with primary antibodies against NP (Abcam) in PBS containing 5% goat serum (dilution 1:1,000), washed and stained with fluorescein isothiocyanate (FITC)-conjugated secondary antibodies (Invitrogen) (dilution 1:150) for 0.5 h. Coverslips were then washed and counterstained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Invitrogen) for nucleus localization and mounted on slides using Prolong Gold antifade mounting medium (Invitrogen) before image analysis by fluorescence microscopy (SPOT Diagnostic Instrument).
Cloning and purification of recombinant influenza A/WSN/33 NP and Y289H variant NP.
The full influenza A/WSN/33 (H1N1) NP or Y289H NP gene was cloned into pET28a vector (Novagen) with C-terminal His-tag provided by the vector, expressed in Escherichia coli BL21 (DE3) cells, and the recombinant NPs purified to homogeneity by HisTrap HP, HiTrap heparin HP and Superdex 200 gel filtration columns (Amersham Biosciences). The purified protein was 95% pure as determined by SDS-PAGE.
Fluorescence spectroscopy.
Fluorescence-quenching method26 was used to examine if nucleozin interacts with purified influenza A NP in vitro. Briefly, a Hitachi F-4800 fluorescence spectrophotometer was used for fluorescence titrations in a 2 ml quartz cuvette at 25 °C. The excitation and emission wavelengths were set at 295 nm and 333 nm, respectively. Samples contained 4 μM purified influenza NP in 20 mM Tris, 150 mM NaCl, pH 7.3. Nucleozin was used at concentrations between 0.03 and 25 μM. For the titrations, additives were not allowed to exceed 1.5% of the total volume of the solution. The experiments were carried out in duplicate and repeated three times for confirmation.
Molecular docking of nucleozin to influenza A NP.
To postulate the potential binding site(s) of nucleozin in the viral NP, molecular modeling for NP-nucleozin interaction was performed using Autodock3.0.5 (ref. 27) and CHARMM force field28. As amino acid residues 73–91, 397–401 and 429–437 do not have defined tertiary structure in the available NP crystal structure of H1N1 virus (Protein Data Bank code: 2IQH), SWISS-MODEL was used to repair the structure of the NP. The three-dimensional structure of nucleozin was then generated by Chem3D (CambridgeSoft). Hydrogen atoms were assigned and Sybyl Mol2 files for Autodock were prepared by InsightII (Accelrys), and partial charge and potential were assigned by CHARMM force field. Autodock3.0.5 Lamarckian Genetic Algorithm and its default parameters were used for docking. After carrying out docking, models of the complexes of nucleozin interacting with influenza NP were obtained. CHARMM force field was used to optimize the docked complexes by removing bad contacts between the ligand and the protein. PyMol (Delano) was used to plot the modeling figures.
Generation of escape mutant influenza A virus resistant to nucleozin.
One method of mutant generation29 was followed to raise mutant viruses resistant to compound FA-1 and nucleozin. Briefly, the influenza A/WSN/33 (H1N1) was passaged in MDCK cells in the presence of increasing concentrations of the compounds and the desired resistant viral clone purified by plaque isolation on MDCK cell monolayers. It took five passages to obtain the escape mutant. After plaque purification, we carried out whole genome sequencing for one clone. The escape mutant viral RNA was extracted by TRIZOL Reagent (Invitrogen) and complementary DNA (cDNA) of all eight segments was obtained by reverse transcription using Superscript III reverse transcriptase (Invitrogen) and cDNA amplified by PCR according to standard procedures. After identification of NP as the potential target of nucleozin, we further sequenced the NP genes of ten independent resistant clones to examine other possible mutations leading to nucleozin resistance. All DNA sequencing was carried out in the Genome Research Center (University of Hong Kong).
Generation of recombinant influenza virus by reverse genetics.
The pHW2000 eight-plasmid system was used to generate desired recombinant viruses17. The eight plasmids contain the cDNA of the eight segments of the influenza A/WSN/33 (H1N1) genome: pHW2000-PB2, pHW2000-PB1, pHW2000-PA, pHW2000-HA, pHW2000-NP, pHW2000-NA, pHW2000-M and pHW2000-NS. Standard PCR-based techniques were used to clone the mutation in the NP gene of the escape mutant into the BsmI sites of pHW2000-NP. On the day before transfection, confluent 293T and MDCK cells were trypsinized and co-cultured in a six-well tissue culture plate (TPP). TransIT -Oligo Tranfection Reagent (Mirrus) was used to transfect the co-cultured 293T and MDCK cells according to manufacturer's instructions. The infectious particles from the supernatants were harvested 48 and 72 h post-transfection, and the recombinant virus titer enumerated in MDCK cells by plaque assay.
Luciferase reporter assay for polymerase complex activity.
The luciferase reporter assay was performed as previously reported18. Full-length genomic segments of NP, PA, PB1 and PB2 derived from different virus strains were cloned into pHW2000 vector. RNP complex comprised of WSN wild-type NP or Y289H variant NP, PA, PB1 and PB2 were co-transfected into 293T cells with a luciferase reporter plasmid, pYH-Luci, which contains noncoding sequence from the M segment of influenza A virus and firefly luciferase gene driven by polI. Plasmid phRL-TK (Promega), which expresses Renilla luciferase, was also co-transfected as an internal control for data normalization. At 2 h after transfection, different amounts (final concentrations: 1–25 μM) of nucleozin (in DMSO) were added to the transfected cells. As a solvent control, some volume of DMSO was added to a final concentration of 0.5%. At 24 h after transfection, the luciferase activities were measured using Dual Luciferase Assay System kit (Promega E1910) and Victor3 multilabel plate reader (Perkin Elmer).
NP nuclear import assay in MDCK cells.
The assay was done as described19. MDCK cells were seeded on glass coverslips and were washed with import buffer (20 mM HEPES, pH 7.4, 110 mM potassium acetate, 1 mM EGTA, 5 mM sodium acetate, 2 mM magnesium acetate and 2 mM dithiothreitol) before being permeabilized with 20 μg/ml of digitonin (Sigma) for 5 min at 25° C. Meanwhile, 25 μM of NP with or without 10 μM of nucleozin were preincubated for 5 min at 25° C. After washing off the digitonin with import buffer, the NP-nucleozin mixture was added to the permeabilized cells and incubated for 30 min. The energy-generating system (0.4 mM ATP, 0.45 mM GTP, 4.5 mM phosphocreatine and 18 U/ml phosphocreatine kinase (Merck)), 15% rabbit reticulocyte lysate (Promega), 1.6 mg/ml BSA (Sigma) and protease inhibitors (Roche) were subsequently added to the cells and incubated at 37 °C CO2 incubator for another 30 min. The cells were then rinsed with import buffer and fixed with 4% paraformaldehyde. Cells were immunostained as described above. The images were visualized using confocal microscropy (Carl Zeiss).
Electrophoretic mobility shift assay.
Electrophoretic mobility shift assay was used to examine the effect of nucleozin on the RNA binding activity of the NP proteins. Purified recombinant wild-type WSN or Y289H variant NP proteins were incubated with nucleozin at 25° C for 30 min, then a 24-nucleotide RNA oligomer16 was added and incubated for another 30 min. Nuclease-free water was added, up to 10 μl. Final concentration of small RNA oligomer was 2 μM and molar ratio of NP/RNA was kept at 4:1. After incubation, the samples were mixed with 3 μl 6× DNA loading dye (0.25% bromophenol blue, 0.25% xylene xyanol, 40% sucrose) and loaded into sample wells of nondenaturing 4–12% gradient Bis-Tris NuPAGE gel (Invitrogen) equilibrated by pre-electrophoresis at 50 V in 1× TBE. Samples were separated by electrophoresis at a constant voltage of 150 V for 35 min at 25° C in 1× TBE. The gel was first visualized by ethidium bromide staining for RNA shift patterns followed by staining with Coomassie brilliant blue G-250 for NP shift patterns. For examining the effects of nucleozin-NP interactions in vitro in the absence of RNA, we used NativePAGE 4–16% Bis-Tris gradient gel (Invitrogen) for the separation of NP under native conditions accordingly30.
Animal experiments.
As previously described22, we kept the 5- to 7-week-old BALB/c female mice in biosafety level 3 housing and gave the mice access to standard pellet feed and water ad libitum. All experimental protocols followed the standard operating procedures of the approved biosafety level 3 animal facilities and were approved by the Animal Ethics Committee. One group (nine mice) was injected intraperitoneally (i.p.) with 100 μl of 20 mg/ml of zanamivir (GlaxoSmithKline), a second group (nine mice) was injected with 2.3 mg/ml of nucleozin, and the untreated group (nine mice) was injected with PBS 1 h before inoculating the mice intranasally with 5 LD50 (100 PFU) of the A/Vietnam/1194/04 H5N1 virus in 20 μl 2 mg/ml zanamivir, 0.23 mg/ml of nucleozin or PBS. Two doses per day of i.p. 100 μl of 20 mg/ml zanamivir, 2.3 mg/ml of nucleozin or PBS were given for 7 d. Animal survival and general conditions were monitored for 21 d or until death. Three mice in each group were euthanized randomly on day 6 post-inoculation and lungs were removed for determination of viral titers by plaque assay.
Supplementary information
Supplementary Tables 1–3 and Supplementary Figs. 1–11 (PDF 657 kb)
Acknowledgements
This study was supported in part by the Carol Yu Center for Infection Seed Fund for Basic Research from the University of Hong Kong, the Research Fund for the Control of Infectious Diseases and the Area of Excellence Scheme of the University Grant Council (Grant AoE/M-12/06). The Beckman Coulter Core system is a generous gift from the Hong Kong Sanatorium Hospital Doctors' Donation Fund by Y.-C. Tsao, C.-M. Chan, G. Lo, K.-M. Lai, R.K.Y. Lo, M. Tsao, B.S.S. Tse, T.-F. Tse, S.W.C. Wu, D.Y.C. Yu, R.Y.H. Yu and Y.-K. Tsao. We are grateful to R. Webster for gifts of the pHW2000 plasmids and E. Hoffmann for luciferase reporter system. We thank V. Poon, C. Chan and Q. Zhang for mice studies and K.H. Chan for virus strains. The use of Confocal Systems Core Facility provided by the LKS Faculty of Medicine, HKU, is acknowledged.
Author Contributions
R.Y.K. and K.-Y.Y conceived the study. R.Y.K. designed and performed experiments and analyzed data. D.Y. gave conceptual advice and technical support on chemistry. L.-S.L., W.H.W.T., J.D., M.-P.C., C.-M.C. and P.W. performed experiments. J.S., L.H., and G.C. performed molecular dockings. B.-J.Z. provided animal study data. J.-D.H. gave conceptual advice on protein trafficking. J.M. constructed database and performed HTS data normalization. H.C. and Y.G. provided reverse genetics system. K.-Y.Y. did troubleshooting and provided the grant support. R.Y.K. and K.-Y.Y. supervised the study and wrote the paper.
Competing interests
The authors declare no competing financial interests.
Contributor Information
Richard Y Kao, Email: rytkao@hkucc.hku.hk.
Kwok-Yung Yuen, Email: kyyuen@hkucc.hku.hk.
References
- 1.Webster RG, Govorkova EA. H5N1 influenza – continuing evolution and spread. N. Engl. J. Med. 2006;355:2174–2177. doi: 10.1056/NEJMp068205. [DOI] [PubMed] [Google Scholar]
- 2.Regoes RR, Bonhoeffer S. Emergence of drug-resistant influenza virus: population dynamical considerations. Science. 2006;312:389–391. doi: 10.1126/science.1122947. [DOI] [PubMed] [Google Scholar]
- 3.Moscona A. Global transmission of oseltamivir-resistant influenza. N. Engl. J. Med. 2009;360:953–956. doi: 10.1056/NEJMp0900648. [DOI] [PubMed] [Google Scholar]
- 4.Dharan NJ, et al. Oseltamivir-Resistance Working Group. Infections with oseltamivir-resistant influenza A(H1N1) virus in the United States. J. Am. Med. Assoc. 2009;301:1034–1041. doi: 10.1001/jama.2009.294. [DOI] [PubMed] [Google Scholar]
- 5.Layne SP, Monto AS, Taubenberger JK. Pandemic influenza: an inconvenient mutation. Science. 2009;323:1560–1561. doi: 10.1126/science.323.5921.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lackenby A, Thompson CI, Democratis J. The potential impact of neuraminidase inhibitor resistant influenza. Curr. Opin. Infect. Dis. 2008;21:626–638. doi: 10.1097/QCO.0b013e3283199797. [DOI] [PubMed] [Google Scholar]
- 7.Yuen KY, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet. 1998;351:467–471. doi: 10.1016/S0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- 8.Cumulative Number of Confirmed Human Cases of Avian Influenza A (H5N1) Reported to WHO (http://www.who.int/csr/disease/avian_influenza/country/cases_table_2009_09_24/en/index.html).
- 9.Wong SS, Yuen KY. Avian influenza virus infections in humans. Chest. 2006;129:156–168. doi: 10.1378/chest.129.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kao RY, et al. Identification of novel small-molecule inhibitors of severe acute respiratory syndrome-associated coronavirus by chemical genetics. Chem. Biol. 2004;11:1293–1299. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kao RY, et al. Characterization of SARS-CoV main protease and identification of biologically active small molecule inhibitors using a continuous fluorescence-based assay. FEBS Lett. 2004;576:325–330. doi: 10.1016/j.febslet.2004.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Portela A, Digard P. The influenza virus nucleoprotein: a multifunctional, RNA-binding protein pivotal to virus replication. J. Gen. Virol. 2002;83:723–734. doi: 10.1099/0022-1317-83-4-723. [DOI] [PubMed] [Google Scholar]
- 13.Davey J, Dimmock NJ, Colman A. Identification of the Sequence Responsible for the Nuclear Accumulation of the Influenza Virus. Cell. 1985;40:667–675. doi: 10.1016/0092-8674(85)90215-6. [DOI] [PubMed] [Google Scholar]
- 14.Boulo S, Akarsu H, Ruigrok RWH, Baudin F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007;124:12–21. doi: 10.1016/j.virusres.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 15.Ozawa M, et al. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 2007;81:30–41. doi: 10.1128/JVI.01434-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye Q, Krug RM, Tao YJ. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature. 2006;444:1078–1082. doi: 10.1038/nature05379. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. USA. 2000;97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang P, et al. Nuclear factor 90 negatively regulates influenza virus replication by interacting with viral nucleoprotein. J. Virol. 2009;83:7850–7861. doi: 10.1128/JVI.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu WW, Sun YH, Panté N. Nuclear import of influenza A viral ribonucleoprotein complexes is mediated by two nuclear localization sequences on viral nucleoprotein. Virol. J. 2007;4:49. doi: 10.1186/1743-422X-4-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Digard P, et al. Modulation of nuclear localization of the influenza virus nucleoprotein through interaction with actin filaments. J. Virol. 1999;73:2222–2231. doi: 10.1128/jvi.73.3.2222-2231.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elton D, Medcalf E, Bishop K, Harrison D, Digard P. Identification of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J. Virol. 1999;73:7357–7367. doi: 10.1128/jvi.73.9.7357-7367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng BJ, et al. Delayed antiviral plus immunomodulator treatment still reduces mortality in mice infected by high inoculum of influenza A/H5N1 virus. Proc. Natl. Acad. Sci. USA. 2008;105:8091–8096. doi: 10.1073/pnas.0711942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stockwell BR. Chemical genetics: ligand-based discovery of gene function. Nat. Rev. Genet. 2000;1:116–125. doi: 10.1038/35038557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strausberg RL, Schreiber SL. From knowing to controlling: a path from genomics to drugs using small molecule probes. Science. 2003;300:294–295. doi: 10.1126/science.1083395. [DOI] [PubMed] [Google Scholar]
- 25.Min JY, Krug RM. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA. 2006;103:7100–7105. doi: 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campanacci V, et al. Moth chemosensory protein exhibits drastic conformational changes and cooperativity on ligand binding. Proc. Natl. Acad. Sci. USA. 2003;100:5069–5074. doi: 10.1073/pnas.0836654100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris GM, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- 28.Brooks R, et al. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983;4:187–217. doi: 10.1002/jcc.540040211. [DOI] [Google Scholar]
- 29.Gubareva LV, et al. Characterization of mutants of influenza A virus selected with the neuraminidase inhibitor 4-guanidino-Neu5Ac2en. J. Virol. 1996;70:1818–1827. doi: 10.1128/jvi.70.3.1818-1827.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niepmann M, Zheng J. Discontinuous native protein gel electrophoresis. Electrophoresis. 2006;27:3949–3951. doi: 10.1002/elps.200600172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables 1–3 and Supplementary Figs. 1–11 (PDF 657 kb)